

Abstract
Bcl2-associated athanogene 3 (BAG3) is a 575 amino acid protein that is found predominantly in the heart, skeletal muscle and many cancers. Deletions and truncations in BAG3 that result in haplo-insufficiency have been associated with the development of dilated cardiomyopathy. To study the cellular and molecular events attributable to BAG3 haplo-insufficiency we generated a mouse in which one allele of BAG3 was flanked by loxP recombination sites (BAG3fl/+). Mice were crossed with α-MHC-Cre mice in order to generate mice with cardiac-specific haplo-insufficiency (cBAG3+/−) and underwent bi-weekly echocardiography to assess their cardiac phenotype. By ten weeks of age, cBAG3+/− mice demonstrated increased heart size and diminished left ventricular ejection fraction when compared with non-transgenic littermates (Cre−/−BAG3fl/+). Contractility in adult myocytes isolated from cBAG3+/− mice were similar to those isolated from control mice at baseline, but showed a significantly decreased response to adrenergic stimulation. Intracellular calcium ([Ca2+]i) transient amplitudes in myocytes isolated from cBAG3+/− mice were also similar to myocytes isolated from control mice at baseline but were significantly lower than myocytes from control mice in their response to isoproterenol. BAG3 haplo-insufficiency was also associated with decreased autophagy flux and increased apoptosis. Taken together, these results suggest that mice in which BAG3 has been deleted from a single allele provide a model that mirrors the biology seen in patients with heart failure and BAG3 haplo-insufficiency.
Keywords: Cardiomyopathy (DCM), BAG3, Haplo-insufficiency, Genetic mutation
1. Introduction
Bcl2-associated athanogene 3 (BAG3) is a 575 amino acid protein that is highly conserved in nature.[1] Expressed predominantly in the heart, the skeletal muscle and in many cancers, BAG3 has pleotropic effects in the cell owing to the presence of multiple protein-protein binding domains. For example, it inhibits apoptosis by binding to Bcl2, supports protein quality control (PQC) by serving as a co-chaperone with the heat shock proteins, tethers actin filaments to the Z-disc and facilitates adrenergic responsiveness by coupling the β1-adrenergic receptor and the L-type Ca2+ channel.[1,2]
In 2009, Selcen described three children with severe myofibrillar myopathy, skeletal muscle weakness, giant axon disease and a hypertrophic/restrictive cardiomyopathy that was associated with a non-synonymous single nucleotide polymorphism that resulted in a shift from a proline to a leucine at position 209 (Pro209Leu).[3] Subsequent to the identification of the hypertrophic phenotype, Norton et al identified a family with a dilated cardiomyopathy in the absence of skeletal muscle or neurologic disease that was associated with a large deletion in BAG3.[4] Studies have now identified deletions, truncations and some non-synonymous SNPs as causative in both familial and sporadic cases of dilated cardiomyopathy[5,6,7,8,9,10] and can result in BAG3 haplo-insufficiency.[6,8] In fact, investigators recently posited that BAG3 is a major cardiomyopathy locus.[7]
Unfortunately, there is a paucity of information regarding the mechanisms that regulate BAG3 function and it’s signaling due in part to the absence of viable animal models. Homma reported that mice with homozygous deletion of BAG3 developed a severe non-inflammatory myopathy in both the cardiac and skeletal muscle that resulted in death by 4 weeks of age.[11] However, mice with heterozygous deletion of BAG3 had a normal phenotype. Fang et al recently mitigated the early lethality of homozygous BAG3 disruption by introducing a transgene that was restricted to the myocardium.[12] The resultant progeny had diminished LV function and decreased autophagy flux, but no cardiac BAG3. They found a similar phenotype in mice in which a non-synonymous single nucleotide polymorphism (SNP) found in humans was knocked-in to both alleles; however, BAG3 levels were normal. The present study was undertaken to create a mouse with heterozygous deletion of BAG3, haplo-insufficiency and LV dysfunction in order to mirror the more common phenotype seen in individuals with a BAG3 deletion or truncation.
2. Methods
2.1. Animal protocols and generation of BAG3 haplo-insufficient mice
All experiments were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Temple University Institutional Animal Care and Use Committee (ACUP#4380). Because the effects of female hormones on BAG3 expression and function have not been defined, male mice were used in all studies. The background for all mice was C57BL/6.
The strategy for obtaining mice with deletion of BAG3 is found in Fig. 1. BAG3 (HEPD0556_7_B06) mice were obtained from the MRCHarwell which distributes these mice on behalf of the European Mouse Mutant Archive (www.infrafrontier.eu). The MRC-Harwell is also a member of the International Mouse Phenotyping Consortium (IMPC) which funded the generation of the BAG3 floxed mice. Funding and associated primary phenotypic information may be found at www.mousephenotype.org. Details of the alleles have been previously published. [13,14,15] Gene targeting produced a “knockout first” allele with both a PGK-neomycin cassette and a LacZ reporter gene. loxP sites flank the promoter/neomycin cassette and were removed by action of cre recombinase, thereby ‘knocking out’ BAG3. [16] BAG3 single allele floxed (BAG3fl/+) mice were crossed with αMHC-Cre mice[16] leading to cardiac restricted heterozygous knockdown of BAG3. These mice are designated as cBAG3+/− mice. Non-transgenic littermate controls had the genotype Cre−/− BAG3fl/+ and are designated as cBAG3+/+. Genotyping of mice was done using gel based genotyping on DNA extracted from tails with primers for a WT band (F: TCTGAAGATGCACAGGGGTG, R: TGAGCGGCACTTTAAGGTTT) and primers for a floxed allele (F: TCTGAAGATGCACAGGGGTG, R: GAACTTCGGAATAGGAACTTCG) with the resulting WT band migrating at 300 base pairs and the floxed allele at 139 base pairs.
Fig. 1. Generation of BAG3 haplo-insufficient mouse.
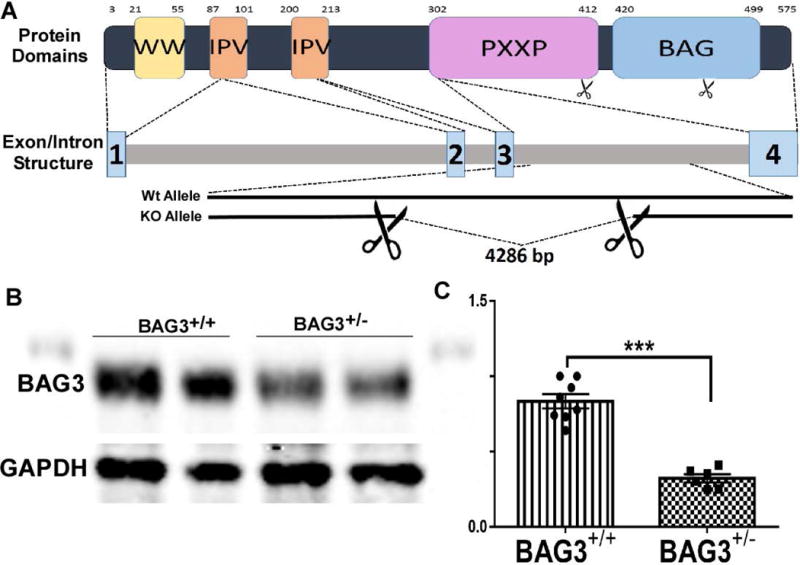
(A) A schematic of generation of heterozygous BAG3 knockout mice using recombination between LoxP sites on the BAG3 gene. Numbers on protein cartoon indicate amino acid positon and numbers 1-4 on transcript indicate intron boundaries. (B) Representative western blot of BAG3 levels in both cBAG3+/+ and cBAG3+/− mice with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the housekeeping protein. (C) Graph showing quantification of BAG3 levels in cBAG3+/+ (n = 8) and cBAG3+/− (n = 6) mice. *** p <0.0001.
2.2. Tissue handling
Mice were weighed, deeply anesthetized with isoflurane, and an incision was made in the abdomen. The inferior vena cava was cannulated, and 30 mmol/L KCl in an isotonic and pH buffered solution was rapidly infused. The heart was then excised, weighed and rinsed in ice-cold 30 mmol/L KCl. Small portions of the left ventricular free wall were removed and placed in ice-cold 10% neutral buffered formalin and the remaining heart tissue was flash-frozen in liquid nitrogen and stored at −70°C for subsequent analysis. The tibia was exposed and measured.
2.3. Echocardiography
Global LV function was evaluated in all mice after light sedation (2% isoflurane) using a VisualSonics Vevo 2100 imaging system and a MS550D scan head (Miami, FL) as described previously.[17] The left ventricular ejection fraction (LVEF) was calculated using the formula EF% = [(LVEDV − LVESV)/LVEDV]×100; where LVEDV and LVESV are left ventricular end-diastolic volume and left ventricular end-systolic volume, respectively. Fractional shortening (FS) was calculated as FS%=[(LVEDD−LVESD)/LVEDD]×100.], where LVEDD and LVESD are left ventricular end-diastolic dimension and end-systolic dimension, respectively.
2.4. Isolation of adult cardiac myocytes, single cell contractility and Ca2+ transients
Cardiac myocytes were isolated from the septum and LV free wall of cBAG3+/− or cBAG3+/+ mice (10-12 weeks old) as previously described[18] and plated on laminin-coated glass coverslips.[19] Coverslips were then mounted in a Dvorak-Stotler chamber and bathed in fresh media before measurements.
Measurements of contraction and [Ca2 +]i in adult cardiac myocytes were obtained as described previously.[20] In brief, myocyte contraction was recorded using a charge-coupled device video camera and myocyte motion was analyzed offline with an edge detection algorithm.[19,21,22,23,24,25] For measurement of calcium transients, Fura-2 loaded myocytes adherent to laminin-coated coverslips were incubated in buffer (1.8 mM [Ca2 +]o) and field stimulated to contract (2 Hz; 37 °C). Myocytes were exposed to excitation light (360 and 380 nm) only during data acquisition. Epifluorescence (510 nm) was measured in steady-state twitches both before and after addition of isoproterenol (Iso; 1 μM).[21,22,23,24,25]
2.5. Immunoblotting
Immunoblots were performed as described previously.[26] In brief, snap frozen left ventricles were lysed in buffer and homogenized in a Bullet Blender. 25μg of reduced protein lysates were run on SDS-PAGE gels and transferred to Odyssey nitrocellulose membranes for blotting. Blots were then incubated with a primary antibody and secondary antibodies prior to scanning with an Odyssey scanner and quantification using Image Studio software. The primary antibodies were BAG3 (Cat. # 10599-1-AP, Protein Tech, Rosemont, IL) and GAPDH (Cat.# sc-137179, Santa Cruz Biotechnology, Dallas TX). Secondary antibodies were goat anti-mouse IRDye 800 (Cat. #926-32210; LiCor Biosciences, Lincoln, NE) and IRDye 680 goat anti-rabbit (Cat. #926-68071; Licor Biosciences, Lincoln, NE).
2.6. Measurement of autophagy flux
Microtubule-associated protein 1A/1B-light chain 3 (LC3) is a soluble protein found ubiquitously in mammalian cells. During autophagy, LC3-1 is conjugated to phosphatidylethanolamine to form LC3-phophatidylethanolamine conjugate (LC3-II) and then is recruited to the autophagasome membrane. When the autophagasome fuses with a lysosome to form an autolysosome, the LC3-II within the autophagalysosome is degraded. Thus, the loss of LC3-II is proportional to the flux of the autophagic complex.[27] Because autophagy is ongoing, the measurement of LC3 levels at any time point may not accurately represent actual flux. The addition of bafilomycin A1; however, blocks the fusion of the lysosome and the autophagasome thereby attenuating LC3 lysis. Increased levels of LC3 in the presence of bafilomycin A1 as compared with levels in the absence of bafilomycin suggests that flux is active whereas a lack of change represents a diminution in autophagy flux.[28]
Two techniques are available for measuring LC3 levels. First, an adenovirus expressing RFP-GFP-LC3 was constructed as previously described.[26] Isolated adult myocytes from cBAG3+/− and Cre−/−BAG3fl/+ controls were infected after isolation with Adv-RFP-GFP-LC3 or Adv-null. Confocal imaging was then performed 48 hours later as described in detail previously (mRFP -red--594 nm ex, 667 nm em: GFP–green-488 nm ex, 543 nm em).[26] Experiments were performed with and without Bafilomycin A1. The puncta of 10 to 14 cells in each condition from three separate isolations were counted after obtaining digital images. When a lysosome fuses with the autophagasome to form an autophagolysosome, the increased acidity in the resulting autophagalysosome quenches the fluorescence of the GFP signal (green) resulting in predominantly red puncta in the autophagalysosome. As noted above, if autophagy flux is active, and autophagasome and lysosome fusion is inhibited by Bafilomycin A1, the number of puncta in both the autophagalysosomes and in the cytoplasm (yellow) increases because the LC3B-BAG3-protein complexes are not degraded in the autophagalysosome. [29,30]
We next corroborated the measurement of autophagy flux in a separate group of experiments, Isolated adult myocytes were plated on six-well plates in the same medium as used for contractility experiments detailed above. Cells were treated for two hours with 50nM bafilomycin A1, lysed in RIPA buffer, vortexed on ice, and spun at 13,000 × g for 6 min at 4°C. The supernatant was removed and protein concentrations were measured as described previously. [26] We then loaded 15μg of protein onto an SDS page gel (4–12%) and blotted using primary and secondary antibodies as described above (Immunoblotting). Levels of LC3I and LC3II were then quantified.
2.7. Measurement of Apoptosis
Apoptosis was measured in cBAG3+/− and cBAG3+/+ cells using a Detection Kit (Abcam, Cambridge, UK #ab176750) according to manufacturer’s instructions. In brief, cells were stained with a red sensor that identifies apoptotic cells by detecting the shift in phosphatidylserine from the inner to the outer surface of the myocyte. (Ex/Em = 630/660 nm). Next, a membrane-impermeable DNA Nuclear Green DCS1 (Ex/Em = 490/525 nm) was used to demonstrate late stage apoptosis(cells both red and green) and necrosis (green alone). Cells were also stained with CytoCalcein Violet 450 (Ex/Em = 405/450 nm) in order to identify living cells (blue) with intact sarcolemma. Live cells were imaged on a Zeiss 510 confocal microscope. The number of viable cells per field was counted by an investigator blinded to the experimental group of each sample to avoid bias and a minimum of 100 cells were counted per group. Results are presented as percentage of apoptotic cells per total cells counted.
2.8. Statistical Analysis
Data are presented as means ± SEM for continuous variables. One-way ANOVA with Tukey multiple comparisons adjustments were used to assess differences across the investigational groups. A Two-way ANOVA was also used for autophagy flux analysis and analysis of [Ca2+]i transient and contraction amplitudes as a function of group and Isoproterenol. Data was analyzed using Graph pad Prizm 6. A p value of <0.05 was considered statistically significant.
3.0 Results
3.1. Cardiac specific and heterozygous deletion of BAG3 results in haplo-insufficiency
When heterozygous floxed mice were crossed with the α-MHC driven Cre mice,(Fig. 1A) the resulting progeny had haplo-insufficiency of BAG3 as seen in the Western blot in Fig 1B and a 51% (p<0.0001) reduction in BAG3 levels when compared with non-transgenic littermate controls (Fig 1C).
3.2. BAG3 haplo-insufficiency causes left ventricular dysfunction by 10 weeks of age
Haplo-insufficiency of BAG3 had no effect on echocardiographic measures of LV function at 4 or 8 weeks after birth. (Fig, 2A and Fig. 2B) However, by 10 weeks of age there was a significant decrease in EF (62.79±6.4 vs 44.53±4.0; p <0.0001) and FS (34.48±4.9 vs 22.05±2.3; p<0.001) in cBAG3+/− mice when compared with non-transgenic littermate controls. The decrease in LV function in the cBAG3+/− mice was sustained as both the ejection fraction (75.26±2.7 vs 43.44±8.9; p = 0.004) and fractional shortening (43.30±2.7 vs 22.89±4.0; p = 0.004) were significantly decreased at 12 weeks. There was also an increase in left ventricular internal systolic dimension (LVIDs) at 12 weeks (1.95±0.04 mm in cBAG3+/+ mice vs 3.12±0.2 mm in cBAG3+/− p = 0.01) and left ventricular systolic volume (LV Vol s) between cBAG3+/+ and cBAG3+/− mice at 12 weeks (12.20±0.6 μl - cBAG3+/+ mice vs 39.83±6.9 μl - cBAG3+/−; p = 0.01). Other echocardiographic parameters such as cardiac output, LVIDd, LVPWd, LVPWs, LV vol d and HR were not significantly different between groups at 12 weeks.
Fig. 2. Effects of BAG3 Haplo-insufficiency on cardiac function.
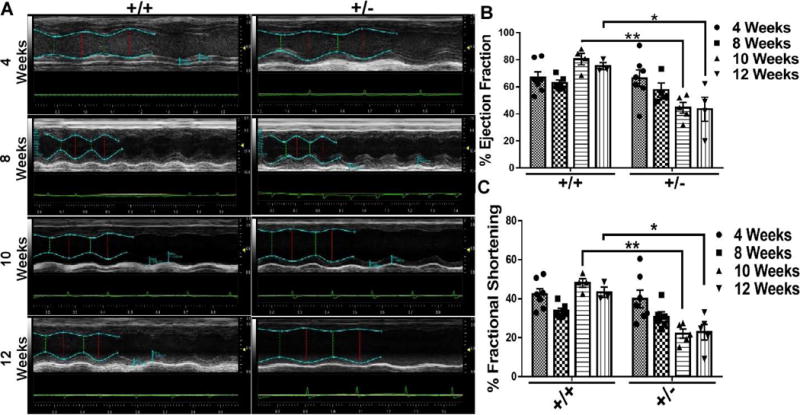
(A) Representative M-mode echocardiograms of cBAG3+/+ and cBAG3+/− mice at 4, 8, 10 and 12 weeks of age. (B & C) Quantification of ejection fraction (B) and percent fractional shortening (C) at 4, 8, 10 and 12 weeks of age. For cBAG3+/+ mice n = 10, 6, 4 and 3 at each time point respectively and for cBAG3+/− mice n = 8, 6, 5 and 4 at each time point respectively. ** p <0.0001, * p = 0.0004
3.3. Single Cell contractility and Ca flux are diminished in the presence of isoproterenol
As seen in Table 1, contractility (% cell shortening) in adult myocytes isolated from 10 to 12 week old cBAG3+/− mice did not differ from that seen in myocytes isolated from cBAG3+/+ control mice. By contrast, in the presence of isoproterenol (1 μM), single cell contractility was significantly (p<0.046) lower in cBAG3+/− mice with haplo-insufficiency then in myocytes isolated from cBAG3+/+ control mice. Similarly, [Ca2+]I transient amplitudes did not differ between cBAG3+/+ and cBAG3+- mice at baseline but were significantly lower (p<0.0001) in myocytes isolated from cBAG3+/− mice in the presence of isoproterenol suggestingBAG3 either modulates β-adrenergic responsiveness of L-type Ca2+ channels therby limiting Ca2+ influx and/or interferes with phospholamban phosphorylation, thereby limiting Ca2+ uptake into the sarcoplasmic reticulum.
Table 1.
Contractility and calcium handling in isolated myocytes from cBAG3+/+ and cBAG3+/− mice.
| Percent Cell Length | Percent Cell Length With ISO | |
|---|---|---|
| cBAG3+/+ | 4.96±0.16 (15) | 11.86±0.41 (11) |
| cBAG3+/− | 4.95±0.27 (16) (NS) | 10.66±0.35 (14)*** |
| Systolic [Ca2+]i (nM) | Systolic [Ca2+]i (nM) With ISO | |
| cBAG3+/+ | 215±14 (11) | 633±43 (15) |
| cBAG3+/− | 202±11 (16) (NS) | 428±28 (12)*** |
| Diastolic [Ca2+]i (nM) | Diastoic [Ca2+]i (nM) With ISO | |
| cBAG3+/+ | 96±7 (11) | 91±7 (15) |
| cBAG3+/− | 98±7 (16) (NS) | 94±6 (12) |
| Transient Amplitude (%) | Transient Amplitude (%) With ISO | |
| cBAG3+/+ | 18.6±1.2 (11) | 58.6±2.5 (15) |
| cBAG3+/− | 17.9±1.0 (16) (NS) | 46.2±2.2 (12)** |
Units of [Ca2+]iI transient amplitude: % increase of fura intensity ratio.
Contractility and Ca2+ handling were not different at baseline (NS) but were reduced in the presence of isoproterenol (ISO). n= number of cells in 3 separate preparations; values are mean ± SE;
p < 0.0001.
p = 0.001.
3.4. Both apoptosis and autophagy are dys-regulated in BAG3 deficient mice
As seen in Figure 3A and 3B, adult myocytes isolated from either cBAG3+/− or cBAG3+/+ mice and transfected with the autophagy flux reporter construct Adv-RFP-GFP-LC3 demonstrated similar amounts of LC3 puncta at baseline. However, inclusion of Bafilomycin A1 in the culture media did not result in an increase in the number of LC3 puncta in adult myocytes isolated from cBAG3+/− hearts. By contrast, there was a significant increase in LC3 puncta in myocytes isolated from cBAG3+/+ when Bafilomycin A1 was included in the culture media suggesting that BAG3 haplo-insufficiency is associated with a significant decrease in autophagy flux (p <0.04). Consistent with the decrease in LC3 puncta in myocytes from cBAG3+/− mice infected with the Adv-RFP-GFP reporter gene, we also failed to find an increase in LC3I and II in adult myocytes from cBAG3+/− mice that had been incubated with Bafilomycin A1 when compared with the obvious increase in LC3I and II seen in myocytes isolated from cBAG3+/+ hearts. (Fig. 4)
Fig. 3. Effects of BAG3 haplo-insufficiency on autophagy and apoptosis.
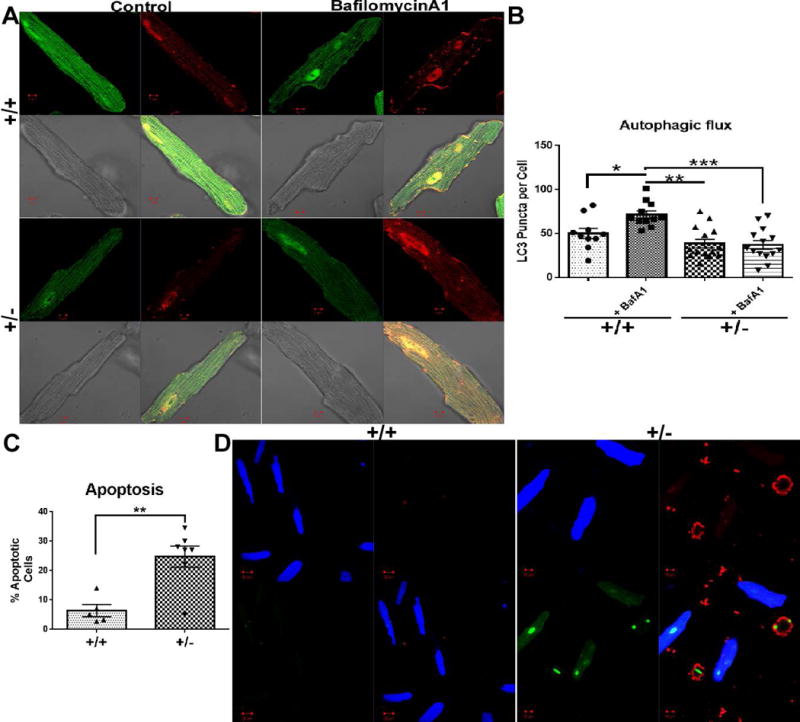
(A) Representative confocal images of isolated adult cardiomyocytes from both cBAG3+/+ and cBAG3+/− mice infected with Ad-GFP-RFP-LC3 reporter construct with and without treatment with Bafilomycin A1. (B) Quantification of LC3 puncta per cell. n= at least 9 cells from 3 separate isolations. * p = 0.03, **p = 0.007, ***p <0.0001. (C) Quantification of percent apoptotic cells per field in cBAG3+/+ and cBAG3+/− cardiomyocytes. n= at least 10 fields from 3 separate isolations. p = 0.006. (D) Representative confocal images of cBAG3+/+ and cBAG3+/− stained for phosphatidylserine (red), compromised nuclei (green) and a cytoplasm dye for viable cells (blue).
Fig. 4. Effects of BAG3 haplo-insufficiency on autophagy flux.
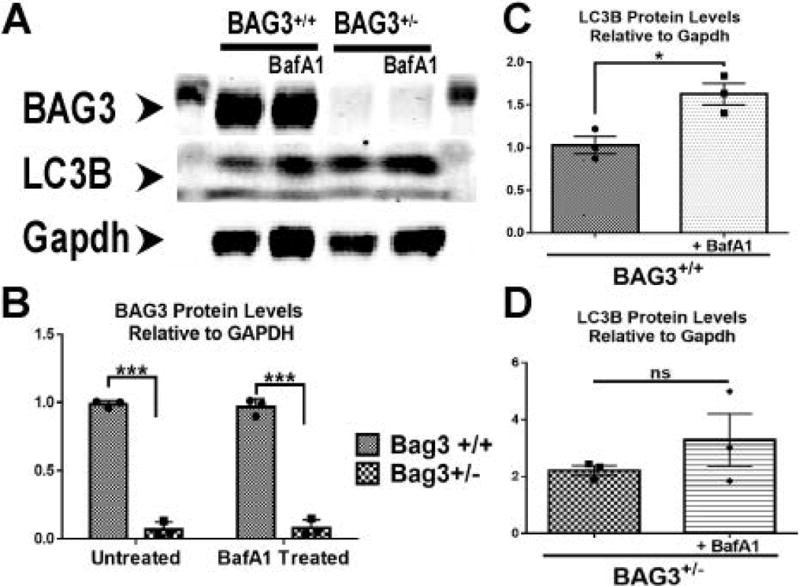
(A) Representative blot of BAG3, LC3 and GAPDH from cardiac myocytes isolated from cBAG3+/+ and cBAG3+/− mice with and without treatment with bafilomycin A1. (B) Quantification of BAG3 levels as measured by western blotting when normalized to GAPDH. (C) Quantification of LC3I and LC3II in BAG3+/+ myocytes when normalized to GAPDH. (D) Quantification of LC3I and LC3II in cBAG3+/− myocytes when normalized to GAPDH. Each experiment was repeated three times. n=3 for all conditions. ***p<0.0001; *p=0,0006.
Similar to the effects on autophagy, BAG3 haplo-insufficiency was associated with a significant increase in apoptosis. As seen in Figure 3C and 3D, the number of apoptotic cells increased significantly (p<0.0001) in myocytes isolated from cBAG3+/− mice when compared with cBAG3+/+ controls. In fact, less than 8% of the total cells from the control mice stained for the apoptosis marker whereas programmed cell death was detected in 24% of the cells isolated from the cBAG3+/− mice with BAG3 haplo-insufficiency.
4.0 Discussion
The BAG3 gene is increasingly being recognized as an important dilated cardiomyopathy locus.[7] However, the mechanisms responsible for the development of the LV dysfunction phenotype have been elusive because of the absence of readily available animal models in which to study the biology of BAG3. Therefore, we used BAG3 single allele floxed (BAG3fl/+) mice and αMHC-Cre mice[16] to generate the first mouse with heterozygous constitutive and cardiac-restricted BAG3 knockdown. The mice showed haplo-insufficiency of BAG3, diminished responsiveness to β-adrenergic stimulation, a modest but significant decrease in autophagy flux and a robust increase in apoptosis. The biologic phenotype of the cBAG3+/− mouse is consistent with our previous observations in mice with left ventricular dysfunction after a myocardial infarction,[31] in mice after ischemia/reperfusion[26] and in isolated adult myocytes in which BAG3 is knocked-down with an siRNA – all of which demonstrate an approximately 50% reduction in BAG3 levels.[20] Thus, the available data suggests that cBAG3+/− mice can provide a useful model for studying the biology of BAG3 mutations that result in haplo-insufficiency.
It is notable that the phenotype of the cBAG3+/− mouse is disparate from recent reports of mice in which BAG3 was knocked out globally or that have a homozygous BAG3 loss-of-function mutation. For example, Homma et al first reported that homozygous deletion of BAG3 led to catastrophic myofibrillar disruption with death by 4 weeks of age; however, mice with heterozygous BAG3 disruption had a normal phenotype.[11] We too found a normal phenotype in mice with haplo-insufficiency of BAG3 at 4 weeks of age; however, by 10 weeks of age they began to demonstrate a significant reduction in LV function. Importantly, young cBAG3+/− mice have significant LV dysfunction in the absence of significant dilatation or hypertrophy, a phenotype that is remarkably different from the profound chamber dilation seen in mice after a myocardial infarction and the marked hypertrophy seen after trans-aortic constriction. Thus these mice provide an interesting window into the early stages of LV dysfunction.
Fang et al recently showed that cardiac-restricted BAG3-KO and Glu455Lys-knockin led to the development of a dilated cardiomyopathy due to decreased binding to HSP70 and a subsequent decrease in autophagy flux.[12] They also showed that decreased coupling with HSP70 in both BAG3-KO and Glu455Lys mice was responsible in part for the phenotype. BAG3-KO also decreased contractility but not [Ca2+]I transient amplitudes in single myocytes. By contrast, we found that 10 week old cBAG3+/− myocytes had normal contractility and Ca2+ amplitudes at rest – but diminished contractility and [Ca2+]I transient amplitudes in response to isoproterenol. Furthermore, while the reduction in autophagy flux found in cBAG3+/− mice was modest, there was a robust increase in apoptosis that was not observed by Fang et al. Thus, cBAG3+/− mice demonstrate a phenotype that accurately mirrors the phenotype seen in individuals with haplo-insufficiency yet differs from the phenotype seen with homozygosity.
Our results provide several important clues regarding the pathobiology of reduced levels of BAG3. First, they support the hypothesis that genetic variants resulting in a loss of BAG3 from a single allele can lead to diminished left ventricular function. Second, the present study demonstrates that a BAG3 variant on a single allele has multiple adverse effects on the heart including decreased β-adrenergic responsiveness, diminished autophagy flux and increased apoptosis and these adverse effects can be seen even early in the course of disease before the ventricle has dilated. In aggregate, these results suggest that cBAG3+/− mice can serve as a useful model for mechanistic studies directed at understanding the molecular and cellular regulation of BAG3 in the heart.
Acknowledgments
This work was supported by the National Institutes of Health grants: RO1-HL123093 (JG &KK) RO1-HL58672, RO1-HL74854 and RO1-HL123-93 (JYC) and PO1-HL91799 (Project 2) and RO1-HL123093 (AMF)
Footnotes
Disclosures are found in the transparency document
References
- 1.Behl C. Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharmacol Sci. 2016 doi: 10.1016/j.tips.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Feldman AM, Gordon J, Wang J, et al. BAG3 regulates contractility and Ca homeostasis in adult mouse ventricular myocytes. J Mol Cell Cardiol. 2016 doi: 10.1016/j.yjmcc.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. 2009;65:83–89. doi: 10.1002/ana.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Norton N, Li D, Rieder MJ, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–282. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villard E, Perret C, Gary F, et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J. 2011;32:1065–1076. doi: 10.1093/eurheartj/ehr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feldman AM, Begay RL, Knezevic T, et al. Decreased levels of BAG3 in a family with a rare variant and in idiopathic dilated cardiomyopathy. J Cell Physiol. 2014;229:1697–1702. doi: 10.1002/jcp.24615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franaszczyk M, Bilinska ZT, Sobieszczanska-Malek M, et al. The BAG3 gene variants in Polish patients with dilated cardiomyopathy: four novel mutations and a genotype-phenotype correlation. J Transl Med. 2014;12:192. doi: 10.1186/1479-5876-12-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toro R, Perez-Serra A, Campuzano O, et al. Familial Dilated Cardiomyopathy Caused by a Novel Frameshift in the BAG3 Gene. PLoS One. 2016;11:e0158730. doi: 10.1371/journal.pone.0158730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chami N, Tadros R, Lemarbre F, et al. Nonsense mutations in BAG3 are associated with early-onset dilated cardiomyopathy in French Canadians. Can J Cardiol. 2014;30:1655–1661. doi: 10.1016/j.cjca.2014.09.030. [DOI] [PubMed] [Google Scholar]
- 10.Arimura T, Ishikawa T, Nunoda S, et al. Dilated cardiomyopathy-associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Hum Mutat. 2011;32:1481–1491. doi: 10.1002/humu.21603. [DOI] [PubMed] [Google Scholar]
- 11.Homma S, Iwasaki M, Shelton GD, et al. BAG3 deficiency results in fulminant myopathy and early lethality. Am J Pathol. 2006;169:761–773. doi: 10.2353/ajpath.2006.060250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang X, Bogomolovas J, Wu T, et al. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest. 2017;127:3189–3200. doi: 10.1172/JCI94310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skarnes WC, Rosen B, West AP, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bradley A, Anastassiadis K, Ayadi A, et al. The mammalian gene function resource: the International Knockout Mouse Consortium. Mamm Genome. 2012;23:580–586. doi: 10.1007/s00335-012-9422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pettitt SJ, Liang Q, Rairdan XY, et al. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat Methods. 2009;6:493–495. doi: 10.1038/nmeth.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agah R, Frenkel PA, French BA, et al. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tilley DG, Zhu W, Myers VD, et al. Beta-adrenergic receptor-mediated cardiac contractility is inhibited via vasopressin type 1A-receptor-dependent signaling. Circulation. 2014;130:1800–1811. doi: 10.1161/CIRCULATIONAHA.114.010434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou YY, Wang SQ, Zhu WZ, et al. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol. 2000;279:H429–436. doi: 10.1152/ajpheart.2000.279.1.H429. [DOI] [PubMed] [Google Scholar]
- 19.Tucker AL, Song J, Zhang XQ, et al. Altered contractility and [Ca2+]i homeostasis in phospholemman-deficient murine myocytes: role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol. 2006;291:H2199–2209. doi: 10.1152/ajpheart.01181.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feldman AM, Gordon J, Wang J, et al. BAG3 regulates contractility and Ca(2+) homeostasis in adult mouse ventricular myocytes. J Mol Cell Cardiol. 2016;92:10–20. doi: 10.1016/j.yjmcc.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Gao E, Rabinowitz J, et al. Regulation of in vivo cardiac contractility by phospholemman: role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol. 2011;300:H859–868. doi: 10.1152/ajpheart.00894.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song J, Zhang XQ, Wang J, et al. Regulation of cardiac myocyte contractility by phospholemman: Na+/Ca2+ exchange versus Na+ -K+ -ATPase. Am J Physiol Heart Circ Physiol. 2008;295:H1615–1625. doi: 10.1152/ajpheart.00287.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song J, Gao E, Wang J, et al. Constitutive overexpression of phosphomimetic phospholemman S68E mutant results in arrhythmias, early mortality, and heart failure: potential involvement of Na+/Ca2+ exchanger. Am J Physiol Heart Circ Physiol. 2012;302:H770–781. doi: 10.1152/ajpheart.00733.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Gao E, Song J, et al. Phospholemman and beta-adrenergic stimulation in the heart. Am J Physiol Heart Circ Physiol. 2010;298:H807–815. doi: 10.1152/ajpheart.00877.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Chan TO, Zhang XQ, et al. Induced overexpression of Na+/Ca2+ exchanger transgene: altered myocyte contractility, [Ca2+]i transients, SR Ca2+ contents, and action potential duration. Am J Physiol Heart Circ Physiol. 2009;297:H590–601. doi: 10.1152/ajpheart.00190.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su F, Myers VD, Knezevic T, et al. Bcl-2-associated athanogene 3 protects the heart from ischemia/reperfusion injury. JCI Insight. 2016;1:e90931. doi: 10.1172/jci.insight.90931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 28.Capasso S, Alessio N, Squillaro T, et al. Changes in autophagy, proteasome activity and metabolism to determine a specific signature for acute and chronic senescent mesenchymal stromal cells. Oncotarget. 2015;6:39457–39468. doi: 10.18632/oncotarget.6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma X, Liu H, Foyil SR, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knezevic T, Myers VD, Su F, et al. Adeno-associated Virus Serotype 9 - Driven Expression of BAG3 Significantly Improves Left Ventricular Function in Murine Hearts with Left Ventricular Dysfunction Secondary to a Myocardial Infarction. JACC Basic Transl Sci. 2016;1:646–653. doi: 10.1016/j.jacbts.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]