

Abstract
The recent X-ray structure of the cytochrome bd respiratory oxygen reductase showed that two of the three heme components, heme d and heme b595, have glutamic acid as an axial ligand. No other native heme proteins are known to have glutamic acid axial ligands. In this work, site-directed mutagenesis is used to probe the roles of these glutamic acids, E445 and E99 in the E. coli enzyme. It is concluded that neither glutamate is a strong ligand to the heme Fe and they are not the major determinates of heme binding to the protein. Although very important, neither glutamate is absolutely essential for catalytic function. The close interactions between the three hemes in cyt bd result in highly cooperative properties. For example, mutation of E445, which is near heme d, has its greatest effects on the properties of heme b595 and heme b558. It is concluded that 1) O2 binds to the hydrophilic side of heme d and displaces E445; 2) E445 forms a salt bridge with R448 within the O2 binding pocket, and both residues play a role to stabilize oxygenated states of heme d during catalysis; 3) E445 and E99 are each protonated accompanying electron transfer to heme d and heme b595, respectively; 4) All protons used to generate water within the heme d active site come from the cytoplasm and are delivered through a channel that must include internal water molecules to assist proton transfer: [cytoplasm]→E107→E99 (heme b595)→E445 (heme d)→oxygenated heme d.
Graphical abstract

1. Introduction
The tri-heme cytochrome bd family of respiratory oxygen reductases (cyt bd’s) are present in many in bacteria and archaea but not in eukaryotes [1]. Included in the cyt bd family are the so-called cyanide-insensitive oxidases or CIOs [1, 2] in which the active-site heme d is replaced by heme b, though the enzymes otherwise appear unchanged. All of the characterized cyt bd’s are quinol:O2 oxidoreductases, catalyzing the 2-electron oxidation of (usually) ubiquinol or menaquinol and the 4-electron reduction of O2 to H2O [3–6]. Cyt bd’s typically have a high affinity for oxygen, and appear to be particularly important for oxygen-scavenging under micro-aerobic growth conditions [7, 8]. Although they are not proton pumps, cyt bd’s generate a proton motive force and are energy-conserving. The oxidation of the quinol molecules releases protons on the periplasmic side of the membrane (electrically positive side) while the protons used to generate water at the enzyme active site are taken from the cytoplasmic side of the membrane (electrically negative). This results in the electrogenic net transfer of 4 protons across the membrane per O2 [9].
Most studies of cyt bd have focused on E. coli cyt bd-I, which is one of two cyt bd’s encoded by E. coli, the other being cyt bd-II [10, 11]. A major advance facilitating research on cyt bd was the recently reported structure of a cyt bd from Geobacillus thermodenitrificans [12], an enzyme that is a homologue of E. coli cyt bd-I. The Gt-cyt bd contains three subunits: CydA and CydB each contain 9 transmembrane helices, and CydS has one transmembrane helix (Figure 1A). These subunits are equivalent to E. coli CydA, CydB and CydX [13–15]. There are three hemes present, all within CydA (Figure 1). Heme b558 is a low spin heme located near the quinol binding site and serves for the input of electrons [4–6]; 2) Heme d is a high spin chlorin that is unique to this family of enzymes and is the site where O2 binds and is reduced to water [3, 16–19]; 3) Heme b595 is a high spin heme which relays electrons from heme b558 to heme d [20] and which is in van der Waal’s contact with heme d [21]. The structures of heme b and heme d are shown in Supplementary Figure S1. Previous evidence of the strong interactions between heme d and heme b595 indicated that the two hemes functioned as a bimetallic active site where O2 is reduced to water [1, 21, 22], analogous to the binuclear center at the active site of cytochrome oxidases in the family of heme-copper respiratory oxygen reductases [23]. The structure, however, showed that the two heme Fe’s do not form a bimetallic center [12].
Figure 1. Views of the three hemes in the crystal structure of cyt bd from Geobacillus thermodenitrificans (PDB code: 5IR6 from [12]).
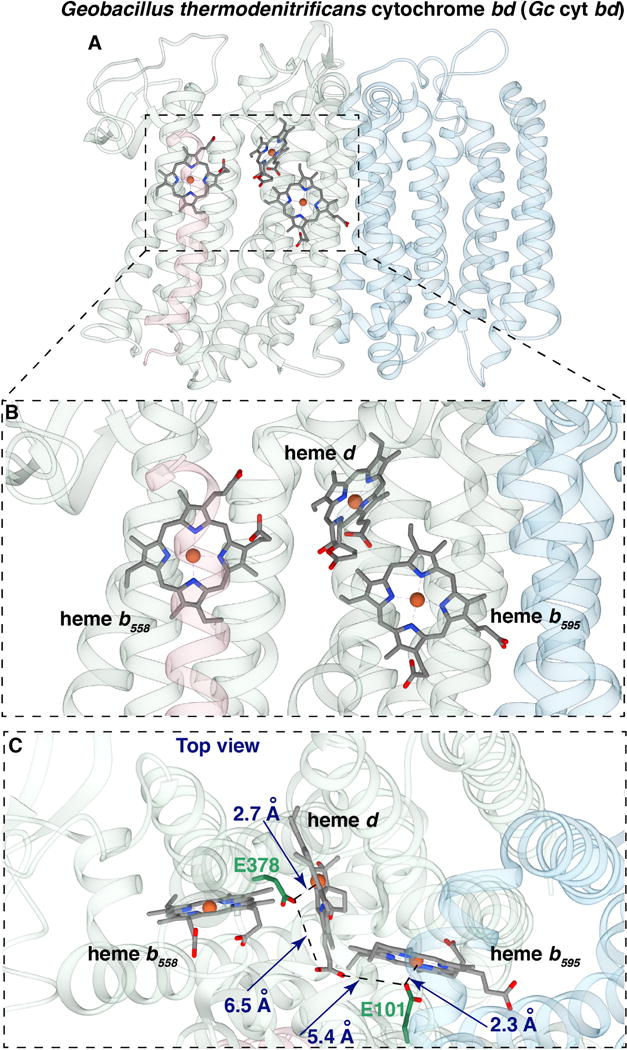
(A) View from the side showing subunit I, containing the hemes, and subunit II (light blue). (B) Closer view from the side of the three hemes. (C) View from the periplasm of the three hemes, also showing the glutamate ligands GtE378 and GtE101, which correspond to EcE445 and EcE99, respectively, from E. coli cyt bd. (Figures made with UCSF package Chimera)
More remarkably, the structure reveals that GtE101 (EcE99) and GtE378 (EcE445) are adjacent to the Fe components of heme b595 and heme d, respectively (Figure 1). (Note that all residues referred to in this work are located within the CydA subunit). In each case, the glutamic acid carboxyl group is close enough to be considered an axial ligand to the respective adjacent heme, 2.1 Å to heme b595 and 2.7 Å to heme d [12]. Although these two hemes are in van derWaal’s contact at their periphery, the heme irons and associated glutamates for heme b595 and heme d are well separated (Figure 1). There are no other examples of native heme proteins in which glutamate or aspartate is a heme axial ligand. Previous studies have suggested that EcE107 (GtE108) and EcE99 (GtE101) play a role in facilitating the proton transfer from the cytoplasm to the catalytic active site, providing protons for the formation of water [9, 24, 25], and it has been proposed that EcE99 is the axial ligand to heme d [26]. However, all the previous studies, performed using Ec-cyt bd-I, were performed prior to the realization that CydX (equivalent to Gt-CydS) is a third subunit in addition to CydA and Cyd B [14, 15]. In the previous studies, recombinant enzyme was obtained using plasmid-encoded cydAB, and the protein was isolated using traditional column chromatography, since affinity tags on CydA or CydB perturb the enzyme or prevent assembly. Quite likely, variable amounts of chromosome-encoded CydX were present in the old preparations of the enzyme, though the isolated enzymes contained the full complement of bound hemes and had reasonable activity. In the current work, the Ec-cyt bd-I is isolated using plasmid-encoded cydABX and isolated using a strep-tag on CydX using a recently developed protocol [15].
The improved protocol results in higher purity, greater stability and higher specific activity of the isolated enzyme, thus justifying a re-examination of previous mutations in both EcE99 and EcE445 as well as examining different mutants not previously studied. Several features not previously noted are discussed, including a salt bridge between EcE445 and EcR448 (GtE378 and GtR381) at the surface of heme d.
2. Materials and methods
2.1 Site directed mutagenesis of cytochrome bd from Escherichia coli
The construct previously prepared for purification of WT cytochrome bd with a strep-tag was used as a template [15]. Mutagenesis primers (Supplementary Information Table S1) were designed for point mutations and the Quikchange mutagenesis kit (Stratagene) was used for PCR amplification. The mutated plasmids were transformed into XL-10 Gold cells and incubated overnight at 37°C to recover colonies. The plasmid sequence was verified by sequencing (ACGT Technologies).
2.2 Cell Growth and protein purification
A single colony was inoculated into 5 ml of LB with 100 μg/ml ampicillin and incubated with shaking at 37 °C. Yeast extract and tryptone were purchased from Acumedia and NaCl from Sigma-Aldrich. The following day, the 5 ml culture was inoculated in 300 ml LB with 100 μg/ml ampicillin and grown overnight at 37 °C. On the third day, 10 ml of the secondary culture was inoculated into twenty four 2-L flasks containing 1 L LB with 100μg/ml ampicillin, each. The flasks were incubated at 37 °C while shaking at 200 rpm, until the OD600 of the culture reached 0.6. IPTG was added to a final concentration of 0.5 mM. The temperature was then lowered to 30°C, and the culture was incubated for 8 hrs or overnight.
The fully grown cultures were pelleted by centrifugation at 8000 rpm for 8 minutes, in 500-ml centrifuge bottles. The harvested cells were resuspended in 100 mM Tris-HCl, 10 mM MgS04, pH 8, with DNaseI and a protease inhibitor cocktail from Sigma. The cells were then homogenized using a Bamix Homogenizer, and passed three times through a Microfluidizer cell at 100 psi to lyse the cells. The soluble fraction of the lysate was separated from the insoluble by centrifugation at 8000 rpm. Membranes were obtained by centrifugation at 42000 rpm using a Beckman 45 Ti rotor in a Beckman ultracentrifuge for 4 hours.
Membranes were resuspended in 50 mM sodium phosphate, 300 mM NaCl, pH 8, and then solubilized with 1% DDM (dodecyl maltoside) or 1% SML (sucrose monolaurate). The solubilized membranes were pelleted by centrifugation at 42000 rpm for 45 minutes to remove unsolubilized membranes. The supernatant was stirred with Strep-tactin resin (QIAGEN) for 1 hr and then loaded onto a column. The column was washed with 20 column volumes of 50 mM sodium phosphate, 300 mM NaCl, pH 8, 0.05% DDM, and the protein was eluted with 2.5 mM desthiobiotin in the same buffer. The fractions with protein were pooled and concentrated using an Amicon concentrator with a 100 kDa molecular weight filter. The concentrated protein was exchanged into 50 mM sodium phosphate, 0.05% DDM, pH 7, using the concentrator. This protein was used for further studies.
2.3 UV-visible spectroscopy
Spectra of the protein were obtained using an Agilent DW-2000 Spectrophotometer in the UV-visible region with a 1-cm pathlength cuvette. The spectrum of this air-oxidized enzyme is referred to as that of the “air-oxidized” or “as isolated” protein. The enzyme was reduced with dithionite to obtain the “reduced” spectrum. It is necessary to incubate the enzyme with an oxidant to obtain the spectrum of the fully oxidized protein.
2.4 Pyridine hemochrome and heme analysis
For the wild type or mutant enzymes, 35 μl of the enzyme solution (1–10 μM) was mixed with an equal volume of 40% pyridine with 200 mM NaOH. The “oxidized” or “reduced” spectra were obtained by adding excess ferricyanide or the dithionite, respectively, and the concentration of heme b was calculated using the method described in [27]. Since the pyridine hemochrome complex of heme d is not stable, the concentration of heme d was estimated, instead, from the spectrum of the dithionite-reduced enzyme using the difference extinction coefficient ε(629–670 nm) = 25 mM−1 cm−1[28]. The “reduced” spectrum is preferred for this purpose rather than the reduced minus oxidized spectrum which has wider peak due to heme d that is more susceptible to perturbations by mutations. The concentration of heme b558 was estimated using the difference extinction coefficient ε(561–580 nm) = 21 mM−1cm−1 for the reduced minus air-oxidized difference spectrum [28]. It was assumed that all the mutants contained one equivalent of heme b558 so, in essence, the concentration of cyt bd is based on the measurement of heme b558.
2.5 Measurement of oxygen reductase activity
Oxygen reductase activity was measured using the Mitocell Miniature Respirometer MT200A (Harvard Apparatus). Ubiquinol-1 (Q1, 250 μM) was used as the electron donor to measure the rate of reduction of O2 by the wild type and mutant enzymes, and the Q1 was maintained reduced by the presence of 5 mM dithiothreitol (DTT). KCN (300 μM) was used to test the cyanide-sensitivity of the enzymes since this will eliminate any activity due to contaminating cytochrome bo3. Such contamination was found not to be an issue.
2.6 Electrochemical measurement of midpoint potential of the hemes
The absorption differences between selected wavelengths [29] were used to determine the redox state of heme d and heme b558 as a function of the solution potential. A potentiometric cell was connected to an Agilent DW-2000 Spectrophotometer to simultaneously measure the solution potential and optical spectra in a set up similar to the one described [30]. A Calomel electrode was connected to a transparent cuvette and the electrode connected to a potentiometer. The cuvette contained 2.5 ml of a solution of the purified enzyme and a cocktail of redox mediators, each at a final concentration of 10 μM: anthraquinone-2-sulfate (Em=−225 mV), duroquinone (Em=+5 mV), p-benzoquinone (Em=+280 mV), (2,6)-dimethylbenzoquinone (Em=+180 mV), hexammine ruthenium (III) chloride (Em=+50 mV), and (α-methyl)ferrocenyl methanol (Em=+430 mV). The solution was poised at a redox potential of 440 mV (SHE) by adding small increments of potassium ferricyanide and then successively reducing the sample using aliquots of a fresh solution of sodium dithionite. At each successive addition of reductant, the UV-visible spectrum was obtained to monitor the reduction of the hemes. The data were analyzed by fitting the absorbance at selected wavelengths to the following function using OriginPro 2016 or Matlab R2015a, based on the Nernst equation.
where y is the absorbance change measured as a function of the fraction of the reduced species; A1 and A0 are parameters related to the absorbance of the fully reduced and fully oxidized heme species; n=1, the number of electrons in the ferric/ferrous transition; X is the solution potential; and Em1 is the midpoint potential of the species being reduced. For simplicity, the redox interactions between the hemes, which are generally small [29], were not included in the analysis.
3. Results
Four mutations of E445 were made and examined: E445A, E445Q, E445H and E445C. The structure [12] shows that the equivalent residue in Gt-cyt bd (GtE378) is about 2.7 Å from the heme d Fe. There is no reliable way to obtain with certainty the values for the heme contents of the isolated proteins. The mutations can alter the UV-visible spectra of heme d and heme b595, and heme d cannot be assayed by extraction of the pyridine hemochrome complex due to instability. The measurement that is most reliable is the pyridine hemochrome measurement of extracted heme b (heme b595 plus heme b558). It is assumed that the mutations do not result in changing the amount of the low spin heme b558 in the preparations, so all enzymes are assigned a content of 1 equivalent of heme b558. This is reasonable since heme b558 is the most stable of the three hemes in the enzyme, and can be present even in the complete absence of both heme d and heme b595 [4]. If the total heme b present is greater than 1 but less than 2 equivalents per heme b558, it is assumed the additional heme b is bound as high spin heme b595. If the total amount of extracted heme b is more than 2 equivalents per heme b558, it is assumed that the excess above 2 equivalents is bound at the site where heme d is normally bound in the wild type enzyme. The heme d content is measured from the UV-visible spectrum of the purified protein, subject to error due to spectroscopic perturbations due to the mutations. This information is shown in Table 1.
Table 1.
Heme content of WT, E99 mutants and E445 mutants of E. coli cyt bd
| Sample | heme b558 | heme b595 | excess heme b | heme d | Quinol oxidase activity (%) |
|---|---|---|---|---|---|
| WT | 1 | 1 | – | 0.92 | 100 |
| E445A | 1 | 1.0 | – | 1.511 | 1 |
| E445C | 1 | 0.85 | – | 1.311 | 3 |
| E445H | 1 | 0.59 | – | 0.74 | <3 |
| E445Q | 1 | 0.81 | – | 0.59 | 15 |
| E99A | 1 | 1.0 | – | 0.43 | 72 |
| E99H | 1 | 1 | 0.653 | < 0.352 | < 3 |
| E99Q | 1 | 1 | 0.633 | < 0.372 | < 3 |
Apparent excess heme d is attributed to perturbation of the extinction coefficients of the protein-bound heme due to the mutations. The pyridine hemochrome of heme d is not stable and quantitation by this approach is unreliable.
The spectra of protein-bound heme d in these mutants is very perturbed making quantitation by the standard extinction coefficients unfeasible. It is assumed that the excess heme b must be bound to the site where heme d normally binds and the amount of heme d bound to that site cannot be more than that needed to account for full occupancy of that binding site.
The amount of heme b extracted from these mutants is more than can be accounted for one heme bound to each of the two heme b binding sites (heme b558 and heme b595). It is assumed that each of these binding sites is fully occupied and the excess heme b binds elsewhere, presumably in the site that normally binds to heme d.
3.1 Estimated heme content of the E445 mutants
Figure 2 shows the absorption spectra of the isolated proteins and indicates that some of the mutants exhibit spectral perturbations. For example, the absorption peak for heme d appears broad in the air-oxidized spectrum of E445A (Figure 2b). Furthermore, an absorption peak at 606 nm is present in the spectra of both the air-oxidized and dithionite-reduced spectra of E445A. This peak is persistent upon removal of oxygen from the sample. (Figure 2i). It was previously reported that in E445A, heme b595 is “locked” in the ferric state (i.e., not reducible by dithionite) [28]. It is, therefore, possible that the 606 nm peak is a perturbed form of ferric b595, and an altered spectrum of heme b595 could, in principle, interfere with the spectroscopic quantitation of heme b558. With these caveats, the estimation of the amounts of each heme in the isolated enzymes are shown in Table 1. Despite the quantitative uncertainties, the data show that all of the E445 mutants have a significant amount of heme d present. Two of the mutants, E445A and E445C appear to have more than 1 mol of heme d per mole of heme b558. Since the heme d peak in the UV-visible spectra is altered and appears wider, it is reasonable to suppose that the measured excess of heme d in these mutants is due to perturbed extinction coefficients for protein-bound ferrous heme d. It appears that E445H and E445Q each have a lower but substantial amount of heme d.
Figure 2. (a-h) Air-oxidized and dithionite-reduced spectra of WT Ec-cyt bd and of EcE445 mutants and EcE99 mutants.
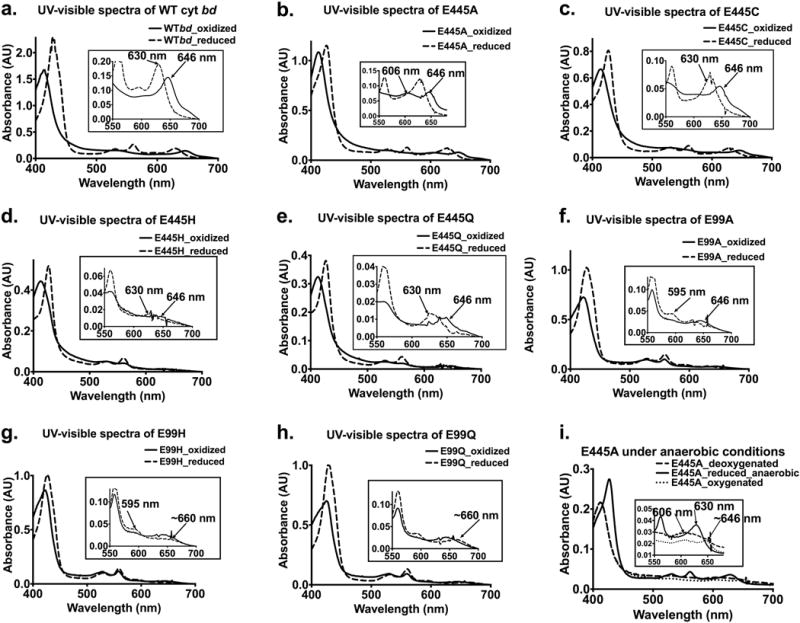
Insets have been used to highlight unique and unexpected spectroscopic features in the UV-visible spectra.
The simplest interpretation is that heme d is perturbed but the amount bound to the enzyme is unchanged when E445 is changed to a small residue (E445A, E445C). Substitution of an isosteric residue, E445Q, or bulkier residue, E445H, both of which can provide nitrogen ligation to heme d, reduces the amount of heme d that is bound, but not drastically. Previous examination of E445Q showed no heme d present [31], but the improved protocols to obtain the enzyme in a more stable form in the current work clearly indicates the heme d remains in this mutant to a significant extent.
Previous studies on E445A [31], on the other hand, reported results similar to those presented here. The presence of heme b595 is not obvious from the spectra of any of the mutants, but the total content of heme b by pyridine hemochrome analysis shows more heme b beyond the 1 equivalent of heme b558. It is reasonably assumed that this “excess” heme b is due to heme b595. By this measure, all the E445 mutants have a significant amount of heme b595, with E445H having the lowest content of heme b595 (0.59) (Table 1).
3.2 Quinol oxidase activity of the E445 mutants
All of the E445 mutants have much lower specific activity (normalized per heme d) than the WT, shown in Table 1. Essentially, E445A, E445C and E445H are inactive. The only mutant with significant activity is E445Q, which retains about 15% of the specific activity. Hence, although the importance of E445 for enzyme function is evident in this and in previous studies, the fact that some activity is retained in the damaged E445Q mutant demonstrates that this residue is not absolutely essential for function.
3.3 Heme content of the E99 mutants
Previous studies of E99 mutants indicated the absence of heme d [25, 26, 29], leading to the conclusion that E99 is the axial ligand to heme d [26]. The current work shows that heme d is present, however, in the E99A, E99H and E99Q mutants. The spectroscopic features of E99A (Figure 2f) are like WT cyt bd so the estimated the content of heme d (0.43) is likely reliable. The spectra of the E99H and E99Q mutants indicate that heme d is present, but the spectrum of the dithionite-reduced form is very red-shifted (peak at 665 nm instead of 630 nm) and not possible to quantify. An upper bound to the amount of heme d present in the E99H and E99Q mutants (Table 1) was estimated by assuming the “excess” heme b (beyond 2 equivalents per mol enzyme) is bound to the heme d-binding site, and assuming the capacity of that site is 1 equivalent (heme b plus heme d). This is discussed further below.
A significant portion of the “as isolated” air-oxidized form of WT cyt bd has O2 bound to ferrous heme d, indicated by a peak in the absorption spectrum at 646 nm. When the air-oxidized form of the WT cyt bd is made anaerobic (e.g., vacuum-treated or after passing Argon over the sample) the amplitude of the peak at 646 nm diminishes (Supplementary Figure S2). The air-oxidized (as isolated) form of both the E99H and E99Q mutants also have a peak at 646 nm, but when the samples are made anaerobic by evacuating the air, the absorption at 646 nm is not diminished. Upon reduction by dithionite, the absorption peak shifts from 646 nm to 665 nm (Supplementary Figure S3). The origin of this spectroscopic feature is not known. Perhaps the oxygen-bound ferrous heme d in the “as isolated” form is not dithionite-reducible in these mutants or perhaps the spectrum is red-shifted due to altered hydrogen bonding.
Both the E99Q and E99H mutants have a large excess of heme b (>2.6) over the expected maximum of 2 equivalents per mol of enzyme. One possible explanation is that a substantial portion of the site that normally binds heme d is bound, instead, to high spin heme b. This is what is found for the so-called cyanide-insensitive oxidases (CIOs), a subset of cyt bd enzymes which lacks heme d and only contains heme b [1, 2]. This is consistent with the hypothesis that heme d is formed in situ from bound heme b by hydroxylation of porphyrin ring (see Supplementary Figure 1). Disruption of proton and/or electron transfer in the E99H and E99Q mutants may inhibit the conversion of heme b to heme d. This proposal is consistent with the observation that the isolated, air-oxidized forms of both the E99H and E99Q mutants have a substantial fraction of heme b558 in the reduced (ferrous) state, suggesting obstruction of electron transfer from heme b595 to heme d. The estimated amounts of heme d (Table 1) in both the E99H and E99Q mutants is based on the assumption that the amount of total heme b that is in excess of that accounted for by full occupancy of the heme b558 and heme b595 sites is due to binding of heme b to the site that normally binds to heme d, and the remaining capacity to bind heme d at that site is limited to a total capacity of one heme at that site. It is noted that the data show that neither E99Q or E99H form strong axial ligands with heme b595 since that would result in a low spin heme that would be optically indistinguishable from heme b558 which is used to measure the enzyme concentration. The consequence would be that the amount of the total extracted heme b beyond that assigned to heme b558 would be very low, which is the opposite of what is observed with the E99Q and E99H mutants (Table 1).
The dramatic influence of the E99 mutants on the stability and properties of the heme d is consistent with previous work [25, 26, 29], which concluded that E99 is the axial ligand to heme d. The X-ray structure [12], however, shows that E99 is adjacent to heme b595.
3.4 Quinol oxidase activity of the E99 mutants
Of the three E99 mutants examined, only E99A has ubiquinol-1 oxidase activity (Table 1). The specific activity is quite high when expressed per equivalent of heme d, about 72% of the wild type value. Previous work also demonstrated that both the E99A and E99L mutants have significant enzyme activity when assayed within E. coli membranes (13% and 7%, respectively) despite the reported content of heme d being too low to be quantified [25].
As concluded for E445, the current work demonstrates that although E99 is very important to the function of cyt bd, it is not absolutely required.
3.5 Perturbation of the midpoint potentials of the hemes by the E445Q, E445A and E445C mutants
Spectro-electrochemical redox titrations were performed to determine the midpoint potentials (pH 7) of heme b558 and heme d due to mutating E445. Previous work has shown that the midpoint potential of heme b558 is sensitive to the environment and depends on the detergent that is present when examining the isolated proteins. Hence, measurements were made with two different detergents, dodecylmaltoside (DDM) and sucrose monolaurate (SML). The midpoint potentials of heme d and heme b558 were resolved, but not that of heme b595, except for the WT cyt bd. The data are shown in Table 2 and in Figure 3.
Table 2. Midpoint potentials of heme d and heme b558 of WT and E445 mutants of E. coli.
cyt bd determined in detergents dodecylmaltoside (DDM) or sucrose monolaurate (SML). Midpoint potentials are vs the Standard Hydrogen Electrode (SHE).
| Sample | heme b558 (mV) | heme d (mV) |
|---|---|---|
| in DDM detergent | ||
| WT | 88 | 246 |
| E445A | 46 | 265 |
| E445Q | 183 | 199 |
| in SML detergent | ||
| WT | 183 | 289 |
| E445C | 156 | 222 |
Figure 3. Spectro-electrochemical titrations of the WT Ec-cyt bd and the EcE445Q and EcE445C mutants.

The redox state of heme d was monitored by the absorption difference (ΔA) as a function of solution potential (panels A, C and D). ΔA was measured by using the following linear combination of wavelengths (A629 nm – 0.5*(A660 nm + A608 nm)). Similar trends were observed with the wavelength combinations proposed in [29]. The redox state of heme b558 was monitored by the absorption difference (ΔA) as a function of solution potential (panel B), where ΔA = A561 nm – 1.6*A573 nm + 0.06*A629 nm). Again, all trends were verified by comparison to midpoint potentials calculated using other wavelength combinations mentioned in [29]. Data were fit to a 1-electron reduction following the Nernst equation and the results are summarized in Table 2.
The most striking observation is that the midpoint potential of heme b558 is sensitive to the mutation of E445, most dramatically for E445Q in the presence of DDM where the mutation results in a 100 mV increase in the midpoint potential of heme b558. The E445A and E445C mutants result in significantly lower midpoint potentials of heme b558. Not surprising, the mutations also perturb the midpoint potential of heme d. The midpoint potential of E445Q in DDM is lowered from 246 mV to 199 mV, which is the expected direction of change if one assumes E445 is deprotonated and E445Q resembles the protonated species. Protonation of E445 is expected to stabilize the reduced form of heme d. Similarly, in SML detergent, the midpoint potential of heme d drops from 289 mV to 222 mV for E445C compared to WT.
4. Discussion
The X-ray structure of cyt bd from G. thermodenitrificans [12] revealed several features that were unexpected from previous biochemical and biophysical studies, mostly on the E. coli homologue. The most unusual feature is that two of the three heme components of the enzyme, heme d and heme b595, each have a glutamic acid located at positions where the carboxyl side chains could ligate to the heme irons: GtE101 (EcE99) adjacent to the Fe of heme b595 and GtE378 (EcE445) adjacent to the Fe of heme d. Considering that no other native heme protein has been reported to have either Asp or Glu as an axial ligand, the current work is aimed at determining the functional roles of these two glutamic acid residues.
4.1 The glutamates are important but not essential for activity
The ubiquinol-1 oxidase activities of the mutants, shown in Table 1 demonstrate that despite the evident importance of these two glutamate residues, they are not absolutely required for activity. The E445Q mutant retains 15% of the activity, normalized per heme d, despite the moderately decreased amounts of both heme d and heme b595. It is also noted that there is a clade of cyt bd in which E445 is replaced by a cysteine, although the E445C mutant of Ec-cyt bd is not functional. Table 1 also shows that the E99A mutant, which appears to have less than half of the heme d present, retains 72% of the ubiquinol-1 oxidase activity (per heme d). The new affinity-tag based protein preparation makes it apparent that although the E99A and E445Q mutants are significantly perturbed, they do maintain substantial ubiquinol-1 oxidase activity.
4.2 The glutamates are not strong ligands to the hemes
The heme analyses of the mutant enzymes are also summarized in Table 1. Despite the ambiguities due to mutation-induced changes in the heme d extinction coefficients, with the reasonable assumption that the content/spectra of heme b558 are unchanged, the data show that none of the mutations completely eliminate either heme d or heme b595. E445A and E445C appear to have excess heme d, which likely is due to altered extinction coefficients, and the larger residue substitutions, E445Q and E445H, have less heme d, 74% and 59%, respectively.
Similarly, the mutations of E99, which is adjacent to heme b595, do not result in eliminating the heme b at this site. E99A has the same total content of heme b as does the wild type, and both E99H and E99Q have excess heme b, possibly indicating that the site that normally binds heme d is partially filled by heme b, as occurs in the so-called “cyanide-insensitive oxidase” variants of cyt bd [1]. The heme d that is bound to the E99H and E99Q mutants have spectra that are highly distorted compared to the WT, precluding quantitation. Oddly, the perturbations due to the E99 mutations appear to have a greater influence on the content and properties of the heme present at the heme d binding site than on the heme b595 site, consistent with previous data that was used to support the assignment of EcE99 (GtE101) as the axial ligand to heme d [26]. Likewise, the most significant consequence of the E445A mutation is not on the properties of heme d, but rather the inability to reduce heme b595, which is locked in the ferric state (i.e., not reduced by dithionite) [28]. The perturbation of the adjacent heme by these glutamate mutations could be mediated either through the protein or directly by the very close interaction of hemes b595 and heme d, which are in van der Waals contact at the porphyrin edges [12] (Figure 4). One manifestation of the intimate electronic coupling between the two hemes is that photolysis of CO from ferrous heme d changes the absorption spectrum of heme b595 within a few picoseconds [22].
Figure 4. Proximity of the three hemes in Gt-cyt bd.
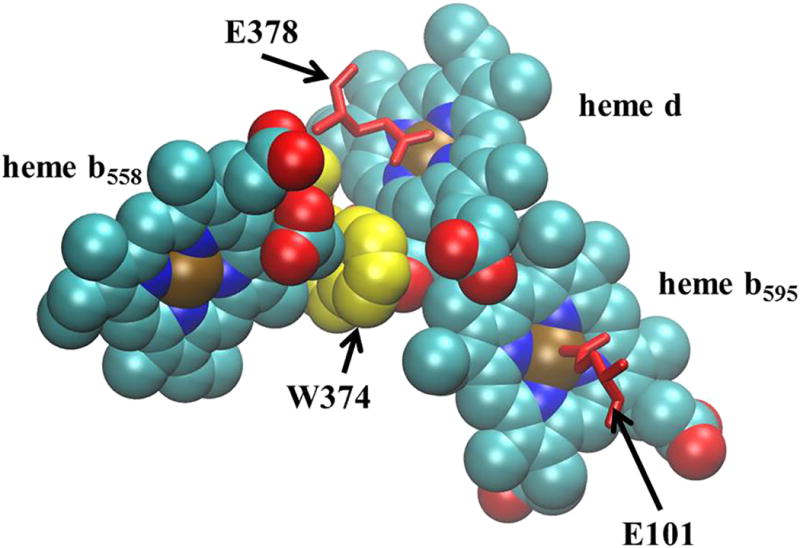
Hemes d and heme b595 are in Van der Waal’s contact. Heme b558 is adjacent to heme d and separated by the indol ring of GtW374 (EcW441). Also shown are GtE101 (EcE99) and GtE378 (EcE445) that form weak axial ligands to heme b595 and heme d, respectively. (Figure made with VMD. [60])
It is concluded that E99 and E445 are not primarily responsible for the stability of the hemes to which they are adjacent. This can be understood in terms of the relatively weak interactions between the heme Fe and carboxylate moiety which is likely to be primarily ionic rather than covalent due to the low basicity of the carboxyl group [32]. These conclusions are entirely consistent with previous spectroscopic studies [33, 34].
4.3 Previous studies show that heme b595 and heme d are high spin
Resonance Raman studies indicate that heme b595 from Ec-cyt bd is 5-coordinate high spin and that His19 is the strong axial ligand to heme b595[34, 35]. In addition, CN− does not interact with heme b595 [34] and only 5% of ferrous b595 binds CO when CO is present in solution [36]. In Gt-cyt bd however, CO can bind to heme b595 and make it 6-coordinate. The extent of excitonic coupling between heme b595 and heme d in Gt-cyt bd is also reduced, indicating structural differences in the vicinity of heme d and heme b595 for the enzymes from these organisms [37]. Hence, heme b595 has one strong histidine ligand on one side of the heme and is blocked from binding to exogenous ligands on the opposite side to varying extents depending on the ligand and the source of the enzyme. The structure suggests that the block against ligand binding to heme b595 is the weak glutamate endogenous ligand.
In both Ec-cyt bd and Gt-cyt bd, heme d has been shown to be high spin and always 5-coordinate [34], while EPR and Raman spectra of Ec-cyt bd indicate that the endogenous ligand to heme d is weak and likely displaced by oxygen [34, 35, 38]. There is no evidence of any ligand on the opposite side of heme d from the weak glutamate ligand.
All of the data lead to the conclusion that E445 and E99 are not strong ligands to heme d and heme b595, respectively, conclusions bolstered by studies examining models of heme proteins with carboxylate-containing compounds as ligands.
4.4 Model studies of glutamate as a heme ligand
The H93G mutant of sperm whale myoglobin has been used to replace the proximal ligand (H93) with several different exogenously added small molecule ligands that can be diffused into the cavity, including acetate[32] and benzoate[33]. The acetate adduct is 6-coordinate high spin with water ligating the heme Fe3+ on the distal side, stabilized by hydrogen bonding to the distal H64. The geometry of the Fe3+-acetate binding is inconsistent with an interaction between an electron pair forming a Fe-O bond. The Fe-O-C angle is 152° and the Fe is not in-plane, defined by the O=C-O carboxylate. The acetate binding is weak, as measured by the dissociation constant. The Fe-O-C bond angle of E99 and heme b595 is also 152° and the carboxylate plane also does not include the heme Fe. The Fe-O bond length is about 2.1 Å for E99 interacting with heme b595, similar to the 2.2 Å for the proximal acetate-heme Fe-O bond in myoglobin. The interaction between E445 and heme d appears to be even weaker, with the Fe-O bond length of about 2.7 Å and an Fe-O-C angle of 87°, indicating that the plane of the carboxylate is nearly parallel to the heme d plane, not consistent with a strong Fe-O bond.
Iron-strapped porphyrin model compounds with carboxylic acid groups hanging over the coordination site have also been examined [39]. A pyridyl residue serves as the proximal ligand to the Fe2+ heme and carboxyl groups are dangling within the distal pocket. Model compounds were examined in which the overhanging carboxyl can directly interact with the Fe2+ as well as compounds where the geometry precludes direct carboxyl interaction with the Fe2+. Compounds with hanging carboxyls which can interact directly with the Fe2+ are 6-coordinate high spin. Of particular interest is that these compounds bind O2 and CO. It is concluded that O2 and CO can displace one of the axial ligands in the 6-coordinate model compounds and that it is the carboxylate that is almost certainly the displaced ligand. Furthermore, it is suggested that the hanging carboxyl in the distal pocket can stabilize bound O2 by either dipolar interactions and/or hydrogen bonding. These studies support a possible role of E445 to stabilize O2 bound to ferrous heme d.
4.5 On which side of heme d does O2 bind?
Although it is clear that O2 binds to heme d, significant questions remain about the nature of O2 binding in the cyt bd active site. The primary question is whether O2 binds on the hydrophilic side of heme d, displacing EcE445, or whether O2 binds on the hydrophobic side of the heme. Cyt bd has a high affinity for oxygen, 0.28 μM for Ec-cyt bd [17] and 0.5 μM for cyt bd from Azotobacter vinlandii (Av-cyt bd) [8], comparable to sperm whale myoglobin (1 μM) but less than Ascaris hemoglobin (3 nM) [40]. The very high affinity of O2 to Ascaris hemoglobin is primarily due to a very slow rate constant for dissociation, 0.004 s−1 vs 15 s−1 for sperm whale myoglobin [40]. The second order rate constant for formation of the O2 adduct is actually slower for Ascaris hemoglobin (1.5 × 106 M−1s−1) than for sperm whale myoglobin (1.7 × 107 M−1s−1). In comparing these cases with cyt bd, it is clear that high affinity for O2 of cyt bd is attributable to the very large apparent rate constant for formation of the complex, 2 × 109 M−1s−1, which is close to the diffusion limit [1, 8, 17, 41] and 100-fold faster than for sperm whale myoglobin. The rate constant for the dissociation of O2 from the mixed valence state of Ec-cyt bd is 78 s−1 [42], 5-times faster than for sperm whale myoglobin (12 s−1) [43]. Effectively, there is no significant barrier impeding O2 from getting from solution to the heme d Fe2+. The extremely rapid rate constant for association is consistent with the short pathway (6 Å to 8 Å) to the surface and the apparently open channel that is observed in the structure [12] (Figure 5). Visually, both sides of heme d appear accessible (Figure 5), aside from the block provided by GtE378 (EcE445) on the hydrophilic face of the heme [12]. The accessibility can be addressed quantitatively using molecular dynamics as it has been applied to other O2 reductases [44].
Figure 5. Views of the oxygen binding site in Gt-cyt bd.
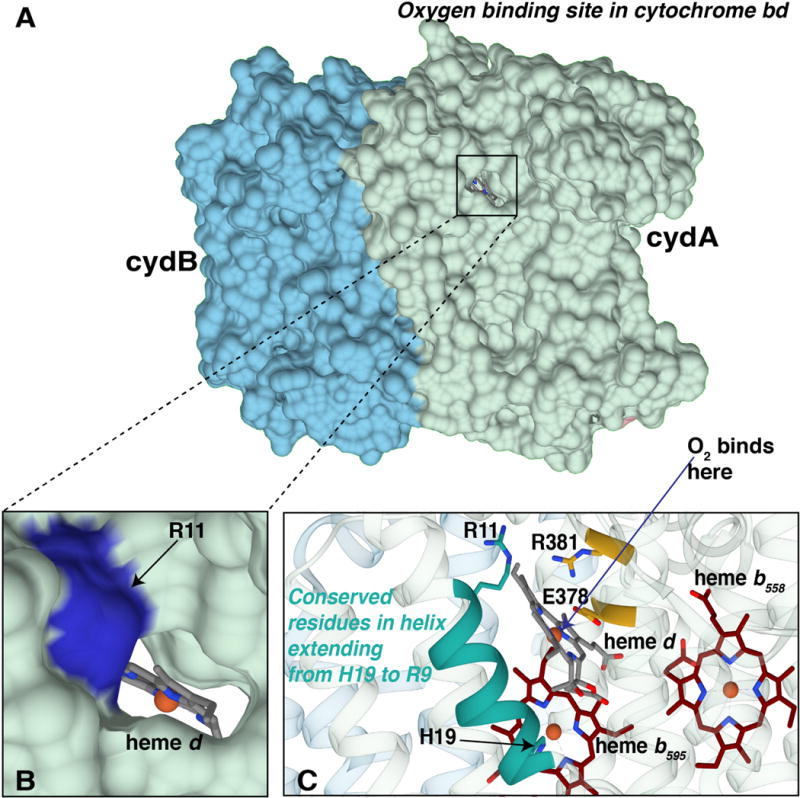
(A) The surface representation of subunits I (light green and II (blue) shows the opening leading from the periplasm to the cavity containing heme d. (B) A more detailed view of the opening, with heme d and its Fe (red) visible and with GtR11 (EcR9) indicated on the rim of the opening. (C) A view showing the α-helix that contains both GtR11 (EcR9) and the axial ligand GtH21 (EcH19) to heme b595. This helix runs over the hydrophobic side of heme d. Also shown are GtR381 (EcR448) and GtE378 (EcE445) that form a salt bridge on the hydrophilic side of heme d. (Figure made with Chimera)
High affinity for O2 can be mediated by hydrogen bond interactions, as is the case in myoglobin where several residues in the distal pocket form hydrogen bonds with O2 when it binds to Fe2+[40, 45]. Genetic mutations to generate a totally hydrophobic distal pocket of myoglobin does not preclude O2 binding, but the affinity is very low [43, 45]. However, the relatively high affinity for O2 exhibited by cyt bd does not in itself exclude the possibility of binding to the hydrophobic side of the heme. The affinity for O2 also depends on the degree to which the Fe2+ is available to react. In myoglobins the plane of the imidazole of the proximal axial histidine ligand eclipses the porphyrin nitrogens of the heme [46]. This restrains the movement of the Fe2+ with respect to the heme plane and substantially reduces the reactivity towards ligands on the distal side because the Fe2+ is pulled away from this side of the heme. The lack of such steric constraints due to the orientation of the proximal histidine is invoked to explain the high O2 affinities of leghemoglobin and the L16A mutant of cyt c′ [47, 48]. Since the glutamate ligand on the opposite face of heme d is a weak ligand, it is unlikely to draw Fe2+ towards itself and limit the access of Fe2+ to O2 on the hydrophobic side.
In the case of cyt c′, the wild type cyt c′ protein does not bind to O2 because the Fe2+ binding site is blocked by the side chain of L16 and, when a space is generated by the L16A mutation, O2 binds with very high affinity [49]. In Gt-cyt bd, there is a leucine (L18) on the hydrophobic side of heme d whose side chain is 3.9 Å from the heme d Fe which could restrict O2 binding to the hydrophobic side of heme d, but this residue is not conserved, however, and is replaced by an alanine in the Ec-cyt bd.
Despite the structure showing a much more open binding site for O2 on the hydrophobic side of heme d and GtE378 (EcE445) blocking access to the Fe on the hydrophilic face of the heme, it is not likely that the catalytic active site is on the hydrophobic side of heme d. There are no residues on the hydrophobic side, conserved or otherwise, to provide protons for the chemistry of reducing O2 to water or to stabilize catalytic intermediates. Accompanying electron transfer, timely and rapid proton delivery to the oxygenated states of heme d that are intermediates during the catalytic cycle is essential. Furthermore, the highly oxidized oxoferryl state of the heme is sufficiently stabilized by the protein that, in the absence of a reductant, the enzyme can be isolated in this state [18, 19]. Cyt bd also has significant quinol peroxidase activity, reducing hydrogen peroxide to water [50]. Considering the conserved sets of catalytically critical residues in the distal pockets of peroxidases, catalases and in other O2 reductases, the expectation that there must be conserved residues involved in the generation and stabilization of transient oxygenated states of heme d during the catalytic cycle is compelling.
If O2 binds to heme d Fe2+ on the same side as EcE445 (GtE378), it must displace EcE445 (GtE378). It is likely that the structure [12] represents the fully oxidized state of cyt bd and it is possible that GtE378 (EcE445) is displaced upon reduction of heme d to the ferrous state. Even if this is not the case, there are numerous examples of O2 binding to hexacoordinate heme proteins by displacement of a weakly bound distal ligand [51, 52], and this is also speculated to be the case for O2 binding in the iron-strapped porphyrin models [39].
4.6 The hydrophilic face of heme d has a conserved GtE378-R381 (EcE445-R448) salt bridge
On the polar side of heme d, Ec E445 (GtE378) is part of a highly conserved region of CydA: 440GWFMT445EFGRQP451W in Ec-cyt bd and 373GWYLA378EVGRQP384W in Gt-cyt bd. The X-ray structure shows (Figure 6) that this region of the protein is a helix that sits on the hydrophilic face of heme d. Besides EcE445 (GtE378), this stretch contains EcW441 (GtW374) which has been suggested [31] as being important for electron transfer between heme b558 and heme d, though direct electron transfer has not been demonstrated. What has not been noted is that GtR381 (EcR448) forms a salt bridge with GtE378 (EcE445) at the surface of heme d (Figure 6A). Although EcE445 is replaced in by a cysteine in a small clade of organisms (e.g., Bacillus cereus), this arginine (EcR448) is invariant. The modification is specific to certain organisms of the order Bacilalles and phylum Cyanobacteria. It is not apparent what function this mutation would serve but in our experiments, this modification is not functional. We suspect that some corresponding mutations that are required to make this mutation viable are missing in our mutations but we are not able to identify what these could be from sequence alignments. The EcE445-R448 (GtE378-R381) salt bridge in cyt bd can be expected to partially neutralize the negative charge on the carboxylate of EcE445 and further weaken the interaction between EcE445 with the heme d Fe. It is also plausible that one role of this arginine (EcR448) is to help to stabilize the negative charge on the oxygen moiety in the oxoferryl intermediates ( and ) [18] that are equivalent to compounds 1 and 2 of peroxidases [53]. In the case of myoglobin from Aplysia limacina, an arginine in the distal pocket provides the stabilizing hydrogen bond for O2 bound to the ferrous heme Fe2+[46]. EcE445 (GtE378) and EcR448 (GtR381) could also interact and provide catalytically important hydrogen bonds, as occurs in the proximal pocket of catalases[53]. Furthermore, the side chain of GtN150 (EcN147), another highly conserved residue, is only 3.8Å from the carboxylate of GtE378 (EcE445) (Figure 6) and could be functionally important upon O2 binding and displacement of GtE378 (EcE445). It is also likely that water molecules are present on the hydrophilic face of heme d and these could participate in important hydrogen bond networks and proton wires. An X-ray structure with improved resolution and of different redox states of the protein will be necessary to clarify this.
Figure 6. Views of GtE378 and GtR381 in Gt-cyt bd.
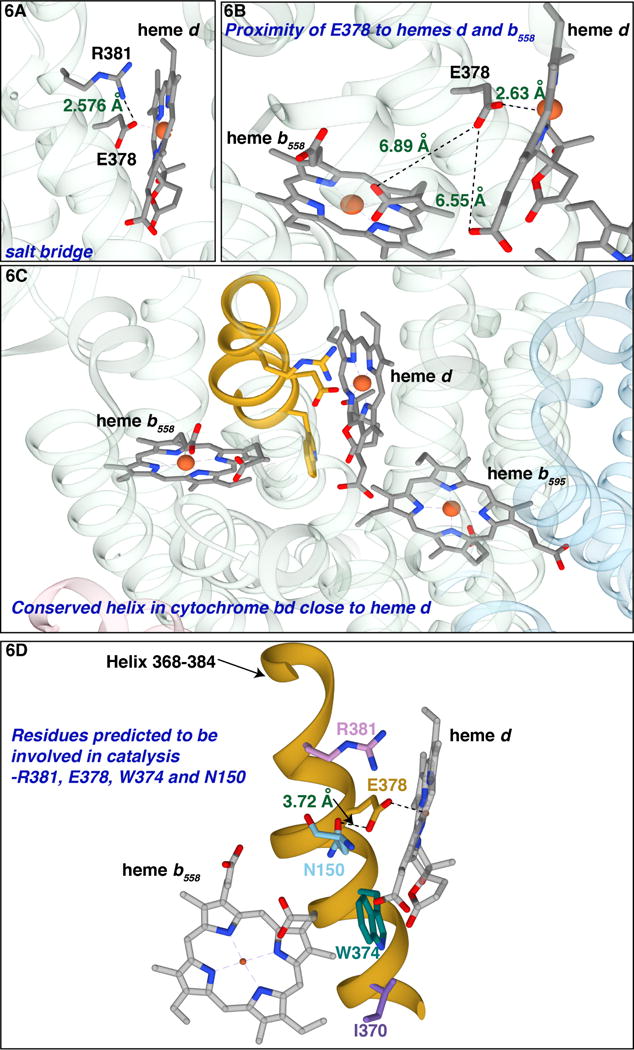
(A) The salt bridge between GtE378 (EcE445) and GtR381 (EcR448) at the hydrophilic side of heme d. (B) GtE378 is 6.89 Å from the propionate of heme b558. (C) The conserved α-helix that runs along the hydrophilic side of heme d, 373GWYLA378EVGRQP384W in Gt-cyt bd. (D) The helix is colored in yellow; GtR381, GtE378, GtW374and GtI370 are indicated along with GtN150 (EcN147). (Figure made with Chimera)
If O2 were to react on the hydrophobic side of heme d, the axial ligation of EcE445 (GtE378) hydrogen bonded to EcR448 (GtR381) would provide little electron donation (push) to stabilize the oxoferryl oxidation states ( and ) of heme d that are intermediates in the reaction catalyzed by the enzyme[18]. In contrast, the “pull” provided by E445/R448 for O2 reacting on the same face of heme d would favor formation of the high oxidation states of the heme d Fe.
Until further data are available, it seems reasonable to model the O2 binding to heme d on the same face as EcE445 by displacing the glutamate, which then plays an important role in hydrogen bonding to oxygenated states of heme d and in proton transfer during the formation of water at the active site.
4.7 Redox state of heme b595 controls O2 access to heme d
While O2 has easy access to heme d, the interaction between heme d and heme b595, adds a level of complexity to gas ligand binding. The crystal structure shows the two hemes are in van der Waals contact [12] (Figure 4), and this close contact is manifested by both the excitonic coupling apparent in CD spectra [37], as well as rapid spectroscopic changes of heme b595 upon photolysis of CO bound to reduced heme d [54]. Interestingly, it was shown with Ec-cyt bd that CO does not bind heme d and heme b595 simultaneously, and CO-photolysis studies suggest negative cooperativity in ligand binding between these sites [55].
An additional manifestation of this heme-heme interaction is the fate of CO that is photolyzed from ferrous heme d. When CO is photolyzed from the fully reduced form ( ) of cyt bd, all of the CO is expelled from the protein and recombines with heme d mono-exponentially with a time constant of 12 μs [54]. When the same experiment is performed with the mixed valence form ( ), 70% of the bound CO is not completely expelled from the protein but, instead, exhibits geminate recombination to one or more sites within the protein with a time constant of 14 ns. These results are explained by the existence of cyt bd in an “open” form and a “closed” form. The fully reduced enzyme is expected to be in “open” form, as demonstrated by the free exchange of gas ligands and fast rate of thermal dissociation of CO and NO. In the mixed valence form, it appears that the enzyme is in equilibrium between the “open” and “closed” states. This supposition is supported by the kinetics of O2 binding to the 30% of the CO-photolyzed mixed-valence form of the enzyme from which CO exits the enzyme [8]. The rate of O2 binding is hyperbolic with respect to the concentration of O2, consistent with a model in which the rate of O2 binding is limited by the equilibration between closed and open states [8]. Experiments with the Av-cyt bd suggest that about 67% of the enzyme is in the “closed” state in the mixed valence form of the enzyme, and that the time for interconversion between the open and closed states is in the microsecond range [8]. Importantly, the geminate CO recombination kinetics of the EcE445A mutant, in which heme b595 is “locked” in the ferric state, resembles the mixed valence state of the enzyme. Hence, it is specifically the redox state of heme b595 that regulates ligand access to heme d.
One possibility is that the opening of the apparent O2 delivery channel [12] is occluded when heme b595 is oxidized. The EcH19 (GtH21) axial ligand to heme b595 is part of an α-helix that includes a highly conserved EcR9 (GtR11) which forms part of the rim of the opening of the putative O2 channel [12], providing a plausible link between the redox state of heme b595 and access of O2 to heme d (Figure 5C). A change in conformation of EcR9 (GtR11) in response to the redox state of heme b595 could alter the access of O2 to heme d. In addition, the same α-helix passes over the hydrophobic face of heme d and contains GtL18 that is about 3.9 Å away from the Fe2+. Movement of this helix due to a redox change in heme b595 could also open a pocket on the hydrophobic side of heme d where photolyzed CO could enter and then rapidly re-bind to heme d at the physiologically relevant hydrophilic side of heme d. This could explain the 70% geminate recombination of CO that is observed [54].
The presence of the conserved arginine EcR9 (GtR11) on the rim of the opening to the O2 binding site on heme d (Figure 5B) also might prevent protons from the bulk solution on the periplasmic side of the membrane gaining access to GtE378 (EcE445). This is a requirement for the enzyme to generate a proton motive force, utilizing protons only from the cytoplasm for the chemistry of reducing O2 to water.
4.8 Proton transfer and the catalytic cycle
The voltage that is generated during turnover of cyt bd is primarily due to proton transfer from the cytoplasmic side of the membrane to the active site at heme d [28]. The proton pathway has been partially sketched out by previous mutagenesis studies [24, 25, 28, 56, 57] as well as by observation of the X-ray structure [12] (Figure 7). The structure shows that EcE445 (GtE378) is the most likely proton donor to oxygenated states during catalysis leading to the formation of water. EcE445 (GtE378) also likely forms a hydrogen bond to stabilize the oxygenated states of heme d (e.g., ferrous-oxy and oxoferryl states).
Figure 7. Proposed proton channel in Gt cyt bd.
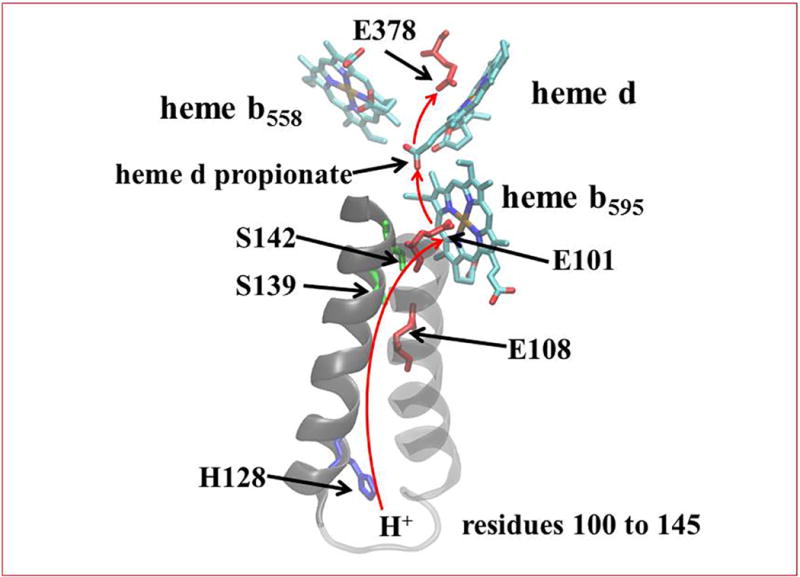
A series of conserved hydrophilic residues along with the propionate of heme d and internal water molecules that are not observed in the X-ray structure line a proton channel conveying protons from the cytoplasm to GtE378 (EcE445) and from there to the oxygenated species at the active site. (Figure made with VMD)
Three conserved glutamatic acids are proposed to be critical components of the proton channel pathway: EcE99 (GtE101), EcE107 (GtE108) and EcE445 (GtE378). EcE107 (GtE108) has been shown to be protonated in both the fully reduced and fully oxidized forms of the enzyme [56] and essential for catalytic activity [57]. It is likely that EcE107 (GtE108) is the proton donor to EcE99 (GtE101), which we propose to become protonated upon reduction of heme b595. Protonated EcE99 (GtE101) then becomes the proton donor to EcE445 (GtE378) accompanying electron transfer from heme b595 to heme d. Protons from Ec445 are used for forming water at the heme d active site. The pathways for proton transfer must involve water molecules that are not resolved in the structure, as well as propionate A of heme d, which lies midway between EcE99 (GtE101) and EcE445 (GtE378), about 7 Å from each in the structure [12] (Figure 7). Once EcE445 (GtE378) is displaced by O2 it could move closer to the propionate, but water molecules, perhaps hydrogen bonded to EcN147 (GtN150) would still be required to provide a pathway for protons to diffuse using the Grotthus-type mechanism, e.g., [58].
It is speculated that during catalytic turnover, the only source of protons to the active site must come through this pathway from the cytoplasm. Since EcE107 (GtE108) has been shown to be protonated in both the oxidized and reduced state of cyt bd [56], it is always prepared to provide a proton to EcE99 (GtE101) whenever heme b595 receives an electron from heme b558. Once heme b558 is reduced, the electron goes preferably to heme b595 because a proton is readily provided to EcE99 (GtE101) to stabilize the ferrous form of heme b595. Presumably, EcE107 (GtE108) is rapidly reprotonated from the cytoplasmic channel. The net proton transfer from the cytoplasm to EcE99 (GtE101) is responsible for the voltage generation accompanying this step of the reaction [9, 28]. Once the proton and electron are at heme b595 and its associated glutamate, EcE99 (GtE101), both the electron and proton can be transferred to heme d and its associate glutamate EcE445 (GtE378), and then to the oxygenated species. In this way, the sequence of electron transfer from heme b558 to heme d is never direct, but requires heme b595, and every electron that goes to heme d is accompanied by a proton that comes through the proton channel from the cytoplasm. Electron transfer between heme b595 and heme d is not electrogenic because the electron is always accompanied by a proton exchanged between EcE99 (GtE101) and EcE445 (GtE378). The reverse electron transfer from heme d following photolysis of the CO adduct of the one-electron reduced cyt bd (
) also goes initially to heme b595 before equilibrating with heme b558 [20, 28] for the same reason. Electron transfer from heme d to heme b595 is accompanied by proton transfer from EcE445 (GtE378) to EcE99 (GtE101) without generating a voltage [28]. Electron transfer from heme b595 to heme b558 requires proton transfer via EcE107 (GtE108) to the cytoplasm, which is slower and does generate a voltage [28]. The very close interaction between heme d and heme b595 is the most probable explanation for the distorted spectra of heme d in the E99H and E99Q mutants. In E99A, it is likely that a water molecule takes the place of E99 in the proton channel, maintaining the supply of protons to the active site.
Figure 8 shows a schematic of the species in the catalytic cycle with the speculated protonation states of the two glutamates included. This builds on previous models that have been proposed [1, 12, 18] and includes speculation based on the considerations presented in this work. Further studies will be required to address the many unanswered questions and test this model.
Figure 8. Proposed catalytic mechanism of cyt bd.
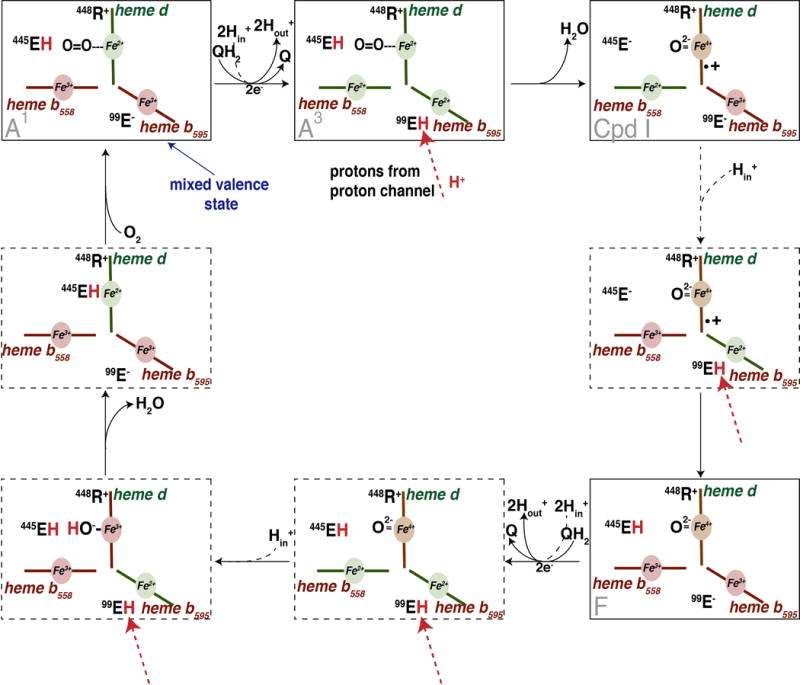
Residue numbers are those of Ec-cyt bd. It is assumed that electron transfer to heme d is always accompanied by proton transfer from EcE99 to EcE445, and electron transfer to heme b595 is always accompanied by proton transfer from EcE107 to EcE99, and EcE107 is rapidly reprotonated by protons delivered through the proton channel from the cytoplasm (in). Protons released by oxidation of ubiquinol (QH2) are released to the periplasm (out). Species A1 is the stable one-electron reduced oxy form; Species A3 is an unobserved, transient 3-electron reduced species with the O2 still bound to ferrous heme d.; Compound I (Cpd I) is the oxoferryl species with a porphyrin cationic radical at heme d; Species F is Compound II, the oxoferryl form following reduction of the porphyrin radical. Species A1, Cpd I and F have been observed along with the mixed valence species (MV).
4.9 Role of E445 in the interaction between heme d and heme b558
In Table 2, it is shown that mutations of EcE445 (particularly E445A and E445Q) affect the midpoint potential of heme b558. It has also been shown that reduction by ubiquinol-1 or by TMPD (N,N,N′,N′-tetramethyl-p-phenylenediamine) via heme b558, of the fully oxidized form of Ec-cyt bd ( ) is 1000-fold slower than reduction of the oxoferryl form ( ), and much too slow to be compatible with the rate of steady state catalysis [59]. The rate of reduction of the mixed valence oxy complex ( or ) by ubiquinol must also be much faster than observed for the all-ferric form of the enzyme to be compatible with steady state turnover since this is also an obligatory step in the catalytic cycle (Figure 8). The implication is that the redox state of heme d controls the rate of reduction of heme b558. Although electrostatics might play a role in some of these observations, conformational coupling appears likely. The structure shows that the edge-to-edge distance between the two hemes is only about 6 Å, with a tryptophan indol ring (GtW374 or EcW441) sandwiched between them (Figure 4). A perturbation of the position or orientation of heme d could be readily sensed by heme b558. The structure suggests two other plausible sources of physical perturbation. The crystal structure places GtE378 (EcE445) 6.9 Å from the propionate group of heme b558 (Figure 6B). Oxygenation of heme d will displace GtE378 (EcE445) in the direction of heme b558 and where it could make a hydrogen bond to its propionate group of heme b558, perhaps with an intervening water. Stabilizing reduced heme b558 with a hydrogen bond could conceivably increase the midpoint potential of heme b558 and accelerate its rate of reduction by a reductant. A second possibility is that any perturbation of GtE378 (EcE445) by either reduction of heme d or by mutation, could be conveyed to heme b558 through the α-helix that contains GtE378 (EcE445) which contains two nearby residues that are in direct contact with heme b558, GtW374 (EcW441) and GtI370 (EcI437) (Figure 6C).
5. Conclusion
The conclusion from the mutagenesis work presented in this study is that neither EcE445 (GtE378) nor EcE99 (GtE101) are strongly ligated to heme d and heme b595, respectively, and they are not primarily responsible for the stability of the bound hemes. It is concluded that it is most likely that O2 binds to the face of heme d on which EcE445 (GtE378) is located and that, therefore, EcE445 (GtE378) must be displaced upon O2 binding. We also point out for the first time the presence of a conserved salt bridge between EcE445 (GtE378) and EcR448 (GtR441) on the hydrophilic face of heme d. Both of these residues are likely important to stabilize oxygenated states of heme d by electrostatics and/or hydrogen bonding. We speculate that reduction of heme d is accompanied by protonation of EcE445 (GtE378), which would break the salt bridge and be linked to a conformational change of both EcE445 (GtE378) and EcR448 (GtR441). We also suggest that EcE445 (GtE378) is the direct source of all protons required to convert O2 to water. The protons all originate from the cytoplasm and reach the active site by a Grotthus-type mechanism that involves EcE107 (Gt108), EcE99 (Gt101) and EcE445 (GtE378) as well as water molecules that are not resolved in the structure.
Cyt bd is unique in utilizing weak axial ligands as proton acceptors for charge compensation when the heme d (EcE445/GtE378) and heme b595 (EcE99/GtE101) act as electron acceptors. These two heme-associated glutamates, in addition of EcE107/GtE108 provide a pathway for the transport of protons, one per electron, to the oxygenated species bound to heme d, including the oxy, oxoferryl and hydroxide-bound species. In this way, EcE445/GtE378 and EcE99/GtE101are critical components for the proton-coupled electron transfer between two hemes and assure that each electron that reaches the active site of the enzyme is delivered by E99 through heme b595 and is accompanied by a proton that originates from the bacterial cytoplasm. Now that a structure is available [12], further experiments can be designed to address the many aspects of this model that are postulated.
Supplementary Material
Highlights.
Cytochrome bd is a respiratory quinol:O2 oxidoreductase present in many pathogens
The enzyme contains 3 hemes in close contact, 2 of which have glutamate axial ligands
Cytochrome bd is the only known heme protein with glutamic acid as a heme ligand
Cytochrome bd contributes to the proton motive force and membrane potential
Glutamate heme ligands are part of the proton-conducting pathway across the membrane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borisov VB, Gennis RB, Hemp J, Verkhovsky MI. The cytochrome bd respiratory oxygen reductases. Biochimica et biophysica acta. 2011;1807:1398–1413. doi: 10.1016/j.bbabio.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunningham L, Pitt M, Williams HD. The cioAB genes from Pseudomonas aeruginosa code for a novel cyanide-insensitive terminal oxidase related to the cytochrome bd quinol oxidases. Molecular microbiology. 1997;24:579–591. doi: 10.1046/j.1365-2958.1997.3561728.x. [DOI] [PubMed] [Google Scholar]
- 3.Miller MJ, Gennis RB. The purification and characterization of the cytochrome d terminal oxidase complex of the Escherichia coli aerobic respiratory chain. Journal of Biological Chemistry. 1983;258:9159–9165. [PubMed] [Google Scholar]
- 4.Lorence RM, Carter K, Green GN, Gennis RB. Cytochrome b558 monitors the steady state redox state of the ubiquinone pool in the aerobic respiratory chain of Escherichia coli. Journal of Biological Chemistry. 1987;262:10532–10536. [PubMed] [Google Scholar]
- 5.Dueweke TJ, Gennis RB. Epitopes of monoclonal antibodies which inhibit ubiquinol oxidase activity of Escherichia coli cytochrome d complex localize functional domain. Journal of Biological Chemistry. 1990;265:4273–4277. [PubMed] [Google Scholar]
- 6.Dueweke TJ, Gennis RB. Proteolysis of the cytochrome d complex with trypsin and chymotrypsin localizes a quinol oxidase domain. Biochemistry. 1991;30:3401–3406. doi: 10.1021/bi00228a007. [DOI] [PubMed] [Google Scholar]
- 7.D’Mello R, Hill S, Poole RK. The cytochrome bd quinol oxidase in Escherichia coli has an extremely high oxygen affinity and two oxygen-binding haems: implications for regulation of activity in vivo by oxygen inhibition. Microbiology. 1996;142(Pt 4):755–763. doi: 10.1099/00221287-142-4-755. [DOI] [PubMed] [Google Scholar]
- 8.Belevich I, Borisov VB, Bloch DA, Konstantinov AA, Verkhovsky MI. Cytochrome bd from Azotobacter vinelandii: evidence for high-affinity oxygen binding. Biochemistry. 2007;46:11177–11184. doi: 10.1021/bi700862u. [DOI] [PubMed] [Google Scholar]
- 9.Jasaitis A, Borisov VB, Belevich NP, Morgan JE, Konstantinov AA, Verkhovsky MI. Electrogenic Reactions of Cytochrome bd†. Biochemistry. 2000;39:13800–13809. doi: 10.1021/bi001165n. [DOI] [PubMed] [Google Scholar]
- 10.Dassa J, Fsihi H, Marck C, Dion M, Kieffer-Bontemps M, Boquet PL. A new oxygen-regulated operon in Escherichia coli comprises the genes for a putative third cytochrome oxidase and for pH 2.5 acid phosphatase (appA) Molecular and General Genetics MGG. 1991;229:341–352. doi: 10.1007/BF00267454. [DOI] [PubMed] [Google Scholar]
- 11.Borisov VB, Murali R, Verkhovskaya ML, Bloch DA, Han H, Gennis RB, Verkhovsky MI. Aerobic respiratory chain of Escherichia coli is not allowed to work in fully uncoupled mode. Proc Natl Acad Sci U S A. 2011;108:17320–17324. doi: 10.1073/pnas.1108217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Safarian S, Rajendran C, Muller H, Preu J, Langer JD, Ovchinnikov S, Hirose T, Kusumoto T, Sakamoto J, Michel H. Structure of a bd oxidase indicates similar mechanisms for membrane-integrated oxygen reductases. Science. 2016;352:583–586. doi: 10.1126/science.aaf2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun YH, de Jong MF, den Hartigh AB, Roux CM, Rolán HG, Tsolis RM. The small protein CydX is required for function of cytochrome bd oxidase in Brucella abortus. Frontiers in Cellular and Infection Microbiology. 2012;2:47. doi: 10.3389/fcimb.2012.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.VanOrsdel CE, Bhatt S, Allen RJ, Brenner EP, Hobson JJ, Jamil A, Haynes BM, Genson AM, Hemm MR. The Escherichia coli CydX Protein Is a Member of the CydAB Cytochrome bd Oxidase Complex and Is Required for Cytochrome bd Oxidase Activity. Journal of Bacteriology. 2013;195:3640–3650. doi: 10.1128/JB.00324-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoeser J, Hong S, Gehmann G, Gennis RB, Friedrich T. Subunit CydX of Escherichia coli cytochrome bd ubiquinol oxidase is essential for assembly and stability of the di-heme active site. FEBS Letters. 2014;588:1537–1541. doi: 10.1016/j.febslet.2014.03.036. [DOI] [PubMed] [Google Scholar]
- 16.Poole RK, Kumar C, Salmon I, Chance B. The 650 nm Chromophore in Escherichia coli is an ‘Oxy-’ or Oxygenated Compound, Not the Oxidized Form of Cytochrome Oxidase d: An Hypothesis. Microbiology. 1983;129:1335–1344. doi: 10.1099/00221287-129-5-1335. [DOI] [PubMed] [Google Scholar]
- 17.Belevich I, Borisov VB, Konstantinov AA, Verkhovsky MI. Oxygenated Complex of cytochrome bd from Escherichia coli: Stability and Photolability. FEBS Letters. 2005;579:4567–4570. doi: 10.1016/j.febslet.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 18.Paulus A, Rossius SGH, Dijk M, de Vries S. Oxoferryl-Porphyrin Radical Catalytic Intermediate in Cytochrome bd Oxidases Protects Cells from Formation of Reactive Oxygen Species. Journal of Biological Chemistry. 2012;287:8830–8838. doi: 10.1074/jbc.M111.333542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kahlow MA, Zuberi TM, Gennis RB, Loehr TM. Identification of a ferryl intermediate of Escherichia coli cytochrome d terminal oxidase by resonance Raman spectroscopy. Biochemistry. 1991;30:11485–11489. doi: 10.1021/bi00113a001. [DOI] [PubMed] [Google Scholar]
- 20.Siletsky SA, Rappaport F, Poole RK, Borisov VB. Evidence for Fast Electron Transfer between the High-Spin Haems in Cytochrome bd-I from Escherichia coli. PloS one. 2016;11:e0155186. doi: 10.1371/journal.pone.0155186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill JJ, Alben JO, Gennis RB. Spectroscopic evidence for a heme-heme binuclear center in the cytochrome bd ubiquinol oxidase from Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:5863–5867. doi: 10.1073/pnas.90.12.5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vos MH, Borisov VB, Liebl U, Martin JL, Konstantinov AA. Femtosecond resolution of ligand-heme interactions in the high-affinity quinol oxidase bd: A di-heme active site? Proc Natl Acad Sci U S A. 2000;97:1554–1559. doi: 10.1073/pnas.030528197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferguson-Miller S, Babcock GT. Heme/Copper Terminal Oxidases. Chemical reviews. 1996;96:2889–2908. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 24.Osborne JP, Gennis RB. Sequence analysis of cytochrome bd oxidase suggests a revised topology for subunit I. Biochim Biophys Acta. 1999;1410:32–50. doi: 10.1016/s0005-2728(98)00171-6. [DOI] [PubMed] [Google Scholar]
- 25.Mogi T, Endou S, Akimoto S, Morimoto-Tadokoro M, Miyoshi H. Glutamates 99 and 107 in Transmembrane Helix III of Subunit I of Cytochrome bd Are Critical for Binding of the Heme b595-d Binuclear Center and Enzyme Activity. Biochemistry. 2006;45:15785–15792. doi: 10.1021/bi0615792. [DOI] [PubMed] [Google Scholar]
- 26.Mogi T. Probing the haem d-binding site in cytochrome bd quinol oxidase by site-directed mutagenesis. J Biochem. 2009;145:763–770. doi: 10.1093/jb/mvp033. [DOI] [PubMed] [Google Scholar]
- 27.Berry EA, Trumpower BL. Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Analytical Biochemistry. 1987;161:1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 28.Belevich I, Borisov VB, Zhang J, Yang K, Konstantinov AA, Gennis RB, Verkhovsky MI. Time-resolved electrometric and optical studies on cytochrome bd suggest a mechanism of electron-proton coupling in the di-heme active site. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3657–3662. doi: 10.1073/pnas.0405683102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bloch DA, Borisov VB, Mogi T, Verkhovsky MI. Heme/heme redox interaction and resolution of individual optical absorption spectra of the hemes in cytochrome bd from Escherichia coli. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2009;1787:1246–1253. doi: 10.1016/j.bbabio.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 30.Hong S, de Almeida WB, Taguchi AT, Samoilova RI, Gennis RB, O’Malley PJ, Dikanov SA, Crofts AR. The Semiquinone at the Qi Site of the bc1 Complex Explored Using HYSCORE Spectroscopy and Specific Isotopic Labeling of Ubiquinone in Rhodobacter sphaeroides via 13C Methionine and Construction of a Methionine Auxotroph. Biochemistry. 2014;53:6022–6031. doi: 10.1021/bi500654y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Hellwig P, Osborne JP, Huang H-W, Moënne-Loccoz P, Konstantinov AA, Gennis RB. Site-Directed Mutation of the Highly Conserved Region near the Q-Loop of the Cytochrome bd Quinol Oxidase from Escherichia coli Specifically Perturbs Heme b595†. Biochemistry. 2001;40:8548–8556. doi: 10.1021/bi010469m. [DOI] [PubMed] [Google Scholar]
- 32.Qin J, Perera R, Lovelace LL, Dawson JH, Lebioda L. Structures of thiolate- and carboxylate-ligated ferric H93G myoglobin: models for cytochrome P450 and for oxyanion-bound heme proteins. Biochemistry. 2006;45:3170–3177. doi: 10.1021/bi052171s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pond AE, Roach MP, Sono M, Rux AH, Franzen S, Hu R, Thomas MR, Wilks A, Dou Y, Ikeda-Saito M, Ortiz de Montellano PR, Woodruff WH, Boxer SG, Dawson JH. Assignment of the heme axial ligand(s) for the ferric myoglobin (H93G) and heme oxygenase (H25A) cavity mutants as oxygen donors using magnetic circular dichroism. Biochemistry. 1999;38:7601–7608. doi: 10.1021/bi9825448. [DOI] [PubMed] [Google Scholar]
- 34.Sun J, Osborne JP, Kahlow MA, Kaysser TM, Hill JJ, Gennis RB, Loehr TM. Resonance Raman Studies of Escherichia coli Cytochrome bd Oxidase. Selective Enhancement of the Three Heme Chromophores of the “As-Isolated” Enzyme and Characterization of the Cyanide Adduct. Biochemistry. 1995;34:12144–12151. doi: 10.1021/bi00038a007. [DOI] [PubMed] [Google Scholar]
- 35.Sun J, Kahlow MA, Kaysser TM, Osborne JP, Hill JJ, Rohlfs RJ, Hille R, Gennis RB, Loehr TM. Resonance Raman Spectroscopic Identification of a Histidine Ligand of b595 and the Nature of the Ligation of Chlorin d in the Fully Reduced Escherichia coli Cytochrome bd Oxidase. Biochemistry. 1996;35:2403–2412. doi: 10.1021/bi9518252. [DOI] [PubMed] [Google Scholar]
- 36.Borisov VB, Sedelnikova SE, Poole RK, Konstantinov AA. Interaction of cytochrome bd with carbon monoxide at low and room temperatures: evidence that only a small fraction of heme b595 reacts with CO. The Journal of biological chemistry. 2001;276:22095–22099. doi: 10.1074/jbc.M011542200. [DOI] [PubMed] [Google Scholar]
- 37.Arutyunyan AM, Borisov VB, Novoderezhkin VI, Ghaim J, Zhang J, Gennis RB, Konstantinov AA. Strong excitonic interactions in the oxygen-reducing site of bd-type oxidase: the Fe-to-Fe distance between hemes d and b595 is 10 A. Biochemistry. 2008;47:1752–1759. doi: 10.1021/bi701884g. [DOI] [PubMed] [Google Scholar]
- 38.Jiang FS, Zuberi TM, Cornelius JB, Clarkson RB, Gennis RB, Belford RL. Nitrogen and proton ENDOR of cytochrome d, hemin, and metmyoglobin in frozen solutions. Journal of the American Chemical Society. 1993;115:10293–10299. [Google Scholar]
- 39.Boitrel B, Hijazi I, Roisnel T, Oohora K, Hayashi T. Iron-Strapped Porphyrins with Carboxylic Acid Groups Hanging over the Coordination Site: Synthesis, X-ray Characterization, and Dioxygen Binding. Inorganic chemistry. 2017;56:7373–7383. doi: 10.1021/acs.inorgchem.7b00343. [DOI] [PubMed] [Google Scholar]
- 40.Draghi F, Miele AE, Travaglini-Allocatelli C, Vallone B, Brunori M, Gibson QH, Olson JS. Controlling ligand binding in myoglobin by mutagenesis. The Journal of biological chemistry. 2002;277:7509–7519. doi: 10.1074/jbc.M109206200. [DOI] [PubMed] [Google Scholar]
- 41.Hill BC, Hill JJ, Gennis RB. The Room Temperature Reaction of Carbon Monoxide and Oxygen with the Cytochrome bd Quinol Oxidase from. Escherichia coli, Biochemistry. 1994;33:15110–15115. doi: 10.1021/bi00254a021. [DOI] [PubMed] [Google Scholar]
- 42.Borisov VB, Forte E, Sarti P, Brunori M, Konstantinov AA, Giuffre A. Redox control of fast ligand dissociation from Escherichia coli cytochrome bd. Biochemical and biophysical research communications. 2007;355:97–102. doi: 10.1016/j.bbrc.2007.01.118. [DOI] [PubMed] [Google Scholar]
- 43.Rohlfs RJ, Mathews AJ, Carver TE, Olson JS. The Effects of Amino Acid Substitution at Position E7(Residue 64) on the Kinetics of Ligand Binding to Sperm Whale Myoglobin. J Biol Chem. 1990;265:3168–3176. [PubMed] [Google Scholar]
- 44.Mahinthichaichan P, Gennis RB, Tajkhorshid E. All the O2 Consumed by Thermus thermophilus Cytochrome ba3 Is Delivered to the Active Site through a Long, Open Hydrophobic Tunnel with Entrances within the Lipid Bilayer. Biochemistry. 2016;55:1265–1278. doi: 10.1021/acs.biochem.5b01255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kiger L, Rashid AK, Griffon N, Haque M, Moens L, Gibson QH, Poyart C, Marden MC. Trematode hemoglobins show exceptionally high oxygen affinity. Biophysical journal. 1998;75:990–998. doi: 10.1016/S0006-3495(98)77587-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Springer BA, Egeberg KD, Sligar SG, Rohlfs RJ, Mathews AJ, Olson JS. Discrimination between Oxygen and Carbon Monoxide and Inhibition of Autooxidation by Myoglobin. J Biol Chem. 1989;264:3057–3060. [PubMed] [Google Scholar]
- 47.Tsai AL, Martin E, Berka V, Olson JS. How do heme-protein sensors exclude oxygen? Lessons learned from cytochrome c′, Nostoc puntiforme heme nitric oxide/oxygen-binding domain, and soluble guanylyl cyclase. Antioxidants & redox signaling. 2012;17:1246–1263. doi: 10.1089/ars.2012.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antonyuk SV, Rustage N, Petersen CA, Arnst JL, Heyes DJ, Sharma R, Berry NG, Scrutton NS, Eady RR, Andrew CR, Hasnain SS. Carbon monoxide poisoning is prevented by the energy costs of conformational changes in gas-binding haemproteins. Proc Natl Acad Sci U S A. 2011;108:15780–15785. doi: 10.1073/pnas.1109051108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marti MA, Capece L, Crespo A, Doctorovich F, Estrin DA. Nitric oxide interaction with cytochrome c′ and its relevance to guanylate cyclase. Why does the iron histidine bond break? Journal of the American Chemical Society. 2005;127:7721–7728. doi: 10.1021/ja042870c. [DOI] [PubMed] [Google Scholar]
- 50.Al-Attar S, Yu Y, Pinkse M, Hoeser J, Friedrich T, Bald D, de Vries S. Cytochrome bd Displays Significant Quinol Peroxidase Activity. Scientific reports. 2016;6:27631. doi: 10.1038/srep27631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rydberg P, Sigfridsson E, Ryde U. On the role of the axial ligand in heme proteins: a theoretical study. Journal of biological inorganic chemistry : JBIC : a publication of the Society of Biological Inorganic Chemistry. 2004;9:203–223. doi: 10.1007/s00775-003-0515-y. [DOI] [PubMed] [Google Scholar]
- 52.Smagghe BJ, Hoy JA, Percifield R, Kundu S, Hargrove MS, Sarath G, Hilbert JL, Watts RA, Dennis ES, Peacock WJ, Dewilde S, Moens L, Blouin GC, Olson JS, Appleby CA. Review: correlations between oxygen affinity and sequence classifications of plant hemoglobins. Biopolymers. 2009;91:1083–1096. doi: 10.1002/bip.21256. [DOI] [PubMed] [Google Scholar]
- 53.Alfonso-Prieto M, Biarnes X, Vidossich P, Rovira C. The molecular mechanism of the catalase reaction. Journal of the American Chemical Society. 2009;131:11751–11761. doi: 10.1021/ja9018572. [DOI] [PubMed] [Google Scholar]
- 54.Rappaport F, Zhang J, Vos MH, Gennis RB, Borisov VB. Heme-heme and heme-ligand interactions in the di-heme oxygen-reducing site of cytochrome bd from Escherichia coli revealed by nanosecond absorption spectroscopy. Biochim Biophys Acta. 2010;1797:1657–1664. doi: 10.1016/j.bbabio.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borisov VB, Verkhovsky MI. Accommodation of CO in the di-heme active site of cytochrome bd terminal oxidase from Escherichia coli. Journal of inorganic biochemistry. 2013;118:65–67. doi: 10.1016/j.jinorgbio.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 56.Yang K, Zhang J, Vakkasoglu AS, Hielscher R, Osborne JP, Hemp J, Miyoshi H, Hellwig P, Gennis RB. Glutamate 107 in subunit I of the cytochrome bd quinol oxidase from Escherichia coli is protonated and near the heme d/heme b595 binuclear center. Biochemistry. 2007;46:3270–3278. doi: 10.1021/bi061946+. [DOI] [PubMed] [Google Scholar]
- 57.Borisov VB, Belevich I, Bloch DA, Mogi T, Verkhovsky MI. Glutamate 107 in subunit I of cytochrome bd from Escherichia coli is part of a transmembrane intraprotein pathway conducting protons from the cytoplasm to the heme b595/heme d active site. Biochemistry. 2008;47:7907–7914. doi: 10.1021/bi800435a. [DOI] [PubMed] [Google Scholar]
- 58.Wineman-Fisher V, Simkovich R, Huppert D, Trujillo K, Remington SJ, Miller Y. Mutagenic induction of an ultra-fast water-chain proton wire. Physical Chemistry Chemical Physics. 2016;18:23089–23095. doi: 10.1039/c6cp05071a. [DOI] [PubMed] [Google Scholar]
- 59.Yang K, Borisov VB, Konstantinov AA, Gennis RB. The fully oxidized form of the cytochrome bd quinol oxidase from E. coli does not participate in the catalytic cycle: direct evidence from rapid kinetics studies. FEBS letters. 2008;582:3705–3709. doi: 10.1016/j.febslet.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of molecular graphics. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.