

Abstract
Type-2 ryanodine receptors (RyR2s) play a pivotal role in cardiac excitation-contraction coupling by releasing Ca2+ from sarcoplasmic reticulum (SR) via a Ca2+ -induced Ca2+ release (CICR) mechanism. Two strategies have been used to study the structure-function characteristics of RyR2 and its disease associated mutations: (1) heterologous cell expression of the recombinant mutant RyR2s, and (2) knock-in mouse models harboring RyR2 point mutations. Here, we establish an alternative approach where Ca2+ signaling aberrancy caused by the RyR2 mutation is studied in human cardiomyocytes with robust CICR mechanism. Specifically, we introduce point mutations in wild-type RYR2 of human induced pluripotent stem cells (hiPSCs) by CRISPR/Cas9 gene editing, and then differentiate them into cardiomyocytes. To verify the reliability of this approach, we introduced the same disease-associated RyR2 mutation, F2483I, which was studied by us in hiPSC-derived cardiomyocytes (hiPSC-CMs) from patient biopsy. The gene-edited F2483I hiPSC-CMs exhibited longer and wandering Ca2+ sparks, elevated diastolic Ca2+ leaks, and smaller SR Ca2+ stores, like those of patient-derived cells. Our CRISPR/Cas9 gene editing approach validated the feasibility of creating myocytes expressing the various RyR2 mutants, making comparative mechanistic analysis and pharmacotherapeutic approaches for RyR2 pathologies possible.
Keywords: ryanodine receptor mutation, CRISPR/Cas9, CPVT, human induced pluripotent stem cells
Graphical abstract
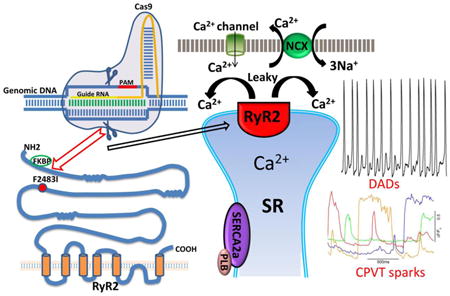
1. Introduction
Cardiac contraction is initiated by small influx of Ca2+ through voltage-gated L-type Ca2+ channels (Cav1.2) during the action potential, triggering much larger release of Ca2+ from sarcoplasmic reticulum (SR) via the type-2 ryanodine receptor (RyR2) [1-4]. The released Ca2+ is both sequestered into the SR by ATP-dependent Ca2+-pump (SERCA2a/PLB /phospholamban complex) and extruded across the membrane by the Na+/Ca2+ exchanger (NCX), causing muscle relaxation. It is well known that dysfunction in this Ca2+ signaling sequence can induce arrhythmia, cardiac hypertrophy and failure.
RyR2 channel is a homotetramer of ∼5000 amino acids polypeptide that is regulated by Ca2+, Mg2+, calmodulin, FKBP12.6 protein, protein kinases and phosphatases [3], suggesting that RyR2 possesses multiple regulatory domains. A number of RyR2 missense mutations have been found to associate with various cardiac pathologies, including catecholaminergic polymorphic ventricular tachycardia (CPVT1, an inherited arrhythmogenic disorder triggered by catecholamine release during physical or emotional stress [5-8]) and long-QT-syndrome [9, 10]. More than 150 missense mutations in human RYR2 gene have been identified to be associated with CPVT1 pathology. Various mechanisms for CPVT1 have been proposed based on heterologous cell expression of recombinant mutant RyR2s and genetically modified mice (knock-in mice), that include hyper-phosphorylation mediated leakiness of RyR2 [4, 11, 12], SR store overload-induced Ca2+ release, SOICR, [13, 14], and RyR2 loss of function resulting in large SR Ca2+ store and release [15]. In all of these mechanisms, the aberrant SR Ca2+ release is thought to activate the electrogenic sarcolemmal NCX to extrude the cytosolic Ca2+ causing membrane depolarization and triggering early or delayed after depolarizations.
While recombinant proteins studies and the mouse models provide valuable insights into RyR2 structure-function relationship and its mutation-associated cardiac pathologies, the recombinant expression system in non-contracting HEK293 cells may be limited in addressing the consequences of RyR2 dysfunction in cardiomyocytes. Similarly, the mouse cardiac pathology is unlikely to mimic human cardiomyopathies not only because of the differences in size of the heart, but also the differences in expression of its ion channels. Recent advances in stem cell technology have made it possible to study RyR2 Ca2+ signaling dysfunctions associated with CPVT1 in human cardiomyocytes derived directly from skin biopsy (hiPSC-CMs) of patients afflicted with this arrhythmia. We have already reported on calcium signaling pathology of F2483I-RyR2 mutation using patient fibroblast-derived cardiomyocytes that showed longer and wandering Ca2+ sparks, smaller SR Ca2+ store, and arrhythmogenesis triggered by isoproterenol [8, 16, 17], as have others on different CPVT1 associated mutations [18-22]. A major disadvantage of patient-derived fibroblast approach, however, is that CPVT1 patient tissues are not readily available as many mutations have had lethal consequences making their characterizations in hiPSC-CMs difficult if not impossible. In this report, we established an alternative research strategy by introducing the F2483I-RyR2 mutation in healthy wild type hiPSCs using CRISPR/Cas9 gene editing and characterizing the Ca2+-signaling pathology of cardiomyocytes differentiated from such cell lines as compared to patient-derived hiPSC-CMs harboring RyR2-F2483I mutation [8, 16, 17]. We report, for the first time, not only on the feasibility of gene-editing approach in creating the selected mutation in RyR2, but also show that the Ca2+-signaling phenotype of gene-edited cardiomyocytes is essentially identical to those cardiomyocytes derived from the patient biopsy carrying F2483I mutation, suggesting that this mutation is causative for CPVT1 pathology.
2. Materials and methods
2.1 Cell Culture
Human induced pluripotent stem cells (hiPSC-K3) [23] were provided from Stephen Duncan at Medical University of South Carolina and were maintained in E8 medium (Gibco) on Matrigel (BD Biosciences) coated plates at 37 °C with 5% (vol/vol) CO2.
2.2 Genome editing in hiPSCs
To introduce CPVT1-associated RyR2 mutation in hiPSC genome, we used CRISPR/Cas9 gene-editing technique according to the established protocol [24]. The guide sequence (5′-CAATCCCATAGACCCTGTCA-3′, Fig. 1A) was designed to digest RYR2 gene near the F2483I mutation site using CRISPR Design Tool (http://crispr.mit.edu) and was cloned into the pSpCas9 (BB)-2A-Puro vector (pX459, Addgene), The pX459 plasmid expresses Cas9 exonuclease, which digest the genome near the guide sequence. Site-directed mutations were introduced by homology-directed repair of the Cas9-digested site using single stranded oligo donor nucleotide (ssODN) (5′-GCAGAAAGCCAACCTCAAGAAGATGCAGGAGGATGTCTTGAACCTCAATCCCATAG ACCCTGTCAAGAAATAAAACCATGGCTGCCTTGTGATCTGGGCAAAACCCCGCAGA CATGTCAGGTTCCACCACATTCCCATCTAAAAGAGAGAAG-3′). In the present study, we designed three mutations (1) a CPVT1 mutation (F2483I); (2) a mutation that creates a new restriction enzyme site to facilitate a process screening positive hiPSC clones; (3) a mutation that eliminates the Cas9 recognized Pam sequence (Fig. 1A) to prevent recurring Cas9 digest. It should be noted that the second and the third mutations were designed not to cause amino acid change.
Figure 1. Introduction of F2483I mutation in RYR2 gene of hiPSC.
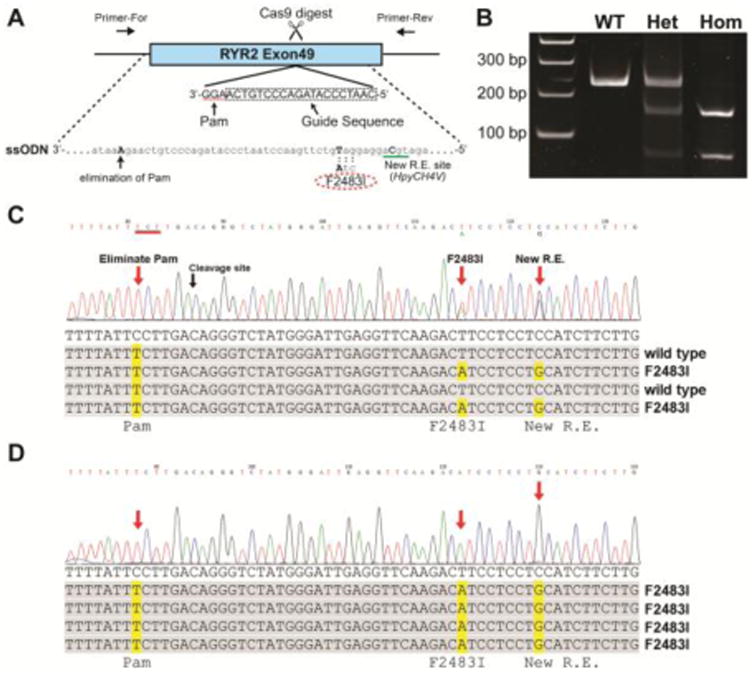
(A) Schematic of gene-editing. Exon49 of RYR2 gene in wild type hiPSC was targeted by CRISPR/Cas9 technique. Guide sequence shown in dotted box was cloned into pX459 vector to express sgRNA guiding Cas9 exonuclease to the targeted Pam sequence (highlighted by dotted line). The ssODN was used to introduce mutations during homology-directed repair of the Cas9-digested genome. The ssODN carried F2483I mutation (TTC to ATC), a mutation creating HpyCH4V restriction enzyme site, and a mutation eliminating Pam sequence (AGG to AGA). Note that Guide sequence and Pam sequence were on the anti-strand of the genome, thus ssODNA was also anti-strand sequence. (B) PCR analysis of the targeted hiPSCs. The gene-edited regions were amplified from genomic DNA by PCR, followed by restriction digest by HpyCH4V. Full size of PCR product was 228 bp (wild type). PCR products from the heterozygous (Het) mutant were partially digested by the enzyme, while ones from homozygous (Hom) mutant were completely digested in to two fragments, 150 and 78 bp. (C) Sequencing of PCR products from the heterozygous mutant. Direct sequencing of the whole PCR products showed double peaks at F2483I and new restriction enzyme (R.E.) sites, indicating that only one allele carries these mutations. Single peak on the Pam sequence mutation site, suggesting that both gene allele has this mutation. Sequencing of cloned PCR products confirmed these mutations (lower panel). (D) Sequencing of PCR products from the homozygous mutant. All three mutation sites showed single peaks in whole PCR sequencing, indicating both allele carries these three mutations. Sequencing of cloned PCR products confirmed this.
The hiPSCs were disassociated into single cells and small clusters with Versene (Gibco) before they reach 70% confluency. About 1×106 cells were resuspended in 100 μl buffer R (Neon™ Transfection Systerm, Thermo), and then co-transfected with the ssODN (0.4 nmol) and the expression pX459 plasmids (20 μg) by electroporation with two 30 msec pulses at 1050 V. The transfected cells were then plated in the 60mm coated dish with 10 μM Y-27632 ROCK inhibitor (Tocris) in the media. Twenty four hours after transfection, the cells were incubated with 1 μg/mL puromycin (Thermo) for 48 h. Surviving cell colonies were harvested 7–10 d after antibiotics treatment. Genomic DNAs were prepared from the harvested cells, and the F2483I RyR2 mutations were screened by PCR (forward primer: 5′-CACAGCCATTGACACCAAA-3′, reverse primer: 5′-GTCCATCGCTCCAATCTCACCGTAT-3′) followed by restriction enzyme (HpyCH4V) digest and direct sequencing of PCR products.
2.3 Differentiation of hiPSCs into cardiomyocytes
Wild type and gene-edited hiPSCs were differentiated into cardiomyocytes as described previously [17, 25]. Briefly, dissociated hiPSCs were plated in 12 well plates and then treated with 12 μM CHIR 99021 (Selleckchem), a GSK3β inhibitor for 24 h in RPMI/B-27 without insulin. Seventy two hours after CHIR99021 treatment, 5 mM IWP2, a Wnt signaling inhibitor, was added to culture with the same media for 48 h. After 48 h continued culture in RPMI/B-27 without insulin, the cells were cultured with RPMI/B-27 medium. Spontaneously contracting clusters were then micro-dissected mechanically into single cardiomyocytes for electrophysiological and Ca2+ imaging experiments.
2.4 Quantitative RT-PCR
Total RNAs of the hiPSC-derived cardiomyocytes were isolated with Trizol reagent (Invitrogen), and cDNAs were synthesized by M-MLV reverse transcriptase and oligo (dT) primer (Thermo Scientific). Quantitative PCR was performed using SYBR Green I (Thermo Scientific) in the CFX Connext Real-Time PCR Detection System (Bio-Rad). The thermal profile for PCR was 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. To obtain a relative quantitation, the results were normalized to those obtained for the corresponding 18S expression levels determined with forward primer 5′-TAGAGGGACAAGTGGCGTTC-3′ and reverse primer 5′-CGCTGAGCCAGTCAGTGT-3′. Each sample was run in at least duplicate.
2.5 Ca2+ imaging
Wild type and F2483I mutant cells were imaged using a multicolor total internal reflection fluorescence (TIRF) imaging system (Leica Microsystems, Buffalo Grove, IL) fitted with a 63× oil-immersion objective lens and an Andor iXon3 camera with 512 × 512 pixels. An argon ion laser was used for excitation at 488 nm and fluorescence emission was measured at wavelengths >515 nm to image Ca2+ signaling by fluorescent Ca2+-indicator dye Fluo-4. Single isolated beating hiPSC-CM was plated on vitronectin (Life Technologies) coated 25 mm glass coverslips and allowed to attach for three days before imaging. Cells were incubated with 1 μM membrane permeable Fluo-4AM (Invitrogen) in normal Tyrode's solution (137 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (titrated to pH 7.4 with NaOH) for 30 min at 37°C. Ca2+ transients and Ca2+ sparks were recorded at 80 Hz with a depth of penetration of 110-150 nm focus on the sub-sarcolemmal Ca2+ release. Sparks data was exported and analyzed with Leica LAS X and a computerized algorithm described previously [26, 27].
SR Ca2+ content and diastolic Ca2+ leakage was measured by a modified approach, reported previously [28]. Spontaneously beating hiPSC-CMs were imaged after 30 min incubation in Fluo-4 AM at 37°C. Global Ca2+ fluorescence changes were recorded at EPI-fluorescence mode of the TIRF microscopy at 30Hz. To measure SR Ca2+ leak, spontaneously beating cells incubated in 2 mM Ca2+ Tyrode control solutions were subjected to rapid application of zero NaCl (replaced with LiCl to disable Na+-Ca2+ exchanger), zero Ca2+, and 1 mM tetracaine plus 1 mM EGTA solution for 3 sec. After removal of tetracaine for 10 sec, SR Ca2+ content was assessed by exposure to 3 mM caffeine. The difference between the baseline fluorescence and the decrease in background fluorescence in the presence of tetracaine was considered as the SR leak.
2.6 Data analysis
Results are indicated as the means ± SEM. Significant differences (p<0.05) were determined using one-way ANOVA followed by Tukey's test or Student's t-test.
Significant differences in the populations of spontaneously pacing cardiomyocytes between two groups (wild type vs heterozygous, wild type vs homozygous, or heterozygous vs homozygous cells) were assessed by chi-square test.
3. Results
Our previous studies had shown that cardiomyocytes derived from dermal fibroblasts of CPVT1 patient harboring F2483I-RyR2 mutation exhibited aberrant intracellular Ca2+ handling, including longer and wandering Ca2+ sparks, smaller SR Ca2+ content, and proclivity to arrhythmogenesis on exposure to isoproterenol [17]. To validate the gene editing approach and gain further insight into dysfunction produced by F2483I-RyR2 mutation, we introduced this mutation in healthy wild type hiPSCs using CRISPR/Cas9 gene editing and characterized their calcium signaling phenotype and compared it with hiPSC-CMs derived directly from patient's dermal fibroblasts.
3.1 Generation of a hiPSC harboring a CPVT1-associated RyR2 mutation
To introduce CPVT1-associated F2483I mutation in RYR2 gene of wild type hiPSCs, the CRISPR/Cas9 gene-editing technique was used. The plasmid DNA to express sgRNA, targeted to the specific site of RYR2 gene, and Cas9 exonuclease was transfected in hiPSCs together with the single stranded oligo nucleotide carrying mutations by electroporation (Fig. 1A). Cells were screened by puromycin resistance, and the correct gene edit and mutations were confirmed by sequencing of PCR products amplified from the mutated RYR2 locus (Fig. 1B-D). We introduced two mutations in addition to F2483I-RyR2: 1) a silent mutation preventing recurring genome digestion by Cas9 (Fig. 1A, also see Methods) and 2) a silent mutation creating a new restriction enzyme site (HpyCH4V), allowing PCR products from the correctly gene-edited clones to be digested by HpyCH4V (Fig. 1B). Two hiPSC clones, one carrying the F2483I mutation in one gene allele (heterozygote mutant), and the other expressing the mutation in both gene alleles of RyR2 (homozygous mutant), were obtained (Fig. 1C and D).
3.2 Differentiation of the F2483I-RyR2 hiPSCs into cardiomyocytes
Both of the targeted hiPSC clones and wild type hiPSCs were differentiated into cardiomyocytes. Twelve days following differentiation 40-60% of cells showed robust spontaneous contractions. We confirmed F2483I RyR2 mutation by sequencing of RT-PCR products amplified from total RNA of the beating hiPSC-derived cardiomyocytes. In both heterozygous and homozygous hiPSC-CMs a short form of the RT-PCR product was also found that lacked 79 nucleotide bases, corresponding to a part of exon-49 of RYR2. This was consistent with the possibility that CRISPR/Cas9 induced mutation(s) may produce a splicing variant. Quantitative PCR (qPCR), to determine the expression level of the intact and the short form of RyR2, showed that the total expression levels of RyR2 in wild-type and mutant cardiomyocytes varied considerably among the three groups but were not statistically significant, Fig 2. Nevertheless, the averaged values in the mutant cells appeared to be smaller than in the wild type. Consistently qPCR products amplified by the primer annealing to the missing 79 nucleotides showed considerable, but not statistically significant, decrease in heterozygous or homozygous hiPSC-CMs. The results suggest that potential splicing variant caused by the CRISPR mutation(s) may reduce the full-length RyR2 mRNA, thereby RyR2 proteins. It should be noted, however, that the short form RyR2 mRNA is unlikely to be translated into the functional RyR2 protein because the missing 79 nucleotide bases causes a translational frameshift.
Figure 2. Quantitative PCR analysis of expression level of RyR2.
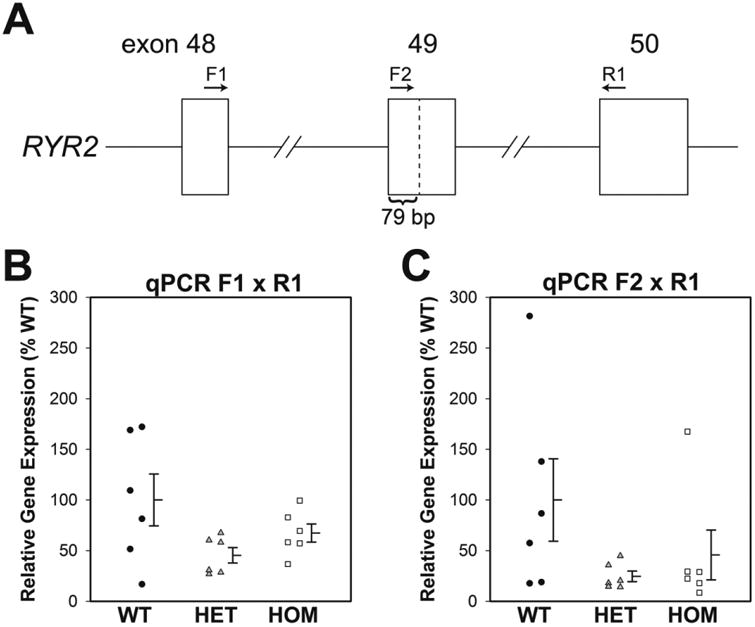
(A) Diagram of RYR2 gene structure. F2483I mutation was introduced in exon 49. Positions of the 79 nucleotides deleted part, and the primers for qPCR are shown. (B) Entire RyR2 expression levels including both intact and deletion forms were determined by qPCR with F1 (5′-TGATTGACCTCTTGGGACG-3′) and R1 (5′-AGAAGCGTGGTGCTCTGTG-3′) primer set. (C) Intact RyR2 expression levels (no 79 nucleotide deletion) were determined with F2 (5′-GAATGTGGTGGAACCTGAC-3′) and R1 primer set. Data are shown as dot plots with mean ± SEM (n=6).
3.3 Calcium sparks in hiPSC-CMs harboring F2483I-RyR2 mutation
To obtain insights into calcium signaling pathology of the mutant hiPSC-CMs expressing F2483I-RyR2, we first measured the characteristics and frequency of occurrence of spontaneously occurring elementary Ca2+ release events in wild type and F2483I-RyR2 mutant hiPSC-CMs, using TIRF-imaging in myocytes dialyzed with Fluo-4 (Fig. 3). The data shows that frequency of occurrence of Ca2+ sparks were not significantly different in the three cell lines (wild type control, heterozygote F2483I mutants, and homozygous mutants). The characteristics of individual Ca2+ sparks, on the other hand, varied significantly between control and mutant cardiomyocytes. Calcium sparks in the mutant myocytes were significantly longer as compared to those of control cells (p<0.05 by ANOVA followed by Tukey's test, see the legend of Fig. 3C), often showing wandering characteristics as to trigger other sites (Fig. 3A and B, see also Fig. 4), consistent with increased diastolic Ca2+ levels.
Figure 3. Calcium sparks in F2483I-RyR2 hiPSC-CMs.
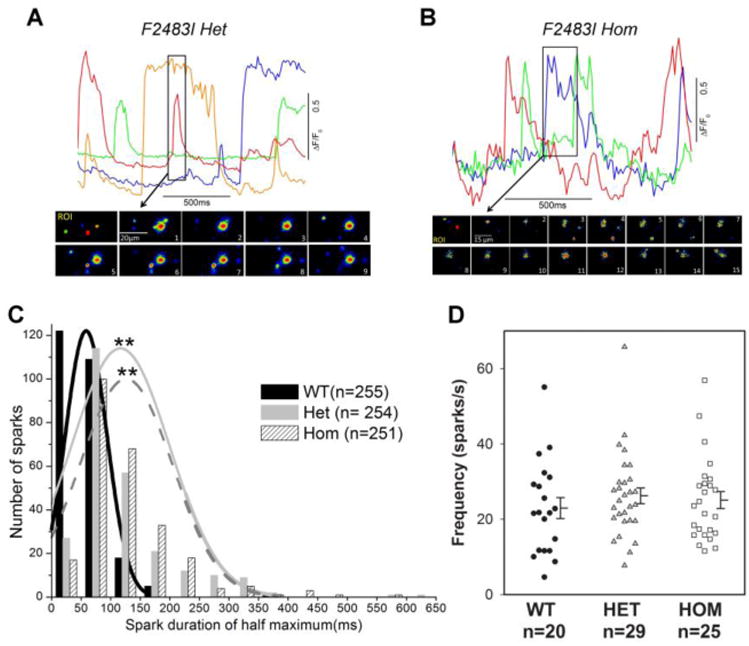
(A and B top panels) The time courses of normalized Ca2+-dependent fluorescence intensity at the selected color-coded regions in) F2483I heterozygous and homozygous cells. (A and B bottom panels) Image sequences at the selected time in the top panels (boxed parts) show evolutions of long-lasting Ca2+ sparks. (C) Histogram of spark durations in wild type and F2483I mutant cells and the fitted curves were generated by ORIGIN8 (Northampton, MA). Averaged spark durations are 58 ± 2 msec (n=255), 117 ± 5 msec ** (n=254), and 126 ± 5 msec ** (n=251) for wild type, heterozygous, and homozygous cells, respectively. ** p<0.001 compared with wild type by one-way ANOVA followed by Tukey's test. (D) Spark frequency measured in the three genotype cells showed no significant differences. Data are shown as dot plots with mean ± SEM.
Figure 4. Aberrant local Ca2+ releases occurring interspersed in between spontaneously active Ca2+-transients in heterozygote and homozygote hiPSC-CMs.
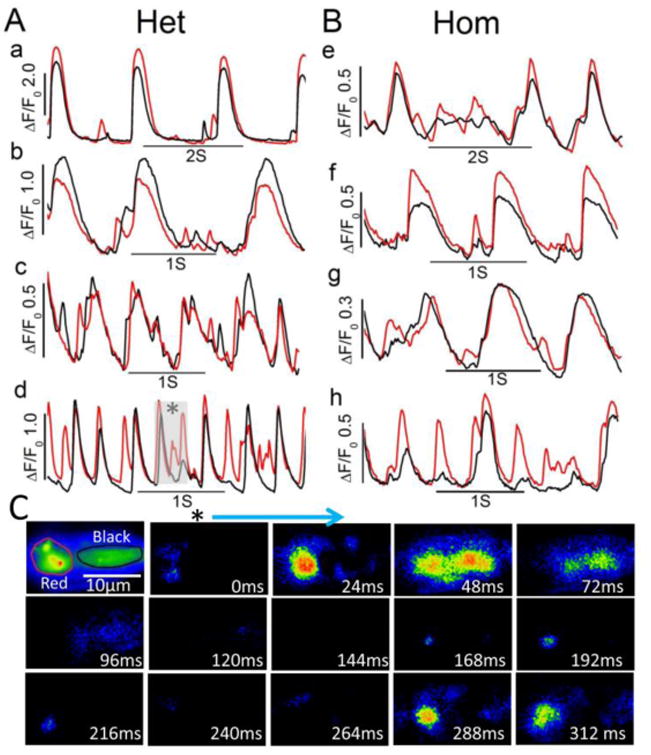
(A and B) time courses of normalized Ca2+-dependent fluorescence intensity at the two regions (red and black in panel C) in four F2483I heterozygous (a-d) and four homozygous (e-h) hiPSC-CMs showing additional Ca2+ release events between the rhythmic Ca2+ releases. (C) Representative image sequences at the selected time in trace d (gray highlighted) showing the evolution of spontaneous beat and aberrant Ca2+ releases. The first panel shows average fluorescence distributions and examples of red and black regions.
The spontaneous beating characteristics of the three cell lines also showed significant variability such that a higher population of F2483I mutant cells contracted spontaneously and had aberrant Ca2+ releases as compared to the wild type cells (wild type: 6 out of 48 cells, 12.5%; heterozygous: 39 out of 100 cells, 39%; homozygous: 17 out of 50 cells 34%). The populations of beating myocytes in the heterozygous and homozygous lines were significantly higher than wild type cells (p<0.05 by chi-square test). Most of the spontaneously beating cells also triggered additional local Ca2+ releases in between the regularly occurring beats (extra-systoles, Fig. 4A and B). Thus, F2483I-RyR2 mutation appears to trigger long-lasting Ca2+ sparks that may underlie their robust automaticity and irregular rhythm.
3.4 SR Ca2+ leak and load in hiPSC-CMs harboring F2483I-RyR2 mutation
In another set of cells, we measured the SR Ca2+ load and leak levels in Fluo4-AM-incubated hiPSC-CMs. Tetracaine, an inhibitor of RyR2 [28] and caffeine, a potent activator of CICR mechanism in RyR2 (in zero calcium and sodium solutions), were used to determine the SR leak and load, respectively. In both heterozygous and homozygous F2483I-RyR2 mutant hiPSC-CMs the SR Ca2+ leak levels were larger (Fig. 5A and B) and the SR Ca2+ load levels smaller (Fig. 5C), consistent with longer sparks and higher diastolic Ca2+ levels.
Figure 5. SR Ca2+ leak and load in F2483I-RyR2 hiPSC-CMs.
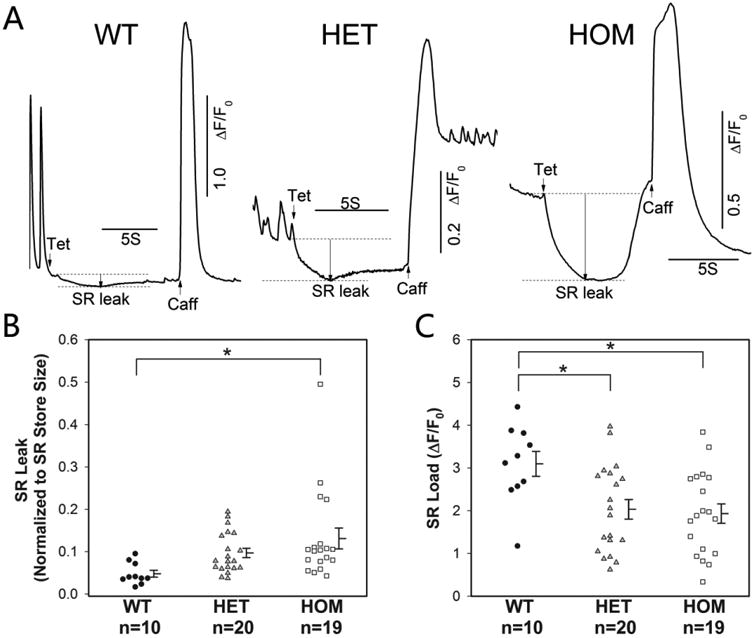
(A) Time courses of normalized Ca2+-dependent fluorescence intensity changes after rapid exposure to 1 mM tetracaine (Tet) followed by application of 3 mM caffeine (Caff) in zero Na+ and Ca2+ solution. Quantified SR leak levels (normalized to the SR store size) (B), and SR load levels (C) in wild type and mutant cells. Data are shown as dot plots with mean ± SEM. * p<0.05 compared with wild type by one-way ANOVA followed by Tukey's test. The difference in SR leak between wild type and heterozygous cells is significant by Student's t-test (p<0.01).
4. Discussion
4.1 A new approach to probe the effect of RyR2 mutations in the cardiomyocytes
The major accomplishment of this study was the establishment of a new research strategy to introduce a RyR2 mutation in human cardiomyocytes by CRISPR/Cas9 gene editing. Our functional studies in heterozygote and homozygote myocytes showed RyR2 mutation dependent aberrant intracellular Ca2+ signaling in the gene-edited cardiomyocytes. The results clearly show that the Ca2+ signaling phenotype of the gene-edited cells mimics our previous findings in patient-derived F2483I-RyR2 hiPSC-CMs [16, 17], suggesting that this mutation is causative rather than associative to the disease. Considering ∼150 CPVT1-associated mutations spread over the length of large RyR2 protein with its multiple regulatory domains, it is likely that pathophysiology of CPVT1 may depend on the RyR2 mutation sites (domains), making gene-editing approach not only unique in determining whether a specific mutation is causative or associative with the disease, but also allow comparative electrophysiological, pharmacological, and mechanistic studies of the various CPVT1 associated mutations in the same cellular genetic background. This approach enables us also to study: (1) structure-function relationship of RyR2 channel in human cardiomyocytes by introducing designed mutations/deletions in the functional regulatory domains; (2) generate human disease-associated mutation without tissue biopsy from patients; (3) carry out comprehensive characterization of Ca2+ signaling pathology, physiology and their pharmacological sensitivities with the same genetic background; (4) validate whether the mutation is causative for the disease; (5) establish homozygote in addition to heterozygote cell-lines carrying the disease-associated mutation. Since most of the disease-associated RyR2 mutations in patients are heterozygous, this approach will provide deeper insights in mutation-related structure/function changes in RyR2.
It should be pointed out that CPVT1 is a complex disease with its etiology having gender [29], and familial or regional components [5]. As such, introducing RyR2 mutations in various wild-type hiPSC lines will shed some light on whether a specific CPVT1 phenotype maybe regulated by the genetic background of the cells. In this respect recombinant protein and mouse models would be somewhat limited to address this issue.
4.2 CRISPR/Cas9 gene editing approach
Recent advances in stem cell technology, especially creation of iPSCs, have enabled characterization of the human congenital cardiac pathologies by differentiating the patient dermal fibroblasts into disease carrying myocytes, allowing characterization of its underlying mechanisms and determining the drug sensitivities of the particular disease in human myocardium. Applying the CRISPR/Cas9 gene editing to hiPSC cell lines provides potentially a comprehensive approach to study of congenital cardiac pathologies created by different mutations of the same gene. Such an approach not only bypasses making iPSCs from skin biopsies of patients, but also allows characterization of even more rare mutations associated with CPVT1 arrhythmia possible. In addition, more basic aspects of structure-function relationship of RyR2 can be addressed by performing site-directed mutagenesis especially focusing on the functional regulatory domains of RyR2 channel.
Our gene-editing approach in hiPSC, unexpectedly, produced a splice variant of RyR2 lacking 79 nucleotide bases. This splice variant was unlikely to produce the full-length RyR2 protein due to the frameshift, or to have caused the aberrant RyR2 function in hiPSC-CMs expressing F2483I mutation, but could have reduced the total amount of RyR2 protein in the heterozygous and homozygous hiPSC-CMs (Fig. 2). This possibility, however, is not compatible with our imaging findings that F2483I gene-edited cells produced long lasting and wandering Ca2+ sparks and higher frequencies of focal Ca2+ release, and had higher SR Ca2+ leaks (Figs. 3-5). If anything, the reduction of RyR2 protein would be expected to generate fewer Ca2+ sparks and smaller diastolic SR Ca2+ leaks, in sharp contrast to our findings. The observed reduction in RyR2 mRNA might even underestimate the observed differences in the calcium signaling pathology between wild type and mutant cardiomyocytes. It is likely that such mRNA differences may arise from our gene editing strategy where we introduce two silent mutations (Fig. 1A) in addition to the CPVT1 mutation.
To address the off-target mutations issue with CRISPR/Cas9 approach, though off-target mutation in hiPSCs has low possibility [30], we used two independent hiPSC-CM clones carrying F2483I mutation and found that both clones showed essentially the same CPVT1 phenotype. Thus, calcium signaling and electrophysiological aberrancies observed in the mutant hiPSC-CMs are most likely due to F2483I-RyR2 mutation.
4.3 Calcium signaling aberrancies
The major Ca2+ signaling findings in gene-edited F2483I-RyR2 hiPSC-CMs are the occurrence of long lasting and wandering Ca2+ sparks, aberrant focal Ca2+ releases in between regularly occurring beats, high diastolic Ca2+ levels, and smaller SR Ca2+ store size. All these findings are consistent with the possibility of “leaky” RyR2, which have been observed in other CPVT1 and human heart failure models [4, 12, 31]. In this scheme, local Ca2+ accumulating in sub-sarcolemmal space from leaky RyR2 activates the membrane electrogenic Na+/Ca2+ exchanger triggering delayed- or early-after depolarizations. This Ca2+ signaling phenotype is quite similar to our previous reports on cardiomyocytes derived directly from patient skin biopsy carrying the F2483I mutation [17]. The unexpected finding that these cells have fairly robust and aberrant Ca2+ release activity (Fig. 4), despite their smaller SR Ca2+ stores, may arise from the higher Ca2+ sequestering activity of SERCA2a /PLB complex.
4.4 Mechanistic and structure/function implications
The data from F2483I-RyR2 mutant expressed in hiPSC-CMs suggests that this mutation produces leaky RyR2 channels. It has been previously proposed that dissociation of a channel stabilizing protein, FKBP12.6 from RyR2 in heart failure and CPVT cardiomyocytes makes the channel leaky, and that RyR2 mutations facilitate the dissociation of FKBP12.6 [4, 12]. Although earlier site-directed mutagenesis studies show RyR1-Val2461 of the central domain (corresponding to RyR2-Ile2427) to be a key amino acid residue for FKBP binding [32], recent crystal structure analysis of the truncated RyR2-SPRY domain suggests that FKBP binds to N-terminal region [33]. High-resolution 3D structural model of RyR2 [34], On the other hand, shows that the position of the F2483 is distinct from such N-terminal domain, suggesting that F2483I mutation may allosterically alter the binding affinity of FKBP12.6 to RyR2. Other CPVT-1 associated RyR2 mutations, which have shown reduced FKBP12.6 binding affinity such as P2328S, S2474S, and Q4201R [11, 12], are also located at positions distant from the proposed N-terminal FKBP12.6 binding domain.
The present experimental approach might provide more critical insights on domain-specific phenotype of CPVT1 and its associated pathophysiology, especially when combined with RyR2 site-directed mutagenesis based on recent near-atomic level cryo-electron microscopic reports on the structure of RyR2 [34]. Thus, the combined stem cell and gene-editing technics provide a powerful and reliable approach in generating different CPVT1-associated mutations in human cell lines, not only allowing characterization of their pathophysiological phenotype and pharmacological sensitivities, but also their structure/function implications.
Acknowledgments
Authors are grateful to Dr. Stephen Duncan for providing us with K3 hiPSC clone and for his unfailing guidance on gene editing.
Funding: The studies were supported by National Institutes of Health Grants R01HL015162 (to MM) and P20GM103499 and UL1TR001450 (to NY).
Footnotes
Author Contributions: MM and NY designed the study, drafted the manuscript and oversaw the electrophysiological/calcium imaging (MM) and molecular/cell biological (NY) experiments. WH, XHZ, and CC performed the experiments. All authors analyzed and interpreted the data and approved the final version of the manuscript.
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morad M, Goldman Y. Excitation-contraction coupling in heart muscle: Membrane control of development of tension. Prog Biophys Mol Biol. 1973;27:257–313. [Google Scholar]
- 2.Näbauer M, Callewaert G, Cleemann L, Morad M. Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science. 1989;244:800–803. doi: 10.1126/science.2543067. [DOI] [PubMed] [Google Scholar]
- 3.Meissner G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium. 2004;35:621–628. doi: 10.1016/j.ceca.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 4.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 5.George CH, Jundi H, Thomas NL, Fry DL, Lai FA. Ryanodine receptors and ventricular arrhythmias: emerging trends in mutations, mechanisms and therapies. J Mol Cell Cardiol. 2007;42:34–50. doi: 10.1016/j.yjmcc.2006.08.115. [DOI] [PubMed] [Google Scholar]
- 6.Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J Am Coll Cardiol. 2009;54:2065–2074. doi: 10.1016/j.jacc.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Venetucci L, Denegri M, Napolitano C, Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012;9:561–575. doi: 10.1038/nrcardio.2012.93. [DOI] [PubMed] [Google Scholar]
- 8.Zhang XH, Morad M. Calcium signaling in human stem cell-derived cardiomyocytes: Evidence from normal subjects and CPVT afflicted patients. Cell Calcium. 2016;59:98–107. doi: 10.1016/j.ceca.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tester DJ, Kopplin LJ, Will ML, Ackermann MJ. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm. 2005;2:1099–1105. doi: 10.1016/j.hrthm.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 10.Kauferstein S, Kiehne N, Erkapic D, et al. A novel mutation in the cardiac ryanodine receptor gene (RyR2) in a patient with an unequivocal LQTS. Int J Cardiol. 2011;146:249–250. doi: 10.1016/j.ijcard.2010.10.062. [DOI] [PubMed] [Google Scholar]
- 11.Lehnart SE, Mongillo M, Bellinger A, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–2245. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehnart SE, Wehrens XH, Laitinen PJ, et al. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004;109:3208–3214. doi: 10.1161/01.CIR.0000132472.98675.EC. [DOI] [PubMed] [Google Scholar]
- 13.Jiang D, Wang R, Xiao B, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–1181. doi: 10.1161/01.RES.0000192146.85173.4b. [DOI] [PubMed] [Google Scholar]
- 14.Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–883. doi: 10.1161/CIRCRESAHA.110.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao YT, Valdivia CR, Gurrola GB, et al. Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function. Proc Natl Acad Sci USA. 2015;112:E1669–1677. doi: 10.1073/pnas.1419795112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fatima A, Xu G, Shao K, et al. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell Physiol Biochem. 2011;28:579–592. doi: 10.1159/000335753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang XH, Haviland S, Wei H, et al. Ca2+ signaling in human induced pluripotent stem cell-derived cardiomyocytes (iPS-CM) from normal and catecholaminergic polymorphic ventricular tachycardia (CPVT)-afflicted subjects. Cell Calcium. 2013;54:57–70. doi: 10.1016/j.ceca.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itzhaki I, Maizels L, Huber I, et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J Am Coll Cardiol. 2012;60:990–1000. doi: 10.1016/j.jacc.2012.02.066. [DOI] [PubMed] [Google Scholar]
- 19.Jung CB, Moretti A, Mederos y Schnitzler M, et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012;4:180–191. doi: 10.1002/emmm.201100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kujala K, Paavola J, Lahti A, et al. Cell model of catecholaminergic polymorphic ventricular tachycardia reveals early and delayed afterdepolarizations. PLoS One. 2012;7:e44660. doi: 10.1371/journal.pone.0044660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Penttinen K, Swan H, Vanninen S, et al. Antiarrhythmic Effects of dantrolene in patients with catecholaminergic polymorphic ventricular tachycardia and replication of the responses using iPSC models. PLoS One. 2015;10:e0125366. doi: 10.1371/journal.pone.0125366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Preininger MK, Jha R, Maxwell JT, et al. A human pluripotent stem cell model of catecholaminergic polymorphic ventricular tachycardia recapitulates patient-specific drug responses. Dis Model Mech. 2016;9:927–939. doi: 10.1242/dmm.026823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Si-Tayeb K, Noto FK, Sepac A, et al. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev Biol. 2010;10:81. doi: 10.1186/1471-213X-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2014;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lian X, Zhang J, Azarin SM, et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc. 2013;8:162–175. doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cleemann L, Wang W, Morad M. Two-dimensional confocal images of organization, density, and gating of focal Ca2+ release sites in rat cardiac myocytes. Proc Natl Acad Sci USA. 1988;95:10984–10989. doi: 10.1073/pnas.95.18.10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woo SH, Cleemann L, Morad M. Spatiotemporal characteristics of junctional and nonjunctional focal Ca2+ release in rat atrial myocytes. Circ Res. 2003;92:e1–11. doi: 10.1161/01.res.0000051887.97625.07. [DOI] [PubMed] [Google Scholar]
- 28.Curran J, Hinton MJ, Ríos E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 29.Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 30.Veres A, Gosis BS, Ding Q, et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobrev D, Wehrens XH. Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res. 2014;114:1311–1319. doi: 10.1161/CIRCRESAHA.114.300568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaburjakova M, Gaburjakova J, Reiken S, et al. FKBP12 binding modulates ryanodine receptor channel gating. J Biol Chem. 2001;276:16931–16935. doi: 10.1074/jbc.M100856200. [DOI] [PubMed] [Google Scholar]
- 33.Yuchi Z, Yuen SM, Lau K, et al. Crystal structures of ryanodine receptor SPRY1 and tandem-repeat domains reveal a critical FKBP12 binding determinant. Nat Commun. 2015;6:7947. doi: 10.1038/ncomms8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng W, Shen H, Wu J, et al. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science. 2016;354:aah5324. doi: 10.1126/science.aah5324. [DOI] [PubMed] [Google Scholar]