

Abstract
Although inflammation is prevalent in many clinical disorders challenging breathing, we are only beginning to understand the impact of inflammation on neural mechanisms of respiratory control. We recently demonstrated one form of respiratory motor plasticity is extremely sensitive to even mild inflammation induced by a single night (8 hours) of intermittent hypoxia (IH-1), mimicking aspects of obstructive sleep apnea. Specifically, phrenic long-term facilitation (pLTF) following moderate acute intermittent hypoxia (AIH) is abolished by IH-1, but restored by high doses of the non-steroidal anti-inflammatory drug, ketoprofen. Since a major target of ketoprofen is cyclooxygenase (COX) enzymes, we tested the involvement of COX in IH-1 suppression of pLTF using the selective COX inhibitor NS-398. Systemic COX inhibition (3 mg/kg, i.p., 3 hrs before AIH) had no effect on pLTF in normoxia treated rats (76±40% change from baseline, n=6), and did not restore pLTF in IH-1 treated rats (−9±7% baseline, n=6). Similarly, spinal COX inhibition (27 mM, 12 μl, i.t.) had no effect on pLTF in normoxic rats (76±34% baseline, n=7), and did not significantly restore pLTF after IH-1 (37±18% baseline, n=7). COX-2 protein is expressed in identified phrenic motor neurons of both normoxia and IH-1 exposed rats, but immunolabeling was minimal in surrounding microglia; IH-1 had no discernable effect on COX-2 immunoreactivity. We conclude that the inflammatory impairment of pLTF by IH-1 is independent of COX enzyme activity or upregulated COX-2 expression.
Keywords: plasticity, inflammation, COX, phrenic motoneurons, respiratory motor plasticity
INTRODUCTION
Although the precise physiological role(s) of respiratory plasticity are still subject to debate (Gonzalez-Rothi et al., 2015; Fuller and Mitchell, 2017; Mateika and Komnenov, 2017), the ability to express respiratory plasticity can be harnessed to recover lost breathing capacity due to disease or injury (Gonzalez-Rothi et al., 2015). Systemic and neural inflammation accompany clinical disorders compromising breathing (Huxtable et al., 2011). Even mild systemic or neuroinflammation undermines an important form of respiratory motor plasticity, phrenic long-term facilitation (pLTF) following moderate acute intermittent hypoxia (AIH; Vinit et al., 2011; Huxtable et al., 2011; Huxtable et al., 2013). For example, mild systemic inflammation elicited by lipopolysaccharide (LPS) (Huxtable et al., 2011; Vinit et al., 2011) or 1 night of intermittent hypoxia (IH-1) (Huxtable et al., 2015) abolishes moderate AIH-induced pLTF. pLTF sensitivity to mild inflammation has significant implications for exploiting respiratory plasticity for therapeutic advantage. Thus, greater understanding of the mechanisms whereby inflammation undermines plasticity are of considerable importance.
Although detailed mechanism(s) whereby inflammation inhibits pLTF are not yet fully understood, plasticity can be restored by high doses of non-steroidal anti-inflammatory drugs (NSAIDs), such as ketoprofen (12.5 mg/kg) (Huxtable et al., 2013; Huxtable et al., 2015). At typical therapeutic doses, most anti-inflammatory actions of NSAIDs are via cyclooxygenase (COX)-1 and/or COX-2 enzyme inhibition (Cashman, 1996; Cabre et al., 1998; Diaz-Reval et al., 2004; Lleo et al., 2007; Kokki, 2010), suggesting that high-ketoprofen doses restore pLTF after systemic inflammation via COX inhibition. We recently demonstrated spinal administration of p38 MAP kinase inhibitors restores pLTF after IH-1 (Huxtable et al., 2015). However, since many p38 MAP kinase inhibitors also inhibit COX activity (Borsch-Haubold et al., 1998) and the specific roles of cyclooxygenases in pLTF inhibition have not been tested, we investigated the impact of selective COX inhibition on pLTF after IH-1.
COX-1 and COX-2 are both expressed in CNS neurons, astrocytes and microglia (Choi et al., 2009). Specifically, COX-2 plays an important pro-inflammatory role, triggering inflammation in response to infection or injury (Bazan, 2001). Further, increased COX-2 activity induced by β-amyloid proteins inhibits other forms of synaptic plasticity such as hippocampal long-term potentiation (LTP); conversely, COX-2 inhibitors restore it (Kotilinek et al., 2008). Since hippocampal LTP shares some mechanistic similarities with AIH-induced pLTF (Mahamed and Mitchell, 2007; Mateika and Sandhu, 2011), we tested the involvement of COX in pLTF impairment after IH-1-induced inflammation, and examined COX-2 expression in phrenic motoneurons, the site of pLTF(Baker-Herman and Mitchell, 2002; Devinney et al., 2015; Dale et al., 2017). Unlike hippocampal LTP, we report here that COX enzymatic activity is not necessary in the mechanism whereby systemic inflammation caused by IH-1 undermines pLTF.
METHODS
All experiments were approved by the Institutional Animal Care and Use Committee at the School of Veterinary Medicine, University of Wisconsin–Madison, and conformed to policies in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experiments were performed on 3–4 month old Harlan male Sprague Dawley rats (colony 217 and 211a) housed under standard conditions, with food and water ad libitum and a 12-hour light/dark cycle.
Experimental Groups
Rats were placed in custom-designed cylindrical, Plexiglas exposure chambers for 8 hrs with ClearH2O® hydrogel (Portland, ME) to provide nutrition and hydration during normoxia or intermittent hypoxia exposures. Gas flow through the chambers was controlled by a customized program (National Instruments, LabVIEW™ 2009, Service Pack 1, version 9.0.1, Austin, TX), and a mass flow controller (Teledyne, Hastings Instruments, Hampton, VA), and O2 levels were continuously monitored by a Gemini Respiratory Gas Analyzer (CWE, Inc., Ardmore, PA). Over an 8-hour period during a standard “dark phase”, rats experienced either intermittent hypoxia (2 min of 10.5% O2, 2 min intervals of 21% O2) or normoxia (21% O2). Upon completion of exposures, rats were returned to their home cages until the next day, and then studied in neurophysiology experiments or perfused for immunohistochemistry.
Prior to electrophysiology experiments, rats were assigned to one of three groups to investigate whether pLTF could be restored by systemic application of the COX inhibitor NS-398 (Sigma Chemical Company, St. Louis, MO): 1) Normoxia (Nx) + NS-398 (n=6); 2) IH-1 + NS-398 (n=6); and 3) NS-398 Time Controls (TC) (3 Normoxia, 2 IH-1; n=5). In a second study to investigate whether pLTF could be restored by spinal COX inhibition, rats were divided into six groups: 1) Nx + NS-398 + AIH (n=6); 2) IH-1 + NS-398 (n=8); 3) NS-398 Time controls (3 Normoxia, 3 IH-1; n=8); 4) Nx + veh + AIH (n=5); 5) IH-1 + veh + AIH (n=4); and 6) Veh TC (n=4).
Electrophysiological Experiments
The protocol used in electrophysiological experiments has been described in detail previously (Bach and Mitchell, 1996; Baker-Herman and Mitchell, 2002; Huxtable et al., 2013; Huxtable et al., 2015). In brief, rats were anesthetized with isoflurane, tracheotomized and pump ventilated (Small Animal Ventilator 683, Harvard Apparatus, Inc., Holliston, MA, USA). After surgery was completed, rats were slowly converted to urethane anesthesia (1.8 g/kg, i.v., Sigma-Aldrich). During a one-hour stabilization period after conversion to urethane, pancuronium bromide (1 mg, i.v.) was given to paralyze the rats. Anesthetic level was assessed by monitoring blood pressure and phrenic nerve responses to toe pinch. Approximately thirty minutes into the stabilization period, an i.v. infusion (1.5–2 ml/hour) began of a solution consisting of Hetastarch (0.3%) and sodium bicarbonate (0.84%) in Lactated Ringers. Infusion rate was adjusted to maintain blood volume, pressure and acid-base balance throughout experiments.
Surgical preparation
Rats were vagotomized and a catheter inserted into the right femoral artery to measure blood pressure and take arterial blood samples. A rectal temperature probe was used to monitor and regulate body temperature. From a dorsal approach, the left phrenic nerve was isolated, cut distally, desheathed and placed on a bipolar silver recording electrode submerged in mineral oil. Nerve activity was amplified (gain ×10K), bandpass filtered (300 Hz to 20 kHz) (A-M Systems, Carlsberg, WA, USA) and integrated (absolute value, Powerlabs 830, AD Instruments, Colorado Springs, CO, USA, time constant 50 ms). The signal was digitized, recorded, and analyzed using Powerlabs 830 (version 7.2.2, AD Instruments).
To examine the effect of spinal COX inhibition, C2 laminectomy was performed and a primed silicone catheter inserted 2 mm caudally so that the catheter tip rested on the C4 dorsal surface, as described previously (Baker-Herman and Mitchell, 2002; MacFarlane and Mitchell, 2009; Huxtable et al., 2015).
Protocol
Baseline nerve activity was established with FIO2 ~0.56 (PaO2 > 300 mmHg) and CO2 added to the inspired gas (balance nitrogen). The CO2 apneic threshold was determined by slowly lowering inspired CO2 until phrenic nerve activity ceased. Inspired CO2 was slowly increased until phrenic nerve activity resumed (recruitment threshold). End-tidal CO2 was set ~2 mmHg above the recruitment threshold to establish baseline nerve activity. End-tidal CO2 was monitored and maintained throughout an experiment using a flow-through capnograph with sufficient response time to measure end-tidal CO2 in rats (Respironics, Andover, MA, USA).
Once phrenic nerve activity was stable, an arterial blood sample was taken to establish baseline conditions; these conditions were maintained for the duration of the experiment. Blood samples (62.5 μl in heparinized plastic catheter) were analyzed for PO2, PCO2, pH and base excess using a blood gas analyzer (ABL 800, Radiometer, Copenhagen, Denmark). After establishing baseline conditions, a moderate AIH protocol began consisting of 3 hypoxic episodes (5 min duration, 9–10.5% O2), separated by 5 min of control oxygen levels. Blood samples were taken during the first hypoxic episode, and 15, 30, and 60 min post-AIH. Data were included in analysis only if they complied with the following criteria: 1) PaO2 during baseline and post-AIH was >180 mmHg; 2) PaO2 during hypoxic episodes was between 35 to 45 mmHg; 3) PaCO2 remained within 1.5 mmHg of baseline throughout the post-AIH period. Maximum hypoxia + hypercapnia was assessed at the end of each experiment. Rats not responding to maximal stimulation were removed from analysis. Phrenic nerve amplitude was evaluated immediately before each blood sample and during minute two of hypoxia. Upon completion of experiments, rats were euthanized with a urethane overdose.
Data Analysis
Peak amplitude of integrated phrenic nerve activity was averaged for 30 bursts at each time point of interest. Changes in phrenic nerve burst amplitude were normalized to baseline values (percent change from baseline).
Statistical comparisons for hypoxic responses were made from data at minute two during the first hypoxic episode using a t-test (systemic NS-398 data), or One-Way ANOVAs on Ranks (spinal NS-398 data). Due to the addition of intrathecal vehicle and NS-398 groups, an ANOVA on Ranks was used for the spinal COX-2 inhibition study to permit appropriate statistical comparisons. Non-parametric analyses were used when data failed normality or equal variance.
Statistical comparisons for phrenic burst amplitude changes after AIH were made using a Repeated Measures Two-Way ANOVA with Fisher LSD post-hoc tests to identify individual differences (Sigma Stat version 11, Systat Software, San Jose, CA). Differences were considered significant if p<0.05; reported values are means ± 1 SEM.
Phrenic motoneuron back-labeling and tissue collection
Separate rat groups were intrapleurally injected with cholera toxin b-subunit (CTB; 25 μg/side, Calbiochem, Billerica, MA) to retrogradely label phrenic motoneurons as described previously (Mantilla et al., 2009; Guenther et al., 2010; Dale-Nagle et al., 2011; Golder et al., 2011; Dale et al., 2012; Lovett-Barr et al., 2012; Huxtable et al., 2015). Three days later, rats were treated with IH-1 (n=6) or normoxia (n=6), and transcardially perfused (16 hours post-IH-1) with cold phosphate buffered saline (PBS, 0.01 M, pH 7.4, Thermo Fisher Scientific, Waltham, MA), followed by 4% paraformaldehyde (PFA, Thermo Fisher Scientific, Waltham, MA) in 0.01 M PBS. After perfusion, brains and spinal cords were immersion-fixed in 4% PFA in 0.01 M PBS overnight at 4° C, saturated in 20% sucrose in 0.01 M PBS at 4°C and then 30% sucrose in 0.01 M PBS at 4°C to prevent tissue damage during the freezing process of tissue sectioning.
Immunohistochemistry
The cervical spinal cord (C3–C6) was cut into 40 μm sections (transverse or coronal) using a freezing microtome (Leica, SM 2000 R) and stored in an antifreeze solution (0.01M PBS, ethylene glycol, glycerol) at 4° C. Tissues were permeabilized with 0.1% Triton in PBS (PBSTx). Non-specific binding sites were blocked with 1% bovine serum albumin (BSA, Research Products International Corporation, Mount Prospect, IL) in PBSTx for one hour. To identify our proteins of interest, slices were placed on an orbital shaker for 16 hrs (room temperature) with the following antibodies: COX-2 (M-19) antibody (rabbit, 1:1000, Santa Cruz Biotechnology, Dallas, TX), anti-cholera toxin B (goat, 1:5000, Calbiochem, Billerica, MA) and CD11b (mouse, 1:200, AbD Serotec, Raleigh, NC) in 0.1% BSA. After incubation, tissues were washed (in PBSTx) and incubated in secondary antibodies (2 hrs, room temperature). Secondary antibodies were: AlexaFluor 488 donkey anti-rabbit (DAR, 1:1000, Invitrogen, Grand Island, NY), AlexaFluor 633 donkey anti-goat (DAG, 1:1000, Invitrogen, Grand Island, NY), and AlexaFluor 555 donkey anti-mouse (DAM, 1:1000, Invitrogen, Grand Island, NY). Sections were then washed in PBSTx and PBS before being mounted on charged microscope slides with anti-fade solution (ProLong Gold antifade reagent, Invitrogen, Grand Island, NY) and stored at 4°C. Negative controls were run concurrently to ensure specific labeling. Negative controls included COX-2 (M-19)-R primary with DAG and DAM secondaries, CD11b primary with DAR and DAG secondaries, anti-cholera toxin B primary with DAR and DAM secondaries were all run on separate sections to examine cross-reactivity between primary and secondary antibodies. Application of only primary antibodies (COX-2 (M-19)-R, CD11b and CTB) with no secondary antibodies was used to examine auto-fluorescence, and all secondary antibodies (DAR, DAM, DAG) with no primary antibodies was used to examine non-specific binding of secondary antibodies. In all cases, no staining was evident in any of the control sections.
Image processing
All immunofluorescent images (1024×1024 pixels, 20× or 100× magnification) were viewed using a Nikon C1 laser scanning confocal microscope with lambda strobing in the Nikon EZ-C1 Gold (Version 3.80) confocal imaging software; 2 μm step increments were used to acquire z-stacks. All image pairs (IH-1 vs. Nx sections) were imaged under identical microscope settings and identically adjusted for contrast/brightness in the Nikon EZ-C1 FreeViewer Gold software to ensure appropriate comparisons.
RESULTS
Systemic COX inhibition does not restore pLTF after IH-1
To begin our investigations of the role of COX enzyme activity in pLTF suppression following IH-1, we inhibited COX activity systemically after normoxia or IH-1 with a selective COX inhibitor (NS-398, 3 mg/kg, i.p., 3 hrs prior to AIH) at a dose previously established to be effective with this delivery method (Mouihate et al., 2010). pLTF was observed in Nx + NS-398 treated rats (76±40% change from baseline, 60 min post-AIH, n=6) (Fig. 1A, D) which is within the normal range for pLTF in Sprague Dawley rats (Baker-Herman and Mitchell, 2008; Baker-Herman and Strey, 2011), demonstrating that NS-398 does not affect pLTF in normal rats. In IH-1 + NS-398 treated rats, pLTF was not evident (−9±7% baseline, n=6), and was significantly reduced versus Nx + NS-398 + AIH treated rats (Fig. 1B, D, p=0.006). In TC rats (n=3 Nx, n=2 IH-1), pLTF was not evident as expected (−11±11% baseline, Fig. 1C, D). TC treated rats were not significantly different from IH-1 + NS-398 + AIH treated rats (p=0.79), but were significantly reduced versus Nx + NS-398 + AIH (p=0.005).
Figure 1.
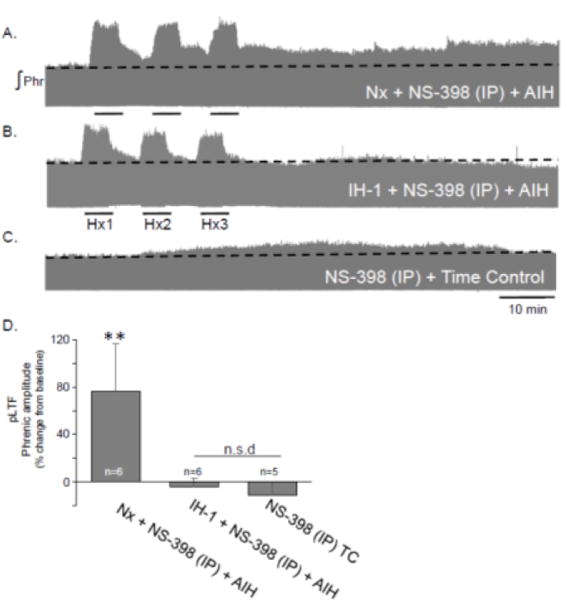
Systemic administration of a COX-2 inhibitor (NS-398, 3 mg/kg, i.p.) did not significantly restore pLTF after IH-1. Representative integrated phrenic neurograms demonstrate changes in phrenic nerve amplitude after acute intermittent hypoxia (3, 5 min Hx) in Nx + NS-398 (A), IH-1 + NS-398 (B), and NS-398 + time control treated rats. pLTF is evident as an increase above baseline (dotted line) in Nx + NS-398. Group data (E) demonstrate systemic treatment of NS-398 did restore pLTF in IH-1 pre-treated rats. (**p<0.01 from all other groups.)
Intrathecal COX inhibition does not restore pLTF after IH-1
Although pLTF following systemic COX inhibition was unaffected, we further tested the idea that spinal COX inhibition may be involved in pLTF impairment after IH-1. We questioned whether pLTF restoration after p38 MAP kinase inhibition (using SB202190; (Huxtable et al., 2015) could have been due to non-specific inhibition of COX activity, since other p38 MAP kinase inhibitors of similar chemical structure inhibit COX activity. Thus, we applied the COX inhibitor intrathecally (27 mM, i.t., 12 μl, 2 μl/30 sec). Spinal COX inhibition had little effect on pLTF in Nx treated rats (76±34% baseline, n=7, Fig. 2A) versus Nx + veh + AIH (55±18%, n=5; p=0.301) (Fig. 2D). While spinal NS-398 appeared to have a small (but insignificant) effect in IH-1 treated rats (37±18% baseline, n=7, Fig. 2B), this response did not reach statistical significance compared to vehicle treated IH-1 rats (−4±3% baseline, n=4; p=0.057, Fig. 2D).
Figure 2.
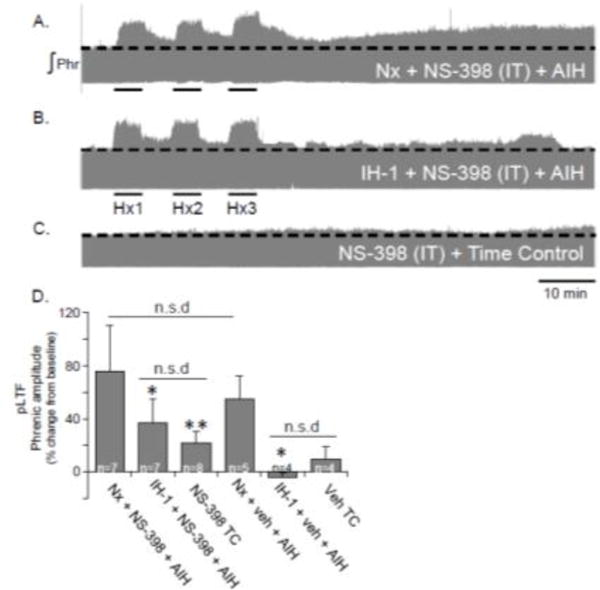
Intrathecal administration of a COX-2 inhibitor (NS-398, 27 mM) did not significantly restore pLTF after IH-1. Representative integrated phrenic neurograms demonstrate changes in phrenic nerve amplitude of after acute intermittent hypoxia in Nx + NS-398 (A), IH-1 + NS-398 (B), and NS-398 + time control treated rats (C). Despite apparent changes in pLTF magnitude, pLTF is not significantly restored after IH-1 + NS-398 (E). (**p<0.01, *p<0.05 significantly different from Nx treated.)
Paradoxically, TC rats treated with spinal NS-398 showed a small increase in phrenic nerve output from baseline (22±9% baseline, n=8, Fig. 2C), although this effect was not significantly different from vehicle time controls (10±9% baseline; p=0.567, Fig. 2D). pLTF was significantly reduced in IH-1 + NS-398 + AIH (p=0.037) and TC + NS-398 (p=0.003) versus Nx + NS-398 + AIH, similar to the vehicle treatment group (Fig. 2D) and previously published work (Huxtable et al., 2015). No significant difference was evident between IH-1 + NS-398 + AIH and TC + NS-398 (p = 0.384).
Short-term hypoxic phrenic responses were unaffected by COX inhibition
The lack of pLTF restoration after systemic NS-398 in IH-1 treated rats was not due to altered short-term hypoxic phrenic responses (Fig. 3) since no significant difference (p=0.637) was evident between the Nx + NS-398 + AIH (106±15% baseline) and IH-1 + NS-398 + AIH (94±15% baseline) groups (Fig. 3A). Similarly, there was no significant difference (p=0.532) between spinal NS-398 and vehicle in Nx or IH-1 treated rats (Fig. 3B). The short-term hypoxic phrenic response was 126±23% in Nx + NS-398 + AIH, 117±15% in IH-1 + NS-398 + AIH, 148±23% in Nx + veh + AIH, and 129±61% in IH-1 + veh + AIH. Thus, spinal COX inhibition had minimal effect on the short-term hypoxic phrenic response.
Figure 3.
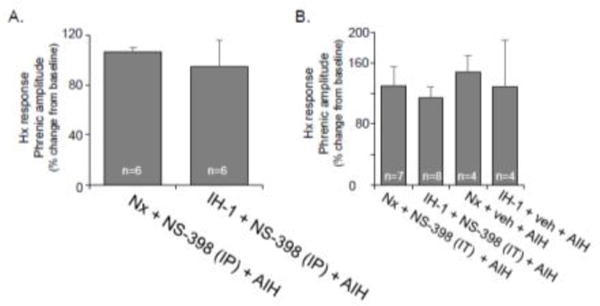
Acute hypoxic ventilatory responses are unaltered by the COX-2 inhibitor (NS-398) given systemically (A) or spinally (B). Systemic NS-398 (3 mg/kg, i.p.) group data (A) demonstrate no significant effect on the hypoxic response. Similarly, intrathecal NS-398 (27 mM) group data (B) show no significant effect on the hypoxic response.
COX-2 protein expression in the phrenic motor nucleus is unchanged by IH-1
COX-2 is constitutively expressed in neurons in the hippocampus, cortex and amygdala, and can be upregulated in neurons, astrocytes, and microglia throughout the CNS in response to inflammation (Yagami et al., 2015). Thus, we hypothesized that COX-2 protein levels in the phrenic motor nucleus would be increased in response to IH-1-induced inflammation. In coronal sections from C3-C6, we observed COX-2 protein expression throughout the phrenic motor pool (Fig. 4). The brightest COX-2 immunofluorescence was colocalized with CTB-labeled phrenic motoneurons. Other small cell gray matter labeling was common, but did not colocalize with CD11b-labeled microglia. No obvious changes in COX-2 protein expression were detected when comparing Nx and IH-1 treated rats (n=6 for each group), suggesting minimal COX-2 protein upregulation after IH-1. These findings are supported by high magnification images, which highlight COX-2 immunostaining in CTB back-labeled phrenic motoneurons, and minimal co-localization with CD11b-labeled microglia (Fig. 5).
Figure 4.
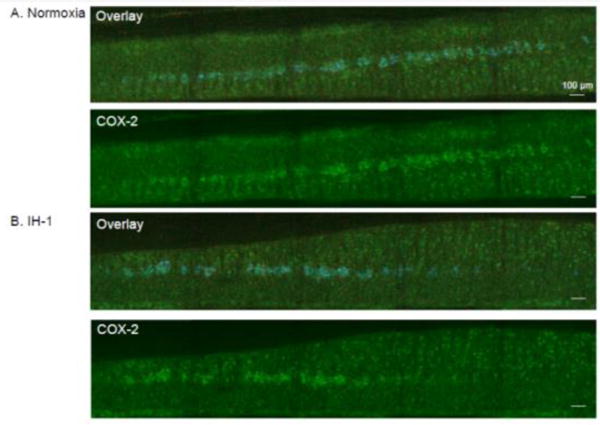
COX-2 immunofluorescence in coronal sections of the ventral cervical spinal cord is similar after systemic inflammation induced by IH-1 compared to normoxia treated. Confocal images (10× magnification) show representative COX-2 staining (green) after normoxia (A, n=6) and IH-1 (B, n=6). Overlay images show phrenic motor neurons identified by CtB back-label (blue) and CD11b labeled microglia (red).
Figure 5.
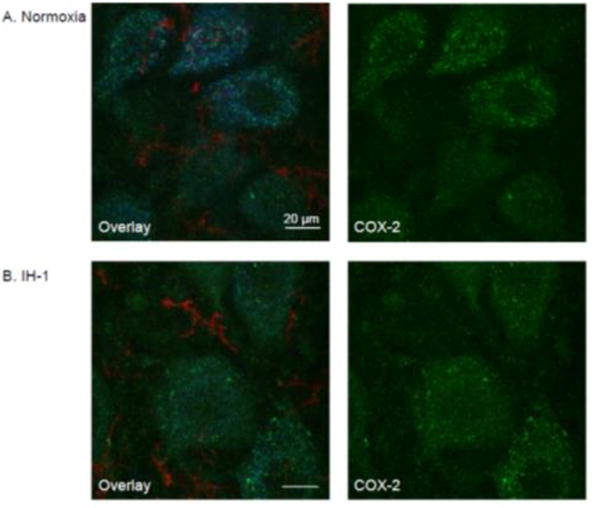
COX-2 immunofluorescence is similar in phrenic motoneurons after systemic inflammation induced by IH-1 compared to normoxia treated. Confocal images at high magnification (100×) show representative COX-2 staining (green) after normoxia (A, n=6) and IH-1 (B, n=6). COX-2 staining is prevalent in phrenic motoneurons (CtB back-labeled, blue) with only minor expression evident in microglia (CD11b, red) after either treatment.
DISCUSSION
To further our understanding of the mechanistic underpinnings of pLTF impairment by inflammation, we investigated the involvement of COX inhibition after IH-1. COX-2 is a bifunctional, rate-limiting enzyme in the production of prostaglandins in the CNS. It reportedly has low basal expression, but is rapidly increased by inflammation (Smith et al., 2000; Kudo and Murakami, 2005; Malcher-Lopes et al., 2008; Ma et al., 2013) which, in turn, further increases inflammation (Yagami et al., 2015). In earlier studies, systemic inflammation induced by LPS or IH-1 abolished pLTF, but was restored by high doses of the NSAID ketoprofen (Huxtable et al., 2013; Huxtable et al., 2015). We also found that a drug inhibiting p38 MAP kinase restores pLTF after IH-1 (Huxtable et al., 2015). However, since other p38 MAP kinase inhibitors of similar chemical structure can have non-selective effects on COX enzymatic activity (Borsch-Haubold et al., 1998), we hypothesized that pLTF restoration by ketoprofen (and potentially the p38 MAP kinase inhibitor) was due to COX inhibition.
Here, we demonstrate that COX inhibition does not restore pLTF. Although a tendency towards pLTF restoration with intraspinal COX inhibition was observed, the same trend was seen with the drug alone independent from AIH, suggesting NS-398 may have unaccounted for effects when delivered to the spinal cord. Importantly, spinal NS-398 fails to significantly increase pLTF compared to vehicle + IH-1 treated rats. Further, we confirm that the day following the last IH episode, IH-1 had no detectable impact on COX-2 protein expression in the cervical spinal cord. These observations are consistent with the interpretation that COX enzymatic activity is not critical in the mechanism whereby inflammation impairs moderate AIH-induced pLTF.
While the immunohistochemical findings do not detect changes in enzymatic activity, previous studies have validated the use of NS-398 to inhibit COX enzyme activity. While it remains possible that NS-398 failed to inhibit COX-2 enzymatic activity, abundant literature supports its effectiveness at the chosen dose and route of administration. Indeed, NS-398 is the most widely used COX-2 inhibitor in animal studies, and a wide range of concentrations/dosages and methods of application have been reported for its delivery in vivo and in vitro. Others using NS-398 at similar concentrations with similar delivery methods report significant COX-2 inhibition (Matsuura et al., 2009). Although NS-398 is described in many studies as a “selective” COX-2 inhibitor (Futaki et al., 1994; Huff et al., 1995; Vecchio and Malkowski, 2011), with an IC50 of 1.77 μM (Matsunaga et al., 2007), it also has an IC50 of 75 μM for COX-1 (Barnett et al., 1994). Since we do not know the spinal drug concentrations used here, after systemic (3 mg/kg) or spinal injection (27 mM), we cannot rule out effects on both COX enzymes at the doses used.
Contrary to other models of inflammation, we found no observable increase in COX-2 protein expression as detected by immunofluorescence in the phrenic motor nucleus after IH-1. However, COX-2 protein expression was evident in labelled phrenic motor neurons in both normoxia and IH-1 treated rats, suggesting COX-2 may play important roles in basal phrenic motoneuron function, similar to hippocampal and cortical glutamatergic neurons (Niwa et al., 2000; Yang and Chen, 2008). Surprisingly, little microglial co-expression was evident in normoxia or IH-1 treated rats, unlike responses to other inflammatory stimuli (Minghetti and Levi, 1995; Fiebich et al., 1996; Slepko et al., 1997; Walsh et al., 2000; Choi et al., 2009). This difference may be the result of the inflammatory stimulus (ie. IH-1 versus LPS), or of the mild inflammation triggered with this stimulus 16 hours after the IH-1 had ended (Huxtable et al., 2015).
These findings support a recent report in humans with chronic, incomplete spinal cord injuries in which ibuprofen (used at clinically relevant doses) failed to enhance AIH-induced motor plasticity based on measurements of ankle flexion strength (Lynch et al., 2017). At the doses used in that study, ibuprofen (an NSAID related to ketoprofen) primarily works by inhibiting COX activity (Auriel et al., 2014). Thus, independent of whether inflammation is present from spinal cord injury or from a single night of IH, COX inhibition does not restore plasticity. While the profile of inflammatory molecules in a chronically injured spinal cord is likely different from 8 hrs of IH, inhibiting COX activity has no effect in either instance.
In conclusion, we suggest increased COX enzyme activity is not involved in inflammation-induced impairment of moderate AIH-induced phrenic motor plasticity. Thus, the actions of the p38 MAP kinase inhibitor SB202190, which restores pLTF after IH-1 (Huxtable et al., 2015), are likely due to direct actions on p38 MAP kinase and are COX independent.
HIGHLIGHTS.
Cyclooxygenase (COX) enzyme activity has been implicated in the restoration of phrenic long-term facilitation (pLTF) following a single night of intermittent hypoxia (IH-1; 8 hours).
Here, we tested the role of COX activity using the pharmacologic inhibitor NS-398.
Neither systemic nor spinal COX inhibition was able to restore pLTF after IH-1.
Further, IH-1 had no discernable effect on COX-2 immunoreactivity in the phrenic motor nucleus.
We conclude that inflammatory impairment of pLTF elicited by IH-1 is independent of COX enzyme activity and likely prostaglandin production.
Acknowledgments
Support: Supported by NIH HL111598, Francis Family Foundation (AGH), Craig H. Neilsen Foundation (BJD)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions:
Huxtable – conception/design of the work, data acquisition and analysis, interpretation of data, drafting and revising the manuscript, final approval of the version to be published, agreeing to be accountable for all aspects of the work
Kopp - data acquisition and analysis, interpretation of data, revising the manuscript, final approval of the version to be published, agreeing to be accountable for all aspects of the work
Dougherty - data acquisition and analysis, interpretation of data, drafting and revising the manuscript, final approval of the version to be published, agreeing to be accountable for all aspects of the work
Watters - conception/design of the work, revising the manuscript, final approval of the version to be published, agreeing to be accountable for all aspects of the work
Mitchell - conception/design of the work, revising the manuscript, final approval of the version to be published, agreeing to be accountable for all aspects of the work
References
- Auriel E, Regev K, Korczyn AD. Nonsteroidal anti-inflammatory drugs exposure and the central nervous system. Handb Clin Neurol. 2014;119:577–584. doi: 10.1016/B978-0-7020-4086-3.00038-2. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Determinants of frequency long-term facilitation following acute intermittent hypoxia in vagotomized rats. Respir Physiol Neurobiol. 2008;162:8–17. doi: 10.1016/j.resp.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Strey KA. Similarities and differences in mechanisms of phrenic and hypoglossal motor facilitation. Respir Physiol Neurobiol. 2011;179:48–56. doi: 10.1016/j.resp.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett J, Chow J, Ives D, Chiou M, Mackenzie R, Osen E, Nguyen B, Tsing S, Bach C, Freire J, et al. Purification, characterization and selective inhibition of human prostaglandin G/H synthase 1 and 2 expressed in the baculovirus system. Biochimica et biophysica acta. 1994;1209:130–139. doi: 10.1016/0167-4838(94)90148-1. [DOI] [PubMed] [Google Scholar]
- Bazan NG. COX-2 as a multifunctional neuronal modulator. Nat Med. 2001;7:414–415. doi: 10.1038/86477. [DOI] [PubMed] [Google Scholar]
- Borsch-Haubold AG, Pasquet S, Watson SP. Direct inhibition of cyclooxygenase-1 and -2 by the kinase inhibitors SB 203580 and PD 98059. SB 203580 also inhibits thromboxane synthase. The Journal of biological chemistry. 1998;273:28766–28772. doi: 10.1074/jbc.273.44.28766. [DOI] [PubMed] [Google Scholar]
- Cabre F, Fernandez MF, Calvo L, Ferrer X, Garcia ML, Mauleon D. Analgesic, antiinflammatory, and antipyretic effects of S(+)-ketoprofen in vivo. J Clin Pharmacol. 1998;38:3S–10S. [PubMed] [Google Scholar]
- Cashman JN. The mechanisms of action of NSAIDs in analgesia. Drugs. 1996;52(Suppl 5):13–23. doi: 10.2165/00003495-199600525-00004. [DOI] [PubMed] [Google Scholar]
- Choi SH, Aid S, Bosetti F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends in pharmacological sciences. 2009;30:174–181. doi: 10.1016/j.tips.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale EA, Satriotomo I, Mitchell GS. Cervical spinal erythropoietin induces phrenic motor facilitation via extracellular signal-regulated protein kinase and Akt signaling. J Neurosci. 2012;32:5973–5983. doi: 10.1523/JNEUROSCI.3873-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale EA, Fields DP, Devinney MJ, Mitchell GS. Phrenic motor neuron TrkB expression is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. Experimental neurology. 2017;287:130–136. doi: 10.1016/j.expneurol.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale-Nagle EA, Satriotomo I, Mitchell GS. Spinal vascular endothelial growth factor induces phrenic motor facilitation via extracellular signal-regulated kinase and Akt signaling. J Neurosci. 2011;31:7682–7690. doi: 10.1523/JNEUROSCI.0239-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinney MJ, Fields DP, Huxtable AG, Peterson TJ, Dale EA, Mitchell GS. Phrenic long-term facilitation requires PKCtheta activity within phrenic motor neurons. J Neurosci. 2015;35:8107–8117. doi: 10.1523/JNEUROSCI.5086-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Reval MI, Ventura-Martinez R, Deciga-Campos M, Terron JA, Cabre F, Lopez-Munoz FJ. Evidence for a central mechanism of action of S-(+)-ketoprofen. European journal of pharmacology. 2004;483:241–248. doi: 10.1016/j.ejphar.2003.10.036. [DOI] [PubMed] [Google Scholar]
- Fiebich BL, Biber K, Lieb K, van Calker D, Berger M, Bauer J, Gebicke-Haerter PJ. Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2a-receptors. Glia. 1996;18:152–160. doi: 10.1002/(SICI)1098-1136(199610)18:2<152::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Mitchell GS. Respiratory neuroplasticity - Overview, significance and future directions. Experimental neurology. 2017;287:144–152. doi: 10.1016/j.expneurol.2016.05.022. [DOI] [PubMed] [Google Scholar]
- Futaki N, Takahashi S, Yokoyama M, Arai I, Higuchi S, Otomo S. NS-398, a new anti-inflammatory agent, selectively inhibits prostaglandin G/H synthase/cyclooxygenase (COX-2) activity in vitro. Prostaglandins. 1994;47:55–59. doi: 10.1016/0090-6980(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Golder FJ, Fuller DD, Lovett-Barr MR, Vinit S, Resnick DK, Mitchell GS. Breathing patterns after mid-cervical spinal contusion in rats. Experimental neurology. 2011;231:97–103. doi: 10.1016/j.expneurol.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rothi EJ, Lee KZ, Dale EA, Reier PJ, Mitchell GS, Fuller DD. Intermittent hypoxia and neurorehabilitation. Journal of applied physiology. 2015;119:1455–1465. doi: 10.1152/japplphysiol.00235.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther CH, Vinit S, Windelborn JA, Behan M, Mitchell GS. Atypical protein kinase C expression in phrenic motor neurons of the rat. Neuroscience. 2010;169:787–793. doi: 10.1016/j.neuroscience.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff R, Collins P, Kramer S, Seibert K, Koboldt C, Gregory S, Isakson P. A structural feature of N-[2-(cyclohexyloxy)-4-nitrophenyl] methanesulfonamide (NS-398) that governs its selectivity and affinity for cyclooxygenase 2 (COX2) Inflammation research: official journal of the European Histamine Research Society [et al] 1995;44(Suppl 2):S145–146. doi: 10.1007/BF01778304. [DOI] [PubMed] [Google Scholar]
- Huxtable AG, Smith SM, Vinit S, Watters JJ, Mitchell GS. Systemic LPS induces spinal inflammatory gene expression and impairs phrenic long-term facilitation following acute intermittent hypoxia. J Appl Physiol. 2013 doi: 10.1152/japplphysiol.01347.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable AG, Smith SM, Peterson TJ, Watters JJ, Mitchell GS. Intermittent Hypoxia-Induced Spinal Inflammation Impairs Respiratory Motor Plasticity by a Spinal p38 MAP Kinase-Dependent Mechanism. J Neurosci. 2015;35:6871–6880. doi: 10.1523/JNEUROSCI.4539-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable AG, Vinit S, Windelborn JA, Crader SM, Guenther CH, Watters JJ, Mitchell GS. Systemic inflammation impairs respiratory chemoreflexes and plasticity. Respir Physiol Neurobiol. 2011;178:482–489. doi: 10.1016/j.resp.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokki H. Ketoprofen pharmacokinetics, efficacy, and tolerability in pediatric patients. Paediatric drugs. 2010;12:313–329. doi: 10.2165/11534910-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Kotilinek LA, Westerman MA, Wang Q, Panizzon K, Lim GP, Simonyi A, Lesne S, Falinska A, Younkin LH, Younkin SG, Rowan M, Cleary J, Wallis RA, Sun GY, Cole G, Frautschy S, Anwyl R, Ashe KH. Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain: a journal of neurology. 2008;131:651–664. doi: 10.1093/brain/awn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo I, Murakami M. Prostaglandin E synthase, a terminal enzyme for prostaglandin E2 biosynthesis. Journal of biochemistry and molecular biology. 2005;38:633–638. doi: 10.5483/bmbrep.2005.38.6.633. [DOI] [PubMed] [Google Scholar]
- Lleo A, Galea E, Sastre M. Molecular targets of non-steroidal anti-inflammatory drugs in neurodegenerative diseases. Cell Mol Life Sci. 2007;64:1403–1418. doi: 10.1007/s00018-007-6516-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barr MR, Satriotomo I, Muir GD, Wilkerson JE, Hoffman MS, Vinit S, Mitchell GS. Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic cervical spinal injury. J Neurosci. 2012;32:3591–3600. doi: 10.1523/JNEUROSCI.2908-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Duffell L, Sandhu M, Srivatsan S, Deatsch K, Kessler A, Mitchell GS, Jayaraman A, Rymer WZ. Effect of acute intermittent hypoxia on motor function in individuals with chronic spinal cord injury following ibuprofen pretreatment: A pilot study. The journal of spinal cord medicine. 2017;40:295–303. doi: 10.1080/10790268.2016.1142137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Matsuwaki T, Yamanouchi K, Nishihara M. Cyclooxygenase-2-related signaling in the hypothalamus plays differential roles in response to various acute stresses. Brain Res. 2013;1508:23–33. doi: 10.1016/j.brainres.2013.02.042. [DOI] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J Physiol. 2009;587:5469–5481. doi: 10.1113/jphysiol.2009.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol. 2007;92:27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- Malcher-Lopes R, Franco A, Tasker JG. Glucocorticoids shift arachidonic acid metabolism toward endocannabinoid synthesis: a non-genomic anti-inflammatory switch. European journal of pharmacology. 2008;583:322–339. doi: 10.1016/j.ejphar.2007.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Zhan WZ, Sieck GC. Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods. 2009;182:244–249. doi: 10.1016/j.jneumeth.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateika JH, Sandhu KS. Experimental protocols and preparations to study respiratory long term facilitation. Respir Physiol Neurobiol. 2011;176:1–11. doi: 10.1016/j.resp.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateika JH, Komnenov D. Intermittent hypoxia initiated plasticity in humans: A multipronged therapeutic approach to treat sleep apnea and overlapping co-morbidities. Experimental neurology. 2017;287:113–129. doi: 10.1016/j.expneurol.2016.05.011. [DOI] [PubMed] [Google Scholar]
- Matsunaga A, Kawamoto M, Shiraishi S, Yasuda T, Kajiyama S, Kurita S, Yuge O. Intrathecally administered COX-2 but not COX-1 or COX-3 inhibitors attenuate streptozotocin-induced mechanical hyperalgesia in rats. European journal of pharmacology. 2007;554:12–17. doi: 10.1016/j.ejphar.2006.09.072. [DOI] [PubMed] [Google Scholar]
- Matsuura T, Takuwa H, Bakalova R, Obata T, Kanno I. Effect of cyclooxygenase-2 on the regulation of cerebral blood flow during neuronal activation in the rat. Neuroscience research. 2009;65:64–70. doi: 10.1016/j.neures.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Levi G. Induction of prostanoid biosynthesis by bacterial lipopolysaccharide and isoproterenol in rat microglial cultures. J Neurochem. 1995;65:2690–2698. doi: 10.1046/j.1471-4159.1995.65062690.x. [DOI] [PubMed] [Google Scholar]
- Mouihate A, Galic MA, Ellis SL, Spencer SJ, Tsutsui S, Pittman QJ. Early life activation of toll-like receptor 4 reprograms neural anti-inflammatory pathways. J Neurosci. 2010;30:7975–7983. doi: 10.1523/JNEUROSCI.6078-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa K, Araki E, Morham SG, Ross ME, Iadecola C. Cyclooxygenase-2 contributes to functional hyperemia in whisker-barrel cortex. J Neurosci. 2000;20:763–770. doi: 10.1523/JNEUROSCI.20-02-00763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepko N, Minghetti L, Polazzi E, Nicolini A, Levi G. Reorientation of prostanoid production accompanies “activation” of adult microglial cells in culture. Journal of neuroscience research. 1997;49:292–300. doi: 10.1002/(sici)1097-4547(19970801)49:3<292::aid-jnr4>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annual review of biochemistry. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Vecchio AJ, Malkowski MG. The structure of NS-398 bound to cyclooxygenase-2. Journal of structural biology. 2011;176:254–258. doi: 10.1016/j.jsb.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinit S, Windelborn JA, Mitchell GS. Lipopolysaccharide attenuates phrenic long-term facilitation following acute intermittent hypoxia. Respir Physiol Neurobiol. 2011;176:130–135. doi: 10.1016/j.resp.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DT, Perry VH, Minghetti L. Cyclooxygenase-2 is highly expressed in microglial-like cells in a murine model of prion disease. Glia. 2000;29:392–396. [PubMed] [Google Scholar]
- Yagami T, Koma H, Yamamoto Y. Pathophysiological Roles of Cyclooxygenases and Prostaglandins in the Central Nervous System. Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9355-3. [DOI] [PubMed] [Google Scholar]
- Yang H, Chen C. Cyclooxygenase-2 in synaptic signaling. Curr Pharm Des. 2008;14:1443–1451. doi: 10.2174/138161208784480144. [DOI] [PMC free article] [PubMed] [Google Scholar]