

Abstract
Current evidence implicates a prothrombotic state in the development of Shiga-toxin (Stx)-mediated hemolytic uremic syndrome (HUS). We recently reported that Stx modulates procoagulant activity by enhancing functional tissue factor (TF) activity on cytokine-activated human glomerular endothelial cells (HGECs). Since angiotensin II (Ang II), the key effector of the renin angiotensin system (RAS), has been shown to increase TF expression in vascular tissue, we examined the possible involvement of Ang II in TF expression in HGECs. HGECs were exposed to tumor necrosis factor (TNF)-α ± Stx-1 ± Ang II. Exogenous Ang II significantly increased TF activity and TF mRNA in TNF-α- ± Stx-1-activated HGECs. This increase was mediated via Ang II type I receptor (AT1R), as losartan, an AT1R inhibitor, attenuated Ang-II-induced TF activity. To study the effect of endogenous Ang II in TF expression by TNF-α ± Stx-1, HGECs were incubated with losartan or an AT2R inhibitor (PD 123319) or an angiotensin-converting enzyme inhibitor (enalapril). Losartan but not PD 123319 decreased TF activity induced by TNF-α ± Stx-1 (P<0.05). Enalapril, also, dose dependently, downregulated TF expression in HGECs exposed to TNF-α ± Stx-1 (P<0.05). AT1R mRNA was upregulated in TNF-α- ± Stx-1-activated HGECs (P<0.05). These data indicate that TF expression in TNF-α- and Stx-1-activated HGECs is enhanced by exogenous Ang II and that endogenous Ang II production may be upregulated by TNF-α ± Stx-1. Hence, local RAS activation may be important in the development of the thrombotic microangiopathy observed in HUS.
Keywords: Angiotensin II, Tissue factor, Shiga-toxin, Hemolytic uremic syndrome, Human glomerular endothelial cells
Introduction
In the circulation, angiotensin II (Ang II), the key effector of the renin angiotensin system (RAS), regulates aldosterone release and blood pressure, whereas locally formed Ang II exerts a variety of nonhemodynamic effects, particularly in the kidney [1]. More specifically, local Ang II can mediate vascular injury and inflammation and can adversely influence the anticoagulant properties of the endothelium, rendering it prothrombotic [2, 3]. Central to a procoagulant phenotype of the endothelium is the induction of tissue factor (TF) on endothelial cell surfaces. TF, a 47,000-kD transmembrane glycoprotein that serves as the nonenzymatic cofactor for factor VII/VIIa, is considered the initiator of the coagulation cascade [4]. Although TF is normally not present on the surface of cells in contact with blood, several agonists, including Ang II, can induce endothelial cells to synthesize and then express TF on their surface membranes, potentially leading to thrombus formation [5–8].
Local thrombosis and platelet-fibrin accumulation are prominent hallmarks of the intrarenal pathology observed in hemolytic uremic syndrome (HUS) [9]. HUS, the leading cause of acute renal failure in children, is most commonly caused by Shiga-toxin (Stx)-producing Escherichia coli infection [10]. The most extensive tissue damage in HUS occurs within the kidney, and Stx-mediated endothelial cell injury is considered the major event triggering the development of HUS [11, 12]. The cytotoxic effects of Stx on the renal endothelium and the importance of various cytokines in potentiating the cytotoxicity of Stx have been studied at length [10].
We have recently demonstrated a role for Stx as a mediator of procoagulant activity by showing that Stx-1 can enhance the expression of functional TF on cytokine-activated human glomerular (microvascular) endothelial cells (HGECs) [13]. We have also observed that cytokine-activated human umbilical vein (macrovascular) endothelial cells (HUVECs) fail to demonstrate further enhancement of the cell-surface TF activity after exposure to Stx, which potentially may explain localization of thrombosis to the microvasculature and account for the specificity of organ involvement in HUS [13]. However, the potential pathogenic or modulatory role of locally produced Ang II in the prothrombotic state that constitutes HUS is largely unknown and has not been addressed in previous in vitro studies. The objective of this study was to examine the effects of local Ang II and the other components of RAS and their interactions with Stx and cytokines on regulation of the TF pathway of coagulation in cultured monolayers of HGECs. Our data suggest that local RAS activation may be important in the development of thrombotic microangiopathy observed in HUS.
Materials and methods
Reagents
Medium 199 with Earle's buffered saline solution (BSS), L-glutamine and 25 mM hydroxyethylpiperazine ethanesulfonic acid (HEPES) buffer (M199), L-glutamine, and penicillin/streptomycin (P/S) were obtained from BioWhittaker (Walkersville, MD, USA). Fetal bovine serum (FBS) was purchased from Hyclone (South Logan, UT, USA). Endothelial cell growth factor (ECGF) was obtained from Biomedical Technologies (Stoughton, MA, USA). Sterile tissue culture plastic ware was purchased from Falcon Plastics (Cockeysville, MD, USA). Fibronectin was obtained from Becton Dickinson Biosciences (San Jose, CA, USA). Endothelial cell attachment factor and passaging reagents were purchased from Cell Systems (Kirkland, WA, USA). Purified Stx-1 was kindly provided by Dr. C. Thorpe (Tufts New England Medical Center, Boston, MA, USA). A monoclonal antibody raised in mice against human TF (anti-TF) was kindly provided by Dr. Y. Nemerson (Mt. Sinai Medical Center, New York, NY, USA). Human coagulation factor (F)VII and FX were obtained from Enzyme Research Laboratories (South Bend, IN, USA). Purified FXa was kindly provided by Dr. J. Hathcock (Mt. Sinai Medical Center). Human recombinant tumor necrosis factor (TNF)-α was purchased from R & D Systems (Minneapolis, MN, USA). Spectrozyme Xa was purchased from American Diagnostica (Greenwich, CT, USA). Ang II, heparin sodium salt, human serum albumin, trypan-blue, acetic acid, enalapril maleate salt, and PD 123319 di(trifluoroacetate) salt were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Losartan was purchased from DuPont Pharmaceuticals (Wilmington, DE, USA). Moloney murine leukemia cirus (MMLV) reverse transcriptase and RNAse inhibitor were purchased from Promega (Madison, WI, USA). Random hexamer primers were obtained from Roche Diagnostics (Indianapolis, IN, USA). Deoxynucleotide triphosphates dNTP were obtained from Fisher Scientific (Hampton, NH, USA). The quantiTest SYBR Green polymerase chain reaction (PCR) kit was purchased from Qiagen (Valencia, CA, USA).
Cell culture
HGECs were obtained from Cell Systems (Kirkland, WA, USA) and were cultured at 37°C in complete medium that consisted of M199 medium supplemented with 10% FBS, 50 μg/ml (equivalent to 0.17 USP U/ml) heparin sodium salt, 50 μg/ml ECGF, 2 mM L-glutamine and 1% P/S in a humidified atmosphere of 5% CO2–95% air. Culture media were changed every 2 days. HGECs were used from the 8th through 12th passages and were verified by indirect immunofluorescence and flow cytometry studies that, whether activated or not, to express von Willebrand factor, confirming that they retained an endothelial cell phenotype. All experiments, except as noted, were performed under the above culture conditions after cells had reached confluence (generally at 6–8 days after passage) as judged by phase-contrast microscopy.
Experimental conditions
HGECs were plated on 12-well tissue culture plates precoated with endothelial cell attachment factor for most studies, except for PCR studies, for which cells were grown in attachment-factor-coated 75 cm2 culture flasks due to the larger number of cells needed per experiment. After reaching confluence, TF production in response to Ang II, TNF-α, Stx-1, or their combination was studied in HGEC monolayers exposed to the following experimental conditions: (1) Ang II (10−12 to 10−6 M) for 1–22 h, (2) TNF-α (20 ng/ml) for 4 or 22 h ± Ang II (10−8 M) for the last 1 or 4 h, (3) TNF-α (20 ng/ml) for 22 h + Stx-1 (10−11 M) for the last 4 h ± Ang II (10−8 M) for the last 1 h.
The concentration of Ang II used in this study ranged from 10−12 to 10−6 M, as Ang II has been shown to exert its biological actions, such as regulation of vascular tone, in the same concentration range. However, for most experiments, 10−8 M of Ang II was used to mimic the tissue levels of Ang II, which are known to exceed those of plasma by far. Seikaly et al. [14] found that 3.28×10−8 M concentration of Ang II is normally present in the glomerular filtrate and the proximal tubule fluid. This concentration approximates known Ang II receptor affinities and is substantially higher than Ang II concentration in the systemic circulation, thus directly supporting the notion that local Ang II can effect regulation of renal biology independently of systemic Ang II.
In order to exclude any potential differences between the effects of Stx-1 and Stx-2 in the TF pathway of coagulation, at the start of our experimental work, we performed a limited number of experiments directly comparing the two toxins with regard to their potency in enhancing TF activity in cytokine-activated HGECs. As we observed no differences between the effect of Stx-1 and Stx-2 in upregulating TF activity in HGECs, we subsequently focused on the effects of Stx-1, which has been used more often in cell culture experiments by other investigators. The concentration of Stx-1 as 10−11 M was selected based both on the observations of Yagi et al. [15], who detected Stx concentration of this order of magnitude in the plasma of patients with HUS, and on our observations that 10−11 M Stx-1 enhanced TF on TNF-α stimulated HGECs [13].
The 18 h incubation with the inflammatory mediator TNF-α prior to the addition of Ang II or Stx-1 was utilized to mimic the cytokine burst that appears early in the development of HUS [10, 11]. In order to determine whether the effect of exogenous Ang II on TF production was mediated via the AT1R, the AT2R, or both, HGECs were exposed to: (1) TNF-α (20 ng/ml) for 22 h, (2) TNF-α (20 ng/ml) for 22 h + Ang II (10−8 M) for the last 4 h, (3) TNF-α (20 ng/ml) + AT1R inhibitor (losartan, 10−6 M) for 22 h + Ang II (10−8 M) for the last 4 h, (4) TNF-α (20 ng/ml) + AT2R inhibitor (PD 123319, 10−6 M) for 22 h + Ang II (10−8 M) for the last 4 h, and (5) TNF-α (20 ng/ml) + losartan (10−6 M) + PD 123319 (10−6 M) for 22 h + Ang II (10−8 M) for the last 4 h. The total incubation time of the experiment was 22 h.
To examine whether blocking the conversion of Ang I to Ang II with an angiotensin-converting enzyme (ACE) inhibitor, and hence the production of endogenous Ang II, could alter TF production by TNF-α and Stx-1, we exposed HGECs to the following conditions: (1) TNF-α (20 ng/ml) ± ACE inhibitor (enalapril, 10−11 to 10−7 M) for 22 h and (2) TNF-α (20 ng/ml) ± enalapril (10−11 to 10−7 M) for 22 h + Stx-1 (10−11 M) for the last 4 h.
We also tested whether Ang II receptor blockers that would block the effect of any endogenously released Ang II could affect TF induction by TNF-α and Stx-1 by using the following experimental conditions: (1) TNF-α (20 ng/ml) for 22 h, (2) losartan (10−6 M) for 44 h + TNF-α (20 ng/ml) for the last 22 h, (3) PD 123319 (10−6 M) for 44 h + TNF-α (20 ng/ml) for the last 22 h, (4) losartan (10−6 M) + PD 123319 (10−6 M) for 44 h + TNF-α (20 ng/ml) for the last 22 h, (5) TNF-α (20 ng/ml) for the last 22 h + Stx-1 (10−11 M) for the last 4 h, (6) losartan (10−6 M) for 44 h + TNF-α (20 ng/ml) for the last 22 h + Stx-1 (10−11 M) for the last 4 h, (7) PD 123319 (10−6 M) for 44 h + TNF-α (20 ng/ml) for the last 22 h + Stx-1 (10−11 M) for the last 4 h, and (8) losartan (10−6 M) + PD 123319 (10−6 M) for 44 h + TNF-α (20 ng/ml) for the last 22 h + Stx-1 (10−11 M) for the last 4 h. The total experimental incubation time was 44 h. The Ang II receptor blockers were added before the activation of the HGECs with TNF-α ± Stx-1 in order to block the total effect of any endogenously produced Ang II by TNF-α and Stx-1.
All experiments were carried out in the presence of complete medium, and dilutions of all experimental reagents were prepared with complete medium. In all experiments, the results were compared to unstimulated HGECs that were exposed only to complete medium with no additives and served as controls. This comparison directly controls for the effect of the medium supplements and eliminates the possibility that these supplements could account for the results obtained.
Determination of FXa production by chromogenic assay
Cell-surface functional TF activity was measured as the rate of generation of activated clotting FX (FXa) using an amidolytic technique as previously described for use with these culture conditions [13, 16]. TF initiates the coagulation cascade by binding FVII, activating it to FVIIa, and forming the TF-FVIIa surface-active enzyme complex. The TF-FVIIa complex, in turn, activates FX to FXa. To measure FXa generation on the cell surface, after exposure of monolayers to different experimental conditions, the medium including any detached cells was removed from each well, adherent cells were washed once with HEPES buffer, and then they were incubated for 60 min at 37°C with a reaction complex (400 μl/well) consisting of HEPES buffer, 10 nM FVII, 100 nM FX, and 0.1% human serum albumin. This HEPES buffer was composed of 0.01 M HEPES with 0.14 M NaCl and 0.005 M CaCl2 and was adjusted to pH 7.3 before use. Aliquots of 360-μl postincubation reaction complex were mixed with an equal volume of 75 mM ethylenediamine tetraacetate (EDTA) to inhibit further activation of FX and then incubated with Spectrozyme Xa (0.5 mM final concentration) at 37°C in a water bath for 30 min. To block further action of FXa on the chromogenic substrate, we then added 30% acetic acid (1 part to 5 parts reaction complex). FXa was measured on control (unstimulated) cells and compared with measurements from stimulated cells. Samples were quantified in a microplate reader (model 450, Bio-Rad Laboratories, Hercules, CA, USA) at 405 nm for increase in absorbance of free chromophore; a calibration curve was constructed with known concentrations of purified FXa. This method was specifically chosen to detect the activity of membrane-associated TF, which regulates the initiation of coagulation [17]. Results are expressed in femtomoles (fmole)/min × cm2, emphasizing the surface area of the culture. Normalizing data by adherent cell count (fmole/min × 100,000 cells), in lieu of surface area, was also routinely calculated; both methods of expressing the data provide useful information concerning TF activity. The surface area approach acknowledges the possibility that cell fragments, subendothelium or microparticles attached to seemingly denuded areas of the tissue culture plate, may contribute to the measured TF activity, whereas normalizing by cell count emphasizes the role of cell number and implicitly accounts for potential loss of adherent cells due to each experimental condition applied. Both modes of expressing the data gave qualitatively similar results.
Anti-TF assay
To demonstrate that FXa production measured on the surface of HGECs specifically originated from TF, additional experiments were performed in which HGEC monolayers, after various experimental conditions, were incubated with either 30 nM anti-TF antibody diluted in complete media or complete media alone for 30 min at 37°C. Following a washing step with HEPES buffer, reaction complex was added, and FXa production was measured as described above.
RNA extraction and quantitative real-time PCR
Total RNA was obtained according to a standard protocol employing guanidine isothiocyanate (GTC) extracts of control and experimental HGECs. Then, 4 μg of total RNA was transcribed into cDNA in each reverse transcription reaction with 400 U of MMLV reverse transcriptase for 60 min at 37°C in 40-μl assays containing 250 μl of 50A260 units of random hexamer primers in the presence of 40 U of RNAse inhibitor and 10 nM (total) dNTP. Real-time PCR was carried out in iCyclerTM iQ, software version 3a for Windows (Bio-Rad Laboratories, Philadelphia, PA, USA) to quantify mRNA levels of selected genes.
PCR primer sequences for the amplification of angiotensinogen, renin, ACE, AT1R and AT2R, TF, TF pathway inhibitor (TFPI), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) amplicons were obtained from Massachusetts General Hospital PrimerBank database and were as follows:
Angiotensinogen: 5′- GGCTCTCTATACCCCTGTGGT-3′ and 5′-TTTGCCTTACCTTGGAAGTGG-3′ (186-bp amplicon); renin: 5′-TCACAAGCTCTTCGATGCTTC-3′ and 5′-GTGATTCCACCCACGGTGATG-3′ (129-bp amplicon); ACE: 5′-CAACCTGCCCCTGGCTAAG-3′ and 5′-CAGGAGCATGGCGTAGCTTC-3′ (169-bp amplicon); AT1R: for the real-time PCR data—5′-CCTTGCCACTAC TAGCAAAAACA-3′ and 5′-ACAAAACAGTGTAACCA CGACA-3′ (193-bp amplicon); for the gel photo presented in Fig. 1: 5′-ACTTTGCCACTATGGGCTGTC-3′ and 5′-GTACAGGTTGAAACTGACGCT-3′ (102-bp amplicon);
Fig. 1.
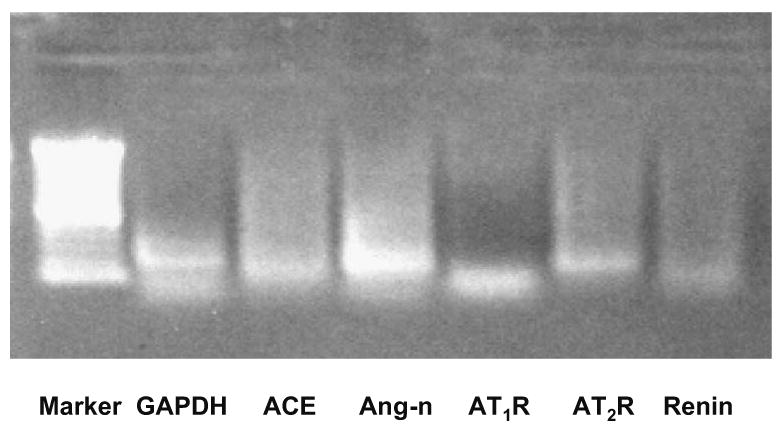
Human glomerular endothelial cells (HGECs) at baseline express components of RAS, as shown in a 1% agarose gel electrophoresis of polymerase chain reaction (PCR) products. As can be seen, there are bands for angiotensin-converting enzyme (ACE, column 3), angiotensinogen (Ang-n, column 4), angiotensin II type 1 receptor (AT1R, column 5), angiotensin II type 2 receptor (AT2R, column 6), renin (column 7). Renin is expressed at very low levels
AT2R: 5′-CCTTGCCACTACTAGCAAAAACA-3′ and 5′-ACAAAACAGTGTAACCACGACA-3′ (193-bp amplicon); TF: 5′-GGCGCTTCAGGCACTCAA-3′ and 5′-GGTGAGGTCACACTCTGTGTC-3′ (96-bp amplicon); TFPI: 5′-TGCGGCTCATATTTACCAAGTC-3′ and 5′-GGCATGAAATGCTATCCAATCCT-3′ (86-bp amplicon); GAPDH: 5′-TGTTGCCATCAATGACCCCTT-3′ and 5′-CTCCACGACGTACTCAGCG-3′ (202-bp amplicon).
cDNA corresponding to 200 ng of total RNA was analyzed in each PCR reaction. Each PCR reaction was run in 50 μl with the QuantiTest SYBR Green PCR Kit, with the MgCl2 concentration being 5 mM for all sets of primers. The cycling program was: (1) initial activation for 15 min at 95°C, (2) 40 amplification cycles with a 30-s denaturing interval at 94°C, and (3) 30-s of annealing at 60°C followed by a 1 min extension at 72°C. Fluorescence was measured at the end of each extension cycle. Melting-curve analysis was carried out for each set of primers to assure absence of secondary products in the reactions. Efficiencies of PCR reactions were determined by using linear regression PCR software from Ramakers et al. [18], and experimental vs. control mRNA ratios were calculated according to Pfaffl [19].
Statistical methods
Experimental data are reported as median, 25th and 75th quartile, and range. N denotes the number of samples per experimental condition obtained in separate experiments. The value for each sample, in turn, represents the average from two tissue culture plate wells or two tissue culture flasks for each experimental condition in each independent experiment. Statistical analysis was performed using the Statistical Package for the Social Sciences (SPSS, Chicago, IL, USA) version 13.0 for Windows. The Mann–Whitney U test was used to test the statistical significance between two groups. Data between more than two different experimental conditions were compared using the Kruskal–Wallis test. Differences were considered significant if the probability (P) value was less than 0.05.
Results
HGECs possess RAS components
To study the role of RAS in our in vitro cell culture system, we first determined whether HGECs express RAS components. As shown in Fig. 1, all components of RAS are present in HGECs: angiotensinogen, renin, ACE, AT1R, and AT2R. This confirms that HGECs constitute a valid and suitable cellular model for assessing RAS interaction with Stx and cytokines on the regulation of the TF pathway of coagulation.
Ang II alone does not induce cell-surface TF activity on HGECs
Cell-surface TF activity, as determined by cell-surface generation of FXa, was measured on intact HGEC monolayers by chromogenic assay. Unstimulated (control) HGECs did not express detectable surface TF activity. In order to assess the effects of Ang II on TF cell-surface activity in HGECs, these cells were exposed to a range of concentrations of Ang II (10−12 M to 10−6 M) for varying times (1–22 h). Ang II by itself did not induce cell-surface TF activity on HGECs at any concentration or timepoint tested. We then examined whether Ang II could enhance TF expression induced by other agonists.
Ang II augments cell-surface TF activity on TNF-α-activated HGECs
TNF-α is a well-known inducer of TF expression in monocytes and endothelial cells and has also been implicated in the pathogenesis of HUS as one of the most important mediators of the observed inflammatory process [10, 11, 20]. Therefore, the possible interaction between Ang II and TNF-α in inducing TF cell-surface activity specifically on HGECs was next studied. Ang II (10−8 M) was added for either the last 1 or 4 h of a 22-h incubation of HGECs with TNF-α (20 ng/ml). The presence of Ang II for the last 1 or 4 h further increased TF expression in TNF-α-activated HGECs by 2.3-fold (P<0.001) and 3-fold (P< 0.05), respectively (Fig. 2a,b).
Fig. 2.

a Exposure of human glomerular endothelial cells (HGECs) to angiotensin (Ang II) (10−8 M) for the last 1 h of a 22-h incubation with tumor necrosis factor (TNF)-α (20 ng/ml) augmented tissue factor (TF) activity in comparison with TNF-α alone (n=6 per experimental condition, median, 25th percentile, 75th percentile, and range, *P< 0.001). b Exposure of HGECs to Ang II (10−8 M) for the last 4 h of a 22-h incubation with TNF-α (20 ng/ml) also augmented TF activity in comparison with TNF-α alone (n=8 per experimental condition, median, 25th percentile, 75th percentile, and range, *P<0.05)
Ang II augments the Stx-induced TF cell-surface activity on TNF-α-activated HGECs
In a previous study, we have shown that Stx-1 enhances TF production in HGECs activated with TNF-α [13]. To investigate whether Ang II could further potentiate this effect, HGECs were exposed to 20 ng/ml of TNF-α for 22 h, 10−11 M of Stx-1 for the last 4 h, and 10−8 M of Ang II for the last 1 h. The addition of Ang II caused a statistically significant augmentation of TF activity on HGECs that had been activated with TNF-α and Stx-1 (Fig. 3).
Fig. 3.

Exposure of human glomerular endothelial cells (HGECs) to angiotensin II (Ang II) (10−8 M) for the last 1 h of a 22-h incubation with tumor necrosis factor (TNF)-α (20 ng/ml) and Shiga-toxin (Stx)-1 (10−11 M) for the last 4 h significantly upregulated tissue factor (TF) activity in comparison with TNF-α and Stx-1 (n=6 per experimental condition, median, 25th percentile, 75th percentile, and range, *P<0.05)
Anti-TF antibody abrogates TF cell-surface activity induced by Ang II
To confirm that the FXa production measured on HGEC surface after Ang II exposure was attributable to the presence of TF, we used an antibody directed against human TF to inhibit FXa production. The addition of 30 nM of a murine monoclonal anti-TF antibody completely abrogated the cell-surface TF activity induced by the combination of TNF-α and Ang II and the combination of TNF-α, Stx-1, and Ang II. In all experimental conditions, TF expression after addition of the anti-TF antibody was nil.
Ang II increases TF cell-surface activity predominantly via the Ang II type 1 receptor
As exogenous Ang II increased TF cell-surface activity in TNF-α- and Stx-1-activated HGECs, we examined whether this effect was mediated via the AT1R or the AT2R. Losartan (10−6 M), a specific antagonist of the AT1R, when present during the incubation period of HGECs with TNF-α (20 ng/ml) for 22 h and Ang II (10−8 M) for the last 4 h, attenuated the increase in TF cell-surface activity caused by Ang II (Fig. 4). PD 123319 (10−6 M), an AT2R antagonist, exerted a smaller effect on Ang II-induced TF activity (Fig. 4). In control HGECs, the addition of AT1R or AT2R inhibitors did not affect TF expression.
Fig. 4.

Human glomerular endothelial cells (HGECs) were exposed to tumor necrosis factor (TNF)-α (20 ng/ml) for 22 h and angiotensin II (Ang II) (10−8 M) for the last 4 h with or without incubation with an Ang II type I receptor (AT1R) inhibitor (losartan, 10−6 M) or AT2R inhibitor (PD 123319, 10−6 M) or their combination (10−6 M) for 22 h. Losartan and losartan in combination with PD 123319, but not PD 123319 alone, caused a statistically significant decrease in tissue factor (TF) activity induced by Ang II (n= 8 per experimental condition, median, 25th percentile, 75th percentile, and range, *P<0.05)
ACE inhibitors downregulate TF activity induced by TNF-α and Stx-1
To determine whether endogenous Ang II production could be involved in TF induction by TNF-α and Stx-1, we used the ACE inhibitor, enalapril, to block the conversion of Ang I to Ang II. We observed that enalapril (10−11 to 10−7 M) decreased TNF-α-induced TF expression as well as the enhanced TF activity caused by Stx-1 in a dose-dependent manner (Fig. 5a,b). Enalapril exerted no effect in unstimulated HGECs.
Fig. 5.

a Blocking endogenous angiotensin II (Ang II) production decreases the tissue factor (TF) activity being induced by tumor necrosis factor (TNF)-α in human glomerular endothelial cells (HGECs). Exposure of HGECs to angiotensin-converting enzyme (ACE) inhibitor (enalapril, 10−9 to 10−7 M) for 22 h dose dependently downregulated TF activity induced by a 22-h incubation with TNF-α (20 ng/ml) (n=6 per experimental condition, median, 25th percentile, 75th percentile, and range, *P<0.05). b Blocking endogenous Ang II production decreases the effect of Shiga-toxin (Stx-1) on TF expression in activated HGECs. Exposure of HGECs to ACE inhibitor (enalapril, 10−11 to 10−7 M) for 22 h dose dependently downregulated TF activity induced by a 22-h incubation with TNF-α (20 ng/ml) and Stx-1 (10−11 M) for the last 4 h (n=6 per experimental condition, median, 25th percentile, 75th percentile, and range, *P<0.05)
Ang II type 1 receptor inhibitors downregulate TF activity induced by TNF-α and Stx-1
As ACE inhibition decreased TF activity induced by TNF-α and TF activity induced by the combination of TNF-α and Stx-1 in HGECs, we next examined whether TF induction could be blocked by AT1R and/or AT2R inhibitors in the absence of exogenous Ang II. Preincubation of HGECs with losartan (10−6 M), the AT1R antagonist, downregulated TF activity induced by TNF-α as well as TF activity induced by TNF-α and Stx-1 (Fig. 6a,b). AT2R inhibitor (PD 123319, 10−6 M) did not cause a similar effect on TF cell-surface activity (Fig. 6a,b). AT1R or AT2R inhibitors did not affect cell-surface TF activity in the absence of HGEC activation with TNF-α and Stx-1.
Fig. 6.

a Blocking the angiotensin II type 1 receptor (AT1R) decreases the effect of tumor necrosis factor (TNF)-α on tissue factor (TF) activity in human glomerular endothelial cells (HGECs). HGECs were exposed to TNF-α (20 ng/ml) for 22 h with or without incubation with an AT1R inhibitor (losartan, 10−6 M) or AT2R inhibitor (PD 123319, 10−6 M) or their combination (10−6 M) for 44 h. Losartan or a combination of losartan and PD 123319, but not PD 123319 alone, downregulated TF activity induced by TNF-α (n=8 per experimental condition, median, 25th percentile, 75th percentile, and range, *P<0.05). b Blocking the AT1R decreases the effect of Shigatoxin (Stx)-1 on TF expression in TNF-α-activated HGECs. HGECs were exposed to TNF-α (20 ng/ml) for 22 h and Stx-1 (10−11 M) for the last 4 h with or without incubation with an AT1R inhibitor (losartan, 10−6 M) or AT2R inhibitor (PD 123319, 10−6 M) or their combination (10−6 M) for 44 h. Losartan or a combination of losartan and PD 123319, but not PD 123319 alone, downregulated TF activity induced by TNF-α and Stx-1 (n=8 per experimental condition, median, 25th percentile, 75th percentile, and range, *P<0.05)
Ang II increases TF mRNA expression in TNF-α- and Stx-1-activated HGECs, and TNF-α and Stx-1 increase Ang II type 1 receptor mRNA expression
In HGECs activated with TNF-α (20 ng/ml) for a 4-h period, the addition of Ang II (10−8 M) for the last 1 h of incubation led to a statistically significant increase in the amount of TF mRNA (Table 1). Moreover, Ang II incubation for the last 1 h was associated with an increase in TF mRNA in HGECs activated with TNF-α for 4 h and Stx-1 for the last 2 h (Table 1). The addition of Ang II in TNF-α- and Stx-1-activated HGECs was also associated with an increase in AT1R mRNA but no change in AT2R mRNA (Table 1). Exogenous Ang II also caused no change in TFPI mRNA in HGECs that were exposed to TNF-α and HGECs that were exposed to the combination of TNF-α and Stx-1 (Table 1).
Table 1.
mRNA for tissue factor (TF), TF pathway inhibitor (TFPI), angiotensin II (Ang II) type 1 receptor (AT1R), and AT2R in human glomerular endothelial cells (HGECs) exposed to combinations of tumor necrosis factor (TNF)-α (20 ng/ml), Shiga-toxin (Stx)-1 (10−11 M), and Ang II (10−8 M)
| Conditions | Ratio of mRNA of gene of interest to GAPDH mRNA, median (25th percentile, 75th percentile) (n) | |||
|---|---|---|---|---|
| TF | TFPI | AT1R | AT2R | |
| TNF-α (20 ng/ml) 4 h + Ang II (10−8 M) last 1 h | 1.46 (1.27, 1.88) (9) * | 1.07 (0.89, 1.29) (11) | 1.44 (1.30, 1.62) (9) * | 1.09 (0.71, 1.46) (10) |
| TNF-α (20 ng/ml) 4 h + Stx-1 (10−11 M) last 2 h + Ang II (10−8 M) last 1 h | 1.80 (1.48, 1.91) (5) * | 1.30 (0.98, 1.80) (8) | 1.46 (1.36, 2.00) (6) * | 1.84 (1.01, 3.03) (5) |
GAPDH glyceraldehyde-3-phosphate dehydrogenase
P<0.05
AT1R mRNA was also upregulated following incubation of HGECs with TNF-α for 4 h and Stx-1 for the last 2 h (Table 2) in the absence of exogenous Ang II. No statistically significant changes in angiotensinogen mRNA, renin mRNA, ACE mRNA, or AT2R mRNA were observed in HGECs following incubation with the combination of TNF-α and Stx-1.
Table 2.
mRNA for renin angiotensin system (RAS) components in human glomerular endothelial cells (HGECs) exposed to combinations of tumor necrosis factor (TNF)-α (20 ng/ml) and Shiga-toxin (Stx)-1 (10−11 M)
| Condition | Ratio of mRNA of gene of interest to GAPDH mRNA, median (25th percentile, 75th percentile) (n) | ||||
|---|---|---|---|---|---|
| ACE | Angiotensinogen | AT1R | AT2R | Renin | |
| TNF-α (20 ng/ml) 4 h + Stx-1 (10−11 M) last 2 h | 1.02 (0.84, 1.14) (12) | 1.28 (0.90, 1.46) (10) | 1.51 (1.22, 1.65) (9) * | 1.07 (0.80, 1.60) (10) | 1.22 (0.78, 1.51) (9) |
GAPDH glyceraldehyde-3-phosphate dehydrogenase
P<0.05
Discussion
In this study, HGECs containing all components of the RAS were used as a cell culture system to explore the possible involvement of RAS in the development of the thrombotic microangiopathy observed in HUS. Control, unstimulated HGECs, did not express cell-surface TF activity. Whereas treatment with exogenous Ang II alone did not induce TF activity in HGECs, Ang II increased TF activity on TNF-α-activated HGECs and also augmented the Stx-enhanced TF cell-surface activity on these cells. The increase in TF expression induced by Ang II was associated in each case with an increase in TF mRNA levels without any apparent change in TFPI mRNA levels. AT1R predominantly mediated the aforementioned effect of Ang II on TF cell-surface activity, as losartan, a specific antagonist of the AT1R, prevented the Ang-II-induced increase of TF production in activated HGECs. Thus, in the presence of exogenous Ang II acting through the AT1R, TF becomes more predominant over TFPI, rendering the renal glomerular endothelial cells more prothrombotic.
The notion that the RAS participates in the pathogenesis of thrombosis and that locally active Ang II may act as a procoagulant factor in many tissues is supported by recent studies [2]. Taubman et al. [7] demonstrated that Ang II rapidly increased both TF mRNA and TF protein in cultured vascular smooth muscle cells. Nishimura et al. [8] also found that Ang II induced TF mRNA expression without changing TFPI mRNA in cultured rat aortic endothelial cells. Furthermore, Ang II was reported to potentiate TF expression by endotoxin in peripheral mononuclear cells and to directly induce TF expression in human monocytes [21, 22]. The thrombogenic effect of Ang II was shown to be exerted through the AT1R [2, 13, 22]. AT1R blockade significantly inhibited Ang-II-stimulated TF expression both at the mRNA and the protein level in monocytes and rat aortic endothelial cells [8, 22]. Our study demonstrates that Ang II enhances TNF-α- and Stx-1-induced TF through the AT1R in a unique vascular bed, the glomerular endothelium.
Evidence supports the presence of a prothrombotic state in HUS [23–25]. The expression of functional TF on the surface of microvascular endothelial cells may constitute a central mechanism in the pathogenesis of HUS [13]. TF may initiate coagulation and platelet activation in the glomerular microvasculature, leading to microthrombus formation, platelet and fibrin deposition, and subsequent microcirculatory incompetence and loss of renal function [13]. Our finding that Ang II augmented TF expression on TNF-α- and Stx-1-activated HGECs strongly supports the concept that there could be an interaction between the local RAS and the TF coagulation pathway in the presence of cytokines and Stx during the development of the microangiopathic changes observed in HUS. A possible association between an activated local RAS, expression of TF, and development of microangiopathy is also suggested by other observations. For example, in a double transgenic rat model that shows microangiopathic changes resembling those seen in HUS, local Ang II overexpression was noted, and TF was upregulated [26]. Pathologic microangiopathic changes are also reported in preeclampsia. Recently, it was shown that AT1R autoantibodies isolated from the serum of preeclamptic patients induced TF expression in vascular smooth muscle cells [27].
In the course of Stx-induced injury in the kidney, the RAS may be activated, potentially amplifying the microangiopathic changes and modulating the clinical course of HUS. Activation of the intrarenal RAS is considered a mechanism by which cyclosporine, which can cause non-Stx-associated HUS, induces nephrotoxicity [28]. An accepted hypothesis is that cyclosporine can upregulate the intrarenal RAS by increasing renin release from juxtaglomerular cells [29]. An initial study by Grunfeld et al. [30] found increased plasma renin activity in children with HUS, but later, Proesmans et al. [31] reported that there was no correlation of plasma renin activity with the course of childhood HUS. However, the peripheral levels of RAS components in mixed venous blood may not accurately reflect a local activation of RAS that may contribute to the pathophysiology of HUS. In our study, an increase in renin mRNA in HGECs activated with TNF-α and Stx-1 was noted, although it did not reach statistical significance.
We also observed AT1R mRNA to be significantly upregulated in HGECs exposed to TNF-α and Stx-1. This implies that in the presence of Stx, there could be an increase in the density of surface AT1R, which may render the glomerular endothelium more sensitive to the effects of endogenously produced Ang II, and therefore, TF expression may be enhanced. Interestingly, the use of losartan, without the presence of exogenous Ang II, was associated with a decrease in the TNF-α-induced TF cell-surface activity in HGECs and a decrease in the TF production induced by the combination of TNF-α and Stx-1. In contrast, AT2R mRNA levels remained unchanged, and PD 123319, a specific AT2R antagonist, did not affect TF production in TNF-α- and Stx-1-activated HGECs. We also noted that the ACE inhibitor enalapril dose dependently downregulated TF activity on HGECs activated with TNF-α and on HGECs activated with the combination of TNF-α and Stx-1. Moreover, enalapril inhibited TF activity on HGECs activated with TNF-α and the combination of TNF-α and Stx-1 to an extent similar to that observed with losartan. These findings suggest that TF expression in activated HGECs may be modulated by endogenous Ang II production that has been stimulated by TNF-α and/or Stx and that AT1R activation by endogenous Ang II is implicated in this process. A similar observation was described by Napoleone et al. [32] who reported that ACE inhibitors downregulated TF synthesis in endotoxin-stimulated monocytes. Moreover, recent studies suggest that several of the beneficial effects of ACE inhibitors and AT1R blockers may be mediated by inhibiting the thrombogenic effects of Ang II on vascular endothelial cells [33, 34].
In conclusion, our data indicate that Ang II, Stx, TNF-α, and the TF pathway of coagulation interact in HGECs. We would suggest that local activation of RAS during the course of HUS may contribute to the thrombotic microangiopathy and may be a significant modulator of disease severity in HUS. The demonstration that the activation of local RAS amplifies microangiopathic changes in HUS, and that blocking RAS effects with ACE inhibitors and/or AT1R antagonists abrogates local pathology, could serve as an important focus for future in vivo studies.
Acknowledgments
This work was supported by National Institutes of Health grants HL 33095, DK 71253 (EFG) and DK 58950 (JRI). The authors would like to thank Dr. Cheleste Thorpe for providing Stx-1, Dr. Yale Nemerson for providing anti-TF antibody, and Dr. James Hathcock for providing purified FXa.
This work was supported by National Institutes of Health grants HL 33095, DK 71253 (EFG) and DK 58950 (JRI).
Footnotes
This paper was presented in part as an abstract at the American Society of Nephrology, 38 Annual Meeting and Scientific Exposition, Philadelphia, PA, USA, November 2005 (Journal of the American Society of Nephrology 2005; 16 Supplement November: PO327) and at the Pediatric Academic Societies' Annual Meeting, San Francisco, CA, USA, May 2006 (E-PAS 2006:59: 2830.32).
Contributor Information
Eirini Nestoridi, Pediatric Nephrology Laboratory, MassGeneral Hospital for Children at Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA; Cardiovascular Thrombosis Laboratory, MassGeneral Hospital for Children at Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
Rafail I. Kushak, Cardiovascular Thrombosis Laboratory, MassGeneral Hospital for Children at Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
Olga Tsukurov, Pediatric Nephrology Laboratory, MassGeneral Hospital for Children at Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
Eric F. Grabowski, Cardiovascular Thrombosis Laboratory, MassGeneral Hospital for Children at Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
Julie R. Ingelfinger, Pediatric Nephrology Laboratory, MassGeneral Hospital for Children at Massachusetts General Hospital, Harvard Medical School, Boston, MA, USAPediatric Nephrology, MassGeneral Hospital for Children at Massachusetts General Hospital, Harvard Medical School, Yawkey 6C, 55 Fruit Street, Boston, MA 02114, USA
References
- 1.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86:747–803. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 2.Brown NJ, Vaughan DE. Prothrombotic effects of angiotensin. Adv Intern Med. 2000;45:419–429. [PubMed] [Google Scholar]
- 3.Galle J, Quaschning T, Seibold S, Wanner C. Endothelial dysfunction and inflammation: what is the link? Kidney Int Suppl. 2003;84:S45–S49. doi: 10.1046/j.1523-1755.63.s84.12.x. [DOI] [PubMed] [Google Scholar]
- 4.Camerer E, Kolsto AB, Prydz H. Cell biology of tissue factor, the principal initiator of blood coagulation. Thromb Res. 1996;81:1–41. doi: 10.1016/0049-3848(95)00209-x. [DOI] [PubMed] [Google Scholar]
- 5.Colucci M, Balconi G, Lorenzet R, Pietra A, Locati D, Donati MB, Semeraro N. Cultured human endothelial cells generate tissue factor in response to endotoxin. J Clin Invest. 1983;71:1893–1896. doi: 10.1172/JCI110945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monroe DM, Key NS. The tissue factor-factor VIIa complex: procoagulant activity, regulation, and multitasking. J Thromb Haemost. 2007;5:1097–1105. doi: 10.1111/j.1538-7836.2007.02435.x. [DOI] [PubMed] [Google Scholar]
- 7.Taubman MB, Marmur JD, Rosenfield CL, Guha A, Nichtberger S, Nemerson Y. Agonist-mediated tissue factor expression in cultured vascular smooth muscle cells. Role of Ca2+ mobilization and protein kinase C activation. J Clin Invest. 1993;91:547–552. doi: 10.1172/JCI116234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishimura H, Tsuji H, Masuda H, Nakagawa K, Nakahara Y, Kitamura H, Kasahara T, Sugano T, Yoshizumi M, Sawada S, Nakagawa M. Angiotensin II increases plasminogen activator inhibitor-1 and tissue factor mRNA expression without changing that of tissue type plasminogen activator or tissue factor pathway inhibitor in cultured rat aortic endothelial cells. Thromb Haemost. 1997;77:1189–1195. [PubMed] [Google Scholar]
- 9.Ruggenenti P, Noris M, Remuzzi G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001;60:831–846. doi: 10.1046/j.1523-1755.2001.060003831.x. [DOI] [PubMed] [Google Scholar]
- 10.Andreoli SP, Trachtman H, Acheson DW, Siegler RL, Obrig TG. Hemolytic uremic syndrome: epidemiology, pathophysiology, and therapy. Pediatr Nephrol. 2002;17:293–298. doi: 10.1007/s00467-001-0783-0. [DOI] [PubMed] [Google Scholar]
- 11.Ray PE, Liu XH. Pathogenesis of Shiga toxin-induced hemolytic uremic syndrome. Pediatr Nephrol. 2001;16:823–839. doi: 10.1007/s004670100660. [DOI] [PubMed] [Google Scholar]
- 12.Proulx F, Seidman EG, Karpman D. Pathogenesis of Shiga toxin-associated hemolytic uremic syndrome. Pediatr Res. 2001;50:163–171. doi: 10.1203/00006450-200108000-00002. [DOI] [PubMed] [Google Scholar]
- 13.Nestoridi E, Tsukurov O, Kushak RI, Ingelfinger JR, Grabowski EF. Shiga toxin enhances functional tissue factor on human glomerular endothelial cells: implications for the pathophysiology of hemolytic uremic syndrome. J Thromb Haemost. 2005;3:752–762. doi: 10.1111/j.1538-7836.2005.01205.x. [DOI] [PubMed] [Google Scholar]
- 14.Seikaly MG, Arant BS, Seney FD. Endogenous angiotensin concentrations in specific intrarenal fluid compartments of the rat. J Clin Invest. 1990;86:1352–1357. doi: 10.1172/JCI114846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yagi H, Narita N, Matsumoto M, Sakurai Y, Ikari H, Yoshioka A, Kita E, Ikeda Y, Titani K, Fujimura Y. Enhanced low shear stress induced platelet aggregation by Shiga-like toxin 1 purified from Escherichia coli O157. Am J Hematol. 2001;66:105–115. doi: 10.1002/1096-8652(200102)66:2<105::AID-AJH1025>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 16.Grabowski EF, Zuckerman DB, Nemerson Y. The functional expression of tissue factor by fibroblasts and endothelial cells under flow conditions. Blood. 1993;81:3265–3270. [PubMed] [Google Scholar]
- 17.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134:1087–1097. [PMC free article] [PubMed] [Google Scholar]
- 18.Ramakers C, Ruijter JM, Deprez RH, Moorman AF. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 2003;339:62–66. doi: 10.1016/s0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- 19.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shebuski RJ, Kilgore KS. Role of inflammatory mediators in thrombogenesis. Pharmacol Exp Ther. 2002;300:729–735. doi: 10.1124/jpet.300.3.729. [DOI] [PubMed] [Google Scholar]
- 21.Di Santo A, Napoleone E, Donati MB, Lorenzet R. Angiotensin II upregulates tissue factor expression by human monocytes. Thromb Haemost SV. 2001:OC215. [Google Scholar]
- 22.He M, He X, Xie Q, Chen F, He S. Angiotensin II induces the expression of tissue factor and its mechanism in human monocytes. Thromb Res. 2006;117:579–590. doi: 10.1016/j.thromres.2005.04.033. [DOI] [PubMed] [Google Scholar]
- 23.Chandler WL, Jelacic S, Boster DR, Ciol MA, Williams GD, Watkins SL, Igarashi T, Tarr PI. Prothrombotic coagulation abnormalities preceding the hemolytic-uremic syndrome. N Engl J Med. 2002;346:23–32. doi: 10.1056/NEJMoa011033. [DOI] [PubMed] [Google Scholar]
- 24.Van Geet C, Proesmans W, Arnout J, Vermylen J, Declerck PJ. Activation of both coagulation and fibrinolysis in childhood hemolytic uremic syndrome. Kidney Int. 1998;54:1324–1330. doi: 10.1046/j.1523-1755.1998.00103.x. [DOI] [PubMed] [Google Scholar]
- 25.Nevard CH, Blann AD, Jurd KM, Haycock GB, Hunt BJ. Markers of endothelial cell activation and injury in childhood haemolytic uraemic syndrome. Pediatr Nephrol. 1999;13:487–492. doi: 10.1007/s004670050644. [DOI] [PubMed] [Google Scholar]
- 26.Theuer J, Dechend R, Muller DN, Park JK, Fiebeler A, Barta P, Ganten D, Haller H, Dietz R, Luft FC. Angiotensin II induced inflammation in the kidney and in the heart of double transgenic rats. BMC Cardiovasc Disord. 2002;2:3. doi: 10.1186/1471-2261-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dechend R, Homuth V, Wallukat G, Kreuzer J, Park JK, Theuer J, Juepner A, Gulba DC, Mackman N, Haller H, Luft FC. AT (1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation. 2000;101:2382–2387. doi: 10.1161/01.cir.101.20.2382. [DOI] [PubMed] [Google Scholar]
- 28.Lassila M. Interaction of cyclosporine A and the renin-angiotensin system; new perspectives. Curr Drug Metab. 2002;3:61–71. doi: 10.2174/1389200023337964. [DOI] [PubMed] [Google Scholar]
- 29.Tufro-McReddie A, Gomez RA, Norling LL, Omar AA, Moore LC, Kaskel FJ. Effect of CsA on the expression of renin and angiotensin type 1 receptor genes in the rat kidney. Kidney Int. 1993;43:615–622. doi: 10.1038/ki.1993.90. [DOI] [PubMed] [Google Scholar]
- 30.Grunfeld B, Gimenez M, Liapchuc S, Mendilaharzu J, Gianantonio C. Systemic hypertension and plasma renin activity in children with the hemolytic-uremic syndrome. Int J Pediatr Nephrol. 1982;3:211–214. [PubMed] [Google Scholar]
- 31.Proesmans W, VanCauter A, Thijs L, Lijnen P. Plasma renin activity in haemolytic uraemic syndrome. Pediatr Nephrol. 1994;8:444–446. doi: 10.1007/BF00856527. [DOI] [PubMed] [Google Scholar]
- 32.Napoleone E, Di Santo A, Camera M, Tremoli E, Lorenzet R. Angiotensin-converting enzyme inhibitors downregulate tissue factor synthesis in monocytes. Circ Res. 2000;86:139–143. doi: 10.1161/01.res.86.2.139. [DOI] [PubMed] [Google Scholar]
- 33.Soejima H, Ogawa H, Yasue H, Kaikita K, Takazoe K, Nishiyama K, Misumi K, Miyamoto S, Yoshimura M, Kugiyama K, Nakamura S, Tsuji I. Angiotensin-converting enzyme inhibition reduces monocyte chemoattractant protein-1 and tissue factor levels in patients with myocardial infarction. J Am Coll Cardiol. 1999;34:983–988. doi: 10.1016/s0735-1097(99)00318-6. [DOI] [PubMed] [Google Scholar]
- 34.Tsikouris JP, Cox CD. Pharmacologic blockade of the renin-angiotensin system: vascular benefits beyond commonly understood pharmacologic actions. Pharmacotherapy. 2003;23:1141–1152. doi: 10.1592/phco.23.10.1141.32763. [DOI] [PubMed] [Google Scholar]