

Abstract
Mitochondria from different organisms can undergo a sudden process of inner membrane unselective leakiness to molecules known as the mitochondrial permeability transition (MPT). This process has been studied for nearly four decades and several proteins have been claimed to constitute, or at least regulate the usually inactive pore responsible for this transition. However, no protein candidate proposed as the actual pore-forming unit has passed rigorous gain- or loss-of-function genetic tests. Here we review evidence for -and against- putative channel-forming components of the MPT pore. We conclude that the structure of the MPT pore still remains largely undefined and suggest that future studies should follow established technical considerations to unambiguously consolidate the channel forming constituent(s) of the MPT pore.
Graphical Abstract
1. The Mitochondrial Permeability Transition: From basic science to clinical trials
Early protocols for isolating mitochondria considered calcium a deleterious agent that promoted mitochondrial uncoupling [1]. Hence, most studies typically included calcium-chelating agents in isolation buffers to minimize the occurrence of such uncoupling [2]. Current views acknowledge the presence of a Mitochondrial Permeability Transition (MPT) pore that shifts the mitochondrial inner membrane permeability [3]. When not reversed, channel opening results in the massive release of calcium alongside other metabolites (for a review see [4]). Further outer mitochondrial membrane rupture may result in the release of medium-sized proteins like cytochrome c, although it is noteworthy to mention that the MPT pore itself is known to present a molecular exclusion threshold of 1.5 kDa, thereby excluding protein movement through this channel [5, 6]. The MPT pore was initially described as a calcium-induced, calcium-release channel [3, 7, 8] and was later shown to be desensitized to calcium with nanomolar amounts of the cyclic undecapeptide cyclosporin A (CsA) [9]. These phenotypes strongly argued for the participation of the mitochondrial calcium uniporter (MCU) and the mitochondrial isoform of a cyclophilin (CypD) in the regulation of the MPT pore. Indeed, treatment of isolated mitochondria with MCU inhibitors prevented against MPT but only before calcium addition, suggesting a requirement for matrix calcium to activate the MPT pore [10]. CsA has been traditionally used to block the immune response following organ transplantation. Its potent effects on graft rejection require inhibition of cytokine production by blocking specific genes in active T cells [11]. To do this, CsA binds to cytoplasmic cyclophilin(s) and blocks calcineurin phosphatase activity. At high doses (~20mg/kg), CsA also suppresses JNK- and p38-dependent signaling pathways following immunogen recognition. During pathological conditions, calcineurin/NFAT signaling also mediates cardiac hypertrophy [12, 13]. Conversely, the use of CsA as MPT desensitizer to calcium requires concentrations in the nanomolar range [14]. This latter feature has allowed a growing cluster of studies to address the participation of the MPT pore in cell death induced by hypoglycemia, ischemia/reperfusion damage as well as on neurodegenerative and neuromuscular disorders (for a review see [15]). These studies also anticipated the MPT pore as a feasible target to ameliorate such diseases despite divergent outcomes in clinical trials and disease models. For example, following a successful phase 2 trial where patients were administered CsA right before primary percutaneous coronary intervention, the trial subjects presented reduced segment elevation myocardial infarct (STEMI) size [16]. Unexpectedly, the subsequent phase 3 trial (also known as CIRCUS) -where patients were injected with a single dose of CsA- resulted in no improvement in clinical outcomes measured at 1 year after STEMI [17]. Some scientists argued that the lack of a positive outcome may have been due to the fact that CsA is not an irreversible MPT pore inhibitor but rather a ‘desensitizer’ [18]. Consequently, whether patients with STEMI can present improved primary outcomes with CypD-independent MPT pore inhibitors remain to be tested [19–21].
Studies assessing mitochondrial dysfunction in a miniature swine model of heart failure with preserved ejection fraction showed exacerbated MPT pore activity, which was reversed by low-intensity aerobic interval training [22]. With this in mind, further studies by the same group aimed to assess the potential cardioprotective role of chronical administration of a non-immunosuppressive dose of CsA in the same miniature swine model [23]. These new experiments showed that CsA alleviated the collapse in mitochondrial function detected in animals experiencing heart failure, as measured by Complex I-dependent mitochondrial respiration and susceptibility to MPT. But quite unexpectedly, the authors detected a markedly decreased cardiomyocyte function consistent with impaired in vivo systolic and diastolic cardiac function. Such impairment was linked to a systemic hypertensive response of chronic CsA administration during heart failure. The authors concluded that chronic CsA treatment is not a viable therapeutic option during heart failure with preserved ejection fraction in miniature swine [23].
Overall, both studies contributed to the current growing notion that MPT pore desensitization with CsA may not be effective enough to suppress potential pore-dependent pathologies without side effects. Examples of such effects include potential risks of immunosuppression and renal damage [24]. These studies further reinforce the necessity to unveil the pore’s core molecular componentry, which would aim in the design of targeted therapies against the aforementioned pathologies. Consequently, it is the purpose of this review to discuss previous and recent efforts to map the channel-forming component(s) of the MPT pore. Specifically, we will focus on the adenine nucleotide translocase (ANT), the mitochondrial phosphate carrier (PiC), ATP synthase and alternative models still awaiting validation.
2. The adenine nucleotide translocase
Genetic studies designed to determine the participation of several proteins as MPT pore constituents have challenged established paradigms on its structure and has involved several groups around the world [25–32]. It was Hunter and Haworth who first proposed a role of the adenine nucleotide translocator (ANT) as a central component of the MPT pore due to the sensitivity of such process to the selective ligands atractyloside (ATR) and bongkrekic acid (BKA), which sensitize and inhibit calcium-induced pore opening respectively [3, 7, 8]. Further reports by the Halestrap laboratory found an in vitro interaction between the fusion glutathione S-transferase-CypD (GST-CypD) and ANT [33]. These experiments highlighted the importance of the redox status of thiol residues for GST-CypD interaction with inner mitochondrial membranes (IMMs). In this work, treatment of isolated mitochondria with diamide (a bifunctional thiol reagent) resulted in an increase in GST-CypD binding to IMMs isolated thereafter, which was absent when diamide was added directly after isolating IMMs [33]. This led the authors to conclude that diamide induced crosslinking of two ANT cysteines required matrix glutathione, which was likely lost during IMM isolation. [14, 34]. Noteworthy is the fact that CsA could also abrogate such binding, which suggests that CypD-MPT pore complex(es) could occur through the CsA-binding domain in a process involving the isomerization of proline residues [33]. Indeed, isomerase-inactive mutant CypD is unable to induce MPT opening as assessed by oxidative stress [35]. Modulation of CypD levels is also pivotal for normal mitochondrial function. For example, heart-specific overexpression of wild type CypD results in a severe hypertrophic phenotype resulting in heart dysfunction. For more insight into the molecular aspects of CypD-dependent MPT, we refer the reader to a previous discussion by our group [36].
Studies by different laboratories have also highlighted the importance of discrete cysteine residues controlling MPT [37–39]. While Costantini and colleagues argued that the MPT pore could be induced at two separate sites involving SH groups, one located either at the outer side of the inner mitochondrial membrane and the second one at the level of regulatory proteins in the inter membrane space or outer membrane, the authors showed that ANT dimerization was not required for MPT onset [37]. Indeed, Kowaltowski and collaborators have proposed that phenylarsine oxide (PAO) can induce MPT through its interaction with an extramitochondrial calcium-binding site [40]. These studies argued against previous working models involving dimerization of ANT [41] or even newer ones involving PAO-induced pore opening [19]. In a similar token, studies by Chavez’s group have shown that MPT induced by copper or calcium can be significantly amplified in the presence of 1,10-phenanthroline or ATR [42]. Thiol group-titration revealed that blockage of 5.9 nmol of SH/mg protein was enough to induce pore opening. Further MPT modulation with ATR, ADP and N-ethylmaleimide was interpreted as ANT being the pore’s core component. In a subsequent study, the same group found that the dithiol reagent mersalyl induced some sort of CsA-insensitive MPT when incubated alongside PAO [39]. Finally, the same group showed that locking the ANT in the “c” conformation facilitated binding of fluorescent eosin maleimide to a ~30kDa protein [39]. Although these studies further reinforced the notion that ANT plays a major role in MPT progression, the authors acknowledged that the role of ANT could be accessory.
However, the role of ANT in cell death appears to be more complex than what can be explained by MPT pore opening alone. For example, ANT overexpression in cardiomyocytes results in cell death [43]. Under these conditions, ANT upregulation can induce apoptosis through the activation of pro-apoptotic Bax. The role of Bax as an MPT pore component has been considered in some studies proposing that Bax and Bak may behave as the mitochondrial outer membrane component of the MPT pore [44]. But this hypothesis -at least for Bax- has also been previously ruled out by others showing the dispensability of this BH-3 protein for mitochondria to undergo MPT [45]. Whether Bax forms part of the MPT pore still merits further investigation, especially if we consider the presence of MPT-like structures in model organisms naturally lacking Bax like Saccharomyces cerevisiae [46]. Nevertheless, results from Madeo’s group suggest hydrogen peroxide may induce cell death through the upregulation of a BH3-only protein in yeast, which can induce CsA-independent swelling in isolated mitochondria (see Section 3 and [47]).
Although informative, the studies addressed above are not conclusive on the possibility that ANT per se can form a calcium-induced calcium-release channel. To address this possibility, Brustovetsky and colleagues showed that purified bovine ANT and recombinant Neurospora crassa ANT could form a 600pS pore reminiscent of half of the MPT pore full conductance [48]. These authors also found that locking ANT in the ‘m’ conformation (with the substrate binding site facing the mitochondrial matrix) with BKA inhibited the channel formed by ANT but not ATR, which blocks the carrier in the ‘c’ conformation (with the substrate binding site facing the cytoplasm). Noteworthy is the fact that the mitochondrial megachannel (MMC) is thought to be the electrophysiological manifestation of the MPT pore and it usually presents conductances near 1.3 nS [49]. On a related note, Rück and colleagues found that liposome-embedded ANT could mediate transport of ATP, malate and AMP upon treatment with calcium, while ATR and HgCl2 also induced a pore-like activity of ANT-containing liposomes [50]. These results further solidified -at the time- the notion that ANT was the central player in MPT pore opening [51]. Indeed, for several years and after several studies, the view where ANT was envisioned as the channel-forming unit of the MPT pore was widely accepted. However, this model was challenged when Kokozska and collaborators generated mice with liver-specific deletion of ANT1 and ANT2 [6]. In this study, knockout mice had the expected ADP-insensitive mitochondrial respiration but could still undergo MPT as induced by calcium, uncouplers or pro-oxidants. Interestingly, isolated organelles required substantially more calcium than their wild type (WT) counterparts, thus suggesting ANT can behave as an indirect pore regulator. A following study from the group led by Brand showed that ANT is responsible of almost half of the basal proton conductance across the inner mitochondrial membrane even when blocked with ATR [52]. This finding suggests ANT is poised to regulate mitochondrial proton gradients directly, and gradients for many other ions indirectly. In fact, Rottenberg and Marbach have proposed that ANT may regulate MPT by altering the surface potential of the inner mitochondrial membrane [53]. Chinopoulos’ group has additionally proposed that ANT1 can sensitize the MPT pore to changes in the electrochemical gradient of the mitochondrial inner membrane [26]. Altogether, the role of ANT on MPT modulation appears to be indirect, which has led several groups to look for other protein candidates (see below).
3. The mitochondrial phosphate carrier hypothesis
A few years after Kokozska and collaborators showed that ANT was dispensable for MPT progression [6], Halestrap’s group published a study showing that the antibody they previously used to detect ANT either in proteoliposomes or solubilized mitochondrial membranes was recognizing the mitochondrial carrier for inorganic phosphate (PiC) [54]. This led to a subsequent working model from his group proposing that the interaction between ANT, PiC and CypD could potentially form the long sought MPT pore [54]. In one of these newer studies, the authors monitored mitochondrial protein binding to a PAO affinity column in the presence of ATR, which prevents ANT binding to PAO. By passing beef heart mitochondrial lysates through such affinity column and eluting bound proteins after extensive washing, only four proteins were detected using mass spectrometry approaches: Ubiquinol-cytochrome c reductase complex core protein 2, PiC, adenylate kinase-2 and NIPSNAP-2. From these proteins, the authors concluded that the most likely MPT pore components would be PiC and adenylate kinase-2. Considering the lack of effects of the adenylate kinase-specific inhibitor 1, P5-di (adeno-side-5′) pentaphosphate on MPT, the authors discarded the notion mitochondrial kinase for further studies [54]. They hypothesized that the role of PiC as a component of the MPT pore would nicely fit with the then reported effects of phosphate on this channel, which is known to inhibit it at low concentrations [55], but markedly activates it at higher levels provided calcium is present [56]. In the study by Leung, the authors also performed “pull-down” experiments where mitochondrial lysates were incubated with GST-CypD. They monitored PiC -but not ANT- binding to the fusion protein in a CsA-sensitive pattern by using different antibodies against PiC. As expected, addition of ATR abrogated ANT binding to the PiC-CypD complex and addition of diamide potentiated detection of the ANT-PiC-GST-CypD complex. Similar interactions were detected through immunoprecipitations using either PiC or ANT antibodies where ANT or CypD detection was decreased with ATR of CsA respectively. From these studies Leung and colleagues concluded CypD binds PiC, while PiC binds ANT but ANT does not bind CypD [54, 57]. To further substantiate a potential relationship between PiC activity and MPT, the authors developed a phosphate transport assay and further showed that NEM (a monothiol reagent) inhibited phosphate transport and MPT-dependent mitochondrial swelling with a similar concentration-dependency, although CsA was without any effect on phosphate transport. It is worth mentioning that this approach presents some limitations, being the most evident that NEM can potentially react with any protein provided it presents an exposed cysteine residue. This means that NEM could inhibit PiC activity on one site and MPT at a different locus, being coincidentally at similar concentrations. The same applies for the previously reported β-amino ketone derivative Ro68-3400 [20], which behaves as a very potent inhibitor of both MPT and phosphate transport, while inducing conformational changes on ANT associated with pore closure [54]. Ro68-3400 was initially described as a potent MPT inhibitor, allegedly acting at the level of VDAC. However, its effects were later shown to persist in mitochondria lacking this protein [27]. In addition, Leung and collaborators showed that Ro68-3400 -just like NEM- reduced the binding of both PiC and ANT to the PAO column [54]. These authors also tested the effects of the previously characterized ubiquinone Ub0 [21] and showed that this molecule successfully suppressed MPT-dependent swelling, but any role as a bona fide PiC inhibitor was not shown. Finally, pretreatment of isolated mitochondria with either Ro68-3400, diamide or Ub0 resulted in no binding of the ANT/PiC complex to the PAO affinity column [54]. Addition of Ro68-3400 or Ub0 to isolated mitochondria suppressed calcium- or calcium plus PAO-dependent swelling but not when PAO was added before these MPT pore inhibitors. Finally, Ub0 and Ro68-3400 were shown to lock ANT in its ‘m’ conformation, which is known to inhibit pore opening, while Ro68-3400 suppressed mitochondrial state 3 respiration being interpreted as ADP translocation inhibition. From this work, the authors concluded that although ANT is still an important regulator for MPT activity, PiC constitutes the main target of PAO-dependent activation of MPT and the locus where CypD binds to execute CsA-dependent pore opening [54, 57]. In line with a potential role of PiC in MPT-dependent cell death, the group led by Pimentel-Muinos showed that PiC silencing significantly attenuated cell death elicited by the general kinase inhibitor staurosporine [58]. This phenotype appears to be conserved in lower eukaryotes, where cell death induced by BH3-only protein overexpression plus H2O2 is halted upon PiC deletion [47].
A few years afterwards, Halestrap’s group published results showing that downregulation of PiC in HeLa cells had no effects on the calcium threshold for MPT [28]. Pore sensitivity to CsA was shown to be independent on whether phosphate was in the incubation media. Specifically, addition of arsenate plus CsA resulted in pore desensitization to calcium. In this study, the authors also argued that PiC silencing may have not been enough to unveil any phenotype and that further PiC knockdown would induce cell death. This phenotype is in agreement with studies by our group showing that phosphate is not an absolute requirement for the inhibitory effects of CypD inhibition with cyclosporin A or CypD deletion on mitochondrial permeability transition [59]. This study shows that MPT can be induced regardless of whether Pi, arsenate or vanadate is co-incubated alongside with calcium. Under these conditions, addition of CsA significantly inhibits calcium-induced mitochondrial swelling. More importantly, attenuation of calcium-induced mitochondrial swelling was still detected in the absence of CypD regardless of whether increasing Pi, arsenate or vanadate levels were used in the incubation media. These results cast insight on whether Pi per se is acting directly to inhibit MPT or if it is acting though a mechanism requiring negative charges, which would make sense considering the effects of vanadate or arsenate.
In the following years, the groups led by Baines and Molkentin generated mouse models of heart-specific PiC downregulation to further elucidate its molecular role on MPT progression [29, 60]. In the first case, the group corroborated previous evidence suggesting CypD binds PiC and further detailed the molecular nature of such interaction. They showed that CypD residues 70 to 110 are required for high affinity binding of recombinant PiC with CypD. To further explore a role of PiC as a component of the MPT pore, two different cardiac-specific PiC transgenic (TG) mouse lines were generated. These lines had PiC expression under control of the α-myosin heavy chain (α-MHC) promoter. The TG lines presented 4- to 6-fold PiC overexpression, but had no differences in terms of heart gravimetrics, fractional shortening and left ventricular end systolic/diastolic dimensions. Mitochondria isolated from these hearts presented similar respiratory control ratios as compared to its non-TG counterpart. MPT pore activity assessed by calcium-induced swelling and calcium-retention capacity (CRC) revealed no changes whatsoever between control non-TG versus PiC TG mouse lines. To understand if PiC upregulation resulted in no differences in MPT pore readouts due to potential stoichiometric differences in terms of PiC versus CypD levels, the authors crossed the PiC TG line overexpressing the carrier 4-fold with the previously described cardiac-specific “26.2” CypD TG mouse to create a double transgenic line (DTG). As expected, the offspring had cardiac-restricted overexpression of PiC and CypD. Since experiments were carried out with 2-month-old mice, the hearts of the 26.2 mouse line still presented no hypertrophy. However, the DTG mouse line presented a decreased sensitivity to calcium-induced swelling but considering the previously reported phenotype of CypD TG mice where mitochondrial ultrastructure was deranged [35], a CRC assay was performed. The results showed that isolated mitochondria presented statistically similar calcium uptake before MPT onset. Interestingly, a decreased CRC response to CsA was monitored at 1μM of this undecapeptide. The authors showed this was probably due to increased CypD since addition of 5μM CsA virtually suppressed such phenotype.
In this study, the authors also generated a mouse model of cardiac-specific PiC knockdown by means of a Tet-off system coupled to an shRNA against PiC. Knockdown mice had 60% silencing of PiC at the protein level and developed mild hypertrophy with decreased fractional shortening. Respirometric analyses showed no differences between mitochondria from WT and PiC knockdown mice. Interestingly, mitochondrial ATP levels were decreased upon PiC knockdown suggesting phosphate deficiency in the mitochondrial matrix. Mice with knockdown of the phosphate carrier presented similar baseline calcium-induced mitochondrial swelling and CRC thus showing similar MPT pore activity as compared with mitochondria fully expressing PiC. In the presence of 1μM CsA, however, PiC knockdown mitochondria presented a decreased CRC threshold. To fully characterize the MPT response to calcium in PiC deficient organelles, the authors isolated cardiomyocytes from both sources and performed a CRC assay in digitonin-permeabilized cells. The results again showed that cardiomyocytes from WT and PiC knockdown mice had identical MPT activity [61].
In parallel, Molkentin’s group published a study showing a detailed characterization of a heart-specific knockout mouse by using a loxP/Cre approach, where Cre transgene overexpression was under control of the alpha myosin heavy chain promoter and induced with tamoxifen injections [29]. In this study, the authors achieved more than 90% of murine cardiac PiC silencing, but not other mitochondrial proteins, which resulted in defects in mitochondrial Pi uptake and ATP production. Interestingly, short term PiC suppression (2 weeks) did not affect cardiac performance and mitochondrial ultrastructure, but longer-term deletion (10 weeks) resulted in severe heart hypertrophy and myopathy. Under conditions of short-term PiC suppression, the extent of MPT-derived mitochondrial swelling was reduced by half alongside exacerbated CRC. As expected, MPT induction with the ANT ligand ATR resulted in no changes on MPT activity as evidenced in mitochondrial swelling experiments. Surprisingly, protection from MPT opening in the absence of PiC was abolished when switching phosphate for arsenate in the swelling buffer. The authors of this study also infected mouse embryonic fibroblasts (MEFs) from Slc25a3fl/fl mice with either a control virus or a Cre adenovirus to deplete PiC. These cells were challenged with the calcium ionophore ionomycin and PiC deficient MEFs had significantly decreased necrotic cell death. In addition, PiC-deficient MEFs had an increased calcein signal upon ionomycin challenge suggesting these cells were somewhat protected against calcium-induced MPT [29].
To further address the role of PiC, calcium and PAO on MPT, we have also downregulated PiC levels in MEFs (Fig. 1A). Treatment with a PiC-specific siRNA decreased PiC levels up to 90% as compared with MEFs treated with a scrambled control siRNA. We performed a CRC assay in these cells and found no changes in the pore sensitivity to calcium among groups irrespective of their PiC levels (Fig. 1A). These results match those previously presented by Halestrap’s group in HeLa cells and are in agreement with our study in PiC knockdown mouse hearts [28, 61]. We next assessed MPT by means of treating MEFs with a single bolus of calcium and subsequent addition of 40μM PAO to the cells. As expected, WT MEFs released calcium indicative of MPT (Fig. 1B). MEFs treated with the PiC siRNA present an almost identical calcium release profile. This suggests that although PiC can bind PAO [19], this prooxidant is targeting calcium release at the level of a still undefined locus. The alternative hypothesis would be that the remaining ~10% PiC can still trigger MPT with the same characteristics. Although this hypothesis seems unlikely, please note that Lemasters has proposed that a single MPT channel “unit” in the nS range can theoretically account for the full pore response [62]. If we consider these results plus those reported previously in a conditional knockout model [29], we can conclude that that current methods for measuring MPT pore activity (i.e. CRC vs. ionomycin/calcein cobalt) may provide divergent results. This may be likely the case for studies assessing a putative role of ATP synthase as a component of the MPT pore (see Section 4). However, another possibility would be that such calcium overload approaches may be triggering cell death under divergent pathways although both involving CypD (please note that ionomycin disrupts calcium gradients on every single organelle in the cell). Evidence for such divergent cell death subroutines has been reported. For example, Moll’s group has convincingly showed that upon noxious insults, p53 translocates to the mitochondria and interacts with CypD to induce necrosis likely by MPT pore opening [63]. But the role of the ROS-p53-CypD-mitochondrial “deadly axis” may also extend to other cell demise subroutines including apoptosis and autophagy, where p53 can bind and induce oligomers of Bax and Bak thus facilitating cytochrome c release [63]. Indeed, Molkentin’s group has recently published evidence showing that both Bax and Bak can induce autophagy by permeabilizing lysosomes [64]. Hence, current working models suggest that CypD can induce cell death activated with either calcium or ROS through parallel pathways but involving different downstream targets (i.e. H2O2 induced CypD-P53 versus Calcium-induced CypD-MPT pore).
Figure 1. The expression levels of PiC in Mouse Embryo Fibroblasts do not modify the MPT pore sensitivity to calcium.

MEFs were isolated from homozygous (Slc25a3+/+) or heterozygous (Slc25a3+/−) mice in the presence or absence of a siRNA targeting PiC from Dharmacon. (A)Ca 2+-retention capacity (CRC) experiments demonstrated an equal MPT pore response to Ca2+ on MEFs in all conditions. CRC was measured by detecting changes in the fluorescence of 1 μM Calcium Green-5N at 530 nm and is represented in nmol Ca2+/1×106 cells using 10mM succinate as respiratory substrate. MEFs were later homogenized in buffer containing 150 mM NaCl, 10 mM Tris(pH 7.4), 1 mM EDTA, and 1% Triton-X100. Proteins (50μg) were resolved by SDS-PAGE using 10% acrylamide, transferred onto PVDF membranes, and immunoblotted usingour previously reported custom-made antibody against PiC (YenZym) or actin as aloading control. We determined PiC levels to be decreased more than 90% through standard densitometry(not shown). (B) Phenylarsine oxide-induced calcium release wasassessed by loading 5 μM CaCl2 and further adding 40μM PAO. Data are presented as mean + SEM from 4 independent replicates.
More recently, Halestrap’s group has proposed that the MPT pore could be formed at the interface of dimers of any mitochondrial carrier member of the SLC25a family. But since ANT and PiC are among the most abundant carriers in the inner mitochondrial membrane, these proteins predominantly participate in MPT pore formation [19]. To substantiate this proposal, the authors again showed that ANT and PiC bind directly to a PAO column and that ANT (but not PiC) binding is abrogated with the MPT pore inhibitor GNX-4975. This provides additional evidence suggesting that PAO activates MPT by binding to a PiC/ANT complex, but PiC per se being not the MPT pore.
On the role of PiC as an MPT pore constituent, we can conclude that 1) it can bind to ANT and CypD to indirectly modulate MPT activity and 2) its participation may be restricted to control pore opening through phosphate levels in the mitochondrial matrix. The latter conclusion can be especially true if we consider that its protective effects are lost when incubating PiC-deficient mitochondria with other anions [29]. Nevertheless, PiC downregulation can protect cells from apoptosis through a still undefined mechanism [58].
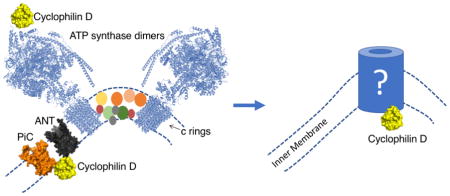
4. The ATP Synthase: c-ring or dimers?
In 2013, three different groups challenged the prevailing notion that the ANT and/or PiC were central components of the MPT pore [31, 32, 65]. In the first study, the authors proposed that ATP synthase C-subunit could mediate MPT [32]. Upon C-subunit overexpression, HeLa cells were more susceptible to the effects of endogenous calcium relocation with histamine, had a decreased basal mitochondrial transmembrane potential (ΔΨ) and mitochondrial fragmentation was significantly exacerbated. When HeLa cells were treated with an siRNA to decrease ATP synthase C-subunit expression, the cells were more resistant to hydrogen peroxide-induced ΔΨ dissipation, cytochrome c release and cell death induced with ionomycin or H2O2. From these experiments, the authors concluded that ATP synthase c-subunit behaves as a core component of the MPT pore complex and further proposed that molecules targeting the c-subunit could be utilized to pharmacologically suppress MPT induced in diverse disease contexts. We have previously raised some questions on the possibility that the c subunit (and the whole ATP synthase complex) can form the MPT pore, based on the likelihood that genetic manipulation of c-rings may distort the ultrastructural features of the mitochondrial network as seen in model organisms [60]. Some colleagues have also raised these concerns on the grounds that genetic silencing of c-rings may alter ATP synthase biogenesis and consequently affect MPT pore readings indirectly due to changes in adenine nucleotide levels [66].
The second group proposing a role of ATP synthase stated that ATP synthase dimers -but not monomers- form the MPT pore [65]. To support this, the authors first defined experimental conditions by which CypD could bind ATP synthase OSCP-subunit. The authors concluded that the CypD-OSCP complex was formed mainly through electrostatic interactions, and such docking was antagonized with increasing concentrations of the ATP synthase inhibitor benzodiazepine 423 (Bz-423). Interestingly, addition of Bz-423 decreased the amount of calcium required for MPT at low -but not high- phosphate concentrations. This result is in agreement with previous studies by the same group showing that Pi can act as an MPT pore inhibitor [55]. Moreover, in the presence of high loads of CsA, Bz-423 lowered the calcium threshold for MPT irrespective of Pi levels. This was interpreted as a CsA-mediated displacement of CypD from its receptor (i.e. OSCP), so that Pi could not excerpt its inhibitory role at the level of CypD and thus Bz-423 could activate MPT in a Pi-independent manner.
This would also imply that Bz-423 activates MPT at a downstream site of CypD-mediated pore desensitization. This authors further explored a potential relationship between ATP synthase activity and pore sensitivity to calcium. In these experiments, the authors established ATP generating or hydrolyzing systems in order to achieve constant ADP or ATP levels for MPT pore activity readouts where the ATP synthase rotor moves either clockwise or counterclockwise. Under these conditions, ATP hydrolyzing mitochondria required twice the amount of calcium as compared to ATP-generating mitochondria in order to achieve MPT. We believe this specific finding per se cannot conclude a direct role of ATP synthase on MPT since earlier experiments showed ADP behaves as a more potent pore inhibitor as compared with ATP even in the presence of oligomycin [67]. In addition, ATP hydrolysis generates phosphate and decreases pH in the mitochondrial matrix, which also inhibits MPT [68].
The third group arguing for a role of ATP synthase as the channel-forming unit of the MPT pore showed that liposome-reconstituted, myc/FLAG-tagged c subunit could mediate conductance values of up to 1.5–2 nS with negative rectification [31]. These values are remarkably similar to those reported for the MMC. In addition, ATP blocked such conductance with an EC50= 660 μM, which led the authors to propose that this molecule was interacting within the c-ring itself to exert its channel-blocking properties. However, ADP as well as AMP inhibited such current with equal potency. Again, this is at odds with earlier results showing inhibition of MPT achieved with ADP but not AMP in isolated mitochondria [67]. The authors also found that calcium markedly increased peak conductance (in SMVs) and that addition of an anti c-subunit antibody readily blocked channel conductance both at baseline or in the presence of calcium. The authors also performed electrophysiology measurements of either purified c-subunit, ATP synthase monomers or SMVs containing CypD and OSCP. The results showed that neither calcium or CsA had any effect on the c subunit-associated conductance, whereas monomeric ATP synthase displayed CsA-sensitive (but unruly) channel activity. Finally, SMV-associated multiple conductance was readily activated with calcium and inhibited with CsA, which led the authors to suggest that the MPT pore is directly related to ATP synthase c subunit. Although the authors did not rule out the possibility that the MMC was activated in such SMV preparations.
The studies addressed above provide evidence for a potential role of the ATP synthase as the actual channel-forming unit during permeability transition, either through the c-ring, which is essentially already a pore in the inner membrane, or through the formation of ATP synthase dimers. In addition, several recent papers have embraced such working model almost as a fact [69–71]. However, two recent studies by Walker’s group seriously challenge all results and models addressed in this Section [72, 73]. In the first study, the authors generated a triple mutant HAP1 cell line lacking transcripts for ATP5G1, ATP5G2, and ATP5G3 genes through Crispr/CAS9-mediated mutagenesis. As expected, this resulted in the lack of c subunits and in a growth defect concomitant with increased mitochondrial DNA levels as compared to isogenic HAP1 cells expressing c subunits. These mutant cells were labeled HAP1-A12 and showed decreased respiration rates but were surprisingly able to generate and sustain similar ΔΨ values as compared to its isogenic control counterpart [73]. Importantly, ΔΨ was insensitive to ADP or oligomycin in HAP1-A12 cells. Intact control and c subunit-deficient intact cells presented an MPT pore as induced with calcium overload upon thapsigargin or the calcium ionophore ferutinin additions. In both cell types, MPT onset was fully reversible with CsA suggesting an intact pore structure. Upon permeabilization with digitonin, both cell types showed identical calcium-retention capacities at baseline and pore desensitization with CsA almost triplicated the amount of calcium used for MPT onset regardless of whether c subunits were expressed or not. Both cell types were equally sensitive to MCU inhibition with ruthenium red and as expected, deletion of CypD in HAP1 cells led to a basal increase in mitochondrial calcium retention. Surprisingly, HAP1-A12 cells were still able to assemble a “vestigial” form of ATP synthase as assessed by SDS-PAGE analysis. The vestigial complex contained an intact set of subunits comprising the F1 sector of the enzyme plus subunits OSCP, b, d and F6, which comprise the peripheral stalk. Moreover, such complex still presented membrane subunits e, f and g while it missed DAPIT and ATP6. In this study, ρ0 cells were compared against HAP1-A12 cells and both cell types presented similar vestigial complexes highlighting the ability of such cells to still assemble several subunits of the enzyme. As shown before by Bernardi’s group, ρ0 cells still present a conserved calcium-induced MPT [74]. An equally important finding of this paper is that HAP1-A12 cells lacked -due to c subunit knockout- dimers and higher oligomeric complexes of ATP synthase. This strongly argues against the possibility that dimers -but not monomers- of ATP synthase form the MPT pore. However, the authors were cautious enough to speculate that the extraction process with digitonin may have artifactually eliminated any potential dimer or higher order aggregate of ATP synthase detected by native electrophoresis. In summary, the authors provided strong evidence against the participation of subunits c, DAPIT and ATP6 as pore forming units of the MPT pore.
The same group published follow up results showing intact MPT activity in HAP1 cells lacking either OSCP or b subunits, which comprise part of enzyme’s static peripheral stalk [72]. In this sequel study, Walker’s group demonstrated equal MPT pore activity in response to thapsigargin or ferrutinin additions to cells depleted of OSCP or b through Crispr/CAS9-mediated gene deletion. Calcium retention capacity and swelling assays revealed no changes whatsoever between isogenic control or mutated cell lines. Finally, a similar vestigial ATP synthase was detected in cells lacking OSCP or b subunits as previously evidenced in HAP1-A12 cells. We believe this result argues against any potential artifactual dimer disruption with digitonin since these new knockout cells still expressed c subunits. Finally, OSCP knockout cells were equally susceptible to MPT-pore desensitization with calcium by using CsA. This means that OSCP is not the target where CypD modulates the calcium-induced MPT.
Thus, there is extensive evidence arguing for a role of ATP synthase on MPT versus very recent loss-of-function studies by He and colleagues alongside atomistic simulations concluding that C-rings (and other ATP synthase subunits) are not responsible for MPT pore formation [75]. To address these divergent results, we decided to measure MPT pore activity in WT mouse embryonic fibroblasts with genetic silencing of the c-subunit by means of treatment with a specific siRNA pool against the three genes encoding c-subunits (Fig. 2). In agreement with He and colleagues [73], we found unchanged MPT pore responsiveness to calcium in fibroblasts with C-subunit depletion using a standard CRC assay regardless of whether complex I- or II-dependent substrates were used (Fig. 2B, C). We also found decreased levels of α subunit but no other proposed MPT pore constituent.
Figure 2. The expression levels of ATP synthase c-subunit (ATPG1-3) in Mouse Embryo Fibroblasts do not modify the MPT pore sensitivity to calcium.

(A) Expression levels of mitochondrial proteins in MEFs treated with either control (scrambled siRNA) or 3 specific siRNA pools against ATP synthase c-ring subunits (ATP5G1,2,3 from Dharmacon). MEFs were treated as in Fig. 1 and immunoblotted using the following commercially available antibodies: anti-ATP synthase c subunit:ab-181243, total oxphos ab110413, anti-CypD, and anti-GAPDH from Abcam. Proteins were immunodetected as follows: (V), ATP synthase; (III), respiratory complex III; (II), succinate dehydrogenase; NDU6, Complex I accessory subunit 6; NDUS3, complex I catalytic core; C1qbp, complement 1 q-binding protein; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase. (B) MEFs were counted (1 × 106 per experiment) and assayed as in Fig. 1 in the presence of 5mM glutamate and malate or (C) 10 mM succinate. Data are presented as mean + SEM from 4 independent biological replicates performed in duplicate.
There is no current clear explanation on the differences between the ‘negative results’ reported by Walker’s group versus the proposed protective effects upon c-subunit silencing. As discussed in Section 3, it is possible that different experimental approaches and cell models may account for the context-specific results addressed herein arguing for a role of ATP synthase. One possibility would involve ROS as a trigger of different cell death subroutines as compared with calcium overload in a c-ring subunit-dependent context. Furthermore, Bonora et al. [32] as well as Alavian and colleagues [31] have only reported potential MPT pore activity upon c-subunit knockdown in cells using the calcein-cobalt technique. Jiuya He and colleagues -as well as our group-have relied on the CRC assay [73]. The former groups have measured MPT pore activity triggered with the calcium ionophore ionomycin, which is known to disrupt intracellular calcium and magnesium gradients [76]. We would also like to mention that ionomycin -alongside with the ionophores ETH129 and A23187- are considered not suitable for inducing MPT opening in cells like murine isolated adult cardiac myocytes [77]. Antithetically, the CRC approach requires calcium to enter the mitochondria through the mitochondrial calcium uniporter, which feeds the mitochondrial calcium cycle and allows accurate titrations of MPT onset. The calcein-cobalt method can provide rates of calcium transients or changes in overall fluorescence intensity but fails to provide information as to the amount of calcium required for MPT. Finally, the experiments addressed herein were performed in different cell lines. This can constitute an additional source of variability given the differing amounts of calcium or other known MPT pore ligands required to regulate permeability transition in mitochondria from different sources[78].
Considering the study by He and colleagues showing a canonical MPT pore in the absence of c-rings, peripheral stalks and OSCP [72, 73], it could be possible that the results published by Bonora et al. as well as Alavian and collaborators rather reflect partial protective effects secondary to changes in mitochondrial ultrastructure due to c-ring-induced modifications in cristae shape and membrane flexibility [31, 32]. Indeed, ATP synthase dimerization can induce strong membrane curvatures [79]. However, the results showing a canonical MPT pore in cells devoid of C-rings, peripheral stalks or OSCP argue against a role of ATP synthase as a channel-forming unit of this unselective pore.
5. Alternative MPT pore working models
Based on the fact that the MPT persists in the absence of ANT, PiC or key subunits of ATP synthase, the need to ‘look elsewhere’ has been recently proposed [80]. On these grounds, one possibility would be to pursue loss-of-function screens in order to identify potential new candidates based on a modified resistance to calcium-induced calcium-release coupled to additional control experiments (see Technical Considerations). Some efforts in this direction have been recently done [81], although it is worth to mention that the results have also been questioned [15, 82, 83]. Another possibility involves looking back at previously proposed pore-forming unit candidates. One of these relatively unexplored working models include pore units formed by misfolded membrane proteins damaged by ROS or HS [84]. Under these conditions, misfolded aggregates can expose hydrophilic residues to the inner mitochondrial membrane phase and hence form water-accessible unselective pores. Such misfolded proteins and chaperones such as HSPs facilitate channel gating while CypD closely associates with such aggregates to provide folding. This hypothesis proposes that low levels of calcium -alongside accessory reagents such as thiol reagents or amphipathic peptides- exacerbates CsA-reversible MPT pore opening. When in the presence of higher amounts of both calcium and/or cystine-inducing reagents, pore opening is CsA-insensitive [84]. This hypothesis was based on the fact that pore induction under both aforementioned conditions presents similar size-exclusion characteristics. Moreover, CypD prevails bound to IMMs under both conditions while this protein is undetectable on IMMs in the presence of CsA after pore induction with high loads of either calcium or cystine-inducing reagents [84]. Evidence supporting this model also shows that HS can effectively block MPT [85]. In addition, HS can protect mitochondria from ROS-induced damage [86] likely resulting in overall protection against lethal systemic and organ damage [87]. Evidence against this model includes the fact that the pore detected by He and Lemasters presents similar size-exclusion properties as compared to the MPT pore, suggesting discrete and not stochastic protein associations form the MPT pore. This could alternatively mean that oxidative stress preferentially modifies a protein or set of proteins probably interacting with previously proposed MPT pore components (i.e. ANT, PiC, ATP synthase, etc.) to form the MPT pore. Another alternative hypothesis requiring further testing is the possibility that components of the mitochondrial PIM constitute the MPT pore [88]. This possibility was first explored by Sokolove and Kinnally by showing that mitochondrial signal peptides from divergent species could increase the permeability of isolated rat liver mitochondria [89]. Following Pfeiffer’s group experiments showing that submicromolar amounts of the cationic wasp peptide mastoparan induced the classic, CsA-reversible MPT, Sokolove and Kinnally decided to test the permeabilization effects of the cationic mitochondrial targeting sequence of cytochrome oxidase subunit IV from Neurospora crassa [89, 90]. Their results showed a concentration-dependent swelling pattern, which was absent when N- or C-terminal sequences from VDAC were used. Interestingly, peptide-induced swelling required energized mitochondria and was slowed down when the uncoupler FCCP was added. This result would match the well-known properties of Tim23-dependent protein import assays performed by Pfanner’s group [91]. Specifically, import of F1-ATPase subunit b, cytochrome c, Fe/S protein of the bcl complex, and Fo-ATPase subunit 9 were inhibited at high FCCP concentrations, while Fo-ATPase subunit 9 import was slowed down at low levels of the uncoupler. Quite strikingly, protein import was reestablished by eliciting an artificial potassium diffusion potential by means of incubating deenergized mitochondria with the ionophore valinomycin in the presence of potassium. If the MPT pore and components of the Tim23-dependent PIM were to share identity, then opening of the MPT pore should affect protein import, while PIM activity could impinge on the MPT. Indeed, Martin and colleagues showed that Fo-ATPase subunit 9 import can be decreased under conditions where MPT should be active (i.e. in the presence of FCCP) [91]. Conversely, an MPT which is partially sensitive to CsA[89], and decylubiquinone [92] can be triggered upon addition of Tim23-specific signal peptides in rat liver and isolated yeast mitochondria respectively [89, 93, 94].
A third alternative model envisions the co-existence of more than one MPT pore channel-forming unit in the inner mitochondrial membrane. In this model, any member of the SLC25a mitochondrial solute carriers would have the potential to form an MPT pore provided calcium triggers conformational changes in these proteins in a cardiolipin-dependent manner (see discussion in [19]). This would explain why in the absence of ANT or PiC, an MPT pore can still be detected albeit with suppressed sensitivity to ANT or PiC agonists. Considering a differential expression profile of SLC25a proteins in several species, the model would also explain why MPT has divergent characteristics in organisms such as plants, fruit flies, yeasts and zebra fish as compared with MPT in rodents or humans [6, 46, 92, 95–99].
Whether any of these (or other) alternative models actually reflect the identity of the MPT pore still merits further testing. For additional analysis on this topic, we refer the interested reader to excellent studies on these (and more) alternative MPT models [62, 100, 101].
6. Technical Considerations
As mentioned above for the ATP synthase studies, we have begun to appreciate that not all experimental techniques measure MPT activity with the same accuracy. For example, why knockdown of ATP synthase c-subunit results in protection against hydrogen peroxide-induced cell death and calcium overload using the calcein/cobalt technique plus calcium ionophores [32], while genetic deletion of this protein results in no changes whatsoever in the calcium retention capacity of the cell [72]? Why PiC knockdown results in protection against staurosorine treatment [58] or calcium overload using the calcein/cobalt technique [29], while the CRC of cells with PiC silencing remains unmodified (Fig. 1)? As determined by Hunter and Haworth and later addressed by Ichas and Mazat [4], the MPT pore can be defined as a calcium-induced, calcium-release channel. This means that any technique measuring a purported downstream MPT activity (i.e. cell death) is not a direct measure of pore opening and should be used in parallel with other techniques plus adequate statistics (i.e. CRC, mitochondrial swelling, electrophysiology and transmembrane potential readouts). Results arising from utilization of oxidative stress as cell death inductor should also be taken with caution considering ROS can generate pro-death signals that may not be related to MPT at all [63]. As mentioned above, calcium ionophores have been recently considered effective for MPT opening only in isolated mitochondria but not in whole cell models [77]. This means that future studies assessing the participation of a given protein as an MPT pore component should adopt as many techniques as possible to avoid false positive (or false negative) results. In addition, the utilization of gain- and loss-of-function models would be highly desirable. This also applies for studies assessing activity of the so-called ‘mitochondrial mega channel’, which is considered the electrophysiological counterpart of the MPT pore [102].
7. Conclusions
Nearly four decades after the groundbreaking discovery by Hunter and Haworth on the fact that the inner mitochondrial membranes harbor a proteinaceous and unselective pore triggered by calcium and regulated by nucleotides, we essentially stand in the same place concerning the still uncertain identity of the channel-forming unit(s) of the MPT pore. That being said, the last decades have been extremely productive in terms of understanding several pore regulatory features including 1) the role of CypD and ANTs, 2) the pathophysiological relevance of the MPT in the context of ischemia/reperfusion, muscular dystrophy, stroke and even cancer as well as 3) its potential physiological roles in mitophagy [103]. Considering the fact that proteins of previously unknown identity playing an indirect role on MPT such as MCU have already been unearthed at the gene, protein and atomic level [104], we are confident that the channel forming unit(s) of the MPT pore will be unveiled more sooner than later. To do so, any potential new candidate should have to fulfill the technical considerations proposed herein.
Highlights.
The mitochondrial permeability transition compromises cellular homeostasis.
Several candidates thought to comprise the channel-forming unit of the mitochondrial permeability transition pore have not passed rigorous loss-of-function genetic tests.
Alternative working models on the channel forming unit(s) of the mitochondrial permeability transition pore still await validation.
Future studies should follow established methodological guidelines to facilitate the identification of the channel forming component(s) of the mitochondrial permeability transition pore
Acknowledgments
This work was supported by the National Institutes of Health grant HL094404 (to C.P.B.) and UNAM-FQ-PAIP 5000-9171 (to M.G.-A.).
Abbreviations
- α-MHC
α-myosin heavy chain
- ΔΨ
mitochondrial transmembrane potential
- ANT
adenine nucleotide translocase
- ATR
atractyloside
- BKA
Bongkrekic acid
- Bz-423
benzodiazepine 423
- CsA
cyclosporin A
- CsABD
cyclosporin A binding domain
- CypD
cyclophilin D
- CRC
calcium retention capacity
- DTG
double transgenic
- GST
glutathione S-transferase
- HS
heat shock
- HSP
heat shock proteins
- IMM
inner mitochondrial membrane
- MCC
multiple conductance channel
- MCU
mitochondrial calcium uniporter
- MEF
mouse embryo fibroblast
- MPT
mitochondrial permeability transition
- NEM
N-ethylmaleimide
- OSCP
oligomycin sensitivity conferral protein
- PAO
phenyl arsine oxide
- PiC
mitochondrial phosphate carrier
- PI
propidium iodide
- PIM
protein import machinery
- STEMI
segment elevation myocardial infarct
- TG
transgenic
- VDAC
voltage dependent anion channel
Footnotes
Conflict of interest
The authors declare having no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hunter FE, Ford L. Inactivation of oxidative and phosphorylative systems in mitochondria by preincubation with phosphate and other ions. The Journal of biological chemistry. 1955;216:357–369. [PubMed] [Google Scholar]
- 2.Vinogradov A, Scarpa A, Chance B. Calcium and pyridine nucleotide interaction in mitochondrial membranes. Archives of Biochemistry and Biophysics. 1972;152:646–654. doi: 10.1016/0003-9861(72)90261-5. [DOI] [PubMed] [Google Scholar]
- 3.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria I. The protective mechanisms. Archives of Biochemistry and Biophysics. 1979;195:453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- 4.Ichas F, Mazat J-P. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1998;1366:33–50. doi: 10.1016/s0005-2728(98)00119-4. [DOI] [PubMed] [Google Scholar]
- 5.Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proceedings of the National Academy of Sciences. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427 doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria II. Nature of the Ca2+ trigger site. Archives of Biochemistry and Biophysics. 1979;195:460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 8.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria III. Transitional Ca2+ release. Archives of Biochemistry and Biophysics. 1979;195:468–477. doi: 10.1016/0003-9861(79)90373-4. [DOI] [PubMed] [Google Scholar]
- 9.Crompton M, Costi A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. European Journal of Biochemistry. 1988;178:489–501. doi: 10.1111/j.1432-1033.1988.tb14475.x. [DOI] [PubMed] [Google Scholar]
- 10.Altschuld RA, Hohl CM, Castillo LC, Garleb AA, Starling RC, Brierley GP. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. The American journal of physiology. 1992;262:704. doi: 10.1152/ajpheart.1992.262.6.H1699. [DOI] [PubMed] [Google Scholar]
- 11.Matsuda S, Koyasu S. Mechanisms of action of cyclosporine. Immunopharmacology. 2000;47:119–125. doi: 10.1016/s0162-3109(00)00192-2. [DOI] [PubMed] [Google Scholar]
- 12.Wilkins BJ, Dai Y-S, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT Coupling Participates in Pathological, but not Physiological, Cardiac Hypertrophy. Circulation Research. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 13.Wilkins BJ, Molkentin JD. Calcium–calcineurin signaling in the regulation of cardiac hypertrophy. Biochemical and Biophysical Research Communications. 2004;322:1178–1191. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 14.Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem. 1997;174:167–172. [PubMed] [Google Scholar]
- 15.Biasutto L, Azzolini M, Szabò I, Zoratti M. The mitochondrial permeability transition pore in AD 2016: An update. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2016;1863:2515–2530. doi: 10.1016/j.bbamcr.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 16.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung T, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, André-Fouët X, Revel D, Kirkorian G, Monassier J-P, Derumeaux G, Ovize M. Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infarction. The New England Journal of Medicine. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 17.Cung T-T, Morel O, Cayla G, Rioufol G, Garcia-Dorado D, Angoulvant D, Bonnefoy-Cudraz E, Guérin P, Elbaz M, Delarche N, Coste P, Vanzetto G, Metge M, Aupetit J-F, Jouve B, Motreff P, Tron C, Labeque J-N, Steg P, Cottin Y, Range G, Clerc J, Claeys MJ, Coussement P, Prunier F, Moulin F, Roth O, Belle L, Dubois P, Barragan P, Gilard M, Piot C, Colin P, Poli F, Morice M-C, Ider O, Dubois-Randé J-L, Unterseeh T, Breton H, Béard T, Blanchard D, Grollier G, Malquarti V, Staat P, Sudre A, Elmer E, Hansson MJ, Bergerot C, Boussaha I, Jossan C, Derumeaux G, Mewton N, Ovize M. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. The New England Journal of Medicine. 2015;373:1021–1031. doi: 10.1056/NEJMoa1505489. [DOI] [PubMed] [Google Scholar]
- 18.Bernardi P, Lisa F. Cyclosporine before PCI in Acute Myocardial Infarction. The New England journal of medicine. 2016;374:89–90. doi: 10.1056/NEJMc1514192. [DOI] [PubMed] [Google Scholar]
- 19.Richardson AP, Halestrap AP. Quantification of active mitochondrial permeability transition pores using GNX-4975 inhibitor titrations provides insights into molecular identity. Biochemical Journal. 2016;473:1129–1140. doi: 10.1042/BCJ20160070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cesura AM, Pinard E, Schubenel R, Goetschy V, Friedlein A, Langen H, Polcic P, Forte MA, Bernardi P, Kemp JA. The Voltage-dependent Anion Channel Is the Target for a New Class of Inhibitors of the Mitochondrial Permeability Transition Pore. Journal of Biological Chemistry. 2003;278:49812–49818. doi: 10.1074/jbc.M304748200. [DOI] [PubMed] [Google Scholar]
- 21.Walter L, Nogueira V, Leverve X, Heitz M-P, Bernardi P, Fontaine E. Three Classes of Ubiquinone Analogs Regulate the Mitochondrial Permeability Transition Pore through a Common Site. Journal of Biological Chemistry. 2000;275:29521–29527. doi: 10.1074/jbc.M004128200. [DOI] [PubMed] [Google Scholar]
- 22.Emter CA, Baines CP. Low-intensity aerobic interval training attenuates pathological left ventricular remodeling and mitochondrial dysfunction in aortic-banded miniature swine. Am J Physiol Heart Circ Physiol. 2010;299:H1348–1356. doi: 10.1152/ajpheart.00578.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hiemstra JA, Gutiérrez-Aguilar M, Marshall KD, McCommis KS, Zgoda PJ, Cruz-Rivera N, Jenkins NT, Krenz M, Domeier TL, Baines CP, Emter CA. A new twist on an old idea part 2: cyclosporine preserves normal mitochondrial but not cardiomyocyte function in mini- swine with compensated heart failure. Physiological Reports. 2014;2 doi: 10.14814/phy2.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klintmalm GB, Iwatsuki S, Starzl TE. CYCLOSPORIN A HEPATOTOXICITY IN 66 RENAL ALLOGRAFT RECIPIENTS. Transplantation. 1981;32:488. doi: 10.1097/00007890-198112000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- 26.Doczi J, Torocsik B, Echaniz-Laguna A, de Camaret B, Starkov A, Starkova N, Gál A, Molnár MJ, Kawamata H, Manfredi G, Adam-Vizi V, Chinopoulos C. Alterations in voltage-sensing of the mitochondrial permeability transition pore in ANT1-deficient cells. Scientific Reports. 2016;6:26700. doi: 10.1038/srep26700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1−/− mitochondria. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2006;1757:590–595. doi: 10.1016/j.bbabio.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 28.Varanyuwatana P, Halestrap AP. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion. 2012;12:120–125. doi: 10.1016/j.mito.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, Wang X, Huang T, Molkentin JD. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death & Differentiation. 2014;21:1209–1217. doi: 10.1038/cdd.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szabo I, Zoratti M. Mitochondrial Channels: Ion Fluxes and More. Physiological Reviews. 2014;94:519–608. doi: 10.1152/physrev.00021.2013. [DOI] [PubMed] [Google Scholar]
- 31.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park H-A, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA, Jonas EA. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proceedings of the National Academy of Sciences. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonora M, Bononi A, Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the FOATP synthase in mitochondrial permeability transition. Cell Cycle. 2013;12:674–683. doi: 10.4161/cc.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woodfield K, RÜCk A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochemical Journal. 1998;336:287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McStay GP, Clarke SJ, Halestrap AP. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochemical Journal. 2002;367:541–548. doi: 10.1042/BJ20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baines CP, Kaiser RA, Purcell NH, Blair SN, Osinska H, Hambleton MA, Brunskill EW, Sayen RM, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 36.Gutiérrez-Aguilar M, Baines CP. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochimica et Biophysica Acta (BBA) - General Subjects. 2015;1850:2041–2047. doi: 10.1016/j.bbagen.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costantini P, Colonna R, Bernardi P. Induction of the mitochondrial permeability transition by N-ethylmaleimide depends on secondary oxidation of critical thiol groups. Potentiation by copper-ortho-phenanthroline without dimerization of the adenine nucleotide translocase. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1998;1365:385–392. doi: 10.1016/s0005-2728(98)00090-5. [DOI] [PubMed] [Google Scholar]
- 38.García N, Martínez-Abundis E, Pavón N, Chávez E. On the Opening of an Insensitive Cyclosporin A Non-specific Pore by Phenylarsine Plus Mersalyl. Cell Biochemistry and Biophysics. 2007;49:84–90. doi: 10.1007/s12013-007-0047-0. [DOI] [PubMed] [Google Scholar]
- 39.García N, Pavón N, Chávez E. The Effect of N-Ethylmaleimide on Permeability Transition as Induced by Carboxyatractyloside, Agaric Acid, and Oleate. Cell Biochemistry and Biophysics. 2008;51:81–87. doi: 10.1007/s12013-008-9016-5. [DOI] [PubMed] [Google Scholar]
- 40.Kowaltowski AJ, Vercesi AE, Castilho RF. Mitochondrial membrane protein thiol reactivity with N-ethylmaleimide or mersalyl is modified by Ca2+: correlation with mitochondrial permeability transition. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1997;1318:395–402. doi: 10.1016/s0005-2728(96)00111-9. [DOI] [PubMed] [Google Scholar]
- 41.Halestrap AP, Woodfield K-Y, Connern CP. Oxidative Stress, Thiol Reagents, and Membrane Potential Modulate the Mitochondrial Permeability Transition by Affecting Nucleotide Binding to the Adenine Nucleotide Translocase. Journal of Biological Chemistry. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- 42.García N, Martínez-Abundis E, Pavón N, Correa F, Chávez E. Copper induces permeability transition through its interaction with the adenine nucleotide translocase. Cell Biology International. 2007;31:893–899. doi: 10.1016/j.cellbi.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 43.Baines CP, Molkentin JD. Adenine nucleotide translocase-1 induces cardiomyocyte death through upregulation of the pro-apoptotic protein Bax. Journal of Molecular and Cellular Cardiology. 2009;46:969–977. doi: 10.1016/j.yjmcc.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng E, Robbins J, Kinnally KW, Molkentin JD. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife. 2013;2 doi: 10.7554/eLife.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marchi U, Campello S, Szabò I, Tombola F, Martinou J-C, Zoratti M. Bax Does Not Directly Participate in the Ca2+-induced Permeability Transition of Isolated Mitochondria. Journal of Biological Chemistry. 2004;279:37415–37422. doi: 10.1074/jbc.M314093200. [DOI] [PubMed] [Google Scholar]
- 46.Carraro M, Giorgio V, Šileikytė J, Sartori G, Forte M, Lippe G, Zoratti M, Szabò I, Bernardi P. Channel Formation by Yeast F-ATP Synthase and the Role of Dimerization in the Mitochondrial Permeability Transition. Journal of Biological Chemistry. 2014;289:15980–15985. doi: 10.1074/jbc.C114.559633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Büttner S, Ruli D, Vögtle FN, Galluzzi L, Moitzi B, Eisenberg T, Kepp O, Habernig L, Carmona-Gutierrez D, Rockenfeller P, Laun P, Breitenbach M, Khoury C, Fröhlich KU, Rechberger G, Meisinger C, Kroemer G, Madeo F. A yeast BH3- only protein mediates the mitochondrial pathway of apoptosis. The EMBO Journal. 2011;30:2779–2792. doi: 10.1038/emboj.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brustovetsky N, Tropschug M, Heimpel S, Heidkämper D, Klingenberg M. A Large Ca2+-Dependent Channel Formed by Recombinant ADP/ATP Carrier fromNeurospora crassaResembles the Mitochondrial Permeability Transition Pore†. Biochemistry. 2002;41:11804–11811. doi: 10.1021/bi0200110. [DOI] [PubMed] [Google Scholar]
- 49.Campello S, Marchi U, Szabò I, Tombola F, Martinou J-C, Zoratti M. The properties of the mitochondrial megachannel in mitoplasts from human colon carcinoma cells are not influenced by Bax. FEBS Letters. 2005;579:3695–3700. doi: 10.1016/j.febslet.2005.05.055. [DOI] [PubMed] [Google Scholar]
- 50.Rück A, Dolder M, Wallimann T, Brdiczka D. Reconstituted adenine nucleotide translocase forms a channel for small molecules comparable to the mitochondrial permeability transition pore. FEBS Letters. 1998;426:97–101. doi: 10.1016/s0014-5793(98)00317-2. [DOI] [PubMed] [Google Scholar]
- 51.Halestrap AP, Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Current medicinal chemistry. 2003;10:1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 52.Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, Cornwall EJ. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochemical Journal. 2005;392:353–362. doi: 10.1042/BJ20050890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rottenberg H, Marbach M. Regulation of Ca2+ transport in brain mitochondria. II. The mechanism of the adenine nucleotides enhancement of Ca2+ uptake and retention. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1990;1016:87–98. doi: 10.1016/0005-2728(90)90010-2. [DOI] [PubMed] [Google Scholar]
- 54.Leung AWC, Varanyuwatana P, Halestrap AP. The Mitochondrial Phosphate Carrier Interacts with Cyclophilin D and May Play a Key Role in the Permeability Transition. Journal of Biological Chemistry. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Basso E, Petronilli V, Forte MA, Bernardi P. Phosphate Is Essential for Inhibition of the Mitochondrial Permeability Transition Pore by Cyclosporin A and by Cyclophilin D Ablation. Journal of Biological Chemistry. 2008;283:26307–26311. doi: 10.1074/jbc.C800132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kowaltowski AJ, Castilho RF, Grijalba MT, Bechara EJH, Vercesi AE. Effect of Inorganic Phosphate Concentration on the Nature of Inner Mitochondrial Membrane Alterations Mediated by Ca Ions A PROPOSED MODEL FOR PHOSPHATE-STIMULATED LIPID PEROXIDATION. Journal of Biological Chemistry. 1996;271:2929–2934. doi: 10.1074/jbc.271.6.2929. [DOI] [PubMed] [Google Scholar]
- 57.Leung A, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2008;1777:946–952. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 58.Alcalá S, Klee M, Fernández J, Fleischer A, Pimentel-Muiños FX. A high-throughput screening for mammalian cell death effectors identifies the mitochondrial phosphate carrier as a regulator of cytochrome c release. Oncogene. 2007;27:1210600. doi: 10.1038/sj.onc.1210600. [DOI] [PubMed] [Google Scholar]
- 59.McGee AM, Baines CP. Phosphate is not an absolute requirement for the inhibitory effects of cyclosporin A or cyclophilin D deletion on mitochondrial permeability transition. Biochemical Journal. 2012;443:185–191. doi: 10.1042/BJ20111881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gutierrez-Aguilar M, Baines CP. Mitochondrial ATP Synthase: Is the Molecular Engine of Life also an Efficient Death Machine. Bioenergetics. 2014;3 [Google Scholar]
- 61.Gutiérrez-Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. Journal of Molecular and Cellular Cardiology. 2014;72:316–325. doi: 10.1016/j.yjmcc.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen A-L. Mitochondrial calcium and the permeability transition in cell death. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marchenko ND, Moll UM. Mitochondrial death functions of p53. Molecular & Cellular Oncology. 2014;1 doi: 10.1080/23723548.2014.955995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karch J, Schips TG, Maliken BD, Brody MJ, Sargent MA, Kanisciak O, Molkentin JD. Autophagic cell death is dependent on lysosomal membrane permeability through Bax and Bak. eLife. 2017;6 doi: 10.7554/eLife.30543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proceedings of the National Academy of Sciences. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Halestrap AP, Richardson AP. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. Journal of Molecular and Cellular Cardiology. 2015;78:129–141. doi: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 67.Novgorodov SA, Gudz TI, Milgrom YM, Brierley GP. The permeability transition in heart mitochondria is regulated synergistically by ADP and cyclosporin A. The Journal of biological chemistry. 1992;267:16274–16282. [PubMed] [Google Scholar]
- 68.Zoratti M, Szabò I. The mitochondrial permeability transition. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes. 1994;1241 doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 69.Niedzwiecka K, Tisi R, Penna S, Lichocka M, Plochocka D, Kucharczyk R. Two mutations in mitochondrial ATP6 gene of ATP synthase, related to human cancer, affect ROS, calcium homeostasis and mitochondrial permeability transition in yeast. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2018;1865:117–131. doi: 10.1016/j.bbamcr.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 70.Beutner G, Alanzalon RE, Porter GA. Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes. Scientific Reports. 2017;7:14488. doi: 10.1038/s41598-017-14795-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gerle C. On the structural possibility of pore-forming mitochondrial FoF1 ATP synthase. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2016;1857:1191–1196. doi: 10.1016/j.bbabio.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 72.He J, Carroll J, Ding S, Fearnley IM, Walker JE. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proceedings of the National Academy of Sciences. 2017;114:9086–9091. doi: 10.1073/pnas.1711201114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proceedings of the National Academy of Sciences. 2017;114:3409–3414. doi: 10.1073/pnas.1702357114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Masgras I, Rasola A, Bernardi P. Induction of the permeability transition pore in cells depleted of mitochondrial DNA. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2012;1817:1860–1866. doi: 10.1016/j.bbabio.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 75.Zhou W, Marinelli F, Nief C, Faraldo-Gómez JD. Atomistic simulations indicate the c-subunit ring of the F1Fo ATP synthase is not the mitochondrial permeability transition pore. eLife. 2017;6 doi: 10.7554/eLife.23781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakashima RA, Dordick RS, Garlid KD. On the relative roles of Ca2+ and Mg2+ in regulating the endogenous K+/H+ exchanger of rat liver mitochondria. The Journal of biological chemistry. 1982;257:12540–12545. [PubMed] [Google Scholar]
- 77.Panel M, Ghaleh B, Morin D. Ca 2+ ionophores are not suitable for inducing mPTP opening in murine isolated adult cardiac myocytes. Scientific Reports. 2017;7:4283. doi: 10.1038/s41598-017-04618-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li B, Chauvin C, De Paulis D, De Oliveira F, Gharib A, Vial G, Lablanche S, Leverve X, Bernardi P, Ovize M, Fontaine E. Inhibition of complex I regulates the mitochondrial permeability transition through a phosphate-sensitive inhibitory site masked by cyclophilin D. Biochim Biophys Acta. 2012;1817:1628–1634. doi: 10.1016/j.bbabio.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 79.Hahn A, Parey K, Bublitz M, Mills DJ, Zickermann V, Vonck J, Kühlbrandt W, Meier T. Structure of a Complete ATP Synthase Dimer Reveals the Molecular Basis of Inner Mitochondrial Membrane Morphology. Molecular Cell. 2016;63:445–456. doi: 10.1016/j.molcel.2016.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chinopoulos C. Mitochondrial permeability transition pore: Back to the drawing board. Neurochem Int. 2017 doi: 10.1016/j.neuint.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 81.Shanmughapriya S, Rajan S, Hoffman NE, Higgins AM, Tomar D, Nemani N, Hines KJ, Smith DJ, Eguchi A, Vallem S, Shaikh F, Cheung M, Leonard NJ, Stolakis RS, Wolfers MP, Ibetti J, Chuprun JK, Jog NR, Houser SR, Koch WJ, Elrod JW, Madesh M. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol Cell. 2015;60:47–62. doi: 10.1016/j.molcel.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bernardi P, Forte M. Commentary: The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Frontiers in Physiology. 2016;7:583. doi: 10.3389/fphys.2016.00583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.König T, Tröder SE, Bakka K, Korwitz A, Richter-Dennerlein R, Lampe PA, Patron M, Mühlmeister M, Guerrero-Castillo S, Brandt U, Decker T, Lauria I, Paggio A, Rizzuto R, Rugarli EI, De Stefani D, Langer T. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Molecular Cell. 2016;64:148–162. doi: 10.1016/j.molcel.2016.08.020. [DOI] [PubMed] [Google Scholar]
- 84.He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Letters. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2. [DOI] [PubMed] [Google Scholar]
- 85.He L, Lemasters JJ. Heat Shock Suppresses the Permeability Transition in Rat Liver Mitochondria. Journal of Biological Chemistry. 2003;278:16755–16760. doi: 10.1074/jbc.M300153200. [DOI] [PubMed] [Google Scholar]
- 86.Polla BS, Kantengwa S, François D, Salvioli S, Franceschi C, Marsac C, Cossarizza A. Mitochondria are selective targets for the protective effects of heat shock against oxidative injury. Proceedings of the National Academy of Sciences. 1996;93:6458–6463. doi: 10.1073/pnas.93.13.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ribeiro SP, Villar J, Slutsky AS. Induction of the stress response to prevent organ injury. New horizons (Baltimore Md) 1995;3:301–311. [PubMed] [Google Scholar]
- 88.Zoratti M, Szabò I, Marchi U. Mitochondrial permeability transitions: how many doors to the house? Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2004;1706:40–52. doi: 10.1016/j.bbabio.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 89.Sokolove PM, Kinnally KW. A Mitochondrial Signal Peptide fromNeurospora crassaIncreases the Permeability of Isolated Rat Liver Mitochondria. Archives of Biochemistry and Biophysics. 1996;336:69–76. doi: 10.1006/abbi.1996.0533. [DOI] [PubMed] [Google Scholar]
- 90.Pfeiffer DR, Gudz TI, Novgorodov SA, Erdahl WL. The Peptide Mastoparan Is a Potent Facilitator of the Mitochondrial Permeability Transition. Journal of Biological Chemistry. 1995;270:4923–4932. doi: 10.1074/jbc.270.9.4923. [DOI] [PubMed] [Google Scholar]
- 91.Martin J, Mahlke K, Pfanner N. Role of an energized inner membrane in mitochondrial protein import. Delta psi drives the movement of presequences. J Biol Chem. 1991;266:18051–18057. [PubMed] [Google Scholar]
- 92.Gutiérrez-Aguilar M, Uribe-Carvajal S. The mitochondrial unselective channel in Saccharomyces cerevisiae. Mitochondrion. 2015;22:85–90. doi: 10.1016/j.mito.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 93.Kushnareva YE, Campo ML, Kinnally KW, Sokolove PM. Signal presequences increase mitochondrial permeability and open the multiple conductance channel. Arch Biochem Biophys. 1999;366:107–115. doi: 10.1006/abbi.1999.1190. [DOI] [PubMed] [Google Scholar]
- 94.Kushnareva YE, Polster BM, Sokolove PM, Kinnally KW, Fiskum G. Mitochondrial precursor signal peptide induces a unique permeability transition and release of cytochrome c from liver and brain mitochondria. Arch Biochem Biophys. 2001;386:251–260. doi: 10.1006/abbi.2000.2201. [DOI] [PubMed] [Google Scholar]
- 95.Uribe-Carvajal S, Luevano-Martinez LA, Guerrero-Castillo S, Cabrera-Orefice A, Corona-de-la-Pena NA, Gutierrez-Aguilar M. Mitochondrial Unselective Channels throughout the eukaryotic domain. Mitochondrion. 2011;11:382–390. doi: 10.1016/j.mito.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 96.von Stockum S, Basso E, Petronilli V, Sabatelli P, Forte MA, Bernardi P. Properties of Ca(2+) transport in mitochondria of Drosophila melanogaster. J Biol Chem. 2011;286:41163–41170. doi: 10.1074/jbc.M111.268375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.von Stockum S, Giorgio V, Trevisan E, Lippe G, Glick GD, Forte MA, Da-Re C, Checchetto V, Mazzotta G, Costa R, Szabo I, Bernardi P. F-ATPase of Drosophila melanogaster forms 53-picosiemen (53-pS) channels responsible for mitochondrial Ca2+-induced Ca2+ release. J Biol Chem. 2015;290:4537–4544. doi: 10.1074/jbc.C114.629766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Azzolin L, Basso E, Argenton F, Bernardi P. Mitochondrial Ca2+ transport and permeability transition in zebrafish (Danio rerio) Biochim Biophys Acta. 2010;1797:1775–1779. doi: 10.1016/j.bbabio.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 99.Scott I, Logan DC. Mitochondrial morphology transition is an early indicator of subsequent cell death in Arabidopsis. New Phytol. 2008;177:90–101. doi: 10.1111/j.1469-8137.2007.02255.x. [DOI] [PubMed] [Google Scholar]
- 100.Zoratti M, Marchi U, Biasutto L, Szabò I. Electrophysiology clarifies the megariddles of the mitochondrial permeability transition pore. FEBS Letters. 2010;584:1997–2004. doi: 10.1016/j.febslet.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 101.Dedkova EN. Inorganic polyphosphate in cardiac myocytes: from bioenergetics to the permeability transition pore and cell survival. Biochemical Society Transactions. 2016;44:25–34. doi: 10.1042/BST20150218. [DOI] [PubMed] [Google Scholar]
- 102.Szabó I, Zoratti M. The mitochondrial megachannel is the permeability transition pore. Journal of Bioenergetics and Biomembranes. 1992;24:111–117. doi: 10.1007/BF00769537. [DOI] [PubMed] [Google Scholar]
- 103.Brenner C, Moulin M. Physiological Roles of the Permeability Transition Pore. Circulation Research. 2012;111:1237–1247. doi: 10.1161/CIRCRESAHA.112.265942. [DOI] [PubMed] [Google Scholar]
- 104.Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, Grabarek Z, Kong L, Liu Z, Ouyang B, Cong Y, Mootha VK, Chou JJ. Architecture of the mitochondrial calcium uniporter, ,jt>Nature. 2016;533:269–273. doi: 10.1038/nature17656. [DOI] [PMC free article] [PubMed] [Google Scholar]