

Abstract
Rationale
Pulmonary arterial hypertension (PH) is a life-threatening condition associated with immune dysregulation and abnormal regulatory T cell (Treg) activity, but it is currently unknown whether and how abnormal Treg function differentially affects males and females.
Objective
To evaluate whether and how Treg-deficiency differentially affects male and female rats in experimental PH.
Methods and Results
Male and female athymic rnu/rnu rats, lacking Tregs, were treated with the vascular endothelial growth factor receptor-2 (VEGFR2) inhibitor SU5416 or chronic hypoxia and evaluated for PH; some animals underwent Treg immune reconstitution (IR) before SU5416 administration. Plasma prostacyclin (PGI2) levels were measured. Lung and right ventricles (RVs) were assessed for the expression of the vasoprotective proteins cyclooxygenase-2 (COX-2), prostacyclin synthase (PTGIS), programmed death ligand-1 (PDL-1), and heme oxygenase-1 (HO-1). Inhibitors of these pathways were administered to athymic rats undergoing Treg IR. Finally, human cardiac microvascular endothelial cells co-cultured with Tregs were evaluated for COX-2, PDL-1, HO-1, and estrogen receptor (ER) expression, and culture supernatants were assayed for PGI2 and IL-10.
SU5416-treatment and chronic hypoxia produced more severe PH in female than male athymic rats. Females were distinguished by greater pulmonary inflammation, augmented RV fibrosis, lower plasma PGI2 levels, decreased lung COX-2, PTGIS, HO-1 and PDL-1 expression and reduced RV PDL-1 levels. In both sexes, Treg IR protected against PH development and raised levels of plasma PGI2 and cardiopulmonary COX-2, PTGIS, PDL-1, and HO-1. Inhibiting COX-2, HO-1, and programmed death-1 (PD1)/PDL1 pathways abrogated Treg protection. In vitro, human Tregs directly upregulated endothelial COX-2, PDL1, HO-1, ERs and increased supernatant levels of PGI2 and IL-10.
Conclusions
In two animal models of PH based on Treg deficiency, females developed more severe PH than males. The data suggest that females are especially reliant on normal Treg function to counteract the effects of pulmonary vascular injury leading to PH.
Subject Terms: Animal Models of Human Disease, Inflammation, Pathophysiology, Pulmonary Biology, Vascular Biology
Keywords: Regulatory T cells, severe pulmonary hypertension, sex, prostacyclin, immune system, lymphocyte
INTRODUCTION
PH is a lethal cardiopulmonary disorder characterized by a narrowing of the terminal pulmonary arterioles that causes right ventricular (RV) failure and death.1 As with an increasing number of cardiovascular diseases2, abnormalities of Treg number and function have also been reported in PH in association with clinical evidence of immune dysregulation.3–8 Preclinical evidence shows that anti-inflammatory Tregs, generally defined as CD4+CD25high forkhead box P3 (FoxP3)+ cells, play a critical role in the resolution of the inflammatory process caused by putative PH disease triggers, such as viral infection or increased shear stress (reviewed in (9)). We and others have previously shown that T-cell deficient rnu/rnu athymic male rats, which lack Tregs, are particularly susceptible to PH.10–15 In these studies, vascular injury caused by treatment of male athymic rats with monocrotaline or the VEGFR2 antagonist, SU5416, resulted in exuberant perivascular inflammation and severe PH. Pathologic changes in the lungs of T cell-deficient animals modeled the microangiopathy observed in the human condition, and this pulmonary vascular disease led to elevated right heart pressure, right ventricular hypertrophy (RVH) and ultimately, death from PH. We showed that restoration of normal Treg activity through IR of athymic rats with Tregs isolated from inbred, MHC-matched, litter-mate euthymic controls prevented PH in males through the induction of an active anti-inflammatory response.14 However, whether Treg-deficiency differentially impacts males and females is currently unknown.
Here, we report that female athymic rats treated with SU5416 or exposed to chronic hypoxia develop more significant PH than their male counterparts. Specifically, females lacking normal Treg activity exhibited greater inflammation, more dramatic RV microvascular dropout and increased periarteriolar fibrosis compared to males with PH. This phenotype was associated with decreased systemic levels of the pulmonary vasodilator, PGI2. We subsequently investigated enzymes responsible for PGI2 synthesis and discovered decreased lung COX-2 and PTGIS expression. Two other proteins associated with vasoprotection and normal Treg function, HO-1 and PDL-1, were also reduced in female lung tissue. Extending this analysis to the heart showed that RV PDL-1 expression was also relatively diminished in Treg-deficient females with PH. Treg IR prevented PH development and was associated with significantly increased levels of plasma PGI2, PTGIS, COX-2, HO-1 and PDL-1 in both sexes. Blocking COX-2, HO-1 and PD-1/PDL-1 signaling abrogated Treg protection in both sexes and, in so doing, demonstrated that these pathways are important for Treg action following a pulmonary vascular insult. To evaluate how Tregs normally mediate protection of vascular endothelium, human Tregs were cocultured with cardiac endothelial cells and were found to specifically upregulate endothelial COX-2, HO-1, PDL-1 and cardioprotective ERs as well as induce the biosynthesis of PGI2 and the anti-inflammatory cytokine, IL-10. These data collectively confirm a special and dominant protective role for Tregs in females at risk for PH.
METHODS
The data and methods that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
Inbred male and female WAG (RT1u) athymic nude rats (rnu/rnu) and euthymic (ET) (rnu/+) rats were obtained from Biomedical Research Models, Inc, (Worcester, MA) and bred in house. ET rats served as MHC-identical controls and as cell donors for IR experiments. To induce PH, 6–8 week-old male and female athymic animals were subcutaneously (s.c) injected with a single dose of SU5416 (20 mg/kg) dissolved in dimethyl sulfoxide (DMSO) (#D2650, Sigma Aldrich, St. Louis, MO) or DMSO alone as vehicle controls. Alternatively, athymic rats were exposed to hypoxia (10% O2) in an airtight plexiglass hypoxia chamber with simulated oxygen controller (Biospherix; ProOx110, Parish, NY). Athymic rats maintained in normoxic conditions (21% O2) served as controls for this experimental group. All animals were sacrificed at 3 weeks post-treatment. The procedures and experimental protocols were approved by the Veterans Affairs Palo Alto Animal Care and Use Committee.
Antibodies and reagents
Antibodies to CD3 (UCHT1) and CD28 (28.2) (#555725 and #555329, respectively) were purchased from BD Pharmingen (San Jose, CA). Additional antibodies from BD Pharmingen were anti-CD25-PE antibody (OX39) (#554866) and anti-COX-2 PE (AS67) (#565125). β-actin antibody (#A5441) and In situ cell death detection kit (Roche #11684795910) was from Sigma Aldrich (St. Louis, MO). The following antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA): anti-PDL-1 (#sc-50298) and Donkey anti-goat HRP (#sc-2056) or from Abcam (Cambridge, MA): anti-PGIS (#ab23668), anti-Reca-1 (#ab9774), anti-Calponin (CALP) (#ab700), anti-alpha SMA (1A4) (#ab7817) and anti-HO-1 (#ab13243). Other antibodies were anti- PDL-1 (#4059, ProSci, Poway, CA), anti- CD68 (ED-1) (#MCA341R, Bio-Rad/AbD Serotec, Hercules, CA), anti-CD274 (B7-H1/PDL-1) APC (MIH1) (#17-5983-42, eBioscience, San Diego, CA), anti-HO-1 FITC (HSP32) (#NBP1-77460F, NovusBiologicals, Littleton, CO); anti-ERalpha Alexa Flour488 (C542) (#NBP1-19318AF488, NovusBiologicals); anti-Ki67(#NB500-170, NovusBiologicals); anti-ERbeta PE (NR342) (#IC7106P, R&D Systems, Minneapolis, MN). The following antibodies were from Cell Signaling Technology, (Beverly, MA): anti-COX-2 (D5H5), Goat anti-mouse HRP (#7076S) and Donkey anti-rabbit HRP (#7074S). Anti-PD1 (RMP1-14), (#BE0146) and isotype control IgG (2A3), (#BE0089) were obtained from BioXCell, West Lebanon, NH). Alexa Fluor 488 donkey anti-mouse IgG (#715-545-151), Alexa Flour 647 donkey anti-mouse IgG (#715-605-150) and Cy3 donkey anti-rabbit IgG (#711-165-152) were obtained from Jackson ImmunoResearch (West Grove, PA). Vascular endothelial growth factor receptor 2 (VEGFR2) inhibitor SU5416 was synthesized utilizing a previously described modified protocol.14 The heme oxygenase -1 (HO-1) inhibitor, Zinc (II) Protoporhyrin IX (Zn(II)PPIX) (#691550) and selective cyclooxygenase -2 (COX-2) inhibitor NS-398, (#70590) was purchased from Cayman Chemical (Ann Arbor, MI).
Tregs (CD4+CD25high) isolation and immune reconstitution (IR)
Rat Tregs were isolated from the inbred ET animals. Dissociated spleens were filtered through a 100 μm cell strainer (#352360, Corning, NY) to obtain a single cell suspension, and the cells were washed with Dulbecco’s phosphate-buffered saline (DPBS, #14190-144, Gibco, Grand Island, NY) supplemented with 2% fetal bovine serum (FBS) (#10438026, Gibco) by centrifugation. The CD4+CD25highTreg subset was isolated by a two-step method using fully automated RoboSep™ (STEMCELL Technologies, Vancouver, Canada). Negative selection was performed using EasySep™ Rat CD4+ T Cell Isolation Kit (#19642, STEMCELL Technologies) followed by labeling CD4+ T cells with anti-CD25-PE antibody; the CD25high cells subset was selected with the EasySep™ PE Selection Kit (#18557, STEMCELL Technologies). This cell fraction was further purified by fluorescent activated cell sorting (FACSAria II; BD Biosciences, San Jose, CA); the resulting CD4+CD25high population was >98% pure. Purified rat Tregs (CD4+CD25high) cells (4–5 × 106 cells) were injected intravenously (i.v.) into AT male and female rats 7d prior to SU5416 injection, and IR was confirmed 7d post SU5416 treatment by flow cytometry.
Western blot analysis
Western blot was performed as previously described. 1477 Briefly, frozen rat lung and heart tissue (−80°C) were homogenized in Pierce protein extraction RIPA buffer (#89901, Thermo Scientific, Rockford, IL) containing Halt protease inhibitor cocktail (#78429, Thermo Scientific) at 4°C. The BCA protein assay kit (#23225, Thermo Scientific) was used to measure protein concentration. Protein samples (30 μg) were loaded onto Bolt 4–12% Bis-Tris Plus precast gels (#NW04125BOX, Invitrogen, Carlsbad, CA) followed by electrophoresis and immunoblot analysis. Primary antibodies were anti-HO-1 (1:500), anti-PDL-1 (1:200), anti-COX-2 (1:500), ant-PGIS (1:200) and anti-βactin (1:2000) antibodies. Immunoblots were imaged with molecular imager Chemidoc XRS+ (Bio-Rad, Hercules, CA) and data are expressed as densitometric units determined using ImageJ (version 1.48) software.
Immunofluorescence imaging
Rat lungs were insufflated with a 1:1 mixture of optimal cutting temperature (OCT) compound and 30% sucrose and embedded in Tissue Tek OCT compound (#4583, Sakura, Torrance, CA) in sub-zero temperature. Rat heart tissue was frozen with OCT in dry ice. Cryosections (7 μm) of lung/heart tissue were placed on superfrost/plus slides (#48311-703, VWR, Radnor, PA). For immunofluorescent staining, the slides were fixed in methanol/acetone (1:1), washed with PBS, and incubated in 0.2% Triton X-100 (#T9284, Sigma Aldrich). The sections were blocked with normal 10% donkey serum (Jackson ImmunoResearch, West Grove, PA), and then exposed for 1 h or overnight to primary antibodies to CD68 (ED-1) (1:50), Reca1 (1:50), Calponin (CALP) (1:50), HO-1 (1:100), COX-2 (D5H5) (1:200), and anti-PDL-1 (1:200), Ki67(1:50), anti-alpha SMA (1:100) followed by Alexa Fluor 488 conjugated donkey anti-mouse IgG, Alexa Flour 647 conjugated donkey anti-mouse IgG, Cy3 conjugated donkey anti-rabbit IgG conjugated secondary antibodies (1:600) and Apoptosis detection kit. The sections were mounted with mounting medium and DAPI (#H-1200, Vector Laboratories, Burlingame, CA). Quantification of Ki67 and TUNEL positive vascular SMC was performed in a blinded fashion in randomly chosen lung tissue sections in each experimental group. (n=4 per group). 10 vessels per animal were counted and in total for each marker two hundred forty small pulmonary vessels were analyzed using Image J (version 1.48) software. Microscopic analysis was performed with the LSM 710META confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
ELISA
ELISA kits were used to measure the levels of Estradiol (E2), (#ES180S-100, Calbiotech, Spring Valley, CA), 6-keto-prostaglandin F1 α (PGF1α, #515211, Cayman Chemical, Ann Arbor, MI), and IL-10 (#100764, Abcam, Cambridge, MA).
Statistics
GraphPad Prism_6.0 was used for statistical analysis. Differences between multiple groups were compared using 1-way analysis of variance with Bonferroni multiple-comparisons test and between two groups, a student’s t-test. For Western blot analyses, for Ki67 and TUNEL positive vascular SMC quantification analyses, two analyses were performed: (1) To compare male to female values with normal distribution, an unpaired t test with Welch’s correction was used and for non-parametric data, the Mann-Whitney test. (2) To compare same-sex vehicle and SU5416+Treg groups to the same-sex SU5416 group, the ANOVA with Bonferroni multiple-comparisons test was employed. A P-value of <0.05 was considered significant. Data are expressed as mean ± SEM.
RESULTS
Female athymic rats have increased disease severity, attenuated by Treg IR
We previously showed that athymic (T-cell deficient) male rats develop significant PH after treatment with the VEGFR2 antagonist, SU5416 due to the absence of anti-inflammatory CD4+CD25high FoxP3+ Tregs.11, 13, 14 To determine whether female athymic rats are similarly susceptible to PH, rats of both sexes were treated with SU5416 (20 mg/kg) or DMSO (vehicle) and evaluated 21d post-treatment. PH severity was greater in female rats as determined by higher right ventricular systolic pressures (RSVP; Figure 1A) and a greater degree of right ventricular hypertrophy (RVH; Figure 1B) and reduced pulmonary artery acceleration times (PAATs; Figure 1C). Increased disease severity was also observed in female athymic rats housed in hypoxic conditions for 3 weeks (10% inspired O2 (PaO2 ≈ 40 mm Hg; Online Figure I). SU5416-treated PH lungs exhibited a greater CD68+ pulmonary macrophage infiltration in females (Online Figure II) and a larger decline in RV capillary density associated with greater RV perivascular and interstitial fibrosis (Online Figure III). Consistent with our previous findings in male rats14, IR with CD4+CD25highTregs (3 × 106 i.v. 7d before SU5416 treatment) prevented PH14 and was equally protective in male and female animals with near-normal RVSPs and RV/LV+S values in both sexes (Figure 1). Not surprisingly, the immunologically-pleiotropic and vasoprotective hormone, estradiol (E2)16 was higher in female serum for all experimental groups evaluated (Online Figure IV); a result which suggested that any role for E2, if present, in PH was necessarily complex given that its expression is higher in both females with PH and in females protected from PH.
Figure 1. Severe PH in athymic female rats.
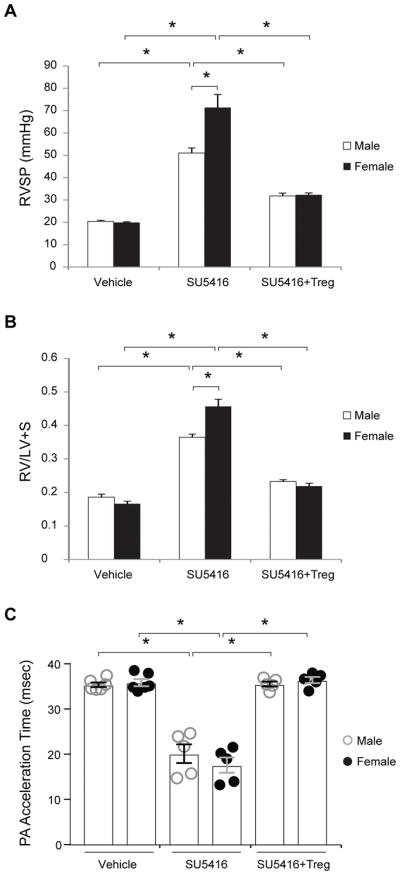
(A) RVSP measurements assessed on d21 post-treatment of athymic male and female rats with vehicle control, s.c. VEGFR2 inhibitor SU5416 (20 mg/kg) single dose, and s.c. SU5416 (20 mg/kg) with Tregs. (B) RVH measurements determined by RV/(LV+S) ratio assessed on d21 post-treatment of athymic male and female rats with vehicle control, s.c. VEGFR2 inhibitor SU5416 (20 mg/kg) single dose, and s.c. SU5416 (20 mg/kg) with Tregs. (C) PAAT by sequential echocardiography observed on d21 in all athymic male and female animal groups prior to pre-terminal right heart catheterization. (n=5–7 per group). Data shown as means with error bars representing SEM (*P<0.05)
Increased disease severity in female rats is associated with decreased PGI2 synthesis
To better understand the factors involved in the increased disease severity of athymic females and begin to elucidate the function of Tregs in PH prevention, we asked whether vasoprotective pathways are altered in a sex-specific fashion in athymic rats and whether there was a differential effect of Treg IR in males versus females. We first focused on PGI2, a potent vasodilator that exerts multiple vasoprotective effects, and the enzymes most responsible for its synthesis.17 PGI2 has a half-life of 42 seconds and is broken down into 6-keto-PGF1,18 a metabolite that is used to estimate systemic levels. PGI2 levels were assessed 21d after treatment with vehicle or SU5416 with or without Treg IR. Plasma PGI2 levels were decreased in females with PH suggesting that reduced PGI2 synthesis may contribute to worse PH in Treg-deficient females (Figure 2A). An ≈4-fold increase in the PGI2 values above the control baseline was observed in both sexes undergoing Treg IR with serum levels of ≈ 1,300 pg/ml in males and females, showing here the special reliance females have for normal Treg function. To further evaluate factors responsible for PGI2 biosynthesis, we evaluated COX-2 and PTGIS. Consistent with lower PGI2 blood levels in females, Western blot analysis of male and female PH lungs analyzed at d21 post SU5416 treatment demonstrated relatively reduced COX-2 and PTGIS levels in female compared to male PH lungs (Figure 2B, C, D). Administration of the COX-2 inhibitor, NS-398, abrogated the IR-associated increase in PGI2 levels. Thus, lower pulmonary COX-2 and possibly lower pulmonary PTGIS, in athymic females provides one explanation of why females have lower circulating PGI2. Collectively, these results show that more severe PH in females, occurring in the absence of normal Treg activity, correlates with relatively reduced pulmonary COX-2 and PTGIS expression and a concomitant decrease in serum PGI2.
Figure 2. Athymic female rats with PH express decreased systemic PGI2 and reduced pulmonary COX-2, PTGIS, HO-1 and PDL-1.

(A) Prostacyclin release in vivo was measured by 6-ketoPGF1α levels in plasma of athymic male and female rats treated with vehicle control, SU5416, or SU5416 + Tregs and analyzed on d21 (n = 4 per group). (B–D) Representative Western immunoblots and densitometric quantitation of COX-2, PTGIS, relative to β-actin in lung lysates from vehicle-, SU5416− and SU5416 + Treg-treated athymic male and female rats assessed on d21 (n = 5 per group). (E–G) Representative Western immunoblots and densitometric quantitation of HO-1, PDL-1, relative to β-actin in lung lysates from athymic male and female rats treated with vehicle control, SU5416, SU5416 + Treg, and assessed on d21 (n = 5 per group). Data are shown as means with error bars representing SEM (*P<0.05)
PDL-1 and HO-1 is decreased in Treg-deficient females with PH
The PD-1/PDL-1 negative costimulatory pathway is critical for Treg immunomodulatory function.19 In immunoblotting assays, PDL-1 expression was reduced in female lungs compared to males (Figure 2E, G). HO-1, another enzyme which helps mediate Treg function20 was strongly expressed in control female lungs but was notably decreased in PH; HO-1 was not significantly expressed in male lungs with or without PH (Figure 2E, F). The data suggested that reduced PDL-1 and HO-1 expression could contribute to increased disease severity in Treg-deficient females. Treg IR significantly increased the expression of both PDL-1 and HO-1 in both males and females, underscoring the apparent contribution of these pathways to Treg activity following vascular injury.
IR with Tregs prior to vascular injury decreases pulmonary arteriolar remodeling with reduced vascular smooth muscle cell proliferation and enhanced apoptotic activity with increased COX-2, PTGIS, HO-1 and PDL1 expression
To assess vascular remodeling, male and female lungs were evaluated for smooth cellular proliferation by Ki67 staining and for apoptosis with TUNEL assay (Figure 3A, B, C). Vascular smooth muscle apoptosis was induced by Treg IR, similar to that observed with estradiol protection21. The occlusive arteriopathy athymic rats is characterized by αSMA+ cells obstructing the vascular lumen (Figure 3D) and enhanced vascular wall thickness (Figure 3E); both processes being prevented by Tregs. Having seen that Treg IR upregulates COX-2, PTGIS, HO-1, and PDL-1 protein expression, we wanted to better understand their tissue localization better in reconstituted animals protected from PH. Rat lungs, from animals that had undergone IR, were evaluated by immunohistochemistry at 21d post-SU5416 administration (Figure 3F). Histologically, Treg-conferred protection was manifested by a prevention of SU5416-induced occlusive vasculopathy and an increased expression of all four proteins in the calponin+ pulmonary arteriolar smooth muscle cells (Figure 3F). Similar results were observed in male and female rats. (Online Figure V).
Figure 3. IR with Tregs prior to vascular injury decreases pulmonary arteriolar remodeling with reduced vascular smooth muscle cell proliferation and enhances apoptotic activity with increased COX-2, PTGIS, HO-1 and PDL1 expression.

(A) Immunofluorescent images of lung tissue sections from female athymic rats treated with vehicle control, SU5416 and SU5416 plus IR with Tregs on d21 and immunolabeled with α –SMA (cyan), Ki67(red), TUNEL (green). Nuclei stained with DAPI (blue). Negative control represents samples exposed only to conjugated secondary antibodies. (n = 4 per group). (B) Percentage of Ki67 positive cells in vascular SMC in all groups of athymic male and female rat lungs. (C) Percentage of TUNEL positive cells in vascular SMCs in all groups of athymic male and female rat lungs. (D) Immunohistochemistry images of vessels stained with α-SMA (brown) in lung tissue at d21 after treatment with vehicle control, SU5416 and SU5416 plus IR with Tregs (n =5/group). (E) Percentage of wall thickness of α-SMA positive vessels < 100 μm in external diameter in all groups of athymic male and female rat lungs. (F) Immunofluorescent images of lung tissue sections from athymic rats treated with SU5416 plus IR with Tregs on d21 and immunolabeled with COX-2, PTGIS, HO-1 and PDL1 in red and Calponin (green) for vessel smooth muscle cells. Nuclei stained with DAPI (blue). Scale bars: (A) 25 μm. (D) 50 μm. (F) 25 μm. Data are shown as means with error bars representing SEM (*P<0.05).
Tregs induce RV expression of COX-2, PTGIS, HO-1, and PDL-1
RV failure is a sentinel event predicting rapid clinical decompensation in PH and is considered a maladaptive response to progressive pulmonary vascular disease.22 In the athymic rat model, RV changes are not readily detectable by echocardiography before d10 post SU5416 administration,14 and it remains unknown to what extent RV changes are due to progressive pulmonary vascular resistance as opposed to processes that may be intrinsic to the RV alone. To examine whether males and females also differ in terms of RV expression of the enzymes involved in PGI2 synthesis, protein extracts from male and female RVs were blotted with antibodies to COX-2 PTGIS, as well as HO-1 and PDL-1. As with the male PH lungs, male RVs exhibited an increase in PDL-1 expression with disease, but this was not the case in female RVs; Treg IR administered before SU5416 increased the RV expression of all four vasoprotective proteins (Figure 4A, B–E). Cardiac histology of athymic rats 21d post-SU5416 administration confirmed COX-2, PTGIS, HO-1 and PDL-1 upregulation in vascular endothelial cells and myocardium from the male and female animals which had undergone Treg IR (Figure 5, Online Figure VI). Real-time PCR of lung and RV tissue for all experimental groups were consistent with the protein expression data (Online Figure VII). Collectively, data suggest that female RVs may be rendered more vulnerable to PH in the absence of normal Treg activity due to relatively less PDL-1 activity than males. However, Tregs protect RVs in both sexes, through COX-2, PTGIS, HO-1, and PDL-1 modulation, further suggesting that these cells play a more dominant role in females to prevent PH following vascular injury.
Figure 4. IR with Tregs operates through activation of COX-2, PTGIS, HO-1, PD1/PDL1 pathways in RV tissue of both male and female athymic rats.

(A) Representative Western immunoblots of COX-2, PTGIS, HO-1 and PDL1 relative to β-Actin in RV lysates from untreated, SU5416−, and SU5416 + Treg-treated athymic male and female rats; all groups analyzed on d21 (n = 5 per group). (B–E) Representative densitometric quantitation of COX-2, PTGIS, HO-1, PDL1 relative to β-Actin in RV lysates assessed in vehicle, SU5416−, and SU5416 + Treg-treated athymic male and female rats; all groups analyzed on d21 (n = 5 per group). Data are shown as means with error bars representing SEM (*P<0.05).
Figure 5. Immune reconstitution with Tregs leads to increased COX-2, PTGIS, HO-1 and PDL1 expression in endothelial cells of RV intramyocardial coronary vessels.

(A–D) Representative immunofluorescent images of RV tissue sections of athymic rats after treatment with SU5416 plus IR with Tregs on d21 and immunolabeled with COX-2 (A), PTGIS (B), HO-1 (C) and PDL1 (D) in red and Reca1 (green) for RV vessel endothelial cells. Nuclei were stained with DAPI (blue). Scale bars: (A–D) 50μm.
Blocking COX-2, HO-1 and PD-1/PDL-1 signaling pathways abrogates Treg-mediated protection from PH
To confirm that the increased PH severity in Treg-deficient females is due to decreased COX-2, HO-1 and PDL-1 signaling, we examined whether blocking these pathways abrogates Treg-mediated protection. Athymic male and female rats treated with NS-398 (a selective COX2 inhibitor) at the time of Treg IR were not protected against PH development as manifested by significantly higher RVSPs and greater RVH assessed 21d after SU5416 (Figure 6A, B). Administration of the HO-1 inhibitor zinc (II) protoporphyrin IX (ZnPPIX), into animals that had undergone Treg IR, at the time of SU5416 treatment, also resulted in a loss of Treg-mediated protection in both sexes, as did treatment with an anti-PD-1 (PDL-1 ligand) antibody. Collectively, these findings support the conclusion that the systemic Treg protection against PH depends on intact COX-2, HO-1, and PD-1/PD-L1 pathways in both male and female rats.
Figure 6. Blocking COX-2, HO-1 and PD-1/PDL-1 signaling pathways abrogates Treg-mediated protection from PH.

(A) RVSP measurements in athymic male and female rats treated with SU5416 + Tregs at d21 and: injected i.p. with NS-398 (a selective COX2 inhibitor)(10mg/kg) daily from day 0 until d21, injected i.p with zinc (II) protoporphyrin IX (ZnPPIX)/HO-1 inhibitor), (40mg/kg) on days 0,2,3,7,14 and 18, injected i.p with anti-PD-1 (PDL-1 ligand) antibody (600mg/kg loading dose) on day 0 with 300mg/kg every other day until d21, injected with isotype control antibody (600mg/kg loading dose) on day 0 and 300mg/kg every other day until d21 (n=4–5 per group). (B) RVH measurements as assessed by RV/(LV+S) ratio in athymic male and female rats treated with SU5416 + Tregs at d21 and: injected i.p. with NS-398 (a selective COX2 inhibitor)(10mg/kg) on day 0 and when daily until d21, injected i.p with zinc (II) protoporphyrin IX (ZnPPIX)/HO-1 inhibitor), (40mg/kg) on days 0,2,3,7,14 and 18, injected i.p with anti-PD-1 (PDL-1 ligand) antibody (600mg/kg loading dose) on day 0 with 300mg/kg every other day until d21, injected with isotype control antibody (600mg/kg loading dose) on day 0 with 300mg/kg every other day until d21 (n=4–5 per group). Data are shown as means with error bars representing SEM (*P<0.05).
Tregs upregulate COX-2, HO-1, PDL-1, PGI2 and ER surface expression in HMCECs and increase culture supernatant concentrations of PGI2 and IL-10
To examine whether Tregs were sufficient to directly induced the upregulation of COX-2, PTGIS, HO-1 and PDL-1 in human microvascular cardiac endothelial cells (HMCECs) and simultaneously assessed the expression of the highly-vasoprotective ERs.23 Purified human CD4+CD25high Tregs were co-cultured with human microvascular cardiac endothelial cells (12 hours; 37°C). HMCECs, co-cultured with CD4+CD25−, T cells served as controls. The HMCECs were subsequently stained with antibodies to COX-2, HO-1, PDL-1, and the E2 receptors, ER-alpha and ER-beta, and analyzed by flow cytometry. Surface expression of COX-2, HO-1 and PDL-1 was significantly upregulated in cardiac endothelial cells co-cultured with Tregs, as compared to those co-cultured CD4+CD25−T cells (Figure 7A–C). Furthermore, a dramatic upregulation of both estrogen receptors was observed in the Treg-co-cultured endothelial cells but not in CD4+CD25−T-co-cultured endothelial cells (64-fold for ER-alpha and 22-fold for ER-beta; Figure 7D, E).
Figure 7. Tregs upregulate COX-2, HO-1, PDL-1, and estrogen receptor expression in human cardiac microvascular cells and increase culture supernatant concentrations of PGI2 and IL-10.

(A–E) Fluorescence histogram from flow cytometric analysis of COX-2, HO-1, PDL-1, ERalpha, ERbeta expression in hCMECs (male donor) after 24h coculture with human (male donor) Tregs. (F–G) Prostacyclin release was measured by 6-ketoPGF1α and IL-10 levels from ELISA of culture supernatants after hCMECs (male donor) 24h co-culture with human (male donor) Tregs, treated with ICI180, 780 (ER inhibitor), (10μM), NS-398 (a selective COX2 inhibitor), (10μM), zinc (II) protoporphyrin IX (ZnPPIX)/HO-1 inhibitor), (10μM), (n=4 per group). Data are shown as means with error bars representing SEM (*P<0.05).
Finally, because RV cardiac endothelial cells appeared to be a site for COX-2 localization, we wanted to know whether Tregs could act on vascular endothelium in concert with E2 (through the ERs) to induce vasodilation through the release of prostanoids, including PGI2.23 HMCECs were cultured with Tregs (12 hrs; 37° C) in the presence or absence of inhibitors for ERs (ICI 180, 780 24), COX-2, and HO-1, and the culture supernatant was assayed for the PGI2 metabolite 6-keto-PGF1. Blocking ERs, COX-2 and HO-1 significantly reduced supernatant PGI2 levels (Figure 7F). Further, because PGI2 is known to stimulate a key anti-inflammatory Treg cytokine, IL-10, we also evaluated its expression under similar conditions.25 Compared to the Treg-endothelial control group, those cultures in which ER signaling, COX-2, and HO-1 were blocked demonstrated significantly reduced IL-10 (Figure 7G). Collectively, the data suggest that Tregs may directly mediate vasoprotection through upregulation of ERs, COX-2, HO-1, PGI2 and IL-10 biosynthesis in cardiac, vascular endothelial cells.
DISCUSSION
Treg abnormalities are a clinical feature of PH conditions.3–8 While increased numbers of Tregs are observed in the blood of PH patients6, reduced Tregs are observed in the lungs of these individuals.26 Idiopathic PH is associated with an expansion of peripheral blood Tregs that are non-suppressive4, and PH associated with connective tissue disorders exhibit increased Th17/Treg ratios.7 The Paris group, led by Marc Humbert, reported a recent clinical study of 62 patients with idiopathic, heritable and connective tissue-associated PH discovered dysfunctional Tregs in all three subsets and implicated defective leptin-signaling as a possible Treg disturbance in idiopathic and connective tissue disease PH, but not heritable PH.3 Tregs limit pulmonary vascular injury that can cause experimental PH.14 However, their ability to differentially affect males versus females, and the involved mechanisms are unknown. We report that in the absence of protective Tregs, PH was more severe in female than male rats using both the SU5416 and chronic hypoxia model systems. SU5416-induced PH is a well-established model of PH27 based on pulmonary artery endothelial injury in contrast to the hypoxia model which occurs as a consequence of sustained pulmonary vasoconstriction. Histologically, female athymic rats with PH exhibited greater pulmonary inflammation, a more significant drop out in RV microvessels, more RV peri-arteriolar fibrosis with Tregs exerting a more dominant protective function in females compared to their male counterparts. This athymic rat model of PH is distinguished from some other disease models in which males have more significant disease. With the frequently-used monocrotaline and chronic hypoxia PH models, female rodents develop unreliable or mild PH compared with males.28–33 These studies have identified E2 (or its 16a-hydroxyestrone metabolite) as either a PH disease mediator or a protective agent.34 A recent study using SU5416 and a different strain of athymic rats showed worse disease in males;35 this study differed from the current one by using genetically-outbred rats, which constrains against IR experiments, and was relatively limited in scope. Variation in strain, even between colonies of the same strain, has a remarkable influence on the nature and severity of the response to SU5416, consistent with an important role for genetic modifiers of the PH phenotype.36
We started this evaluation by comparing systemic PGI2 concentrations in both sexes after PH was established because of its important role as an endogenous pulmonary vasodilator and discovered relatively decreased concentrations in females. Two enzymes implicated in PGI2 biosynthesis, COX-2, and PTGIS, were also diminished in female lungs. PGI2 is an important endogenous vasoactive factor that acts through adenylyl cyclase and cyclic AMP to cause vasodilation and limit pulmonary artery smooth muscle cell proliferation. PGI2 synthesis is reduced in PH patients who also demonstrate lower pulmonary artery endothelial cell PTGIS.37, 38 It is possible that the lower PGI2 levels observed in clinical PH is attributable to the results being obtained in a predominantly female and immune-dysregulated population. Estrogen, acting mainly through ER-alpha, is known to acutely activate PGI2 synthesis in the vascular endothelium of females.23, 39, 40 Yet, even though female athymic rats have higher estrogen levels than males in all experimental conditions, female PGI2 systemic levels were relatively low in disease. This finding raises the possibility that, in the absence of Tregs, poor estrogen signaling is occurring in the cells mainly responsible for PGI2 synthesis. With normal immunity, estrogens have been shown to increase the production of nitric oxide (NO) and PGI2, and attenuate both vasoconstriction, vascular remodeling, and cardiac fibrosis.39, 41–44 Our in vitro findings suggest that Tregs dramatically upregulate ER isoform expression and provide one explanation of how, in the absence of normal Treg function, estrogen is rendered less effective for promoting PGI2 synthesis.
To further examine why females developed more severe PH than males in the absence of Tregs, we evaluated two other vasoprotective proteins which also direct Treg immunomodulatory function, HO-1 and PDL-1. HO-1 is an anti-inflammatory molecule that also helps mediate Treg function20 and may confer cardioprotection in females.45 Decreased HO-1 activity has been established to exacerbate pulmonary inflammation and RV hypertrophy.46, 47 We found that pulmonary HO-1 protein levels decreased in female PH lungs suggesting that HO-1 may also confer protection in females. Next, we evaluated PDL-1. PDL-1 and its ligand, PD-1, are both detected on Tregs and control the development, maintenance, and function of induced Tregs.32 In the current study, PDL-1 was significantly reduced in the lung and RV tissues of females athymic rats with PH. Why males upregulate pulmonary PDL-1 to a greater degree than females in the absence of Tregs is not clear. However, it is relevant that, while most research on PD-1/PDL-1 signaling focuses on controlling T cell immunity, this pathway is also involved in limiting inflammation caused by other cells, such as NK cells, and therefore reduced PDL-1 expression in females could enhance inflammation even in the absence of T cells.19 Collectively, the reduced expression of these related vasoprotective proteins (PDL-1, HO-1, COX-2 and PTGIS), may help explain why females that lack normal Treg activity may develop more severe PH.
Given the differences in responses of athymic male and females, we were interested in determining whether differences between the sexes could also be detected when they were protected from PH. Treg IR was equally effective in male and female rats in preventing SU5416-induced PH, limiting pulmonary inflammation, diminishing RV fibrosis, upregulating systemic PGI2 biosynthesis and enhancing cardiopulmonary expression of COX-2, PTGIS, HO-1, and PDL-1. The importance of these vasoprotective pathways in mediating Treg protection against PH was demonstrated when selective inhibition of COX-2, HO-1 and PD-1 abrogated the conferred benefit of IR. The mechanism by which Tregs induce PGI2 synthesis is unknown with both direct and indirect interactions between Tregs and PGI2-producing endothelial cells being potentially involved. This possibility is supported by the finding that vascular COX-2 is a major contributor to systemic PGI2 and a promoter of Treg function.48–51 We found that Treg-induced COX-2 expression in SU5416-treated athymic rats as well as in endothelial cells in vitro and augmented PGI2 production. These findings collectively demonstrate that Treg deficiency more profoundly reduces the vasoreparative upregulation of vascular COX-2 in females compared to males and thus confirm a dominant function that Tregs play in females when disease is prevented.
Lung-specific HO-1 upregulation prevents the development of hypoxia-induced PH and inhibits pulmonary vascular remodeling and pulmonary inflammation.52, 53 Cardiac HO-1 helps regulate myocardial cell energetics to limit cell death, pathological remodeling, and fibrosis and may confer this cardioprotection by decreasing oxidant projection and limiting endothelial cell damage. While HO-1 can help mediate Treg immunomodulatory activity20, we found that Tregs induce endothelial and myocardial HO-1 surface expression in vitro and in vivo with cardiopulmonary HO-1 expression being significantly elevated by Treg IR in both male and female athymic rats. The PD-1/PDL-1 pathway delivers inhibitory signals for controlling immune responses and helps maintain central and peripheral tolerance.54 Estrogen normally induces the expression of PD-1 55, 56 and may serve to enhance the anti-inflammatory action of normally-functioning Tregs. The vulnerability of athymic females to more severe PH may, in part, relate to the absence of Treg-induced HO-1 and PDL-1 expression in the vasculature and myocardium.
Our in vitro studies in Treg-HCMEC coculture studies indicate that CD4+CD25hiFoxP3+Tregs, but not CD4+CD25− T cells, induce PGI2 biosynthesis in an ER-, COX-2- and HO-1-dependent manner. This is particularly interesting because estrogen promotes the expansion and frequency of Treg cells and upregulates the expression of FoxP3, PD-1, and CTLA-4 via ER-alpha signaling.55, 57 Indeed, it has been suggested that the protective effects of estrogen in autoimmune conditions may be due to estrogen-mediated Treg expansion and activation.57 However, the effect of sex hormones on immune cells in health may differ from that occurring during vascular injury and disease.55, 58 Conversely, Treg-mediated upregulation of ERs on HCMECs could enhance the cardioprotective effect of E2 following vascular injury. While the direct mechanism of this contribution is still unclear, our data suggest that, in animals lacking Tregs, the potentially beneficial role played by E2 in limiting vascular inflammation may be significantly restricted. The relationship between Tregs, vascular endothelium, PGI2, COX-2, HO-1, PD-1/PDL-1 and ER signaling is schematically modeled in Figure 8.
Figure 8. Speculative model of Treg signaling in the vascular wall.

Tregs exert paracrine and direct effects on vascular endothelial and smooth muscle cells. Treg PD-1/PDL-1 interactions help quell inflammation in concert with estrogen interacting with ERs. Tregs act on the vascular intima and smooth muscle layer to promote COX-2 expression and PGI2 biosynthesis. Tregs similarly enhance vascular HO-1, IL-10, Areg and TGF-β concentrations which have pronounced anti-inflammatory and tissue repair properties. Treg produce and respond to PGE2, which increases intracellular cAMP that, in turn, upregulates FOXP3 expression. Immunosuppressive activity may be mediated by intercellular transfer of cAMP from Treg to endothelial cells via gap junctions presumably formed by Cx43, which is the connexin found in T cells. These cumulative actions limit vascular injury that would otherwise lead to pulmonary vascular remodeling.
The increased severity of female PH in the current athymic rat study, using inbred WAG animals, either exposed to SU5416 or chronic hypoxia, indicates that Treg deficiency as the basis of PH pathogenesis (relative to other models not relying on immune dysregulation). Beyond Treg anomalies, other forms of immune dysregulation are also likely important in PH pathogenesis, such as those attributable to abnormal regulatory natural killer cells. There are several caveats to the current studies. Clinical PH is more prevalent in women but more severe in men59, and thus our animal model can only draw inferences about the relative importance of Tregs as one of many components (e.g., BMPR2 mutation status, hormonal differences, environmental factors) in disease pathogenesis in both sexes. Our experiments are costly and time-consuming spanning more than 5 years for the current study, with 3–4 Treg donors being required per athymic recipient; not surprisingly, a number of key questions remain unaddressed. While female RVs demonstrated a higher microvascular density in health, and correspondingly, the apparent vascular dropout with PH development was more profound in females, there is a potential bias in the capillary density measurements due to the anisotropic orientation of the RV vasculature. Without using a proper stereologic methodology, which evaluates the entire volume of the ventricle, it’s not possible to say with certainty whether there was an actual dropout of vessels in PH. The mechanism by which Tregs induce endothelial PGI2 biosynthesis is currently unknown. We also don’t know why females differ from males in the expression of vasoprotective proteins in PH; a question that could be addressed by evaluating the specific impact on E2 in males and females through hormonal supplementation, antagonism and oophorectomies. In the studies involving Treg- HCMEC cocultures, the media were supplemented with FCS containing picomolar levels of E2, and it will be informative to understand how titrating concentrations of E2 would affect PGI2 levels. Finally, because of the many possible Treg interactions, the current study was unable to evaluate every cell-cell interaction nor evaluate Treg migration in vivo. In light of the current findings, it will be interesting to determine how Tregs directly influence PASMCs and pulmonary artery adventitial fibroblasts and track their movements to various in vivo compartments. Determining the differential effects of prostacyclin therapy in males versus females is also warranted in light of the current findings. Ongoing studies are designed to address these questions and will evaluate animals after longer periods of observation to ascertain the durability of the differences between the sexes observed at 3 weeks.
Abnormal Treg function is a clinical feature of PH and may be responsible for the immune dysregulation observed in the disease.60, 61 Sexual differences are present in various PH manifestations, and the study of how regulatory immunity differentially impacts men and women with this condition is a highly promising new avenue of scientific investigation. The experimental findings of the current study suggest that Tregs play a dominant role quelling vascular injury in females. In IR groups, a notable pulmonary infiltration of Tregs occurs, and by this action, PH is prevented through distinct anti-inflammatory and vasoprotective immunity.14 This finding has led to an interest in using autologous, conditioned Tregs as a basis for treating patients. Treg immunotherapy has been criticized for its requirement of potentially billions of cells.62 Further, in the rat model of PH, Treg immunotherapy (albeit with far fewer cells) loses efficacy if administered after SU5416 dosing.14 Clinical efforts are underway to overcome these problems by expanding Tregs ex vivo to optimize Treg dose, improve antigen specificity, and utilize adjunct therapies to potentiate Treg therapeutic effects.63 As Treg therapy for a variety of immunological disorders begin to enter the clinic,63, 64 vulnerable PH patients represent a particularly promising target population for this and other immunomodulatory therapies.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Pulmonary hypertension (PH) is a potentially fatal condition associated with abnormal regulatory T cells (Tregs) and dysregulated immunity.
Abnormal Treg activity may predispose patients to PH.
Replacing Tregs in athymic rats, which are Treg-deficient, prevents PH.
What New Information Does This Article Contribute?
Female athymic rats that lack Tregs develop more severe PH than males.
PH in Treg-deficient females is associated with greater inflammation and right ventricular (RV) fibrosis, lower plasma prostacyclin (PGI2), decreased lung cyclooxygenase-2 (COX-2), PGI2 synthase (PTGIS), hemoxygenase-1 (HO-1) and programmed death ligand-1 (PDL-1) expression and reduced RV PDL-1 levels.
Tregs prevent experimental PH through several signaling pathways including COX-2, HO-1, and PDL-1.
Tregs upregulate endothelial estrogen receptors (ERs) and PGI2 production.
Dysregulated immunity contributes to the development of PH, but the effect of sex on vascular inflammation is unknown. Given the implications for clinical disease, we sought greater insight into how protective immunity works to maintain pulmonary vascular health. Treg immune reconstitution (IR) restores protective immunity in athymic rats and prevents PH, thus providing a platform to better understand these processes. In this study, male and female athymic rats were treated with the vascular endothelial growth factor receptor-2 (VEGFR2) inhibitor, SU5416, or chronic hypoxia to induce PH. Females developed more severe PH than males. Blood levels of the pulmonary vasodilator, PGI2, were lower in females than males, possibly due to lower levels of the enzymes responsible for PGI2 biosynthesis (COX-2, PTGIS). Females exhibited diminished pulmonary HO-1 and PDL-1 expression, but Treg IR significantly upregulated these proteins, as well as COX-2, PTGIS, and systemic PGI2. Inhibiting COX-2, HO-1, and programmed death-1 (PD1)/PDL1 pathways eliminated Treg protection from disease. Tregs directly upregulated endothelial COX-2, PDL1, HO-1, ERs and increased PGI2 and IL-10 production. These experiments suggest that females are notably reliant on Tregs counteracting vascular injuries which cause PH. As cellular therapies are considered for PH, autologous Treg administration is a promising approach.
Acknowledgments
The authors thank Dr. Davis McKinley for help with managing the inbred athymic rat breeding colony and Dr. Lusijah Sutherland for assistance with cell sorting. The authors would also like to acknowledge Biomedical Research Models, Inc, (Worcester, MA) which generously provided the inbred athymic rats used to populate a breeding colony in Palo Alto.
SOURCES OF FUNDING
This study was supported by NHLBI HL122887 (MRN), HL014985 (MRN), HL125739 (MRN), HL120001 (MRN), HL138473 (MRN), 1T32HL098049 (JQ)
Nonstandard Abbreviations and Acronyms
- COX-2
Cyclooxygenase-2
- E2
Estradiol, estrogen
- ER
Estrogen Receptor
- FoxP3
Forkhead box P3
- hMCEC
Human microvascular cardiac endothelial cell
- HO-1
Heme oxygenase-1
- IR
Immune reconstitution
- LV
Left ventricle
- PAAT
Pulmonary artery acceleration time
- PCNA
Proliferating cell nuclear antigen
- PDL-1
Programmed death ligand-1
- PGI2
Prostacyclin
- PH
Pulmonary arterial hypertension
- PTGIS
Prostacyclin synthase
- RV
Right ventricular (or right ventricle)
- RVH
Right ventricular hypertrophy
- S
Septum
- Treg
Regulatory T cell
- VEGFR2
Vascular endothelial growth factor receptor-2
Footnotes
DISCLOSURES
None.
References
- 1.Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard HJ, Nicolls MR. Pathobiology of pulmonary arterial hypertension and right ventricular failure. European Respiratory Journal. 2012;40:1555–1565. doi: 10.1183/09031936.00046612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, Chen W, Zhang C, Zhang Y. Regulatory t cells in cardiovascular diseases. Nat Rev Cardiol. 2016;13:167–179. doi: 10.1038/nrcardio.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huertas A, Phan C, Bordenave J, Tu L, Thuillet R, Le Hiress M, Avouac J, Tamura Y, Allanore Y, Jovan R, Sitbon O, Guignabert C, Humbert M. Regulatory t cell dysfunction in idiopathic, heritable and connective tissue-associated pulmonary arterial hypertension. Chest. 2016;149:1482–1493. doi: 10.1016/j.chest.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Sada Y, Dohi Y, Uga S, Higashi A, Kinoshita H, Kihara Y. Non-suppressive regulatory t cell subset expansion in pulmonary arterial hypertension. Heart Vessels. 2016;31:1319–1326. doi: 10.1007/s00380-015-0727-4. [DOI] [PubMed] [Google Scholar]
- 5.Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: A perspective. Eur Respir J. 2005;26:1110–1118. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- 6.Ulrich S, Nicolls MR, Taraseviciene L, Speich R, Voelkel N. Increased regulatory and decreased cd8+ cytotoxic t cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration. 2008;75:272–280. doi: 10.1159/000111548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaowa S, Zhou W, Yu L, Zhou X, Liao K, Yang K, Lu Z, Jiang H, Chen X. Effect of th17 and treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediators of inflammation. 2014;2014:247372. doi: 10.1155/2014/247372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jasiewicz M, Moniuszko M, Pawlak D, Knapp M, Rusak M, Kazimierczyk R, Musial WJ, Dabrowska M, Kaminski KA. Activity of the kynurenine pathway and its interplay with immunity in patients with pulmonary arterial hypertension. Heart. 2016;102:230–237. doi: 10.1136/heartjnl-2015-308581. [DOI] [PubMed] [Google Scholar]
- 9.Tamosiuniene R, Nicolls MR. Regulatory t cells and pulmonary hypertension. Trends Cardiovasc Med. 2011;21:166–171. doi: 10.1016/j.tcm.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyata M, Sakuma F, Ito M, Ohira H, Sato Y, Kasukawa R. Athymic nude rats develop severe pulmonary hypertension following monocrotaline administration. International archives of allergy and immunology. 2000;121:246–252. doi: 10.1159/000024324. [DOI] [PubMed] [Google Scholar]
- 11.Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, Gera L, Farkas L, Rabinovitch M, Zamanian RT, Inayathullah M, Fridlib M, Rajadas J, Peters-Golden M, Voelkel NF, Nicolls MR. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Science translational medicine. 2013;5:200ra117. doi: 10.1126/scitranslmed.3006674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian J, Tian W, Jiang X, Tamosiuniene R, Sung YK, Shuffle EM, Tu AB, Valenzuela A, Jiang S, Zamanian RT, Fiorentino DF, Voelkel NF, Peters-Golden M, Stenmark KR, Chung L, Rabinovitch M, Nicolls MR. Leukotriene b4 activates pulmonary artery adventitial fibroblasts in pulmonary hypertension. Hypertension. 2015;66:1227–1239. doi: 10.1161/HYPERTENSIONAHA.115.06370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absence of t cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med. 2007;175:1280–1289. doi: 10.1164/rccm.200608-1189OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, Long CS, Voelkel NF, Nicolls MR. Regulatory t cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res. 2011;109:867–879. doi: 10.1161/CIRCRESAHA.110.236927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ormiston ML, Deng Y, Stewart DJ, Courtman DW. Innate immunity in the therapeutic actions of endothelial progenitor cells in pulmonary hypertension. Am J Respir Cell Mol Biol. 2010;43:546–554. doi: 10.1165/rcmb.2009-0152OC. [DOI] [PubMed] [Google Scholar]
- 16.Straub RH. The complex role of estrogens in inflammation. Endocrine reviews. 2007;28:521–574. doi: 10.1210/er.2007-0001. [DOI] [PubMed] [Google Scholar]
- 17.Ricciotti E, Yu Y, Grosser T, Fitzgerald GA. Cox-2, the dominant source of prostacyclin. Proc Natl Acad Sci U S A. 2013;110:E183. doi: 10.1073/pnas.1219073110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cawello W, Schweer H, Muller R, Bonn R, Seyberth HW. Metabolism and pharmacokinetics of prostaglandin e1 administered by intravenous infusion in human subjects. Eur J Clin Pharmacol. 1994;46:275–277. doi: 10.1007/BF00192562. [DOI] [PubMed] [Google Scholar]
- 19.Francisco LM, Sage PT, Sharpe AH. The pd-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia ZW, Zhong WW, Xu LQ, Sun JL, Shen QX, Wang JG, Shao J, Li YZ, Yu SC. Heme oxygenase-1-mediated cd4+cd25high regulatory t cells suppress allergic airway inflammation. J Immunol. 2006;177:5936–5945. doi: 10.4049/jimmunol.177.9.5936. [DOI] [PubMed] [Google Scholar]
- 21.Yuan P, Wu WH, Gao L, Zheng ZQ, Liu D, Mei HY, Zhang ZL, Jing ZC. Oestradiol ameliorates monocrotaline pulmonary hypertension via no, prostacyclin and endothelin-1 pathways. Eur Respir J. 2013;41:1116–1125. doi: 10.1183/09031936.00044112. [DOI] [PubMed] [Google Scholar]
- 22.Ryan JJ, Huston J, Kutty S, Hatton ND, Bowman L, Tian L, Herr JE, Johri AM, Archer SL. Right ventricular adaptation and failure in pulmonary arterial hypertension. The Canadian journal of cardiology. 2015;31:391–406. doi: 10.1016/j.cjca.2015.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sobrino A, Oviedo PJ, Novella S, Laguna-Fernandez A, Bueno C, Garcia-Perez MA, Tarin JJ, Cano A, Hermenegildo C. Estradiol selectively stimulates endothelial prostacyclin production through estrogen receptor-{alpha} J Mol Endocrinol. 2010;44:237–246. doi: 10.1677/JME-09-0112. [DOI] [PubMed] [Google Scholar]
- 24.Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: An oestrogen receptor antagonist with a novel mechanism of action. Br J Cancer. 2004;90(Suppl 1):S2–6. doi: 10.1038/sj.bjc.6601629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaffar Z, Wan KS, Roberts K. A key role for prostaglandin i2 in limiting lung mucosal th2, but not th1, responses to inhaled allergen. J Immunol. 2002;169:5997–6004. doi: 10.4049/jimmunol.169.10.5997. [DOI] [PubMed] [Google Scholar]
- 26.Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, Scheed A, Ritter C, Dahal BK, Vater A, Klussmann S, Ghofrani HA, Weissmann N, Klepetko W, Banat GA, Seeger W, Grimminger F, Schermuly RT. Immune/inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012 doi: 10.1164/rccm.201202-0335OC. [DOI] [PubMed] [Google Scholar]
- 27.Nicolls MR, Mizuno S, Taraseviciene-Stewart L, Farkas L, Drake JI, Al Husseini A, Gomez-Arroyo JG, Voelkel NF, Bogaard HJ. New models of pulmonary hypertension based on vegf receptor blockade-induced endothelial cell apoptosis. Pulm Circ. 2012;2:434–442. doi: 10.4103/2045-8932.105031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McMurtry IF, Frith CH, Will DH. Cardiopulmonary responses of male and female swine to simulated high altitude. Journal of applied physiology. 1973;35:459–462. doi: 10.1152/jappl.1973.35.4.459. [DOI] [PubMed] [Google Scholar]
- 29.Rabinovitch M, Gamble WJ, Miettinen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Physiol. 1981;240:H62–72. doi: 10.1152/ajpheart.1981.240.1.H62. [DOI] [PubMed] [Google Scholar]
- 30.Burton RR, Besch EL, Smith AH. Effect of chronic hypoxia on the pulmonary arterial blood pressure of the chicken. The American journal of physiology. 1968;214:1438–1442. doi: 10.1152/ajplegacy.1968.214.6.1438. [DOI] [PubMed] [Google Scholar]
- 31.Smith P, Moosavi H, Winson M, Heath D. The influence of age and sex on the response of the right ventricle, pulmonary vasculature and carotid bodies to hypoxia in rats. The Journal of pathology. 1974;112:11–18. doi: 10.1002/path.1711120104. [DOI] [PubMed] [Google Scholar]
- 32.Farhat MY, Chen MF, Bhatti T, Iqbal A, Cathapermal S, Ramwell PW. Protection by oestradiol against the development of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br J Pharmacol. 1993;110:719–723. doi: 10.1111/j.1476-5381.1993.tb13871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parker TA, Ivy DD, Galan HL, Grover TR, Kinsella JP, Abman SH. Estradiol improves pulmonary hemodynamics and vascular remodeling in perinatal pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2000;278:L374–381. doi: 10.1152/ajplung.2000.278.2.L374. [DOI] [PubMed] [Google Scholar]
- 34.Lahm T, Tuder RM, Petrache I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014;307:L7–26. doi: 10.1152/ajplung.00337.2013. [DOI] [PubMed] [Google Scholar]
- 35.Guihaire J, Deuse T, Wang D, Fadel E, Reichenspurner H, Schrepfer S. Sex differences in immunology: More severe development of experimental pulmonary hypertension in male rats exposed to vascular endothelial growth factor receptor blockade. BioMed research international. 2015;2015:765292. doi: 10.1155/2015/765292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang B, Deng Y, Suen C, Taha M, Chaudhary KR, Courtman DW, Stewart DJ. Marked strain-specific differences in the su5416 rat model of severe pulmonary arterial hypertension. American journal of respiratory cell and molecular biology. 2016;54:461–468. doi: 10.1165/rcmb.2014-0488OC. [DOI] [PubMed] [Google Scholar]
- 37.Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, Loyd JE. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. The New England journal of medicine. 1992;327:70–75. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- 38.Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, Badesch D, Voelkel NF. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925–1932. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- 39.Sherman TS, Chambliss KL, Gibson LL, Pace MC, Mendelsohn ME, Pfister SL, Shaul PW. Estrogen acutely activates prostacyclin synthesis in ovine fetal pulmonary artery endothelium. Am J Respir Cell Mol Biol. 2002;26:610–616. doi: 10.1165/ajrcmb.26.5.4528. [DOI] [PubMed] [Google Scholar]
- 40.Egan KM, Lawson JA, Fries S, Koller B, Rader DJ, Smyth EM, Fitzgerald GA. Cox-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306:1954–1957. doi: 10.1126/science.1103333. [DOI] [PubMed] [Google Scholar]
- 41.Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, Justice MJ, Brown MB, Van Demark M, Trulock KM, Dieudonne D, Reddy JG, Presson RG, Petrache I. 17beta-estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med. 2012;185:965–980. doi: 10.1164/rccm.201107-1293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lahm T, Crisostomo PR, Markel TA, Wang M, Weil BR, Novotny NM, Meldrum DR. The effects of estrogen on pulmonary artery vasoreactivity and hypoxic pulmonary vasoconstriction: Potential new clinical implications for an old hormone. Crit Care Med. 2008;36:2174–2183. doi: 10.1097/CCM.0b013e31817d1a92. [DOI] [PubMed] [Google Scholar]
- 43.Hara A, Yuhki K, Fujino T, Yamada T, Takayama K, Kuriyama S, Takahata O, Karibe H, Okada Y, Xiao CY, Ma H, Narumiya S, Ushikubi F. Augmented cardiac hypertrophy in response to pressure overload in mice lacking the prostaglandin i2 receptor. Circulation. 2005;112:84–92. doi: 10.1161/CIRCULATIONAHA.104.527077. [DOI] [PubMed] [Google Scholar]
- 44.Kaneshige T, Saida Y, Tanaka R, Soda A, Fukushima A, Ida N, Takenaka M, Yamane Y. Effect of long-term administration of a prostacyclin analogue (beraprost sodium) on myocardial fibrosis in dahl rats. J Vet Med Sci. 2007;69:1271–1276. doi: 10.1292/jvms.69.1271. [DOI] [PubMed] [Google Scholar]
- 45.Posa A, Kupai K, Menesi R, Szalai Z, Szabo R, Pinter Z, Palfi G, Gyongyosi M, Berko A, Pavo I, Varga C. Sexual dimorphism of cardiovascular ischemia susceptibility is mediated by heme oxygenase. Oxidative medicine and cellular longevity. 2013;2013:521563. doi: 10.1155/2013/521563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goto J, Ishikawa K, Kawamura K, Watanabe Y, Matumoto H, Sugawara D, Maruyama Y. Heme oxygenase-1 reduces murine monocrotaline-induced pulmonary inflammatory responses and resultant right ventricular overload. Antioxidants & redox signaling. 2002;4:563–568. doi: 10.1089/15230860260220058. [DOI] [PubMed] [Google Scholar]
- 47.Issan Y, Kornowski R, Aravot D, Shainberg A, Laniado-Schwartzman M, Sodhi K, Abraham NG, Hochhauser E. Heme oxygenase-1 induction improves cardiac function following myocardial ischemia by reducing oxidative stress. PLoS One. 2014;9:e92246. doi: 10.1371/journal.pone.0092246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z, Landesberg G, Crichton I, Wu W, Pure E, Funk CD, FitzGerald GA. Vascular cox-2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;4:132ra154. doi: 10.1126/scitranslmed.3003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharma S, Zhu L, Yang SC, Zhang L, Lin J, Hillinger S, Gardner B, Reckamp K, Strieter RM, Huang M, Batra RK, Dubinett SM. Cyclooxygenase 2 inhibition promotes ifn-gamma-dependent enhancement of antitumor responses. J Immunol. 2005;175:813–819. doi: 10.4049/jimmunol.175.2.813. [DOI] [PubMed] [Google Scholar]
- 50.Ha TY. The role of regulatory t cells in cancer. Immune network. 2009;9:209–235. doi: 10.4110/in.2009.9.6.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pidgeon GP, Tamosiuniene R, Chen G, Leonard I, Belton O, Bradford A, Fitzgerald DJ. Intravascular thrombosis after hypoxia-induced pulmonary hypertension: Regulation by cyclooxygenase-2. Circulation. 2004;110:2701–2707. doi: 10.1161/01.CIR.0000145613.01188.0B. [DOI] [PubMed] [Google Scholar]
- 52.Minamino T, Christou H, Hsieh CM, Liu Y, Dhawan V, Abraham NG, Perrella MA, Mitsialis SA, Kourembanas S. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci U S A. 2001;98:8798–8803. doi: 10.1073/pnas.161272598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christou H, Morita T, Hsieh CM, Koike H, Arkonac B, Perrella MA, Kourembanas S. Prevention of hypoxia-induced pulmonary hypertension by enhancement of endogenous heme oxygenase-1 in the rat. Circ Res. 2000;86:1224–1229. doi: 10.1161/01.res.86.12.1224. [DOI] [PubMed] [Google Scholar]
- 54.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH, Sharpe AH. Tissue expression of pd-l1 mediates peripheral t cell tolerance. J Exp Med. 2006;203:883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polanczyk MJ, Carson BD, Subramanian S, Afentoulis M, Vandenbark AA, Ziegler SF, Offner H. Cutting edge: Estrogen drives expansion of the cd4+cd25+ regulatory t cell compartment. J Immunol. 2004;173:2227–2230. doi: 10.4049/jimmunol.173.4.2227. [DOI] [PubMed] [Google Scholar]
- 56.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. Pd-1 and its ligands in tolerance and immunity. Annual review of immunology. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Offner H, Polanczyk M. A potential role for estrogen in experimental autoimmune encephalomyelitis and multiple sclerosis. Annals of the New York Academy of Sciences. 2006;1089:343–372. doi: 10.1196/annals.1386.021. [DOI] [PubMed] [Google Scholar]
- 58.Khan D, Ansar Ahmed S. The immune system is a natural target for estrogen action: Opposing effects of estrogen in two prototypical autoimmune diseases. Frontiers in immunology. 2015;6:635. doi: 10.3389/fimmu.2015.00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ventetuolo CE, Praestgaard A, Palevsky HI, Klinger JR, Halpern SD, Kawut SM. Sex and haemodynamics in pulmonary arterial hypertension. Eur Respir J. 2014;43:523–530. doi: 10.1183/09031936.00027613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nicolls MR, Voelkel NF. The roles of immunity in the prevention and evolution of pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2017;195:1292–1299. doi: 10.1164/rccm.201608-1630PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Riley JL, June CH, Blazar BR. Human t regulatory cell therapy: Take a billion or so and call me in the morning. Immunity. 2009;30:656–665. doi: 10.1016/j.immuni.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang Q, Bluestone JA. Regulatory t-cell therapy in transplantation: Moving to the clinic. Cold Spring Harbor perspectives in medicine. 2013:3. doi: 10.1101/cshperspect.a015552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singer BD, King LS, D’Alessio FR. Regulatory t cells as immunotherapy. Front Immunol. 2014;5:46. doi: 10.3389/fimmu.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.