

Abstract
INTRODUCTION
The stereotypical progression of Alzheimer’s (AD) pathology is not fully understood. The selective impact of AD on distinct regions has led the field to question if innate vulnerability exists. This study aims to determine if the causative factors of regional vulnerability are dependent on cell-autonomous or transneuronal (non-cell autonomous) processes.
METHODS
Using mathematical and statistical models, we analyzed the contribution of cell-autonomous and non-cell-autonomous factors to predictive linear models of AD pathology.
RESULTS
Results indicate gene expression as a weak contributor to predictive linear models of AD. Instead, the Network Diffusion Model acts as a strong predictor for observed AD atrophy and hypometabolism.
DISCUSSION
We propose a convenient methodology for identifying genes and their role in determining AD topography, in comparison with network-spread. Results reinforce the role of trans-neuronal network spread on disease progression, and suggest innate gene expression plays a secondary role in seeding and subsequent disease progression.
Keywords: Alzheimer’s Disease, Brain Network Models, Regional Vulnerability, Regional Gene Expression, Alzheimer’s Pathology, Network Diffusion Model, Cell-Autonomous
1. INTRODUCTION
Alzheimer’s Disease (AD) is a neurodegenerative disease that affects millions of adults over the age of 65 [1]. Since the early 1900’s the pathological characteristics of AD are known to involve neuronal death, amyloid plaques and tau tangles [2]. AD tau pathology sequentially spreads from the entorhinal cortex to hippocampus, temporal cortices, posterior cingulate, precuneus, and eventually frontal cortices [3], in roughly that order. The observed stereotypical progression has led to a discussion on the innate vulnerability of regions affected by the disease.
The subject of selective regional vulnerability is extremely broad and widely reviewed [4,5,6,7]. Although the etiology and causative mechanisms behind observed selective vulnerability in AD are not fully known, current hypotheses have focused on two general concepts: a) cell type, cytoarchitecture, genetic or molecular factors, here referred to as cell-autonomous factors [6] and b) processes dependent on cell-cell communication, network connectivity and topology, here referred to as non-cell autonomous factors [4]. Warren et al. proposed the term ‘molecular nexopathy’ to refer to a coherent conjunction of pathogenic protein and intrinsic neural network characteristics [8]. They enumerated a diverse set of potential mechanisms by which molecular dysfunction might interact with the neural architecture to produce observed disease topography in neurodegenerative diseases, including dysfunction of synaptic function or maintenance, axonal transport or repair, or a result of downstream trophic or cell–cell signaling. Protein transmission was highlighted as a prime mechanistic example of non-cell autonomous factors, whereas regional gene expression was considered a potential source of intrinsic vulnerability. Specifically, local profiles of protein expression were thought to confer selective vulnerability of network elements to particular neurodegenerative diseases, and their functional phenotypic signature [8].
In this paper, we address this issue rigorously using mathematical and statistical modeling. We use healthy regional gene expression of AD-implicated genes, obtained from the public Allen Brain Atlas (ABA) [9], as a surrogate of factors governing cell-autonomous (innate) vulnerability. Our study does not require our ABA cohort to have AD already since previous explorations of gene expression from healthy brains reveal a strong spatial clustering [10,11,12]. Therefore, if innate regional vulnerabilities drive AD pathology, the spatial patterns of healthy gene expression from ABA should recapitulate the spatial patterning of AD atrophy. To impart AD-specificity, we only considered genes or groups of genes that are associated with AD pathophysiology. ABA’s young healthy subjects have no AD-related comorbidities or evidence of AD pathology; hence their gene expression data putatively represents an innate vulnerability free from the influence of activity-, age-, environment- or transport-dependent expression of proteins.
Next, we capture cell non-autonomous processes using a general model of inter-regional communication, called the Network Diffusion Model (NDM) [13]. The NDM was proposed as a model of trans-neuronal spread of AD pathology and successfully recapitulated both cross-sectional and longitudinal patterns of AD atrophy and metabolism measured from neuroimaging data [13,14]. Its effectiveness suggests a disease mechanism that stereotypically progresses along brain connectivity networks via cell-to-cell transmission.
We accordingly investigated whether cross-sectional MRI-derived regional atrophy measured from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database [15], is better explained by trans-neuronal models like the NDM, or by an innate vulnerability governed by healthy regional gene expression. A sequence of linear regression models was built, each having two covariates: a common NDM predictor, and a gene expression vector corresponding to specific implicated genes or a reduced dimensionality (PCA) vector of subsets of genes involved in a coherent functional domain. Although the network-centric NDM is based on pathology spread, here we apply it to model patterns of atrophy since (tau) pathology distribution is closely related to atrophy distribution, and the latter may be considered a direct consequence of the former [16,17]. However, atrophy is considered to represent a later stage of AD pathology than tau pathology, and in order to provide additional context we also used FDG-PET-derived regional metabolism maps as outcome variables in a separate analysis. Like the previous applications of the NDM model, we continue to use a static healthy connectome, under the assumption that the structural network serves merely as a conduit for transmitting proteinopathies, rather than being the first to be impaired itself (14).
We aim to address the effects of cell-autonomous and non-cell autonomous factors on the disease mechanism that drives AD by using linear models to quantify their statistical significance as predictive variables of atrophy and metabolism. We hypothesize that our application of both imaging and gene expression data will provide consolidated support for the importance of transneuronal spread in neurodegenerative diseases like AD.
2. METHODS
The overall pipeline of our statistical approach is depicted in Figure 1, where expression patterns of implicated genes are tested alongside the prediction of the NDM. In order to ensure regionally unbiased analysis, we repeatedly (computationally) seeded the NDM at all brain regions, and chose the seed site that gave the best match against empirical atrophy data. The map of each region’s plausibility as a seed is referred to as the “seeding pattern” and is simply the maximum Pearson correlation it achieves against empirical data (see Figure 2). The gene and NDM patterns were used in a sequence of linear statistical models whose outcome variable was the cross-sectional atrophy of ADNI patients. The same process was performed on FDG-PET data of the same ADNI patient cohort for metabolism as the predicted outcome. Visual representations of regional atrophy, metabolism and gene expression patterns are shown with glass brains that were generated with an open source visualization tool called Brainography developed by our group [18].
Figure 1.
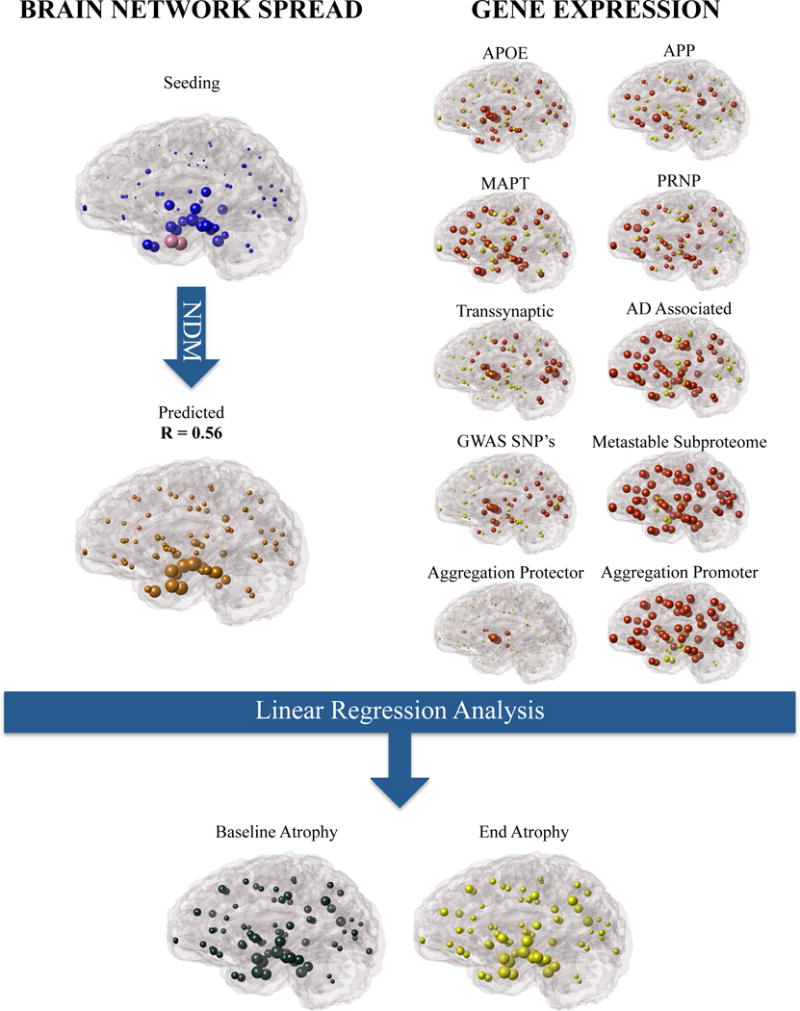
The presented methodology in which brain network connectivity models like the NDM and healthy gene expression can be used as proxies in the analysis of non-cell autonomous and cell-autonomous processes as contributors to the observed regional AD atrophy. Selected genes were compiled (AD Associated) and separated into functional subsets including (a) single genes APOE, MAPT, APP, PRNP (b) genes that were obtained from a regionally analyzed genome wide analysis (RAGWAS) (c) genes that play a role in cell to cell transmission (Transsynaptic) (d) genes that have a role in cell clearance (Metastable Subproteome) (e) genes that protect from aggregation (Aggregation Protector) and (f) genes that promote aggregation (Aggregation Promoter).
Figure 2.
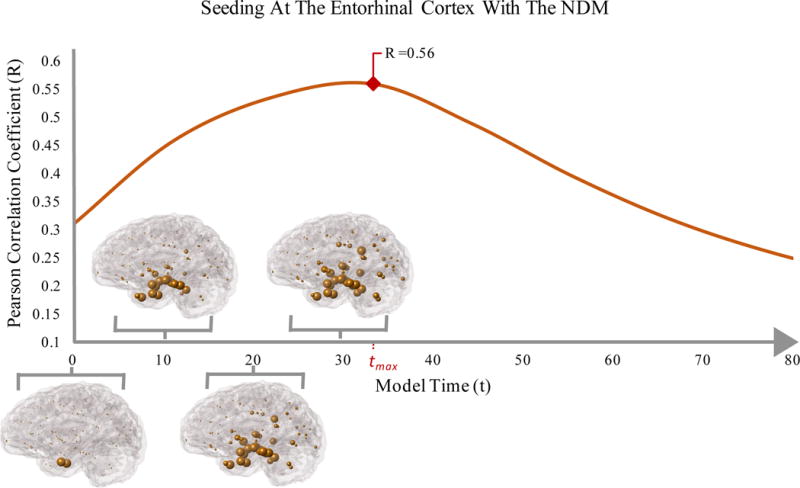
Glass Brain representation of NDM predicted atrophy over time (t). The blue spheres depict the affected regions and indicate a propagation of AD pathology that can be partially recapitulated by a single regional seed at the entorhinal cortex of the NDM. Regional seeding with the NDM propagates into atrophy patterns that have a high correspondence to cross-sectional atrophy of AD patients. Seeding for all regions were conducted; however, the figure represents seeding at the Entorhinal Cortex which resulted in a high correlation strength (Rmax = 0.56, p = 2.03 E-08).
2.1 ADNI Patient Data and Image Processing
ADNI provides subjects’ diagnosis labels at each longitudinal time point, into AD, MCI or normal control, based on natural history and cognitive assessment. T1-weighted images from the ADNI-2 database were obtained at 1.5 Tesla, using volumetric 3D MPRAGE or equivalent protocols with resolution of 1.25×1.25 mm in-plane spatial resolution and 1.2 mm-thick sagittal slices. Acquisition parameters were standardized and reported elsewhere [15]. We compared matched controls and AD patient groups (Table 1) whose regional atrophy was mapped to the same 86 regions as the gene expression data. Automated cortical and subcortical volume measures were performed with FreeSurfer software package, version 5.3 (http://surfer.nmr.mgh.harvard.edu/fswiki) [19,20]. Group wide T-statistics were calculated for baseline atrophy measurements and were used in linear models.
Table 1.
ADNI Demographic info
| Disease Stage | Group Size (Age) | M (Age) | F (Age) | MMSE |
|---|---|---|---|---|
| Control | 232 (75.14 ± 6.92) | 110 (76.37 ± 7.00) | 122 (74.03 ± 6.68) | 28.79 ± 1.14 |
| AD | 153 (75.06 ± 8.07) | 87 (76.19 ± 7.90) | 66 (73.57 ± 8.12) | 20.92 ± 4.08 |
| EMCI | 327 (71.85 ± 7.46) | 179 (72.47 ± 7.20) | 148 (71.09 ± 7.73) | 27.96 ± 1.71 |
| MCI | 2 (85.45 ± 6.01) | 1 (89.7 ± 0) | 1 (81.2 ± 0) | 16 ± 12.73 |
| LMCI | 201 (73.59 ± 7.77) | 113 (75 ± 7.52) | 87 (71.66 ± 7.71) | 26.06 ± 3.09 |
None of the ADNI patients have confirmed pathology. Patients who did not pass Freesurfer analysis were not included in the study. ADNI demographic info can be found at: http://adni.loni.usc.edu/wp-content/uploads/2012/08/ADNI_Enroll_Demographics.pdf
2.2 Allen Brain Atlas Gene Data
Gene data was obtained from six healthy brains available from the ABA [9]. Of these six brains, four had data pertaining to only the right hemisphere while the other two contained information for both hemispheres (Table 2). This data is unrelated to our AD patient data, and is meant to convey information about only healthy gene expression. Although it would be revealing to observe spatial gene expression and AD pathology in the same set of subjects, the current experimental design is appropriate for testing the role of innate, as compared to disease-induced, gene expression in AD. Gene expression data initially consisted of 926 brain regions, each one having a set of 58,692 probes that correspond to 29,181 distinct genes. The 926 brain regions were mapped to 86 regions of the Desikan atlas. The ABA and Desikan are in the same stereotactic space, this was done by collecting all sample locations from ABA that fell within the same Desikan region, using the sample co-ordinates supplied by the ABA. White matter tracts were not mapped and were excluded from analysis as done in previous research that successfully linked gene expression to AD vulnerability [21]. ABA samples that were within a 1 voxel margin of GM were assigned to the nearest GM region. All samples within the same region were averaged for each gene, and all probes corresponding to a single gene were averaged. Gene expression values were averaged across the 6 individuals. Our final regional expression data was an 86 × 29181 table.
Table 2.
ABA Gene Data Information
| Subject | Number of Sampling Locations | Half or Whole Brain |
|---|---|---|
| H0351.1009 | 363 | Half |
| H0351.1012 | 529 | Half |
| H0351.1015 | 470 | Half |
| H0351.1016 | 501 | Half |
| H0351.2001 | 946 | Whole |
| H0351.2002 | 893 | Whole |
2.3 Selecting AD-implicated genes
Following Freer et al., 2016 [21], we selected several genes that are closely associated with AD including: APP, amyloid precursor protein; MAPT, microtubule associated protein tau, which codes for tau protein, whose misfolding is a hallmark of AD [22]; PRNP, prion protein, which has been studied for insights on the prion-like transmission of non-prion proteins like amyloid and tau but has also been associated to some AD cases [23]; and Apolipoprotein, APOE, which is the most common genetic risk factor of AD [24].
Following Freer et al., 2016 [21], these and several other genes were enumerated into separate gene lists corresponding to specific functional domains: Metastable Subproteome, Aggregation Promoters, and Aggregation Protectors. An additional “AD Associated” gene list was compiled which encompassed all the genes from the above-mentioned lists including genes related to tau expression. A separate “Transsynaptic” gene list was compiled from an extensive literature search that focused on cell-to-cell communication factors that were implicated in neurodegenerative diseases like AD. Lastly, a short list of genes implicated from a regionally analyzed Genome Wide Associated Study (RAGWAS) was also assembled [25]. A list of all the genes used in this study can be found in the supplementary data.
We added the expression of these genes as a single predictor of empirical cross-sectional atrophy to a linear regression model. Each test corresponded to gene expression as a predictor (g) and cross sectional atrophy or metabolism t-statistics (aAD) as the outcome variable. The R-squared value for the whole model and p-values for the t-statistic of the gene predictor variable were calculated with MATLAB’s LinearModel.Fit() function (Table 3).
Table 3.
Gene Vector Predictors of cross sectional End-of-Study Atrophy
| Model aAD = a + b * g |
APOE | APP | MAPT | PRNP |
|---|---|---|---|---|
| R2 | 0.006 | 0.070 | 0.13 | 0.060 |
| p-value | 0.48 | 0.014 | 0.00063 | 0.023 |
| Model aAD = a + b * gPCA |
Transsynaptic (T) | AD Associated (ADA) | RAGWAS |
|---|---|---|---|
| R2 | 0.16 | 0.11 | 8.22E-04 |
| p-value | 0.00012819 | 1.97E-03 | 0.79325 |
|
Model aAD = a + b * gPCA |
Metastable Subproteome (MS) | Aggregation Promoter (APM) | Aggregation Protector (APT) |
| R2 | 0.07 | 0.0014 | 0.056 |
| p-value | 0.018 | 0.73 | 0.028 |
*Variable g is meant to represent a given gene or PCA of genes. Red font denotes results which did not pass a significance threshold (p = 0.05); no multiple comparisons correction.
2.4 Principle Component Analysis of AD Genes of Interest
Dimensionality reduction was performed on gene data sets via principle component analysis (PCA). The first principle component of our gene lists portrays the expression of genes with the highest variance (Percentage of variance: Transsynaptic PCA = 57%; AD Associated PCA = 39%; RAGWAS PCA = 54%), which reduces the influence of multicollinearity. Given that the first component captures a large amount of variance, it was used in subsequent analysis as a dimensionality-reduced stand-in for the entire gene set it was computed from. The first principle components were calculated with MATLAB’s PCA() function and were covaried against imaging-derived regional statistics in linear regression analysis. Linear models included the first principle component as a predictor with the dependent variable being the observed atrophy or metabolism as calculated from imaging data.
2.5 Predicting Atrophy with The Network Diffusion Model
The NDM can be mathematically represented by Equation 1, which describes the spread dynamics of a pathologic entity given by the 86×1 vector x(t), starting from its initial pattern x0, also 86×1 vector. Elements of x(t) and x0 represent the amount of pathology or atrophy at each of the brain’s 86 GM regions, as predicted by a model that assumes diffusion on the network defined by the (86×86) structural connectivity matrix. The pattern and rate of spread is governed by the diffusion kernel (e−βHt) [13,14]. Here H is the network Laplacian matrix [13].
| (1) |
Let aAD be the 86×1 atrophy vector whose elements contain the t-statistic of a t-test that compares cross sectional regional volume of AD subjects to age-matched healthy subjects. As the NDM proceeds following Eq (1) for t>0, at some point the NDM predicted pattern x(t) will begin to resemble the empirical atrophy pattern aAD, and at t = tmax it will give the model time at which the best match of the NDM model is observed against empirical atrophy data, as the model evaluates network diffusion following the seed configuration x0. Hence
| (2) |
The NDM models a trans-neuronal spread of AD pathology along the entire brain network connectivity. Eqs (1) and (2) were repeated 43 times, each time allowing x0 to be 1 at a single brain region (both hemispheres), and zero elsewhere. The correlation strength Rmax = corr(x(tmax),aAD), of repeated bilateral seeding followed by NDM was tabulated to determine the region of origin that best explains AD onset [14] (Supplementary Table 1). Then, the NDM predicted atrophy vector x(tmax) was used as a covariate in linear regression analysis that predicts cross sectional end-of-study atrophy. Lastly, in order to address another form of observed AD phenotype at an earlier stage than atrophy, we repeated the above-mentioned NDM and linear models on metabolism maps from FDG-PET data.
3. RESULTS
3.1 Single Regional Seeding of The NDM Indicates Heterogeneous Onset of AD Pathology Amongst Patients
Bilateral seeding was repeated for all 86 regions of interest and the predicted atrophy pattern, x(t), was calculated with the NDM over time (Figure 2). The Entorhinal Cortex (EC) emerged as the seeding region that best predicted cross-sectional atrophy patterns, with the highest Pearson’s correlation coefficient (Rmax = 0.56, p = 2.03E-08) (Figure 3a). The Pearson correlation coefficients for seeding at all 86 regions is shown in Supplementary Table 1. Although the EC’s correlation strength of Rmax = 0.56 is high enough to distinguish important regions of onset like the EC from other regions; it also implies that single regional seeding of the NDM only partially recapitulates the observed pathology in AD patients, suggesting a heterogeneous onset of AD pathology. However, since the subsequent analysis requires group averages, we chose not to allow individual subjects to have a different seed location even if they would have given better Rmax compared to the canonical seed at EC. This way, a common group averaged NDM vector x(tmax), seeded at the EC, could be used as a canonical NDM predictor in further analyses. This design choice probably under-estimates the effect of network-driven effects, as heterogeneous onset would have been better captured by subject-specific seeds. Multiple comparisons correction was not used in reporting R and p during these repeated seeding experiments. The reason is that repeated seedings are not independent from each other, and we are not interested in determining significance amongst these seedings. We are only interested in obtaining the most plausible seed region (here, EC), after which point we discard all other seeding results. Hence multiple comparisons correction was not deemed necessary.
Figure 3.
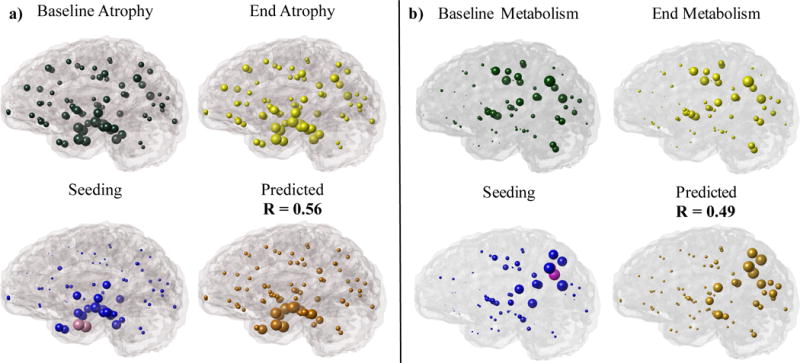
Glass Brain representations of (a) cross-sectional atrophy and (b) metabolism for ADNI patients at the start and end of study are indicated in green and yellow, respectively. Regional seeding of the NDM at 86 regions of interest are indicated in pink and blue spheres. Pink spheres represent bilateral seeding of the (a) EC and (b) Precuneus with the highest correlation strength while blue spheres indicate other regions with lower correlation strengths. Orange indicates the NDM predicted pathology from the best time point, x(tmax), which highly correlated with observed (a) cross-sectional atrophy measurements, R = 0.56 (p = 2.03E-08) and (b) metabolism from FDG-PET measurements, R = 0.49 (p = 2.01E-06).
3.2 Regional expression patterns of implicated genes are weak predictors of regional atrophy
The regional expression pattern of genes APOE, APP, MAPT, and PRNP is displayed in “glass brain” rendering to provide a visual depiction of brain regions encased within a semi-transparent rendering of the cortical surface (Figure 4). Correlation strengths for linear models that predict empirical end-of-study atrophy from a single gene predictor variable ranged from R2 = 0.006 to 0.13 with only APP, MAPT and PRNP genes being statistically significant predictor variables (p < 0.05) (Table 3). This suggests that cell-autonomous processes driven by healthy gene expression have weak relative contribution to regional atrophy patterns. In particular, APOE, whose protein product is known to be highly involved in upstream pathogenesis, gives very weak and non-significant predictors. However, APP, MAPT and PRNP present a low (R2 = 0.07, R2 = 0.13 and R2 = 0.06, respectively) but significant association. Given that MAPT and PRNP genes are involved in transport and cell-cell communication, this could also indirectly highlight the contribution of both cell-autonomous and non-cell-autonomous processes.
Figure 4.
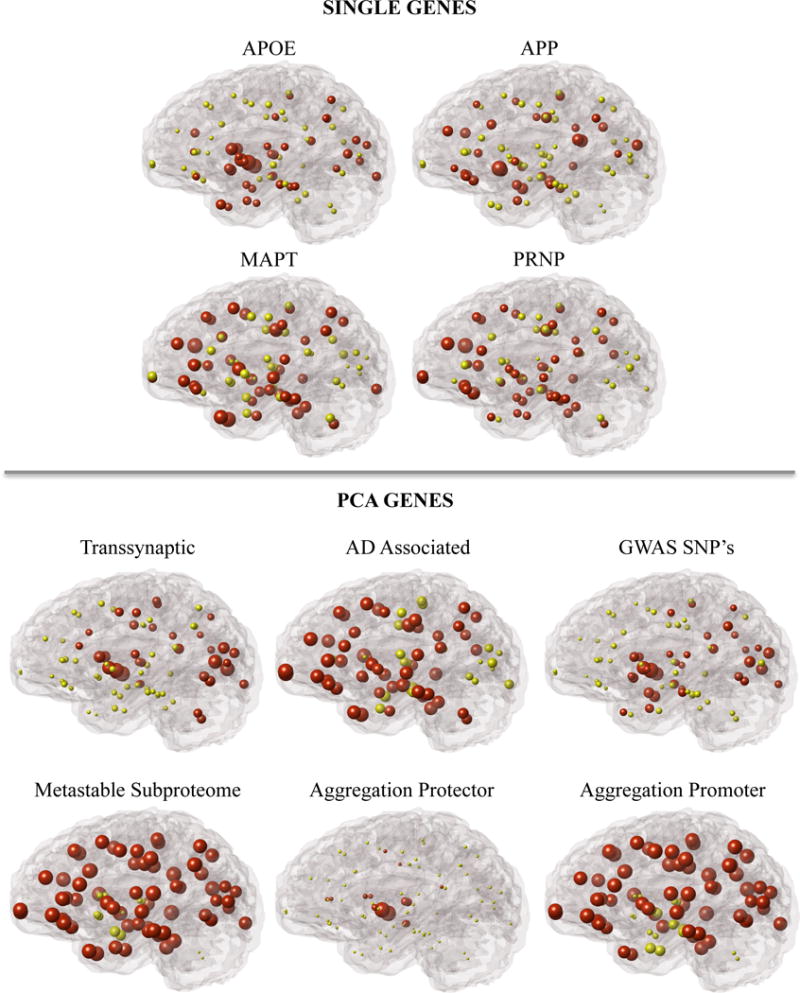
Glass brain representation of the regional gene expression of single genes and the first principle component of gene lists among 86 regions of interest. Gene lists including: Transsynaptic, AD Associated, RAGWAS, Metastable Subproteome, Aggregation Promoter, and Aggregation Protector genes were all selected from literature that implicates their functional roles in AD. Red spheres represent regions whose gene expression levels are above the mean. Yellow spheres represent regions whose gene expression are below the mean.
3.3 The NDM Model Explains AD Pathology Better Than Single Gene Predictors
We establish the ability of the NDM model to predict regional atrophy in AD by testing a statistical model with the NDM predicted atrophy as the single predictor of empirical end-of-study atrophy. The model, aAD = a + c * x(tmax) resulted in an R-squared value of 0.34 (p = 3.84e-09, for the t-statistic of the NDM predictor in the model described).
We next explored whether linear models that include both regional gene expression patterns and the NDM seeded at the EC as predictors can improve the above associations with observed AD pathology. This test evaluates the relative contribution to regional atrophy patterns of cell-autonomous processes (putatively driven by healthy gene expression) and of non-cell-autonomous processes driven by cell-to-cell communication, modeled by the NDM. In all cases, the NDM predictor was a much stronger and statistically more significant predictor (p < 0.05) with p-values ranging from 1.27e-09 to 1.90e-08 compared to single gene expression with p-values ranging from 0.00046 to 0.84 (Table 4).
Table 4.
NDM and Gene predictors of AD End-of-Study Pathology
| Model | Predictors | Atrophy | Metabolism | ||
|---|---|---|---|---|---|
| R2 | p-value for t-stat | R2 | p-value for t-stat | ||
| aAD = a + b * gAPOE + c * x(tmax) | NDM | 0.32 | 1.66E-08 | 0.29 | 1.59E-06 |
| APOE | 0.39 | 0.017 | |||
| aAD = a + b * gAPP + c * x(tmax) | NDM | 0.41 | 1.28E-09 | 0.27 | 1.09E-06 |
| APP | 0.00080 | 0.044 | |||
| aAD = a + b * gMAPT + c * x(tmax) | NDM | 0.41 | 1.70E-08 | 0.36 | 1.58E-07 |
| MAPT | 0.00061 | 0.00015 | |||
| aAD = a + b * gPRNP + c * x(tmax) | NDM | 0.38 | 5.94E-09 | 0.37 | 5.25E-08 |
| PRNP | 0.0068 | 0.000080 | |||
| aAD = a + b * gADA + c * x(tmax) | NDM | 0.41 | 4.42E-09 | 0.41 | 4.60E-08 |
| AD Associated (ADA) | 0.00046 | 3.93E-06 | |||
| aAD = a + b * gRAGWAS + c * x(tmax) | NDM | 0.32 | 1.90E-08 | 0.29 | 4.67E-07 |
| RAGWAS | 0.84 | 0.0113 | |||
| aAD = a + b * gT + c * x(tmax) | NDM | 0.44 | 8.93E-09 | 0.45 | 1.18E-08 |
| Transsynaptic (T) | 6.55E-05 | 2.13E-07 | |||
| aAD = a + b * gMS + c * x(tmax) | NDM | 0.40 | 1.27E-09 | 0.36 | 3.95E-06 |
| Metastable Subproteome (MS) | 0.0010 | 0.00020 | |||
| aAD = a + b * gAPM + c * x(tmax) | NDM | 0.33 | 1.26E-08 | 0.26 | 1.27E-05 |
| Aggregation Promoter (APM) | 0.35 | 0.12 | |||
| aAD = a + b * gAPT + c * x(tmax) | NDM | 0.36 | 5.27E-09 | 0.30 | 4.20E-06 |
| Aggregation Protector (APT) | 0.0070 | .0071 | |||
Since our weak finding for APOE is somewhat inconsistent with its known role as the highest risk factor for AD, we probed deeper into this aspect by comparing atrophy patterns between those subgroups in ADNI that have 0, 1 or 2 e-4 alleles of APOE. We found indistinguishable differences between APOE subgroups, indicating that allele status does not change atrophy patterns in patients (Supplementary Figure 1). A linear model with the APOE gene and NDM as predictors of AD pathology for different APOE subgroups also did not indicate the APOE gene as a significant predictor of atrophy for any APOE subgroup in comparison to the NDM (Supplementary Table 3).
3.4 NDM Explains AD Pathology Better Than PCA of multiple Genes
We reinforce the above results by considering a larger set of genes. We chose to investigate the contributions of multiple gene sets associated with AD by regional gene expression from lists including Transsynaptic, AD Associated, RAGWAS, Metastable Subproteome, Aggregation Promoter, and Aggregation Protector genes (Figure 4).
A series of linear models were used with the first principal components of each gene list (gPCA) as a predictor variable and cross sectional atrophy t-statistics (aAD) as the outcome variable (Table 3). The regression analysis resulted in low correlation strengths, ranging from R2 = 8.22e-04 – 0.16; with Transsynaptic, AD Associated, Metastable Subproteome, and Aggregation Protector gene lists being statistically significant (p < 0.05, for t-statistic of gene predictors). Results also indicate that Aggregation Promoter and RAGWAS genes have no statistically significant contribution to the observed cross-sectional atrophy in AD patients.
We investigated the contribution of the NDM to these linear models by adding the NDM model prediction x(tmax) as a second predictor variable to above models (Table 4). Results for the PCA of gene lists were similar to what was observed for single gene predictor models in that the addition of the NDM increased the correlation strength of the whole model. These results suggest the NDM has a greater contribution to regional AD atrophy than regional expression of single genes and multiple gene sets. The best overall model was a combination of NDM and the transsynaptic genes, followed by NDM combined with the subproteome genes.
3.5 Network Transmission of Statistically Significant Gene Predictors
Even though regional patterns of single genes, and their domain-specific principal components are weak contributors, it is possible that the proteins they code are themselves subject to network-based spread, and hence strictly local associations might hide certain gene-driven effects. In order to investigate this, we use regional gene expression as the initial pattern, x0, in the NDM equation (1), and assess the ability of this joint model to predict cross-sectional AD. If the correlation coefficient improves as model time increases from zero, it would suggest that the expression of these genes causes local production of disease factors that then propagates in a trans-neuronal fashion and recapitulates AD topography. Such a finding would support the idea that while early selective vulnerability of brain regions may be explained by healthy gene expression of the statistically significant genes, the observed patterns of AD atrophy are better explained by subsequent axonal transport. However, our results show the correlation coefficients of the gene-seeded NDM model do not improve over model time for any of the selected genes in this study (Figure 5a). Hence, neither do these genes directly cause selective regional vulnerability in AD, nor do they lead to protein products whose subsequent network transmission recapitulates regional vulnerability in AD.
Figure 5.
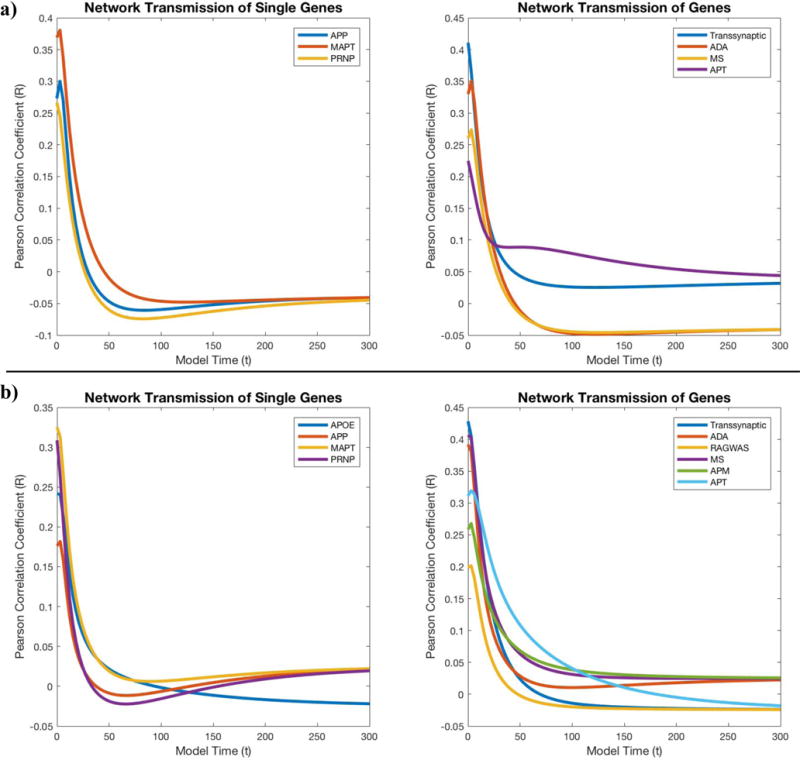
Analyzing the contribution of the NDM to the local production of selected gene expression with Pearson correlation coefficients that assess the interdependence between gene product diffusion and (5a) empirical atrophy or (5b) metabolism. Neither single genes (APP, MAPT, PRNP) or gene list (Transsynaptic, AD Associated, Metastable Subproteome, Aggregation Protector) appears to benefit from the contribution of the NDM.
3.6 Gene Expression are Better Predictors of Hypometabolism Than Atrophy
As was done for atrophy, single regional seeding of the NDM was conducted for FDG-PET-derived hypometabolism from the ADNI AD patient group (Figure 3b). The Precuneus had the highest Pearson’s correlation coefficient (Rmax = 0.49, p = 2.01e-06), indicating its importance as a region linked to AD hypometabolism. However, seeding at the Precuneus only partially recapitulates the observed pathology in AD patients. Linear models of observed AD hypometabolism indicated that the NDM was a more statistically significant predictor with p-values ranging from 1.18e-08 to 1.27e-05, in comparison to gene expression whose p-values ranged from 2.13e07 to 0.12 (p < 0.05) (Table 4). On the other hand, gene expression was more statistically significant for metabolism (p = 2.13e-07 - 0.12) than what we observed for atrophy (p varied in the range 6.55e-05 - 0.84) (Table 4). The APOE gene’s predictive role specifically for regional FDG-PET metabolism is improved in patient groups with one or no e4 allele. Nevertheless, the NDM still outperforms gene expression. However, linear models indicated some differences in gene expression’s predictive capabilities between APOE groups (Supplementary Table 3). Similar to AD atrophy, we used the NDM to predict network transmission of gene predictors used in our linear models of metabolism. Again, we found that the correlation coefficients of the gene-seeded NDM model do not improve over model time for any of the selected genes (Figure 5b).
3.7 Selective vulnerability of seed regions does not arise from innate gene expression
Since our results indicate that the EC and Precuneus are the most likely seeds for atrophy and metabolism, respectively, we investigated whether their specific vulnerability to initial insult has a molecular basis. Hence we assessed whether these regions stand out from other regions in terms of gene expression by computing Z-scores of the EC and Precuneus compared to the other 85 regions. We found that no single gene or subset of genes used in this study showed a significant difference of expression in the seed region in comparison to other regions, with z-scores ranging from −0.73 to 0.38 for the EC and −0.28 to 0.96 for the Precuneus (Supplementary Table 4). No result gave p < 0.05. Therefore, the role of EC or Precuneus as seed regions is not rooted in innate gene expression.
4. DISCUSSION
The goal of our study was to explore the molecular basis of regional variation in AD vulnerability and compare it against an alternative hypothesis that relies on network-based spread of disease. The study was designed to answer specifically: what role does healthy (as compared to environment- or age-induced) gene expression, as a proxy for innate molecular properties of brain regions, plays in governing their selective vulnerability to AD? The key assumption underlying this work is if cell-autonomous factors dependent on innate biomolecular composition of vulnerable regions is responsible for observed AD topography, then the genetic proxy for such a composition in the healthy brain must be able to track the spatial patterning of observed AD topography. Otherwise, non-cell-autonomous processes unrelated to innate vulnerability must be partially or wholly responsible. Here we specifically explore the role of cell-cell interaction based on network connectivity, using a mathematical model of transneuronal spread called the Network Diffusion Model (NDM). Our goal was not primarily to uncover the genetic correlates of AD; although we report several prominent genes and their effect on regional vulnerability, these are of secondary concern, meant to establish convergence with prior studies on specific genes and to propose targets for future in-depth interrogation. A full exploration of the genetic correlates of AD would require a much more comprehensive study design and genomic data on patients that are not yet available.
4.1 Summary of Main Results
Our main contribution is showing that selective regional vulnerability in AD can be tested using preferential gene expression in healthy brains. By selecting AD associated genes, we maintain pathological relevance of our investigation without suffering from high dimensionality, where conventional dimensionality reductions are ineffective [26]. The statistically significant single gene predictors were APP, MAPT and PRNP (p < 0.05), indicating a more relevant functional role of these proteins, which are involved in protein misfolding, over APOE in AD pathology. The NDM was implemented on the healthy connectome, and was repeatedly seeded at each region in turn. The best result was found after seeding the EC, hence EC-seeded NDM was used in subsequent analysis. When the selected genes were entered into a linear model along with EC-seeded NDM, in all cases the NDM predictor was a much stronger and statistically more significant predictor (p < 0.05) with p-values ranging from 1.27e-09 to 1.90e-08 compared to single gene expression with p-values ranging from 0.00046 to 0.84 (Table 4). The use of PCA on larger gene sets allows us to examine a large set of functionally related genes, without requiring correction for multiple comparisons. Dimensionality-reduced gene sets (AD Associated, RAGWAS, Transsynaptic. Metastable Subproteome, Aggregation Promoter, Aggregation Protector) also did not perform better as predictors of AD atrophy (Table 4). However, adding the NDM as a second predictor variable greatly improved the linear model’s correlation strength (Table 4), making the NDM a far better predictor of AD atrophy.
The same procedure was repeated for FDG-PET-derived hypometabolism from the ADNI AD patient group (Figure 3b), and the best seed was the Precuneus (Rmax = 0.49, p = 2.01e-06), which is known as one of the earliest regions to display hypometabolism in AD (27); and individual genes’ contributions were also somewhat different and generally stronger (p varied in the range 2.13e-07 to 0.12) than for atrophy (p-range: 6.55e-05 - 0.84) (Table 4). This is expected, as the molecular mechanisms leading to hypometabolism are likely to be different from that for atrophy. While Precuneus-seeded NDM only partially recapitulates observed hypometabolism, the conclusion that molecular vulnerability is a poor predictor of AD topography in comparison to NDM, is as true for hypometabolism as it is for atrophy. The exact role of hypometabolism in the AD trajectory is controversial, but our data suggest that it may have a stronger underpinning in cell-autonomous processes compared to atrophy.
4.2 Selective vulnerability does not appear to arise from innate gene expression
Taken together, our results favor the contribution of non-cell-autonomous processes (NDM predicted atrophy), over cell-autonomous processes (healthy gene expression), when identifying key players in the development and progression of AD regional patterns. Intriguingly, the best overall model for both atrophy and hypometabolism was a combination of NDM and the trans-synaptic genes (R2 = 0.44, 0.45, resp. in Table 4). The latter are thought to be involved in cell-cell interactions that mediate protein transmission between cells. Therefore, their involvement indirectly supports the network-spread hypothesis. We also tested a joint model, whereby protein products of selected genes would subsequently transmit trans-neuronally through brain circuits. This approach did not fit empirical data, suggesting that sporadic or heterogeneous processes underlie regional vulnerability. Although we showed that NDM is a far better predictor of atrophy and hypometabolism than innate gene expression, perhaps the empirically identified seed regions (EC and Precuneus, respectively) are themselves selectively vulnerable to initial pathological insult and infiltration due to innate genetic or molecular factors. To our surprise, we found that neither EC nor Precuneus stands out from the other 85 regions in expression of any single gene or subset of genes used in this study, with z-scores ranging from -0.73 to 0.38 for the EC and −0.28 to 0.96 for the Precuneus (Supplementary Table 4). No result gave p < 0.05. Essentially, the privileged role of the Entorhinal cortex or Precuneus as the most plausible seeding location cannot be explained by innate genetic or molecular factors. However, several caveats apply, as enumerated in Limitations section.
4.3 Comparison with literature on AD genetics and imaging genetics
APP and MAPT have been targets of intensive study due to their pathological aggregation and transmission in AD [28,22]. However, PRNP has been studied less for its direct contribution to AD and more for insight into the prion-like transmission of Amyloid beta and Tau [23]. The implication of our results that PRNP is a functionally relevant AD gene is supported by the role of PRNP in previously studied AD cases [29,30] and the essential role of PRNP gene variants in genetic interactions that mediate signaling, survival and synapse loss in AD transgenic model mice [31].
The weak finding for APOE as a predictor of AD atrophy is somewhat surprising, as it is known to be the highest risk factor for AD. A possibility is that gene expression from cohorts at highest risk of AD (two e4 alleles) might have emerged as better predictors than was observed in our study. This possibility cannot be directly tested since APOE allele status is absent in gene expression data from ABA. However, the ADNI AD cohort provides allele status, hence we compared atrophy patterns between APOE subgroups in ADNI. We found indistinguishable differences between APOE subgroups, indicating that allele status does not change atrophy patterns in patients (Supplementary Figure 1). Additional linear models were tested with the APOE gene and NDM as predictors of AD pathology for different APOE subgroups. Findings indicate the APOE gene is a weak predictor of atrophy for all APOE subgroups in comparison to the NDM.
Due to the sporadic nature of most AD cases, an exclusively deterministic role for genetics is not expected in AD, where environmental factors [32] or aging [33] can be important. Recent work has begun to uncover the role of gene expression and its connection to gray matter atrophy, cognition, and topological activity, with imaging tools like MRI and PET [34,35,36,25]. A sophisticated statistical framework was developed by Zhao et al. [25] to tackle the numerically and statistically challenging problem of combining high dimensional data from GWAS and image voxels. However, no previous study has sought to determine the relative contribution to selective regional AD vulnerability of gene expression against network models like NDM.
4.4 Implications
This study supports the hypothesis of trans-neuronal transmission of AD pathology, which is a complex mechanism that is not yet fully explained. The role of statistically significant gene predictors like MAPT, PRNP, and transsynaptic signaling genes in transportation and synaptic dysfunction [37], provide support for relevance of trans-neuronal communication and network models in neurodegenerative diseases like AD. Other statistically significant genes involved with clearance (Metastable Subproteome) and aggregation protection (Aggregation Protector) support a prominent role for protein misfolding, aggregation and clearance: processes that are highly relevant to trans-neuronal spread in AD progression.
Our approach can provide new insights and treatments for diseases beyond AD, such as Parkinson’s Disease, various tauopathies, Epilepsy, and Creutzfeldt-Jakob. If the above data are reproduced in AD and related dementias, it might help to rule out innate molecular composition of vulnerable regions as the sole mediator of selective vulnerability in neurodegeneration and would encourage the exploration of alternate explanations like aging-associated phenomena like lifetime [38,39], oxidative stress [6], and spread of AD pathology along white matter connectivity pathways [40,13,25].
Our results are concordant with the “molecular nexopathy” paradigm [8] in that a central role is noted for network spread, but also discordant in that we find no evidence for selective vulnerability based on cell-type, architectonic and other intrinsic properties of brain regions that may be reflected in innate gene expression. It is noteworthy however that the nexopathy paradigm enumerates a far more diverse set of potential mechanisms by which molecular dysfunction might interact with the neural architecture (e.g. see below), than is possible to explore in a single study. Possibly, the seed regions inferred by our NDM approach (EC for atrophy and Precuneus for hypometabolism) may have specific molecular vulnerability, e.g. to tau and amyloid, that remained undiscovered by our study design, despite Table S4 that shows no gene-related effect.
4.5 Limitations
Many other genetic influences that could potentially play a role in AD pathogenesis and in increasing a person’s susceptibility to AD remain outside the purview of this study. Gene expression in different tissues can change with age [41], stress and environment, such as epigenetic effects like DNA methylation. There can be other sources of regional variability, e.g. oxidative stress, vascular effects, etc. Epigenetic or post-translational modifications caused by these regionally variable causes will produce gene expression in diseased groups that are different from healthy subjects studied here, and therefore are not possible to study using the current approach. However, the primary goal of uncovering the molecular versus network correlates of AD topography may be largely unaffected by these additional factors, since they are either unlikely to have spatial gradients (e.g. age, environment) or have non-AD-like spatial patterning (oxidative/vascular stress).
Related to this is the possibility that our results are influenced by significant group differences in size or background of ABA and ADNI cohorts. Ideally, we would like to have access to regional gene expression at the SNP level in a large number of patients and age-matched controls. In practice, ABA is the only publicly available gene expression data that is regionally sampled at sufficient number of brain regions. As and when new regionally sampled data becomes available on genetic, epigenetic, metabolomic and other -omics, some of these questions will become possible to answer. To date, ABA represents our best chance of using regional expression data to address an important issue in a substantive if not complete manner.
Our weak finding for APOE as a predictor of AD atrophy may be due to the absence of APOE allele status in gene expression data from ABA, as noted earlier. Although our suggest that e4 allele status does not change atrophy patterns in patients (Figure S1), further investigation with SNP data would be needed to fully address this matter.
Another possible limitation of this study is our reliance on atrophy data, which are late-stage in the AD trajectory in comparison to other biomarkers. The use of atrophy is appropriate for our goal of understanding the basis for spatial patterning in AD. It is well known that regional atrophy is strongly associated with regional pathology [16,17], which is an early event. We partially addressed this limitation by showing analogous results for FDG-PET hypometabolism, which is thought to be an earlier event than atrophy. Other imaging data like functional MRI could provide insight on earlier disease stages that might be closer to the intrinsic pathophysiology.
Supplementary Material
Supplementary Figure 1: Glass Brain representation of NDM predicted atrophy for varying APOE allele groups. Indistinguishable difference in Atrophy patterns indicate that allele status does not change atrophy patterns in patients.
Highlights.
A methodology that uses regional imaging and gene expression data as proxies for transneuronal and cell-autonomous processes and as predictors of Alzheimer’s pathology in simple linear models is proposed.
Onset of Alzheimer’s pathology is heterogeneous but can be best partially recapitulated by seeding at the Entorhinal Cortex
Contrary to APOE, single gene predictors like APP, MAPT, and PRNP are statistically significant predictors of cross-sectional atrophy in Alzheimer’s
Regional expression of genes whose functional roles are involved in transsynaptic transmission, are better predictors of cross-sectional atrophy than other Alzheimer’s associated genes
Subsequent network transmission of protein products from selected gene’s regional expression does not recapitulate regional vulnerability in Alzheimer’s
Research in Context.
Systematic review: Authors reviewed previous literature that combines imaging and gene expression data to provide valuable insight on known neurodegenerative disease pathologies. From previous literature, which are appropriately cited, gene lists were formed and regional gene expression and atrophy measurements were mapped to study the role of transneuronal and cell-autonomous processes on innate vulnerability.
Interpretation: Our findings led to the hypothesis that simple linear models and statistics could identify transneuronal and cell-autonomous processes as predictors of observed regional atrophy in Alzheimer’s.
Future Directions: We propose a methodology in which two powerful pieces of information compiled from MRI and gene expression data can be regionally assessed and used to predict pathology in neurodegenerative diseases. Future directions rule out innate molecular composition of vulnerable regions as the sole mediator of selective vulnerability in neurodegeneration to explore alternate explanations including 1) Aging-associated phenomena or 2) oxidative stress, which could explain Alzheimer’s topography without assuming innate vulnerability.
Acknowledgments
We thank ADNI and the ABA institute for their invaluable data. As well as all members of the Raj lab for their teamwork and guidance.
Funding: We wish to acknowledge the following grants. FP was funded by a predoctoral grant from the Ford Foundation. AR and FP were funded by the NIH grant R01 NS092802. Data for this project were obtained from the ADNI study, which was funded by NIH grant U01 AG024904.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts: The authors declare no conflict of interest.
References
- 1.Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12:459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Zilka N, Novak M. The tangled story of Alois Alzheimer. Bratisl Lek Listy. 2006;107:343–5. [PubMed] [Google Scholar]
- 3.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 4.Wang M, Roussos P, McKenzie A, Zhou X, Kajiwara Y, Brennand KJ, et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Medicine. 2016;8:104. doi: 10.1186/s13073-016-0355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grothe MJ, Gonzalez-Escamilla G, Teipel SJ. MOLECULAR PROPERTIES UNDERLYING REGIONAL VULNERABILITY PROFILES FOR AMYLOID DEPOSITION AND NEURODEGENERATION IN ALZHEIMER’S DISEASE. Alzheimer’s & Dementia. 2016;12:P274–5. doi: 10.1016/j.jalz.2016.06.501. [DOI] [Google Scholar]
- 6.Saxena S, Caroni P. Selective Neuronal Vulnerability in Neurodegenerative Diseases: from Stressor Thresholds to Degeneration. Neuron. 2011;71:35–48. doi: 10.1016/j.neuron.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 7.Jackson WS. Selective vulnerability to neurodegenerative disease: the curious case of Prion Protein. Dis Model Mech. 2014;7:21–9. doi: 10.1242/dmm.012146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warren JD, Rohrer JD, Schott JM, Fox NC, Hardy J, Rossor MN. Molecular nexopathies: a new paradigm of neurodegenerative disease. Trends Neurosci. 2013;36:561–9. doi: 10.1016/j.tins.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–9. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bohland JW, Bokil H, Pathak SD, Lee C-K, Ng L, Lau C, et al. Clustering of spatial gene expression patterns in the mouse brain and comparison with classical neuroanatomy. Methods. 2010;50:105–12. doi: 10.1016/j.ymeth.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Ko Y, Ament SA, Eddy JA, Caballero J, Earls JC, Hood L, et al. Cell type-specific genes show striking and distinct patterns of spatial expression in the mouse brain. Proc Natl Acad Sci USA. 2013;110:3095–100. doi: 10.1073/pnas.1222897110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goel P, Kuceyeski A, LoCastro E, Raj A. Spatial patterns of genome-wide expression profiles reflect anatomic and fiber connectivity architecture of healthy human brain. Hum Brain Mapp. 2014;35:4204–18. doi: 10.1002/hbm.22471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raj A, Kuceyeski A, Weiner M. A network diffusion model of disease progression in dementia. Neuron. 2012;73:1204–15. doi: 10.1016/j.neuron.2011.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raj A, LoCastro E, Kuceyeski A, Tosun D, Relkin N, Weiner M. Network Diffusion Model of Progression Predicts Longitudinal Patterns of Atrophy and Metabolism in Alzheimer’s Disease. Cell Reports. 2015;10:359–69. doi: 10.1016/j.celrep.2014.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller SG, Weiner MW, Thal LJ, Petersen RC, Jack C, Jagust W, et al. The Alzheimer’s disease neuroimaging initiative. Neuroimaging Clin N Am. 2005;15:869–877. xi–xii. doi: 10.1016/j.nic.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitwell JL, Dickson DW, Murray ME, Weigand SD, Tosakulwong N, Senjem ML, et al. Neuroimaging correlates of pathologically defined subtypes of Alzheimer’s disease: a case-control study. Lancet Neurol. 2012;11:868–77. doi: 10.1016/S1474-4422(12)70200-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia C, Makaretz SJ, Caso C, McGinnis S, Gomperts SN, Sepulcre J, et al. Association of In Vivo [18F]AV-1451 Tau PET Imaging Results With Cortical Atrophy and Symptoms in Typical and Atypical Alzheimer Disease. JAMA Neurol. 2017;74:427–36. doi: 10.1001/jamaneurol.2016.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LoCastro E, Kuceyeski A, Raj A. Brainography: an atlas-independent surface and network rendering tool for neural connectivity visualization. Neuroinformatics. 2014;12:355–9. doi: 10.1007/s12021-013-9206-1. [DOI] [PubMed] [Google Scholar]
- 19.Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–55. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 20.Fischl B, Salat DH, van der Kouwe AJW, Makris N, Ségonne F, Quinn BT, et al. Sequence-independent segmentation of magnetic resonance images. Neuroimage. 2004;23(Suppl 1):S69–84. doi: 10.1016/j.neuroimage.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 21.Freer R, Sormanni P, Vecchi G, Ciryam P, Dobson CM, Vendruscolo M. A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimer’s disease. Science Advances. 2016;2:e1600947. doi: 10.1126/sciadv.1600947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B, et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. Journal of Experimental Medicine. 2016 doi: 10.1084/jem.20160833. jem.20160833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee S-J, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol. 2010;6:702–6. doi: 10.1038/nrneurol.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms, and therapy. Nat Rev Neurol. 2013;9:106–18. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Fei Z, Zhaohua L, Knickmeyer RC, Hongtu Z. Bayesian Feature Selection for Ultrahigh Dimensional Imaging Genetics Data. In: Dalca A, et al., editors. Imaging Genetics. Elsevier Science; 2017. [Google Scholar]
- 26.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. PNAS. 1998;95:14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailly M, Destrieux C, Hommet C, Mondon K, Cottier J-P, Beaufils E, et al. Precuneus and Cingulate Cortex Atrophy and Hypometabolism in Patients with Alzheimer’s Disease and Mild Cognitive Impairment: MRI and 18F-FDG PET Quantitative Analysis Using FreeSurfer. BioMed Research International. 2015 doi: 10.1155/2015/583931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bekris LM, Yu C-E, Bird TD, Tsuang DW. Genetics of Alzheimer Disease. J Geriatr Psychiatry Neurol. 2010;23:213–27. doi: 10.1177/0891988710383571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang W, Jiao B, Xiao T, Pan C, Liu X, Zhou L, et al. Mutational analysis of PRNP in Alzheimer’s disease and frontotemporal dementia in China. Sci Rep. 2016;6:38435. doi: 10.1038/srep38435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Del Bo R, Scarlato M, Ghezzi S, Martinelli-Boneschi F, Fenoglio C, Galimberti G, et al. Is M129V of PRNP gene associated with Alzheimer’s disease? A case-control study and a meta-analysis. Neurobiology of Aging. 2006;27:770.e1–770.e5. doi: 10.1016/j.neurobiolaging.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 31.Haas LT, Salazar SV, Kostylev MA, Um JW, Kaufman AC, Strittmatter SM. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain. 2016;139:526–46. doi: 10.1093/brain/awv356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuelsdorff ML, Engelman CD, Friedman EM, Koscik RL, Jonaitis EM, Rue AL, et al. Stressful events, social support, and cognitive function in middle-aged adults with a family history of Alzheimer’s disease. J Aging Health. 2013;25:944–59. doi: 10.1177/0898264313498416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panigrahi PP, Singh TR. Computational studies on Alzheimer’s disease associated pathways and regulatory patterns using microarray gene expression and network data: revealed association with aging and other diseases. J Theor Biol. 2013;334:109–21. doi: 10.1016/j.jtbi.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 34.Goñi J, Cervantes S, Arrondo G, Lamet I, Pastor P, Pastor MA. Selective brain gray matter atrophy associated with APOE ε4 and MAPT H1 in subjects with mild cognitive impairment. J Alzheimers Dis. 2013;33:1009–19. doi: 10.3233/JAD-2012-121174. [DOI] [PubMed] [Google Scholar]
- 35.Adamczuk K, De Weer A-S, Nelissen N, Chen K, Sleegers K, Bettens K, et al. Polymorphism of brain derived neurotrophic factor influences β amyloid load in cognitively intact apolipoprotein E ε4 carriers. Neuroimage Clin. 2013;2:512–20. doi: 10.1016/j.nicl.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bai F, Liao W, Yue C, Pu M, Shi Y, Yu H, et al. Genetics pathway-based imaging approaches in Chinese Han population with Alzheimer’s disease risk. Brain Struct Funct. 2016;221:433–46. doi: 10.1007/s00429-014-0916-4. [DOI] [PubMed] [Google Scholar]
- 37.Xu W, Tan L, Yu J-T. The Role of PICALM in Alzheimer’s Disease. Mol Neurobiol. 2015;52:399–413. doi: 10.1007/s12035-014-8878-3. [DOI] [PubMed] [Google Scholar]
- 38.Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, et al. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–17. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Haan W, Mott K, van Straaten ECW, Scheltens P, Stam CJ. Activity Dependent Degeneration Explains Hub Vulnerability in Alzheimer’s Disease. PLOS Computational Biology. 2012;8:e1002582. doi: 10.1371/journal.pcbi.1002582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci USA. 2004;101:4637–42. doi: 10.1073/pnas.0308627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J, Huang T, Petralia F, Long Q, Zhang B, Argmann C, et al. Synchronized age-related gene expression changes across multiple tissues in human and the link to complex diseases. Scientific Reports. 2015;5:srep15145. doi: 10.1038/srep15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Glass Brain representation of NDM predicted atrophy for varying APOE allele groups. Indistinguishable difference in Atrophy patterns indicate that allele status does not change atrophy patterns in patients.