

Abstract
Mass spectrometry (MS)-based isobaric labeling has developed rapidly into a powerful strategy for high throughput protein quantification. Sample multiplexing and exceptional sensitivity allow for the quantification of tens of thousands of peptides and by inference thousands of proteins from multiple samples in a single mass spectrometry experiment. Accurate quantification demands a consistent and robust sample preparation strategy. Here, we present a detailed workflow for SPS-MS3-based quantitative abundance profiling of Tandem Mass Tag (TMT)-labeled proteins and phosphopeptides, which we have termed the Streamlined (SL)-TMT protocol. We describe a universally-applicable strategy that requires minimal individual sample processing and permits the seamless addition of a phosphopeptide enrichment step (“mini-phos”) with little deviation from the deep proteome analysis. To showcase our workflow, we profile the proteome of wild-type S. cerevisiae yeast grown with either glucose or pyruvate as the carbon source. Here, we have established a streamlined TMT protocol that enables deep proteome and medium-scale phosphoproteome analysis.
Keywords: Sample preparation, Multi-Notch, SPS, Synchronous precursor selection, Tandem mass tag, Orbitrap Fusion Lumos, Phosphoproteome
TOC image
INTRODUCTION
Mass spectrometry-based technology is at the forefront of proteomics research. Bottom-up proteomics is among the most commonly adopted proteome profiling strategies (1). This technique characterizes peptide sequences from proteolytic digests by mass spectrometry analysis and subsequently assembles peptides into protein identifications. Coincidently, various technologies have emerged, such as isobaric labeling, that can quantitatively profile these proteins in perturbed systems.
Tandem Mass Tag (TMT) reagents are NHS ester-based isobaric labels that are designed for concurrent analysis of multiple samples (2). Each sample is differentially labeled with a specific TMT tag, having the same total mass, but with mass deviations in the reporter and balancer regions of the molecule (3). The TMT signal-to-noise values of sample-specific reporter ions represent the relative abundance of each protein, allowing comparisons across all pooled samples. Although the technique has the advantages of high-throughput sample processing and multiplicity, one inherent limitation of TMT in MS2-based quantification is reporter ion interference from the co-fragmentation of different co-isolated ion species (4). However, synchronous precursor selection (SPS)-MS3 technology (4, 5), has been successful in dampening this drawback. Performing SPS-MS3 divorces identification (MS2) and quantification (MS3) into separate scans that can be optimized individually to comprehensibly profile peptide and protein abundance.
Numerous sample preparation protocols are readily available and diverse instrument settings have been published detailing TMT-SPS-MS3 strategies. However, little consistency in methodology exists that can provide a bona fide reference protocol to researchers. For example, certain protocols suggest desalting each sample prior to labeling (6), which adds time, cost, and the potential for sample loss to the procedure. Differences in digestion buffer (7) and precipitation techniques (8) are also common. In addition, up to 10 to 20 mg of protein starting material is frequently recommended for phosphopeptide enrichment, which is often unobtainable in sample-limited assays (9, 10).
Here, we describe a simple, yet universally applicable sample preparation strategy, which incorporates an optional phosphopeptide enrichment step, thereby allowing for deep proteome and medium-scale phosphoproteome analysis. We provide a detailed protocol, which in our hands, has improved data quality and increased proteome depth and throughput.
As an application to showcase our method, we compared the proteome of the budding yeast S. cerevisiae grown in glucose- or pyruvate- containing medium. We determined the subset of significantly altered proteins, which we annotated using gene ontology classifications. Physiology, glucose is an important carbon source and changes in its availability affects levels of certain metabolites, mRNAs, and proteins in the cell. Glucose targets the repression or activation of several genes, such that a set of canonical proteins will be activated, inactivated or degraded in presence of glucose (11). To analyze further these data, we explored the behavior of canonical up- and down-regulated proteins in response to growth in glucose or pyruvate. By altering the carbon source, we expected metabolic changes which should be reflected in the tricarboxylic acid cycle (12, 13), which we indeed observed. Our data showed that this optimized and streamlined SPS-MS3 protocol is a robust protein and phosphopeptide quantification strategy, and as such is an asset to any proteomics toolbox.
EXPERIMENTAL SECTION
Materials
Tandem Mass Tag (TMT) isobaric reagents, Pierce/BCA Protein Concentration Kit, Pierce Quantitative Colorimetric Peptide Assay Kit, Trypsin, SOLA-HRP desalting columns, and High-Select Fe-NTA Phosphopeptide Enrichment Kit were from ThermoFisher Scientific (Rockford, IL). StageTip Empore-C18 material was purchased from 3M (St. Paul, MN). Waters Sep-Pak cartridges (100 mg) were from Waters (Milford, MA). Lys-C protease was from Wako (Boston, MA). Water and organic solvents were from J.T. Baker (Center Valley, PA). cOmplete protease and PhosStop phosphatase inhibitors were from MilliporeSigma (St. Louis, MO). The yeast strain was used was BY4716 from ThermoFisher Scientific (Waltham, MA). Yeast synthetic complete media was from Sunrise Science (San Diego, CA). Unless otherwise noted, all other chemicals were from ThermoFisher Scientific (Waltham, MA).
Yeast growth and sample processing
We showcased our method using a TMT10-plex of yeast (S. cerevisiae wild-type strain BY4716) grown in synthetic complete media supplemented with 2% glucose (n=5) or 2% pyruvate (n=5) as the carbon source. We harvested the cells at OD600nm=0.8. Cells were lysed by bead-beating in 8 M urea 200mM EPPS (4-(2-Hydroxyethyl)-1-piperazinepropanesulfonic acid), pH 8.5 and with protease and phosphatase inhibitors. Protein concentration was determined with the BCA assay. The BCA assay was performed according to manufacturer’s instructions with samples that were diluted at least 1:20, to ensure that the 8M urea has been diluted far below its compatibility limit. Samples were reduced with 5mM TCEP, alkylated with 10 mM iodoacetamide that was quenched with 10 mM DTT. A total of 100 μg of protein were chloroform-methanol precipitated. Protein was reconstituted in 200 mM EPPS pH 8.5 and digested by Lys-C overnight and trypsin for 6 h, both at a 1:100 protease-to-peptide ratio. Directly to the digest, we added a final volume of 30% acetonitrile and labelled 100 μg of peptide with 200 μg of TMT. To check mixing ratios, 2 μg of each sample were pooled, desalted, and analyzed by mass spectrometry. Using normalization factors calculated from this “label check,” samples were mixed 1:1 across all channels and desalted using a 100 mg Sep-Pak solid phase extraction column. The Pierce High-Select Fe-NTA Phosphopeptide Enrichment Kit was used to enrich phosphopeptides from the pooled TMT-labeled mixture. The unbound fraction and washes from this enrichment were combined and fractionated with basic pH reversed-phase (BPRP) HPLC, collected in a 96-well plate and combined down to 12 fractions prior to desalting and subsequent LC-MS/MS processing (14, 15).
Mass spectrometry analysis
Mass spectrometric data were collected on an Orbitrap Fusion Lumos mass spectrometer in-line with a Proxeon NanoLC-1200 UHPLC. The 100 μm capillary column was packed with 35 cm of Accucore 150 resin (2.6 μm, 150Å; ThermoFisher Scientific). Spectra were converted to mzXML using a modified version of ReAdW.exe. Database searching included all entries from the Saccharomyces Genome Database (SGD; August 21, 2017). This database was concatenated with one composed of all protein sequences in the reversed order. Searches were performed using a 50-ppm precursor ion tolerance for total protein level profiling. The product ion tolerance was set to 0.9 Da. These wide mass tolerance windows were chosen to maximize sensitivity in conjunction with SEQUEST searches and linear discriminant analysis (16, 17). TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification. For phosphorylation analysis, deamidation (+0.984) on asparagine and glutamine and phosphorylation (+79.966) on serine, threonine, and tyrosine were set as variable modifications. Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR) (18, 19). PSM filtering was performed using a linear discriminant analysis, as described previously (17) and then assembled further to a final protein-level FDR of 1% (19). Phosphorylation site localization was determined using the AScore algorithm (13). AScore is a probability-based approach for high-throughput protein phosphorylation site localization. Specifically, a threshold of 13 corresponded to 95% confidence in site localization. Proteins were quantified by summing reporter ion counts across all matching PSMs, as described previously (20). Reporter ion intensities were adjusted to correct for the isotopic impurities of the different TMT reagents according to manufacturer specifications. The signal-to-noise (S/N) measurements of peptides assigned to each protein were summed and these values were normalized so that the sum of the signal for all proteins in each channel was equivalent, to account for equal protein loading. Lastly, each protein was scaled such that the summed signal-to-noise for that protein across all channels was greater than 100, thereby generating a relative abundance (RA) measurement. A detailed description of the methods in a step-by-step outline is available in the Supplementary Materials.
RESULTS AND DISCUSSION
We describe an efficient and streamlined sample preparation strategy that can be adopted easily to virtually any TMT-based quantitative proteomics experiment (Figure 1). Each sample was processed in a one-pot format in which digestion and TMT labeling shared a common buffer (200mM EPPS, pH 8.5). The method required only one C18 desalting step from cell lysis to HPLC fractionation. We seamlessly incorporated centrifugation-based phosphopeptide enrichment into this protocol to analyze alterations in protein phosphorylation under the different carbon sources. This procedure was summarized in the Methods section and step-by-step details are in the Supplementary Methods. Supplemental Figure 1 outlined the major steps in the protocol, and included the approximate time required for each step. Below we showcased our protocol by comparing yeast cultures grown in synthetic complete media with 2% glucose or 2% pyruvate as the carbon source. The TMT10-plex experiment was organized in a five-versus-five sample arrangement (Supplemental Figure 2).
Figure 1. General TMT protocol overview.
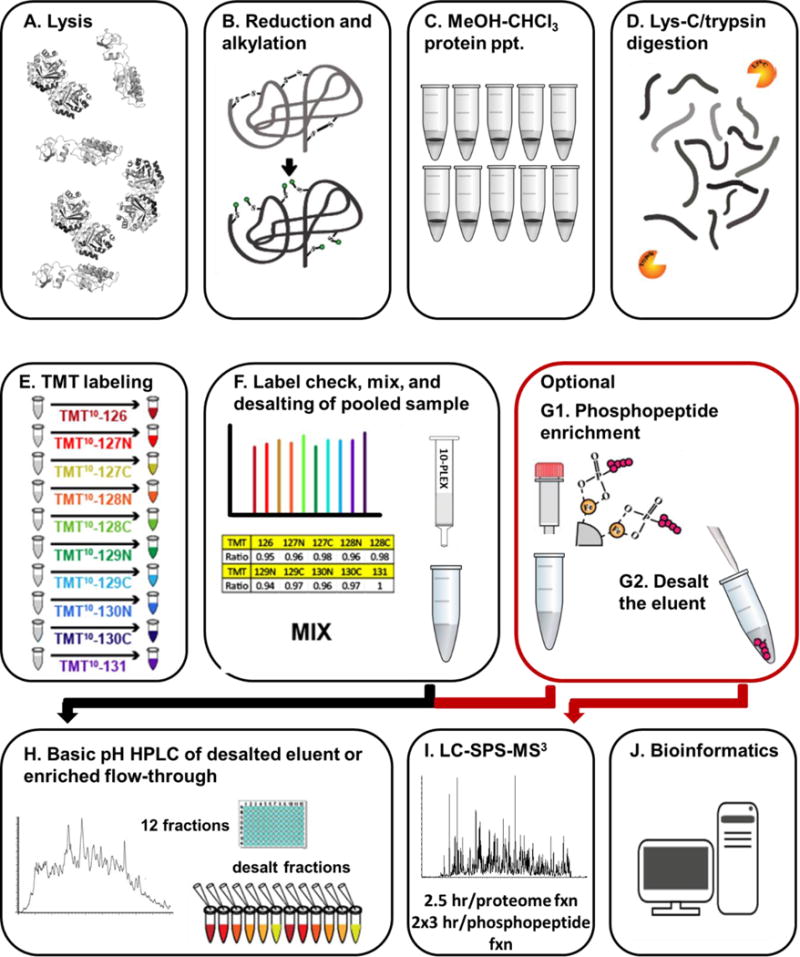
A) Cells were lysed after which B) cysteine bonds were reduced and alkylated. C) Methanol-chloroform precipitation was performed to extract proteins which were D) digested using Lys-C followed by trypsin. E) The resulting peptides were labeled with TMT and F) a “label check” ensured that samples will be mixed 1:1 across all channels and a single desalting step is performed. G) Optionally, the dried, mixed, desalted sample was subjected to centrifugation-based phosphopeptide enrichment and desalted for SPS-M3 analysis. The flow-through from this enrichment was desalted and H) fractionated by basic pH reversed-phase (BPRP) HPLC. The fractions were desalted by StageTip and I) analyzed by SPS-MS3. J) Database searching, and reporter ion quantification was performed. In addition, bioinformatics analysis extracted meaningful biological information from the protein and phosphopeptide data.
Cell lysis, cysteine bond reduction and alkylation, and protein concentration measurements were performed in a urea-containing buffer
Efficient cell lysis is essential for deep coverage of cellular proteomes (Figure 1A). Lysis may be performed using 8M urea 200 mM EPPS pH 8.5, as described herein. However, virtually any other buffer, such as RIPA buffer or 2% SDS, was compatible with our protocol. We used urea as it was effortlessly removed for downstream mass spectrometry applications. The buffer was supplemented with protease and phosphatase inhibitors. We estimated lysis buffer volume that would result in a protein concentration of 1-3 mg/mL, which we empirically determined to be ideal for protein precipitations. For example, adding 1 mL of lysis buffer to a cell pellet of a 100 mL S. cerevisiae culture having an OD600nm reading of 0.8 resulted in ~3 mg/mL of protein. Here we used yeast, but for mammalian cell culture, we typically add 1 mL of lysis buffer to a 15-cm 80% confluent culture dish of cells and 500 μL to a 10-cm dish. It was important to note that this amount of lysis buffer used varies with respect to cell type and confluency and must be determined empirically for a specific experiment. The bicinchoninic acid (BCA) protein concentration assay was incompatible with reducing reagent, thereby requiring the reduction and alkylation of samples after determining protein concentration. Proteins solubilized in 8M urea-containing buffers were prone to carbamylation if exposed to elevated temperatures. To avoid this caveat, we reduced disulfide bonds with neutralized TCEP, alkylated with iodoacetamide (IAA), and quenched with DTT, all at room temperature (Figure 1B). The protein was then extracted from the lysed cells prior to digestion to ensure high efficiency of TMT labeling.
For each sample, 100 μg of protein was methanol-chloroform precipitated and then digested
Between the time of lysis and methanol-chloroform precipitation, the sample is stored at room temperature. As the actual precipitation step is also performed at room temperature, we have no urea precipitation throughout the protocol. We aliquoted 100 μg of protein which were diluted in a total of 100 μL of lysis buffer for methanol-chloroform precipitation (Figure 1C). Using 100 μg of protein at a 1 mg/mL concentration was key to streamlining the methanol-chloroform precipitation protocol. Following the first centrifugation of a methanol-chloroform precipitation procedure, a protein interface formed between the aqueous and organic layers. Interestingly, 75-100 μg of protein generally produced a thin uniform disk at this interface in a standard 1.5 mL microfuge tube. To eliminate one washing step, we aspirated both layers, carefully rotating the microfuge tube so that the protein disk attached to the tube wall, thereby allowing access to, and aspiration of, the bottom layer. Traditional methanol-chloroform precipitation includes two methanol washing steps after the protein disk is formed. As we remove both liquid layer and leave only the pellet in the tube, we can omit the second methanol wash as only trace residue remains, which has no effect on digestion efficiency and reduces sample loss. Moreover, the pellet was not allowed to dry completely following the wash, so as to not hinder protease digestion. Typically, ~10 μL of methanol remained following protein precipitation. Digestion was performed in a 100 μL volume at a protein concentration of approximately 1 mg/mL. We used 200 mM EPPS pH8.5 for digestion and TMT labeling. Although HEPES and TEAB are often recommended and can be used for TMT labeling, pH 8.5 is beyond the buffering capacity of HEPES, and we have observed contaminant peaks when TEAB buffer is used. Also, using a denaturant-free buffer eliminated an extraneous desalting step prior to TMT labeling. Lys-C was added at a 1:100 protease: protein ratio overnight at room temperature, shaking gently on a vortex (Figure 1D). Trypsin was then added at a 1:100 ratio and the sample was digested further on a 37°C shaker at a 45° angle (to avoid particulates settling at the bottom of the tube) for 6 hrs.
A single desalting step was performed on the pooled TMT10-plex sample after TMT labeling and protein-loading normalization
TMT labeling was performed directly in the digestion buffer (Figure 1E). We avoided buffer exchange on individual samples as was outlined in other TMT protocols (6), thereby sidestepping variable sample losses due to separate desalting steps. As such, no buffer exchange or desalting was required until all samples were combined (i.e., prior to HPLC fractionation). We added anhydrous acetonitrile to a final volume of 30% and 5 μL of a 20 μg/μL dilution of the appropriate TMT label to 100 μg of sample. The reaction was incubated for 60 minutes, with a quick vortex every 10 minutes. Prior to quenching the TMT reaction, a “label check” was performed to ensure that the total amount of protein in each channel was equal, the labeling efficiency was high, and the digestion continued to completion. The TMT-compatible Pierce Quantitative Colorimetric Peptide Assay kit can also verify that equal amounts of total protein was analyzed in each sample. Under certain experimental conditions, however, such as immunoprecipitations, equal loading may not be expected and normalization to total protein is counterproductive. In such instances, not normalizing, or normalizing to a specific protein (e.g., a bait from an affinity precipitation) would be recommended. Nonetheless, for proteome profiling experiments, slight adjustments were required to equate the total amount of protein in each channel, due to slight variability in protein precipitation. The “label check” revealed that the TMT labeling efficiency (>98%) and missed cleavage rate (<9%) were acceptable, thus we quenched unreacted TMT with 0.03% hydroxylamine and the samples were pooled (Figure 1F). Hydroxylamine also reversed undesired labeling of tyrosine residues (21). Quenching of unreacted TMT is not performed until after the “label check” has been analyzed. As such, more label can be added if necessary without requiring buffer exchange. The labeled samples are stored at −80°C until the “label check” has been analyzed. Once samples are pooled, desalting with solid phase extraction (SPE) was performed using C18-based SEP-PAK or SOLA-HRP cartridges prior to the phosphopeptide enrichment (optional) and basic pH reversed-phase (BPRP) fractionation.
Fe-NTA centrifugation-based columns were used to enrich phosphopeptides after sample pooling and before proteome fractionation
Previously-published protocols simply desalted and fractionated the pooled sample via basic pH reversed-phase chromatography prior to analysis by mass spectrometry (13–15). Recently, simple and efficient centrifugation-based phosphopeptide enrichment has been introduced commercially as the Pierce Fe-NTA Phosphopeptide Enrichment Kit (Figure 1G), which has been shown to have a very high recovery rate (22). These convenient IMAC spin columns can process 50 μg to 5 mg of peptide and can yield up to 150 μg of phosphopeptides. Incorporation of this phosphopeptide enrichment step (which we term “mini-phos”) into a standard TMT workflow required only a single additional sample preparation step. The phosphopeptides from 1 mg of pooled, TMT-labeled peptides were enriched, while the unbound peptides and washes were saved for deep proteome analysis. The enriched phosphopeptides were desalted via StageTip and two separate 3 hr SPS-MS3 analyses were performed. Two distinct mass spectrometry methods were used, each exploiting differences in phosphopeptide fragmentation, with one method using collision-induced dissociation coupled to multistage activation (CID-MSA) and another using higher energy collisional dissociation (HCD) for the MS2, as well as the MS3, scan. Details of the specific parameters are available in the Supplementary Methods.
Basic pH reversed-phase (BPRP) fractionation allowed for deep proteome analysis
Following enrichment, the unbound fraction plus the washes were combined, desalted, and fractionated via basic pH reversed-phase chromatography (Figure 1H), as described previously (13) and outlined in the Supplementary Methods. For this full proteome analysis, a total of 96 fractions were collected and concatenated so that every 24th fraction was pooled (i.e., samples in wells A1, C1, E1, and G1 were combined) and only alternating pooled fractions (a total of 12) were analyzed (13). Alternatively, if <300 μg of total peptide was to be fractionated, comparable proteome depth may be achieved using Pierce high-pH reversed-phase spin columns according to manufacturer’s instructions (23). Each peptide fraction was analyzed using a 2.5 hr SPS-MS3 method (Figure 1I).
Database searching, peptide quantification, and protein assignments may be performed with various analysis tools
Following data acquisition, database searching can be performed using various open source and commercial search engines (Figure 1J). We used an in-house SEQUEST-based suite, most aspects of which have been implemented in ProteomeDiscoverer (ThermoFisher Scientific). Other software packages, such as MaxQuant (24, 25) or PEAKS (26), can also analyze SPS-MS3 data.
Here we describe briefly our data processing strategy. Database searching was performed with SEQUEST. Our peptide false discovery rate was set at 1%. For peptide quantification, we extracted the TMT signal-to-noise and column normalized each channel to correct for equal protein loading. We implemented two peptide cut-off filters: isolation purity (>0.7) and summed signal-to-noise (>100 S:N summed over all channels in a TMT10-plex). Isolation purity is the percentage of the signal in the MS1 isolation window that originated from the peptide of interest. The TMT signal-to-noise (S:N) value for peptides passing these criteria were summed, thereby giving more weight to the most intense peptides. The data were scaled such that the signal-to-noise value of the reporter ions for each protein across all 10 channels was 100. We summarized the data for protein and phosphorylation sites acquired for the yeast experiment in Figure 2A and Figure 3A. Our TMT10-plex proteome analysis quantified a total of 71,160 peptides, of which 44,499 were unique. These peptides were assigned to a total of 4,613 non-redundant proteins at a 1% false discovery rate (FDR). From our medium-scale TMT10-plex phosphoproteome analysis, we quantified a total of 4,589 phosphorylation events.
Figure 2. Summary of deep proteome data set.
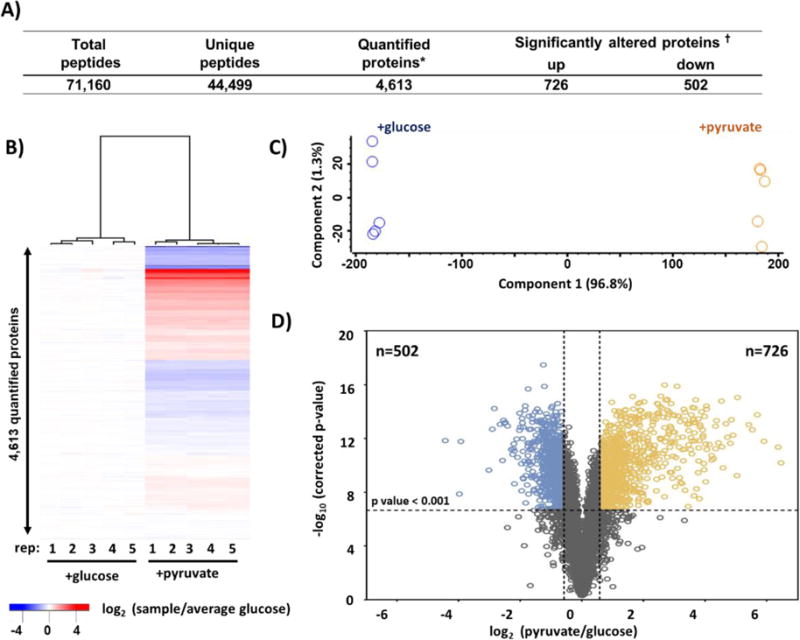
A) Table of quantified proteins in this data set. B) Heatmap of hierarchical clustering and B) PCA (principal components analysis) of the replicate samples in the TMT10-plex that plots principal component 1 versus principal component 2. C) Volcano plot displaying the −log10 (p – value) versus log2 (average pyruvate/average glucose) for all quantified proteins. * Quantified across all 10 channels. † Benjamini-Hochberg-corrected p-value<0.001 and a fold change beyond ± 1.75.
Figure 3. Summary of medium-scale phosphoproteomics data set.
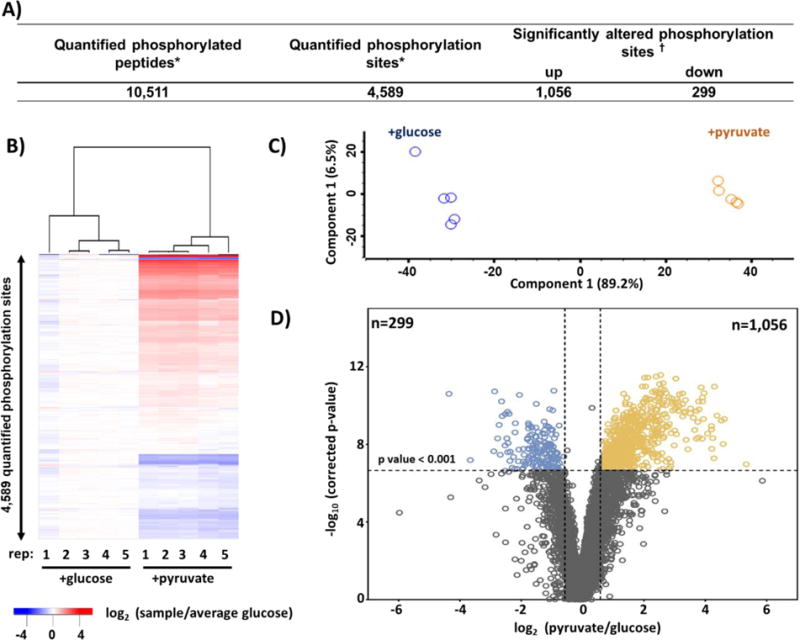
A) Table of quantified phosphopeptides in this data set. B) Heatmap of hierarchical clustering and C) PCA (principal components analysis) of the replicate samples in the TMT10-plex that plots principal component 1 versus principal component 2. D) Volcano plot displaying the −log10 (p – value) versus log2 (average pyruvate/average glucose) for all quantified phosphopeptides. * Quantified across all 10 channels. † Benjamini-Hochberg-corrected p-value<0.001 and a fold change beyond ± 1.75.
Bioinformatic tools can extract meaningful biological information and aid in drawing insightful conclusions from these data
Many methods are available to determine proteins demonstrating statistically significant changes in abundance, including linear modeling (27, 28) or simply the application of a two-sided t-test and fold change threshold (29). Statistical analysis may be performed in R with Bioconductor, for example, or other software packages such as MatLab and/or Mathematica. Orthogonal validation by other means, such as protein microarrays, western blotting, or parallel reaction monitoring, for example should be performed for targets to be pursued further regardless of the statistical method chosen.
Here, student T-tests are performed for each protein or phosphorylation site measurement and a p-value is chosen a priori. In our case, a p-value of 0.001 was chosen that implies 0.1% chance of false positive. However, this chance of a false positive was per test, as such if one performs more than one test, the overall false positive rate for the entire dataset increases (30). As thousands of statistical tests are performed within an experiment, multiple testing corrections - such as the Benjamini-Hochberg false discovery rate (31) or more stringent Bonferroni correction (32) - should be applied to ensure that the false positive rate was low. In addition to the FDR correction, a fold change cut off is used to remove proteins (or phosphorylation sites) that are statistically different, but with absolute differences that are small. The fold change cut-off was estimated after computing the coefficient of variation (CV) of each protein in the data set, as suggested previously (29). Only proteins with CVs greater than the 97th percentile were considered significantly changing. To be deemed a significantly altered protein, criteria included a Student’s t-test with a Benjamini-Hochberg-corrected p-value<0.001 and a fold-change beyond +/− 1.75. Of the 4,613 proteins identified in our dataset, 502 and 726 were significantly low or high, respectively, in abundance (Figure 2A). Likewise, of the 4,589 phosphorylation events quantified, we determined 299 and 1,056 of these phosphorylation events to be significantly low or high, respectively, again accepting a Student’s t-test with a Benjamini-Hochberg corrected p-value<0.001 and a fold change beyond +/− 1.75 (Figure 3A).
Data from proteomic workflows may also be analyzed with software suites originally used for genetic research, such as MeV (Multi-experiment viewer) (33) or GSEA (gene set enrichment analysis) (34). In addition, the widely-used Perseus (35) is a proteomics-centric stand-alone software program that can be used for the analysis of data obtained from virtually any mass spectrometry platform. These programs are also useful to assess data quality. For example, heat maps, principal components analysis (PCA), and hierarchical clustering under the appropriate conditions will reveal the agreement among replicates, cell type and/or response to exogenous perturbations. In addition, data are commonly displayed in two dimensions as a volcano plot (−log10 p-value vs. log2 fold change).
In our example of yeast grown in glucose versus pyruvate, we showed for the protein level analysis: a heatmap of hierarchical clustering (Figure 2B), PCA plot (Figure 2C), and a volcano plot displaying the −log10 of p-value for all quantified proteins versus log2 (pyruvate/glucose) (Figure 2D). PCA can reduce multidimensional data in efforts to interpret the results by discovering clusters and important patterns (36). The heatmap and associated dendrogram show the expected clustering of replicates. Similarly, the PCA analysis shows that 96.8% of the variance can be explained by the first principle component which separates the samples by carbon source. The volcano plot illustrated the 502 proteins with significantly lower abundance (top left quadrant) and the 726 proteins with significantly higher abundance (top right quadrant). Similarly, for the phosphoproteome analysis, we showed: a heatmap (Figure 3B), PCA plot (Figure 3C), and a volcano plot (Figure 3D). The heatmap and associated dendrogram revealed the expected clustering of replicates, similar to the protein level analysis. Likewise, the PCA illustrated that 89.2% of the variance can be explained by the first principle component which separated the samples by carbon source. The volcano plot illustrated the 299 phosphorylation events with significantly lower abundance (top left quadrant) and 1,056 phosphorylation events with significantly higher abundance (top right quadrant).
Discriminating between differences in signaling events and alterations in protein expression is imperative when profiling phosphoproteome changes. In this data set, for example, of the 1,094 phosphorylation sites that were significantly different and had corresponding protein level quantification. Of these, approximately one third (356) showed significantly different levels in protein quantification as well. We provided several examples of protein and phosphopeptide abundance ratios overlaid in a single plot (Figure 4). These proteins have different regulation patterns between cells grown in glucose or pyruvate. Specifically, we showcased the abundance profiles of 12 phosphorylation sites that are each associated with one of five proteins, specifically: NUP159 (NUclear Pore 159), DCP2 (mRNA DeCaPping 2), RGA1 (Rho GTPase Activating Protein 1), PCK1 (Phosphoenolpyruvate CarboxyKinase 1), and DDR48 (DNA Damage Responsive 48). Except for PCK1 and DDR48, these proteins had consistent protein abundance levels between carbon sources but patterns for phosphorylation sites differ. NUP159 is a nucleoporin component of the nuclear pore complex and contributes directly to nucleocytoplasmic transport. Here, two sites increased similarly in the pyruvate-grown cultures, while another peptide remained constant with the protein level (Figure 4A-C). Also, for DCP2, a transcription initiating protein, two sites increased differentially in the pyruvate-grown cultures, while one remained unchanged (Figure 4C-F). For RGA1, a GTPase-activating protein, the phosphorylation at one site increased, while two other sites remained constant, like the corresponding protein profile (Figure 4G-I). However, we note that a site on PCK1 (an enzyme involved in gluconeogenesis) was among the most highly altered phosphorylation sites (Figure 4J). However, the phosphorylation site profile was well correlated with the protein itself, likely meaning that changes observed in the phosphorylation profiles may have been due to increased protein abundance rather than signaling. A phosphorylation site on DDR48, a DNA damage-response protein, had a distinctly different profile when compared to its protein level. Whereas the protein abundance was elevated when the cultures were grown in pyruvate, phosphorylation of this site decreased. Additional studies will be needed to determine the role of this site regarding its altered profile when cultures are grown on pyruvate. The examples highlighted in Figure 4 illustrated the importance of determining the protein abundance profiles that corresponded to altered phosphorylation sites, as was integrated into our method when analyzing the unbound fraction from the phosphopeptide enrichment column.
Figure 4. Examples of phosphorylation sites and associated protein levels.
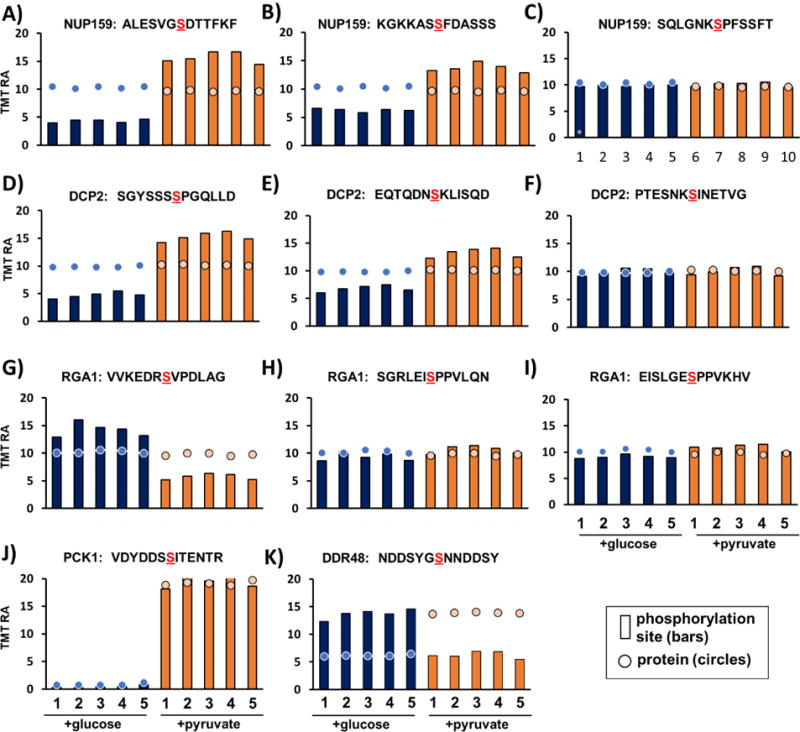
Phosphorylation site profiles (bars) were overlaid on its associated protein profiles (circles) showing phosphorylation event profile for yeast culture with glucose (purple) and pyruvate (orange) as the carbon source. We provide examples of the protein and phosphorylation site relative abundance profiles for: A) - C) NUP159, D) - F) DCP2, G) - I) RGA1, J) PCK1, and K) DDR48. TMT RA, tandem mass tag relative abundance.
In addition, statistically significant proteins or phosphorylation events can be explored further using a wide array of on-line tools. Web server-based software such as Panther (37), GOrilla (38), DAVID (39), Bioplex (40), and KeGG (41) allowed us to extract biological information from the collected data. Information gained from these databases allowed for the classification of proteins based on their molecular function, cellular compartment, associated biological pathways and protein-protein interactions, among other characteristics. For phosphorylation site analysis, several on-line programs were also available to explore further phosphorylation sites, related kinases, and associated networks, including Signor (42), Networkin/Kinforest (43), and PhosphoSitePlus (44). In particular, PhosphoSitePlus incorporates an expansive array of tools, such as motif searching and a sequence logo generator.
In addition, we used PantherGo-slim biological processes (Fisher’s Exact Test with FDR multiple test correction threshold of 5%) to illustrate the gene ontology enrichment categories for all quantified proteins in our data set (Figure 5A). As expected, when changing the culture’s carbon source, the majority of the significantly altered proteins participate in metabolic processes. In addition, we showed the canonical down- and up-regulated proteins (Figures 5B and C) in response to pyruvate versus glucose as the carbon source follow predictable patterns. Again, we observed that many of the proteins with altered abundance fell under the metabolic process category. As such, we explored further the relative abundance profiles of Krebs Cycle proteins in response to glucose as the carbon source (Figure 5D). The heatmaps compared the abundance profiles of Krebs Cycle proteins from cultures grown with pyruvate or glucose as the carbon source. Once again, as expected, enzymes that metabolize pyruvate were up-regulated generally in samples in which pyruvate was used as the carbon source (45).
Figure 5. Bioinformatics data analysis.
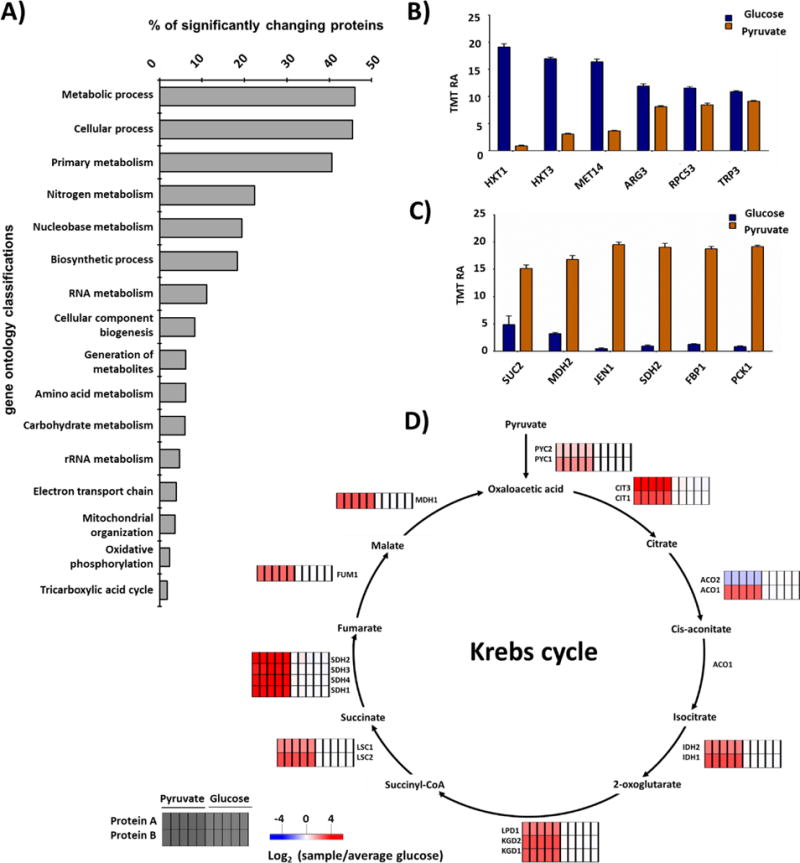
A) Gene ontology enrichment categories for all quantified proteins showing significant difference in protein abundance in this data set. The S. cerevisiae genome is used as the background. Canonical B) down- and C) up-regulated proteins in response to pyruvate versus glucose as the carbon source. Bars represent mean ± S.E.M, n=5. D) Krebs Cycle diagram showing the relative abundance of associated enzymes. S.E.M., standard error of the mean.
Conclusion
We have outlined our streamlined sample preparation workflow, SL-TMT, for SPS-MS3 analysis, which now seamlessly incorporated a medium-scale phosphopeptide enrichment. Our methodology allowed for the completion of an end-to-end TMT-based deep proteome analysis and an overview of the major phosphoproteome alterations in less than a typical workweek. This protocol has evolved over time, as demonstrated by a series of previous publications (13, 15, 46–48), all of which resulted in among the deepest TMT-based proteome analyses at that time. The single desalting step in our protocol was performed on the combined sample, thereby decreasing the variability among samples. As such, following the BCA protein assay, only the methanol-chloroform precipitation step had any associated sample-to-sample processing variability. Moreover, the incorporation of FPD (49) or FASP (50) into this workflow may help eliminate the variability due to precipitation. Although we implemented a phosphorylation enrichment step to our already streamlined TMT workflow, further improvements may enhance this analysis. Relatively few phosphorylation sites were identified by this “mini-phos” method compared to other published works (48, 51). Several enrichment strategies have been performed without prior sample fractionation including those using metal oxides or ions (52–57) and using motif-specific antibodies (58). However, we begin with less than 1 mg of total protein, representing 10 to 100 times less starting material than used in other methods. In addition, fractionation directly from the final StageTip desalting, for example with steps of 10%, 20%, and 70% acetonitrile or using the TAFT (59) technique may increase further the number of quantified phosphopeptides. In addition, application of MS3-based identification and quantification (MS3-IDQ) may be beneficial if specific peptides are targeted (60, 61). As with any methodology, room for improvement persists, yet our streamlined TMT protocol offered a solid scaffold upon which others may build. In summary, we have presented an optimized TMT-based workflow (SL-TMT) that required minimal individual sample processing but enabled high-coverage proteomic and medium-scale phosphoproteomic analyses.
Supplementary Material
Supplemental Table 1: Proteins quantified in this dataset. Columns include: SGD protein identifier (proteinID), gene symbol (Gene Symbol), protein description/name (Description), number of peptides identified per protein (peptides), the normalized summed signal-to-noise for each of the 10 channels (126 to 131).
Supplemental Table 2: Peptides quantified in this dataset. Columns include: SGD protein identifier (proteinID), gene symbol (Gene Symbol), protein description/name (Description), redundancy, whether peptide is a unique or razor peptide, peptide sequence, and the summed signal-to-noise for each of the 10 channels (126 to 131). For each peptide, we have included the cross-correlation score (XCorr), the delta correlation (ΔCN), and peptide mass difference (PPM).
Supplemental Table 3: Phosphorylation sites quantified in this dataset. Columns include: type of site (single or composite), SGD protein identifier (proteinID), gene symbol (Gene Symbol), protein description/name (Description), site position, motif, AScore (>13 is considered localized), redundancy, peptide sequence, number of quantified peptides, and the summed signal-to-noise for each of the 10 channels (126 to 131; >50). For each best-scoring peptide of a given phosphorylation site, we included the cross-correlation score (XCorr), the delta correlation (ΔCN), and peptide mass difference (PPM).
Supplemental Figure 1: Workflow timeline. This graphic illustrates the experimental steps and the approximate time required at each stage of the workflow, as described in the text. MS, mass spectrometry.
Supplemental Figure 2: Yeast experimental set-up. Experimental set-up used in the yeast proteomics/phosphoproteomics analysis to showcase the SL-TMT protocol.
Acknowledgments
We would like to thank the members of the Gygi Lab at Harvard Medical School. This work was funded in part by an NIH/NIDDK grant K01 DK098285 (J.A.P.) and GM97645 (S.P.G.).
Footnotes
CONFLICT OF INTEREST DISCLOSURE
The authors declare no competing financial interest.
SUPPORTING INFORMATION
The following information is available free of charge at ACS website: http://pubs.acs.org):
Supplemental Methods: Detailed Methodology for liquid chromatography/tandem mass spectrometry and data analysis.
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Rauniyar N, Yates JR., 3rd Isobaric labeling-based relative quantification in shotgun proteomics. J Proteome Res. 2014;13(12):5293–309. doi: 10.1021/pr500880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75(8):1895–904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 4.Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods. 2011;8(11):937–40. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem. 2014;86(14):7150–8. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edwards A, Haas W. Multiplexed Quantitative Proteomics for High-Throughput Comprehensive Proteome Comparisons of Human Cell Lines. Methods Mol Biol. 2016;1394:1–13. doi: 10.1007/978-1-4939-3341-9_1. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Elias JE. Relative Protein Quantification Using Tandem Mass Tag Mass Spectrometry. Methods Mol Biol. 2017;1550:185–198. doi: 10.1007/978-1-4939-6747-6_14. [DOI] [PubMed] [Google Scholar]
- 8.Vetter DE, Basappa J. Multiplexed Isobaric Tagging Protocols for Quantitative Mass Spectrometry Approaches to Auditory Research. Methods Mol Biol. 2016;1427:109–33. doi: 10.1007/978-1-4939-3615-1_7. [DOI] [PubMed] [Google Scholar]
- 9.Lee LS, Banks PA, Bellizzi AM, Sainani NI, Kadiyala V, Suleiman S, Conwell DL, Paulo JA. Inflammatory protein profiling of pancreatic cyst fluid using EUS-FNA in tandem with cytokine microarray differentiates between branch duct IPMN and inflammatory cysts. J Immunol Methods. 2012;382(1–2):142–149. doi: 10.1016/j.jim.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paulo JA, Lee LS, Banks PA, Steen H, Conwell DL. Proteomic analysis of formalin-fixed paraffin-embedded pancreatic tissue using liquid chromatography tandem mass spectrometry. Pancreas. 2012;41(2):175–85. doi: 10.1097/MPA.0b013e318227a6b7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gancedo JM. The early steps of glucose signalling in yeast. FEMS Microbiol Rev. 2008;32(4):673–704. doi: 10.1111/j.1574-6976.2008.00117.x. [DOI] [PubMed] [Google Scholar]
- 12.Paulo JA, O’Connell JD, Gaun A, Gygi SP. Proteome-wide quantitative multiplexed profiling of protein expression: carbon-source dependency in Saccharomyces cerevisiae. Mol Biol Cell. 2015;26(22):4063–74. doi: 10.1091/mbc.E15-07-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paulo JA, O’Connell JD, Everley RA, O’Brien J, Gygi MA, Gygi SP. Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J Proteomics. 2016;148:85–93. doi: 10.1016/j.jprot.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paulo JA. Nicotine alters the proteome of two human pancreatic duct cell lines. JOP. 2014;15(5):465–74. doi: 10.6092/1590-8577/2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paulo JA, Gygi SP. Nicotine-induced protein expression profiling reveals mutually altered proteins across four human cell lines. Proteomics. 2017;17(1–2) doi: 10.1002/pmic.201600319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24(10):1285–92. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 17.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143(7):1174–89. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. doi: 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 20.McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, Gygi SP. Increasing the Multiplexing Capacity of TMTs Using Reporter Ion Isotopologues with Isobaric Masses. Analytical Chemistry. 2012;84(17):7469–7478. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Böhm G, Prefot P, Jung S, Selzer S, Mitra V, Britton D, Kuhn K, Pike I, Thompson AH. Low-pH Solid-Phase Amino Labeling of Complex Peptide Digests with TMTs Improves Peptide Identification Rates for Multiplexed Global Phosphopeptide Analysis. Journal of Proteome Research. 2015;14(6):2500–2510. doi: 10.1021/acs.jproteome.5b00072. [DOI] [PubMed] [Google Scholar]
- 22.Paulo JA, Navarrete-Perea J, Erickson AR, Knott J, Gygi SP. An Internal Standard for Assessing Phosphopeptide Recovery from Metal Ion/Oxide Enrichment Strategies. J Am Soc Mass Spectrom. 2018 doi: 10.1007/s13361-018-1946-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paulo JA, Jedrychowski MP, Chouchani ET, Kazak L, Gygi SP. Multiplexed isobaric tag-based profiling of seven murine tissues following in vivo nicotine treatment using a minimalistic proteomics strategy. Proteomics. 2018:e1700326. doi: 10.1002/pmic.201700326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016;11(12):2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 25.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Xin L, Shan B, Chen W, Xie M, Yuen D, Zhang W, Zhang Z, Lajoie GA, Ma B. PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteomics. 2012;11(4):M111 010587. doi: 10.1074/mcp.M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Brien JJ, O’Connell JD, Paulo JA, Thakurta S, Rose CM, Weekes MP, Huttlin EL, Gygi SP. Compositional Proteomics: Effects of Spatial Constraints on Protein Quantification Utilizing Isobaric Tags. J Proteome Res. 2018;17(1):590–599. doi: 10.1021/acs.jproteome.7b00699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim MY, O’Brien J, Paulo JA, Gygi SP. Improved Method for Determining Absolute Phosphorylation Stoichiometry Using Bayesian Statistics and Isobaric Labeling. J Proteome Res. 2017;16(11):4217–4226. doi: 10.1021/acs.jproteome.7b00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Podwojski K, Stephan C, Eisenacher M. Important issues in planning a proteomics experiment: statistical considerations of quantitative proteomic data. Methods Mol Biol. 2012;893:3–21. doi: 10.1007/978-1-61779-885-6_1. [DOI] [PubMed] [Google Scholar]
- 30.Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19(3):368–75. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- 31.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57(1):289–300. [Google Scholar]
- 32.Dunnett CW. A Multiple Comparison Procedure for Comparing Several Treatments with a Control. Journal of the American Statistical Association. 1955;50(272):1096–1121. [Google Scholar]
- 33.Chu VT, Gottardo R, Raftery AE, Bumgarner RE, Yeung KY. MeV+R: using MeV as a graphical user interface for Bioconductor applications in microarray analysis. Genome Biol. 2008;9(7):R118. doi: 10.1186/gb-2008-9-7-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007;23(23):3251–3. doi: 10.1093/bioinformatics/btm369. [DOI] [PubMed] [Google Scholar]
- 35.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13(9):731–40. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 36.Lever J, Krzywinski M, Altman N. Principal component analysis. Nature Methods. 2017;14:641. [Google Scholar]
- 37.Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016;44(D1):D336–42. doi: 10.1093/nar/gkv1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiao X, Sherman BT, da Huang W, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics. 2012;28(13):1805–6. doi: 10.1093/bioinformatics/bts251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, Ordureau A, Rad R, Erickson BK, Wuhr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar RA, Harris T, Artavanis-Tsakonas S, Sowa ME, De Camilli P, Paulo JA, Harper JW, Gygi SP. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 2015;162(2):425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27(1):29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perfetto L, Briganti L, Calderone A, Cerquone Perpetuini A, Iannuccelli M, Langone F, Licata L, Marinkovic M, Mattioni A, Pavlidou T, Peluso D, Petrilli LL, Pirro S, Posca D, Santonico E, Silvestri A, Spada F, Castagnoli L, Cesareni G. SIGNOR: a database of causal relationships between biological entities. Nucleic Acids Res. 2016;44(D1):D548–54. doi: 10.1093/nar/gkv1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horn H, Schoof EM, Kim J, Robin X, Miller ML, Diella F, Palma A, Cesareni G, Jensen LJ, Linding R. KinomeXplorer: an integrated platform for kinome biology studies. Nat Methods. 2014;11(6):603–4. doi: 10.1038/nmeth.2968. [DOI] [PubMed] [Google Scholar]
- 44.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–20. doi: 10.1093/nar/gku1267. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fendt SM, Sauer U. Transcriptional regulation of respiration in yeast metabolizing differently repressive carbon substrates. BMC Syst Biol. 2010;4:12. doi: 10.1186/1752-0509-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paulo JA, Mancias JD, Gygi SP. Proteome-Wide Protein Expression Profiling Across Five Pancreatic Cell Lines. Pancreas. 2017;46(5):690–698. doi: 10.1097/MPA.0000000000000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paulo JA, Gaun A, Gygi SP. Global Analysis of Protein Expression and Phosphorylation Levels in Nicotine-Treated Pancreatic Stellate Cells. J Proteome Res. 2015;14(10):4246–56. doi: 10.1021/acs.jproteome.5b00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paulo JA, McAllister FE, Everley RA, Beausoleil SA, Banks AS, Gygi SP. Effects of MEK inhibitors GSK1120212 and PD0325901 in vivo using 10-plex quantitative proteomics and phosphoproteomics. Proteomics. 2015;15(2–3):462–73. doi: 10.1002/pmic.201400154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stepanova E, Gygi SP, Paulo JA. Filter-Based Protein Digestion (FPD): A Detergent-Free and Scaffold-Based Strategy for TMT Workflows. J Proteome Res. 2018;17(3):1227–1234. doi: 10.1021/acs.jproteome.7b00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5):359–62. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 51.Sharma K, D’Souza RC, Tyanova S, Schaab C, Wisniewski JR, Cox J, Mann M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8(5):1583–94. doi: 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 52.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002;20(3):301–5. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 53.Pinkse MW, Uitto PM, Hilhorst MJ, Ooms B, Heck AJ. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal Chem. 2004;76(14):3935–43. doi: 10.1021/ac0498617. [DOI] [PubMed] [Google Scholar]
- 54.Bodenmiller B, Mueller LN, Mueller M, Domon B, Aebersold R. Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat Methods. 2007;4(3):231–7. doi: 10.1038/nmeth1005. [DOI] [PubMed] [Google Scholar]
- 55.Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci U S A. 2004;101(33):12130–5. doi: 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Villen J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc. 2008;3(10):1630–8. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kettenbach AN, Gerber SA. Rapid and reproducible single-stage phosphopeptide enrichment of complex peptide mixtures: application to general and phosphotyrosine-specific phosphoproteomics experiments. Anal Chem. 2011;83(20):7635–44. doi: 10.1021/ac201894j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Possemato AP, Paulo JA, Mulhern D, Guo A, Gygi SP, Beausoleil SA. Multiplexed Phosphoproteomic Profiling Using Titanium Dioxide and Immunoaffinity Enrichments Reveals Complementary Phosphorylation Events. J Proteome Res. 2017;16(4):1506–1514. doi: 10.1021/acs.jproteome.6b00905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ren L, Li C, Shao W, Lin W, He F, Jiang Y. TiO2 with Tandem Fractionation (TAFT): An Approach for Rapid, Deep, Reproducible, and High-Throughput Phosphoproteome Analysis. J Proteome Res. 2018;17(1):710–721. doi: 10.1021/acs.jproteome.7b00520. [DOI] [PubMed] [Google Scholar]
- 60.Berberich MJ, Paulo JA, Everley RA. MS3-IDQ: Utilizing MS3 Spectra beyond Quantification Yields Increased Coverage of the Phosphoproteome in Isobaric Tag Experiments. J Proteome Res. 2018;17(4):1741–1747. doi: 10.1021/acs.jproteome.8b00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang X, Bomgarden R, Brown J, Drew DL, Robitaille AM, Viner R, Huhmer AR. Sensitive and Accurate Quantitation of Phosphopeptides Using TMT Isobaric Labeling Technique. J Proteome Res. 2017;16(11):4244–4252. doi: 10.1021/acs.jproteome.7b00610. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Proteins quantified in this dataset. Columns include: SGD protein identifier (proteinID), gene symbol (Gene Symbol), protein description/name (Description), number of peptides identified per protein (peptides), the normalized summed signal-to-noise for each of the 10 channels (126 to 131).
Supplemental Table 2: Peptides quantified in this dataset. Columns include: SGD protein identifier (proteinID), gene symbol (Gene Symbol), protein description/name (Description), redundancy, whether peptide is a unique or razor peptide, peptide sequence, and the summed signal-to-noise for each of the 10 channels (126 to 131). For each peptide, we have included the cross-correlation score (XCorr), the delta correlation (ΔCN), and peptide mass difference (PPM).
Supplemental Table 3: Phosphorylation sites quantified in this dataset. Columns include: type of site (single or composite), SGD protein identifier (proteinID), gene symbol (Gene Symbol), protein description/name (Description), site position, motif, AScore (>13 is considered localized), redundancy, peptide sequence, number of quantified peptides, and the summed signal-to-noise for each of the 10 channels (126 to 131; >50). For each best-scoring peptide of a given phosphorylation site, we included the cross-correlation score (XCorr), the delta correlation (ΔCN), and peptide mass difference (PPM).
Supplemental Figure 1: Workflow timeline. This graphic illustrates the experimental steps and the approximate time required at each stage of the workflow, as described in the text. MS, mass spectrometry.
Supplemental Figure 2: Yeast experimental set-up. Experimental set-up used in the yeast proteomics/phosphoproteomics analysis to showcase the SL-TMT protocol.