

Abstract
Alzheimer’s disease (AD) is a chronic and progressive neurodegenerative disease of central nervous system (CNS). Nowadays, increasing evidence suggests that immune system plays a significant role in the mechanisms of AD’s onset and progression. Microglia, the main participator in the immune system of CNS, is always regarded as a protector of our brain in a healthy state and also has a beneficial role in maintaining the homeostasis of CNS microenvironment. However, chronic and sustained stimulation can push microglia into the state termed priming. Primed microglia can induce the production of amyloid β (Aβ), tau pathology, neuroinflammation and reduce the release of neurotrophic factors, resulting in loss of normal neurons in quantity and function that has immense relationship with AD. The therapeutic strategies mainly aimed at modulating the microenvironment and microglial activity in CNS to delay progression and alleviate pathogenesis of AD. Overall, in this review, we highlight the mechanism of microglial priming, and discuss the profound relationship between microglial priming and AD. Besides, we also pay attention to the therapeutic strategies targeting at microglial priming.
Keywords: Alzheimer’s disease (AD), amyloid beta (Aβ), microglial priming, neuroinflammation, tau-protein
Introduction
Alzheimer’s disease (AD) has become the most common type of dementia among elders, especially those who are more than 65 (1). Now, it is known that the two main hallmarks of AD are the extracellular deposition of amyloid β (Aβ) in the form of neuritic plaques and the intracellular accumulation of abnormal tau proteins in neurofibrillary tangles (NFTs). Those pathological changes, directly or indirectly, induce loss of synaptic function, mitochondrial damage, microglial activation and neuronal cell death as consequences (2,3). However, the pathogenesis of AD is not clear and the treatment measures are not perfect. Currently, plenty of studies have been done and a wealth of evidence indicates that the activation and priming of microglial cells contribute to AD pathogenesis and progression.
Microglia, accounting a small part about 10–15% of all the glial cells, generally belong to resident innate immune system of central nervous system (CNS) and have an indispensable impact on brain (4). Microglial cells contribute to the detection of and the response to changes in the physiological and pathological condition by altering their morphology, phenotype and function (5). In a normal physiological state, microglial cells serve as sentinels to continuously monitor their surroundings. Once the homeostasis of the brain was disrupted, microglia will change into the formation of amoeba-like shape actively, expressing various antigens and becoming more phagocytic (6). If the imbalance of homeostasis in CNS is continuous, the microglial cells will be activated in a much stronger state, which is called “priming”. Microglial priming, as mentioned above, is one step further in activation. Primed microglia are more sensitive to second stimulating factors and have a much stronger potential for reacting to the stimulating factors and even normal cells. The nature of primed microglia is a “double-edged sword” (5,7). In fact, they are derived from diverse phenotypes of microglia, and the phenotypes are context-dependent, which is related to the sequence and duration of their exposure to various stimuli in different pathologies (8,9) In this review, we highlight the effect of primed microglial cells on CNS, especially in the progression of AD.
The role of microglia in CNS
Microglia are the resident macrophages of the CNS, which are considered as the most versatile cells in brain (8). They are derived from mesoderm bone marrow precursor cells in the mesodermal yolk sac (10), and distributed throughout the brain of different densities from one site to another, much obvious in midbrain substantia nigra compacta, which has close relation to cognition. Microglia keep in a ‘quiescent’ or ‘resting’ state when their microenvironment is stable. They have a small cell body and morphological branches extending to all directions so as to survey and maintain the normal function of CNS actively (11) and slight changes in their microenvironment can translate microglia into ‘activated’ state. In a nutshell, a wealth of evidence indicates that microglia have an irreplaceable role in brain development and are important in synaptic pruning and clearing cell debris (12-14). Moreover, microglia form immune surveillance system of human brain and regulate critical processes associated with AD pathology, including the clearance and uptake of Aβ and abnormal tau protein as well as the production of neurotrophic factors and neuroinflammatory factors.
The priming of microglia
Microglial priming is a second interruption in brain microenvironment which induces an exaggerated or heightened microglial response compared to the first interruption referring to “microglial activation”, and primed microglia in CNS are more sensitive to potentially minor stimuli. This amplified response involves the change of microglial proliferation, morphology, physiology and biochemical markers (phenotype) (15,16). In consequence, these changes will lead to higher cytokines and inflammation mediator production which have immense impact on synaptic plasticity, neuronic survival, individual cognitive and behavioral function.
Mechanisms of microglial priming
A major element regulating microglia is the CNS microenvironment, for example, age (17-19). Increased oxidative stress, lipid peroxidation and DNA damage are associated with brain aging (17). Another factor for microglial priming is traumatic brain injury; significant evidence indicates that traumatic CNS injury causes prolonged activation of microglia and the development of the primed microglial profile. Massive studies show that both focal and diffuse traumatic brain injury induce formidable inflammatory process in the brain mediated by resident microglia and astrocytes (20,21). Then, the CNS infection is also a facet of the priming of microglia and viruses are the main causative agents of CNS infection. Both DNA and RNA viruses can trigger the priming of glial cells including microglia and astrocytes (22). Besides, some new studies show that complement dysfunction can change the expression of complement receptors of the immunoglobulin family on special tissue-resident macrophages and then trigger microglial priming following the activation by taking part in innumerable processes including synapse maturation, immune product clearance, hematopoietic stem/progenitor cells (HSPC) mobilization, lipid metabolism and tissue regeneration (23,24). In addition, there is also increased priming of microglia in pre-symptomatic neurodegenerative disease. For example, microglia with a morphologically activated phenotype are found in large numbers in brain (8). In the past several years, emerging lines of evidence have suggested that intricate neuroinflammation as a significant contributor to the priming of microglia, and all the above situations are closely related to neuroinflammation (17,21-23). In the context of the above situations, microglia are primed though a series of proinflammatory stimuli, such as lipopolysaccharide (LPS) (25), pathogenetic proteins (e.g., Aβ) (26,27), αsynuclein (28), human immunodeficiency virus (HIV)-Tat (29,30), mutant huntingtin (31), mutant superoxide dismutase 1 (32) and chromogranin A (27).
There are various signaling pathways and it is well known that different kinds of cells express special pattern recognition receptors (PRRs) which can prime inflammatory signaling pathways. For example, some signaling pathways termed pathogen-associated molecular patterns (PAMPs) that customarily accumulate in infected tissue could monitor microbial molecules. In addition, aggregated peptides or mislocalized nucleic acids have been identified as the misfolded proteins via a series of pathways named danger-associated molecular patterns (DAMPs) (33). Toll-like receptors (TLRs) and carbohydrate binding receptors mainly work in these pathways. Besides, there are massive different receptors existing in microglia, for example, triggering receptors expressed on myeloid cells (TREM), Fcγ receptors (FcγRs), CD200 receptor (CD200R), receptor for advanced glycation end products (RAGE), chemokine receptors (CX3CR1, CCR2, CXCR4, CCR5, and CXCR3) (34), and they can be recognized and combined in other signaling pathways but some pathways are still not clear.
Consequences of microglial priming
Microglia show a low rate of mitosis in the ‘quiescent’ or ‘resting’ state and a high rate of proliferation after being primed, indicating that microglia possess the ability to counteract cell turnover and resist pro-inflammation stimuli (35). Under the persistent or chronic stimulation, microglia undergo dramatic alterations from resting, ramified ones into activated, amoeboid ones in morphology (13). But the changes of shape cannot distinguish the characteristics of microglial activation (36), and the performance of primed microglia depends on their phenotypes which are associated with receptors on the surface and molecules that they secrete and recognize. The vast majority of tissue macrophages, under micro-environmental impetus, are able to differentiate M1 and M2 phenotypes (37,38). M1 polarization (classical activation) requires interferon-γ (IFN-γ) combined with TLR4 signaling, which promotes the excretion of inducible nitric oxide syntheses (iNOS), reactive oxygen species (ROS), proinflammatory cytokines and reduces the release of neurotrophic factors, thus inducing the exacerbating inflammation with increased markers of main histocompatibility complex II (MHC II), interleukin-1β (IL-1β) and CD68 (39,40). M2 polarization (alternative activation) is considered to contribute to tissue-supportive in the situation of wound healing, tending to depress inflammation and promote tissue repairment of collagen form. They occur in response to IL-4 and IL-13 in vivo and predominate. M2 polarization is characterized by increased expression of neurotrophic factors, proteases, enzymes arginase 1 (ARG1), IL-10 transforming growth factor-β (TGF-β), scavenger receptor CD206 and coagulation factors, and it enhances phagocytic activity (41). In fact, there is no clear boundaries between the polarizations, and M1 phenotype shares many characteristics with M2 phenotype (42). Recently, another phenotype of primed microglia called acquired deactivation has been identified. This new phenotype overlaps with M2 and has the ability for anti-inflammatory and functional recovery (43). In addition, a team conducted ultra-structural analyses and discovered a bran-new phenotype named “dark microglia” which is seldom seen in stationary state instead of in aging or AD (44).
In healthy individuals, systemic inflammation translates microglia into a transient activated state in order to promote tissue and cell recovery and make a turn to homeostasis. However, microglial priming is the second interruption in CNS microenvironment, which is much more complex than the initial activation. The primed microglia is a double-edged sword (45) for brain stability and health. On one hand, considerable researches in vivo and in vitro have demonstrated that neuronal injury is associated with microglial activation (46,47).The inflammatory phenotypes (mainly about M1) of microglia release neurotoxic factors, mediators and ROS that are detrimental to CNS (46). On the other hand, primed microglia (mainly about M2) have an important and beneficial role in neuronal regeneration, repair and neurogenesis (41,48). Primed microglia respond much vigorously to brain injury, inflammation and aging challenge, and boost the activation by switching from an anti-inflammation, potentially protective phenotype (M2) to a pro-inflammation destructive phenotype (M1) (42) (Figure 1).
Figure 1.
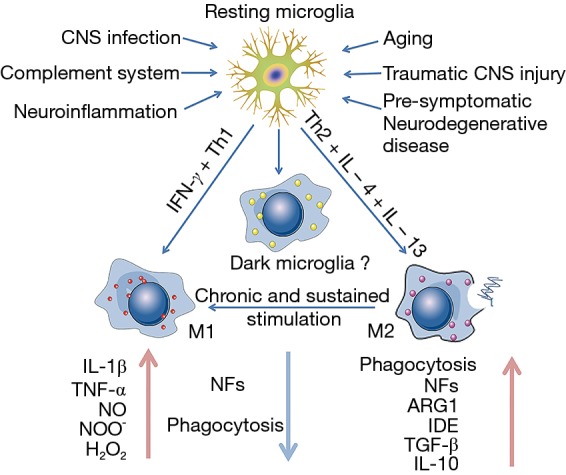
Microglial priming and altering. In the absence of disease, microglia are in a resting state with a small cell body and morphological branches extending to all directions so as to survey and maintain the normal function of CNS actively. With long-term stimuli from different kinds of inducement, the homeostasis of CNS is bankrupt and through different signaling pathways the microglia are changing their morphology and profile, altering their phenotypes that have different kinds and density of receptors. M1 and M2 are not separated complete though M1 is regarded as bad cell and M2 as good cell. Both the primed cells have a change in their phagocytosis and the release of NFs and pro-inflammatory mediators (IL-1β, TNF-α). Under chronic and sustained stimulation M2 can turn into M1. NFs, neurotrophic factors; CNS, central nervous system; TGF, transforming growth factor; IL-1β, interleukin-1β; TNF, tumor necrosis factor.
In the early stage of priming, the ability and activity to phagocytize cell debris, misfolded proteins, and inflammatory medium are increased, and more protecting molecules like IL-4, IL-13, IL-1RA, and scavenging receptors (45) are secreted. Thus, those changes can hasten normal progression of wound healing, even damage tissue repairment, neuron protection and homeostasis recovery (49). Over time, classical activated microglia (M1) account for a large proportion in all of the microglia and induce an intensive excretion of neurotoxic factors, such as IL-1β, TNF-α, NO and H2O2 (6), and more microglia are primed soon afterwards. This amplified and prolonged neuroinflammation induced by primed microglia can participate in the formation and clustering of protein tau and Aβ, furthermore it can lead to loss of neurons, decline of cognition and memory, for example, in AD patients (8,50). Although the mechanisms are not clear enough, people have reached an agreement that primed microglia incline to a chronic proinflammatory response and a self-perpetuating cycle of neurotoxicity. And this is a key factor to enhance the process of neuron damage, resulting in neurodegenerative diseases (6).
Microglial priming in AD
Aging of the CNS relate to a proinflammatory status including changes in the function of microglia. Extracellular Aβ deposition and phosphorylated tau protein accumulation in NFTs are the main neuropathological hallmarks in AD (51). Both of the neuropathological hallmarks are closely associated with activated microglia and astrocytes that are linked to neurodegenerative disease by the synthesis and secretion of inflammatory mediators, such as iNOS, ROS and proinflammatory cytokines. However, microglial priming is also mainly induced by the inflammation in CNS. Thus, whether microglial priming is the consequence or the cause of inflammation is still controversial (52). Microglial priming mainly induces a boost of Aβ, tau protein, neuroinflammation and reduction of neurotrophic factors, resulting in loss of normal neurons and formation of neuritic plaques and neurofibrillary tangles that have immense relationship with AD. With the progression of AD, changes from neuronal dysfunctions (still have no obvious signs and symptoms) to memory loss and cognitive impairment are becoming more visible.
Microglial priming in Aβ
Aβ is the main component of neuritic plaques, meanwhile Aβ is a series of 37–43 amino acid peptides, which are generated in the procedure of sequential enzymatic cleavage of amyloid precursor protein (APP) by β-secretase and λ-secretase (53). Microglia have a variety of receptors on the surface combined with Aβ, such as the RAGE and SR (54). Evidence confirms the notion that microglial priming caused by inflammation is an inducer of the accumulation of Aβ in AD rather than a primary cause of neurodegeneration (55,56). And from many studies, we summarize a conclusion that the activation of microglial cells is enhanced (microglial priming) under proinflammatory conditions, indicating that activated microglia responding to Aβ related proteins can critically rely on the priming of glial cells by proinflammatory factors (52). However, microglia and astrocytes have been found by studies to play important roles in engulfing and purging Aβ plaques as well as in remodeling cerebrovascular Aβ deposits in a cerebral amyloidosis mouse model that has been transformed in the gene (57). In the last few years, a point has been accepted by most people about the accumulation and clearance of Aβ plaques. For one thing, Aβ adheres to microglia and promotes their synthesizing and secreting of inflammatory mediators, which accelerates the progress of AD. For another, Aβ plaques are eliminated by activated microglia via phagocytosis and clearance (50).
Like other amyloid proteins, two subtypes of Aβ (Aβ1–40 and Aβ1–42) with different performances, higher aggregation degree Aβ (58) and soluble Aβ (sAβ) oligomers. And Aβ inclines to aggregate and change its conformation to become an unsolvable form named fibrillar Aβ (fAβ) (59). And it has been identified that oligomeric sAβ is more neurotoxic than fAβ. The oligomers alone are not toxic and stimulate a distinct secretary profile for IL-6 and monocyte chemoattractant protein-1 when compared with fibrils. Finally, soluble oligomers stimulate the loss of cultured cortical neurons which was accelerated via priming of microglia (60). There is also another statement that oligomers contribute to the early phase of AD, while Aβ in the fiber form has a main role in lasting the inflammatory response (61). One experiment with triple florescence labeling indicates that activated microglia participate in Aβ engulfing in microglial somata. Taking into account the intense relation between Aβ deposition and increased microglial cell density, we believed that Aβ aggregation might play a key role in the core mechanism for enhancing microglial density (2). Activated microglia were shown to migrate to newly formed amyloid plaques rapidly (62), indicating that they might transform their shape into an amoeba-like form aiming at Aβ plaque deposits and might contribute to pathology. In recent times, numerous imaging studies made the presence and progression of Aβ plaques out of water (63,64). Increased plaques may be hampered by priming and removing of microglia. Studies in transgenic mouse models indicated that primed microglia become dysfunctional in late-stage AD with decrease in the expression of Aβ-binding receptors and continuous reduction of enzymes, while maintaining even increasing the production of proinflammatory cytokines (65). Although it has long been believed that activated microglia are associated with Aβ plaques, and their explicit impact on the AD progression is still not entirely distinct at present (66).
Microglial priming in tau
Tau becomes a research hot spot in study of neurodegenerative disease after Aβ (50,67). Tau is a kind of microtubule-associated protein playing a significant role in neuronal microtubular stabilization and in axonal outgrowing promotion. Nevertheless, tau aggregation is recognized as tauopathies the common characteristic in many degenerative diseases (68).
Tau oligomers and fibrils are prompted via arachidonic acid, and then they prime the microglia with significant changes in morphology (69). Many experiments suggest the release of extracellular tau by neurons and cell lines via diverse pathways, including cell death (70). Emerging lines of study show that human extracellular tau collection is internalized by microglia. The extracellular tau can be internalized by other cells, including glial cells and neurons. The pattern from cell to cell of tau spreading has been proposed as one of the mechanisms underlying the progression of tau pathology (71,72). Wild type of human tau protein is transforming from ventral hippocampus to next neurons that connects to each other discounting the distance, and the example indicating a trans-synaptic protein transfer is olfactory and limbic systems (71,73).
Activated microglia also plays an opposing role in tau pathogenesis after priming in AD. On one side, they result in the beginning of the disease via liberation neurotoxins, such as proinflammatory mediators and ROS (46,74,75). Meanwhile, activated microglia standing by NFTs participating in the tangle formation show the envelopment of activated microglia in tau pathology. On the other side, triggered microglia restrain the progression of AD by phagocytosing abnormal cumulated, highly phosphorylating tau protein. In addition, activated microglia can synthesize and release numerous neurotrophic factors and antioxidant to resist the pathology of AD (48,75).
Microglial activation contributes to neurodegeneration because immunosuppression depresses the intracellular tangles and neuronal loss in mice expressing mutant tau (76). In the last few years, increasing devotion in modeling and genetics in AD mice has indicated that a defective or abnormal response of microglia has an indispensable role in the synergistic interaction motivating between intracellular NFTs and extracellular Aβ plaques. Thus, this balance is disrupted, leading to the pathology of tau spreading and accelerating the progression to AD (1). Cognitive decline and memory impairment keep in pace with the pathology progression of tau spreading to all cortical regions. In all these advanced stages, it has been discovered that tau pathology is always accompanied with ‘neuritic’ amyloid plaques combined with NFTs that are composed of aggregated hyperphosphorylated tau (77). Microgliosis might be the earliest manifestation of neurodegenerative tauopathies, and abrogation of tau-induced microglial activation could retard the progression of CNS disorders (76,78). Though considerable endeavor and progress have been made in recent years about the role of tau in degenerative diseases, the detail and accurate mechanisms of tau pathology associated with microglia and tau mediated disease are still under water.
Microglial priming in neuroinflammation
Though the precise and detailed role of microglial cells remains to be ascertained and elucidated, there is a consensus on that primed microglia are involved in AD-associated inflammation response of the CNS (66,79). It has been verified that neuroinflammation induced by microglial priming mainly about aging, systemic inflammation, gene regulation and blood brain barrier impairment.
Aging
Aging is the main risk factor for the onset and progression of AD, especially for the sporadic forms (80) and is accompanied by chronic, systemic up-regulation of the proinflammatory factors and a relative decrease in anti-inflammatory answer (81,82). This shift from homeostasis to an inflammatory state is via age-linked elements which induce an imbalance between anti-inflammatory and proinflammatory networks (83). Microglia is primed into an activated state that can amplify the persistent neuroinflammation and inflammatory reactivity in the aged brain (81). A vast of studies have indicated that microglia in the brain of rodent model act in a triggered phenotype during aging characterized with increased expression of CD11b, CD11c and CD68 (84).
Systemic inflammation dependent
Recently, considerable evidence has emerged that the neuroinflammation cascades deduced from primed microglia cells also lead to the AD pathogenesis (85,86). Prolonged activation of microglia can enhance the synthesis and secretion of proinflammatory cytokines and trigger a proinflammatory cascade, consequently resulting in neuronal damage and losses (8,87). Neuroinflammation is not only an early event but a hand pushing the progression of AD. And microglia have an indispensable impact on the inflammation of human brain (88). The CNS inflammation and sickness behaviors can be deduced from systemic inflammation through molecular pathways. A study of Spencer et al. indicates that ROS production of primed microglia reduces the levels of intracellular glutathione and increases nitric oxide production in NADPH oxidase subunit NOX2. Moreover, they identify that these simultaneously occurring processes lead to the production of more neurotoxic peroxynitrite (89). And this has been verified many times in rodent models that are challenged before with peripheral LPS or administration of one or more of the proinflammatory cytokines such as TNF-α, IL-1β and IL-6, IL-33 (38,81,90). The results of immense researches come to an agreement on the opinion that systemic inflammation can lead to microglial activation (91). The consequence of this research highlights the variability of inflammatory response in brain associated with AD patients and the underlying relationship between systemic inflammatory and neuroinflammation (Table 1). Besides, MAPK (mitogen activated protein kinase) signaling pathways are highly evolutionarily and wide-spreading conserved the function of regulate mechanisms of the eukaryotic cell, and microglial MAPK can also lead to an inflammatory response to the aged brain with AD (92). Overall, chronic or prolonged systemic inflammation induces neuroinflammation, leading to the onset and accelerating the progression of AD (93,94).
Table 1. Effects of inflammatory cytokines in Alzheimer’s disease pathology.
| Inflammatory cytokines | Effect on Aβ | Effect on tau | Effect on neurons | References |
|---|---|---|---|---|
| Pro-inflammatory cytokines | ||||
| IL-1β | Increase Aβ production | Increase tau phosphorylation | Increase neuronal death and damage | Andersson, 2012 |
| Reduce Aβ clearance | Increase tau pathology | – | Ghosh, 2013 | |
| IL-6/ TNF-α | Reduce Aβ clearance | Increase tau phosphorylation | Rescue neurons | Luo, 2012 |
| IL-12/IL-23 | Reduce Aβ clearance | Increase tau phosphorylation | Reduce neuronal loss | Vom Berg, 2012 |
| Anti-inflammatory cytokines | ||||
| IL-10 | Reduce Aβ production | Decrease tau phosphorylation; increase Aβ clearance | Rectify neuron dysfunction | He, 2017 |
| IL-33 | Reduce Aβ production; increase Aβ clearance | No significant change | Indirect damage and death | Weitz, 2012 |
Aβ, amyloid β; IL-1β, interleukin-1β; TNF, tumor necrosis factor.
Genetic regulation dependent
In the ageing human brain, gene regulation associated with the innate immune response has been observed. Recently, preclinical, bioinformatics and genetic data have revealed that activation of the brain immune system relates to AD pathology and leads to the pathogenesis of this disease (95). Genome wide association studies (GWAS), functional genomics (96,97) and even proteomic analysis of cerebrospinal fluid (CSF) and blood (98,99) have identified that dysfunctional immune pathways deduced from genic mutation as indispensable susceptibility factors in LOAD (100), which is the vast majority of AD. Recently, GWAS have become a powerful tool in screening genes and several new risk genes associated with AD have been identified. Apolipoprotein E (APOE) ε4allele is the most significant and common risk gene for sporadic AD, and this mutation exacerbates the risk of disease onset by 15 times in homozygous carriers and by three times in heterozygous carriers (101,102). Two further studies have plunk for the presumption of compromised microglial function through distinguishing rare mutations that have an increased risk of AD (97,103). An extracellular domain mutation of TREM2 gene has a similar extent with APOEε4 in increasing the risk of AD. While TREM2 is highly expressed on the surface of microglia (104) and mediates phagocytosis and the removing of neuronal debris (105). In addition, some other genes, such as PICALM, Bin1, CLU, CR1, MS4A and CD33 have been identified as risk genes for AD (106,107). Most of the risk mutation genes are expressed by microglial cells (108).
Blood-brain barrier (BBB) impairment dependent
BBB is between blood and brain, and it is a special barrier mainly formed by tight liner sheets, specific endothelial cells and tight junctions—structures that bring those cells together (51,109). The CNS is important for our body, and BBB is indispensable for CNS. The BBB combined with blood-nerve barrier form a frontier defense system to restrict the communications of cells and soluble factors between blood and the neural tissue, and it has a significant role in maintaining the homeostasis of CNS and peripheral nervous system (110). With development, sustained inflammation can cause damage to the BBB. During the insult and depending on the time, this damage could lead to loss of hypersensitive neurons, and neuroinflammatory sites and focal white matter impairment emerge after the damage (86,100,111). Besides, compromised BBB allows more infiltrating leukocytes to enter into CNS (112); an immune response was aggravated by brain microglia under the condition of peripheral inflammation. These processes are under the control of chemokine and cytokine signaling and have an effect on brain microglial cells (112). For instance, it has been verified that TNF-α, IL-17A and IL-1β can loosen the tight junctions and destroy the BBB. Loss of BBB integrity and altered expression of tight junctions are associated with neuroinflammatory response (51,113). Several studies have indicated that in the animal model of AD the vulnerability of BBB to inflammation is increased and current knowledge indicates that the BBB integrity is more important than previous and modulation of the BBB may provide a new therapeutic target for the treatment of AD (114) (Figure 2).
Figure 2.

Microglial priming and AD. At early stage, primed microglia enhances the ability of phagocytosis and releases more NFs to protect neurons and keep the homeostasis of the CNS. Later, the primed microglia are boosted to release pro-inflammation mediators, such as cytokine and chemokine, leading to a high level neuroinflammation which accelerate the activation of microglial cells in turn. Under this disrupted situation, β-amyloid peptides accumulate and change into amyloid plaque; meanwhile, dissociated tau is phosphorylated and aggregated to form neurofibrillary tangles in neurons especially in those whose gene is associated with APOEε4 mutation. Thus, primed microglia, directly and indirectly, result in neuron damage and loss, the main cause of AD onset or progression. AD, Alzheimer’s disease; CNS, central nervous system; AS, abnormal substances (cell debris, misfolded protein); NFs, neurotrophic factors.
Microglial priming in surrounding cells
The relationship between microglial activation and neighbor cells, especially neurons, is omnifarious and still not completely clear. However, studies have identified that the sustained release by primed microglia of proinflammatory mediators has an effect on the suppression of neurogenesis in adults and axonal transport (115,116). In addition, neuroinflammation deduced from activated microglia restricts the supplement of neurotrophic factors to surrounding cells (116,117)and may affect interneuronal protein handling process which is important for brain physiology. Activated microglial cells affect surrounding neurons and even cause detrimental feedback on microglia in degenerative disease, especially in AD (116). Neurodegeneration starts on distal axonal segments usually, and affects neuron progression proximally to culminate in the death in a dying-back process. Combined with microglial priming, synaptic pathology is the earliest neurotoxic consequence in progressive neurodegeneration (76). Microglial priming deduced from chronic or prolonged microglial activation increases the level of proinflammatory cytokines by accelerating the synthesis and release, and also triggers a proinflammatory cascade leading to neuronal damage and losses later (8,87). There is amplified axon impairment which is associated with cytokines and iNOS released by primed microglial cells (118). Thus, activated microglia probably play a detrimental role in surrounding tissue mainly through the expression of proinflammatory mediators.
Therapeutic interventions
In a study, the risk factors of AD in old people are randomly allocated to a combined life-style of diet, exercise, individual recognized training, standard care and strict control over the risk factors of cardiovascular diseases (119). And in pairs, a meta-analysis has identified that effective interventions in diet, exercise, medications, psychological condition, biochemical exposures, preexisting disease and lifestyle may reduce new incidence rate of AD (120,121). It has been identified that microglial activation has a close relationship with the progression of tau pathology and considerable studies in animal models of AD have approved that modulating the activation of microglial cells, by and large, is a powerful and effective method to prevent the main pathological events including aggradation of Aβ plaque, tau pathology and neuronal loss. And some researches also demonstrate that it is destructive rather than protective of microglia in the condition of undirected or uncontrolled activation (76,122). Modulating microglial priming can be implemented by improving the microenvironment in CNS, altering activated microglial phenotype.
Nowadays, several kinds of drugs are mainly utilized to treat the patients with AD: rivastigmine, galantamine, cholinesterase inhibitors donepezil and glutamate antagonist memantine (119). And a wide range of clinical trials have identified that anti-inflammation drugs such as NASIDs (123), statins (124) and TNF blockers (81) can reduce the incidence of AD in a long time. Those drugs can reduce the detrimental effect of proinflammatory cytokines on neuronal function and of proinflammatory cytokines on the aggradations of Aβ plaques (81). Recently, GWAS studies have confirmed that microglial receptors such as CD36 and RAGE are greatly up- or down-regulated after being activated in the brain of AD patients with genic mutation (108). Besides, neuroinflammation is greatly aggravated in Rag-5xf AD mice (an immune-deficient mouse model with AD that lacks B cells, T cells, and natural killer cells) as indicated by a shift in microglial phenotype, reducing phagocytic capacity and increasing proinflammatory mediators production, and immune cell populations are also important in the progression of AD pathology (125). Current researchers are devoting their energy into identifying the pathways in special that can be targeted efficiently and practically. For instance, it has been demonstrated that blocking the signaling pathway of IL-1β can ameliorate the pathology of CNS in mouse models with AD (122,126). In addition, a new study has verified that administration of TNF-α monoclonal antibody (infliximab) of AD or regulating glucose metabolism can reduce amyloid plaques and phospho-tau (127). Restricting the priming and overactivation of microglia can be a powerful targeting method to treat AD. Treatment strategies designed in the future to counteract the adverse effects of overactivation in these cells should be researched more than ever (128).
Conclusions and future perspectives
Microglia play a beneficial role in maintaining the homeostasis of CNS micro-environment; if the balance of the brain homeostasis is disturbed, microglial cells can be activated and do their best to restore the balance in CNS by defending against the destructors and protecting the aboriginal roles. However, chronic and sustained stimulation can push microglia into a state named priming, which is more sensitive to potentially minor stimuli. Microglial priming mainly induces the boost of Aβ, tau protein, neuroinflammation and reduction of neurotrophic factors, resulting in loss of normal neurons and formation of neuritic plaques and neurofibrillary tangles that have immense relationship with AD. This “double-edged sword” inclines to a detrimental role and can accelerate the progression of abnormal protein production and aggravation as well as neuronal loss and dysfunction. Although we have a clear view that aging has great relevance to AD, we can do nothing about it. Thus, the therapeutic strategies mainly aimed at modulating the microenvironment in CNS by reducing inflammation to delay progression and alleviate pathogenesis of AD and modulating microglial reactivity could be equal to a meaningful approach. Besides, primed microglia also can improve the abilities in engulfing abnormal substances and releasing neurotrophic factors. However, the detailed mechanisms of microglial priming and its functions are not completely clear. But as more and more scientists and researchers focus on it, the method to ameliorate even cure AD is not far away from us.
Acknowledgements
This work was supported by grants from the Shandong Provincial Outstanding Medical Academic Professional Program, Qingdao Key Health Discipline Development Fund, Qingdao Outstanding Health Professional Development Fund, and Shandong Provincial Collaborative Innovation Center for Neurodegenerative Disorders.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Mhatre SD, Tsai CA, Rubin AJ, et al. Microglial Malfunction: The Third Rail in the Development of Alzheimer's Disease. Trends Neurosci 2015;38:621-36. 10.1016/j.tins.2015.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodriguez JJ, Noristani HN, Verkhratsky A. Microglial response to Alzheimer's disease is differentially modulated by voluntary wheel running and enriched environments. Brain Struct Funct 2015;220:941-53. 10.1007/s00429-013-0693-5 [DOI] [PubMed] [Google Scholar]
- 3.Mildner A, Schmidt H, Nitsche M, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci 2007;10:1544-53. 10.1038/nn2015 [DOI] [PubMed] [Google Scholar]
- 4.Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol 2014;32:367-402. 10.1146/annurev-immunol-032713-120240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlachetzki JC, Hull M. Microglial activation in Alzheimer's disease. Curr Alzheimer Res 2009;6:554-63. 10.2174/156720509790147179 [DOI] [PubMed] [Google Scholar]
- 6.Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010;7:354-65. 10.1016/j.nurt.2010.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hensley K. Neuroinflammation in Alzheimer's disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis 2010;21:1-14. 10.3233/JAD-2010-1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol 2010;6:193-201. 10.1038/nrneurol.2010.17 [DOI] [PubMed] [Google Scholar]
- 9.Eggen BJ, Raj D, Hanisch UK, et al. Microglial phenotype and adaptation. J Neuroimmune Pharmacol 2013;8:807-23. 10.1007/s11481-013-9490-4 [DOI] [PubMed] [Google Scholar]
- 10.Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol 2010;8:e1000527. 10.1371/journal.pbio.1000527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salter MW, Beggs S. Sublime microglia: expanding roles for the guardians of the CNS. Cell 2014;158:15-24. 10.1016/j.cell.2014.06.008 [DOI] [PubMed] [Google Scholar]
- 12.Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012;74:691-705. 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norden DM, Muccigrosso MM, Godbout JP. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 2015;96:29-41. 10.1016/j.neuropharm.2014.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science 2013;339:156-61. 10.1126/science.1227901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michell-Robinson MA, Touil H, Healy LM, et al. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015;138:1138-59. 10.1093/brain/awv066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 2015;7:56. 10.1186/s13195-015-0139-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol 2013;39:19-34. 10.1111/j.1365-2990.2012.01306.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia 2013;61:71-90. 10.1002/glia.22350 [DOI] [PubMed] [Google Scholar]
- 19.Wong WT. Microglial aging in the healthy CNS: phenotypes, drivers, and rejuvenation. Front Cell Neurosci 2013;7:22. 10.3389/fncel.2013.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol 2013;4:18. 10.3389/fneur.2013.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lifshitz J, Witgen BM, Grady MS. Acute cognitive impairment after lateral fluid percussion brain injury recovers by 1 month: evaluation by conditioned fear response. Behav Brain Res 2007;177:347-57. 10.1016/j.bbr.2006.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furr SR, Marriott I. Viral CNS infections: role of glial pattern recognition receptors in neuroinflammation. Front Microbiol 2012;3:201. 10.3389/fmicb.2012.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricklin D, Hajishengallis G, Yang K, et al. Complement: a key system for immune surveillance and homeostasis. Nat Immunol 2010;11:785-97. 10.1038/ni.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramaglia V, Hughes TR, Donev RM, et al. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc Natl Acad Sci U S A 2012;109:965-70. 10.1073/pnas.1111924109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pei Z, Pang H, Qian L, et al. MAC1 mediates LPS-induced production of superoxide by microglia: the role of pattern recognition receptors in dopaminergic neurotoxicity. Glia 2007;55:1362-73. 10.1002/glia.20545 [DOI] [PubMed] [Google Scholar]
- 26.Rojanathammanee L, Floden AM, Manocha GD, et al. Attenuation of microglial activation in a mouse model of Alzheimer's disease via NFAT inhibition. J Neuroinflammation 2015;12:42. 10.1186/s12974-015-0255-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Z, Sun L, Hashioka S, et al. Differential pathways for interleukin-1beta production activated by chromogranin A and amyloid beta in microglia. Neurobiol Aging 2013;34:2715-25. 10.1016/j.neurobiolaging.2013.05.018 [DOI] [PubMed] [Google Scholar]
- 28.Rocha EM, Smith GA, Park E, et al. Sustained Systemic Glucocerebrosidase Inhibition Induces Brain alpha-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid Redox Signal 2015;23:550-64. 10.1089/ars.2015.6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paris JJ, Singh HD, Carey AN, et al. Exposure to HIV-1 Tat in brain impairs sensorimotor gating and activates microglia in limbic and extralimbic brain regions of male mice. Behav Brain Res 2015;291:209-18. 10.1016/j.bbr.2015.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turchan-Cholewo J, Dimayuga FO, Gupta S, et al. Morphine and HIV-Tat increase microglial-free radical production and oxidative stress: possible role in cytokine regulation. J Neurochem 2009;108:202-15. 10.1111/j.1471-4159.2008.05756.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crotti A, Benner C, Kerman BE, et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci 2014;17:513-21. 10.1038/nn.3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mesci P, Zaidi S, Lobsiger CS, et al. System xC- is a mediator of microglial function and its deletion slows symptoms in amyotrophic lateral sclerosis mice. Brain 2015;138:53-68. 10.1093/brain/awu312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol 2014;14:463-77. 10.1038/nri3705 [DOI] [PubMed] [Google Scholar]
- 34.Hu X, Liou AK, Leak RK, et al. Neurobiology of microglial action in CNS injuries: receptor-mediated signaling mechanisms and functional roles. Prog Neurobiol 2014;119-120:60-84. 10.1016/j.pneurobio.2014.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ajami B, Bennett JL, Krieger C, et al. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 2007;10:1538-43. 10.1038/nn2014 [DOI] [PubMed] [Google Scholar]
- 36.Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol 2009;40:139-56. 10.1007/s12035-009-8077-9 [DOI] [PubMed] [Google Scholar]
- 37.Perry VH. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol 2010;120:277-86. 10.1007/s00401-010-0722-x [DOI] [PubMed] [Google Scholar]
- 38.Ghosh S, Wu MD, Shaftel SS, et al. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer's mouse model. J Neurosci 2013;33:5053-64. 10.1523/JNEUROSCI.4361-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fenn AM, Gensel JC, Huang Y, et al. Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry 2014;76:575-84. 10.1016/j.biopsych.2013.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sudduth TL, Schmitt FA, Nelson PT, et al. Neuroinflammatory phenotype in early Alzheimer's disease. Neurobiol Aging 2013;34:1051-9. 10.1016/j.neurobiolaging.2012.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McPherson CA, Kraft AD, Harry GJ. Injury-induced neurogenesis: consideration of resident microglia as supportive of neural progenitor cells. Neurotox Res 2011;19:341-52. 10.1007/s12640-010-9199-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perry VH, Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol 2013;35:601-12. 10.1007/s00281-013-0382-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol 2005;5:953-64. 10.1038/nri1733 [DOI] [PubMed] [Google Scholar]
- 44.Bisht K, Sharma KP, Lecours C, et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016;64:826-39. 10.1002/glia.22966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo XG, Chen SD. The changing phenotype of microglia from homeostasis to disease. Transl Neurodegener 2012;1:9. 10.1186/2047-9158-1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levesque S, Wilson B, Gregoria V, et al. Reactive microgliosis: extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain 2010;133:808-21. 10.1093/brain/awp333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knoch ME, Hartnett KA, Hara H, et al. Microglia induce neurotoxicity via intraneuronal Zn(2+) release and a K(+) current surge. Glia 2008;56:89-96. 10.1002/glia.20592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diestel A, Troeller S, Billecke N, et al. Mechanisms of hypothermia-induced cell protection mediated by microglial cells in vitro. Eur J Neurosci 2010;31:779-87. 10.1111/j.1460-9568.2010.07128.x [DOI] [PubMed] [Google Scholar]
- 49.Chen Z, Jalabi W, Hu W, et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun 2014;5:4486. 10.1038/ncomms5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu Y, Ye RD. Microglial Abeta receptors in Alzheimer's disease. Cell Mol Neurobiol 2015;35:71-83. 10.1007/s10571-014-0101-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang F, Jiang L. Neuroinflammation in Alzheimer's disease. Neuropsychiatr Dis Treat 2015;11:243-56. 10.2147/NDT.S75546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.von Bernhardi R. Glial cell dysregulation: a new perspective on Alzheimer disease. Neurotox Res 2007;12:215-32. 10.1007/BF03033906 [DOI] [PubMed] [Google Scholar]
- 53.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem 2008;283:29615-9. 10.1074/jbc.R800019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilkinson K, El Khoury J. Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer's disease. Int J Alzheimers Dis 2012;2012:489456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol 2007;28:138-45. 10.1016/j.it.2007.01.005 [DOI] [PubMed] [Google Scholar]
- 56.Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2013;493:674-8. 10.1038/nature11729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A 2009;106:1261-6. 10.1073/pnas.0805453106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee YJ, Han SB, Nam SY, et al. Inflammation and Alzheimer's disease. Arch Pharm Res 2010;33:1539-56. 10.1007/s12272-010-1006-7 [DOI] [PubMed] [Google Scholar]
- 59.Sondag CM, Dhawan G, Combs CK. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation 2009;6:1. 10.1186/1742-2094-6-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meda L, Cassatella MA, Szendrei GI, et al. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 1995;374:647-50. 10.1038/374647a0 [DOI] [PubMed] [Google Scholar]
- 61.Li Y, Tan MS, Jiang T, et al. Microglia in Alzheimer's disease. Biomed Res Int 2014;2014:437483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meyer-Luehmann M, Spires-Jones TL, Prada C, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature 2008;451:720-4. 10.1038/nature06616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCarter JF, Liebscher S, Bachhuber T, et al. Clustering of plaques contributes to plaque growth in a mouse model of Alzheimer's disease. Acta Neuropathol 2013;126:179-88. 10.1007/s00401-013-1137-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burgold S, Bittner T, Dorostkar MM, et al. In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol 2011;121:327-35. 10.1007/s00401-010-0787-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Orre M, Kamphuis W, Osborn LM, et al. Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiol Aging 2014;35:2746-60. 10.1016/j.neurobiolaging.2014.06.004 [DOI] [PubMed] [Google Scholar]
- 66.Rodriguez JJ, Witton J, Olabarria M, et al. Increase in the density of resting microglia precedes neuritic plaque formation and microglial activation in a transgenic model of Alzheimer's disease. Cell Death Dis 2010;1:e1. 10.1038/cddis.2009.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ando K, Brion JP, Stygelbout V, et al. Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer's brains. Acta Neuropathol 2013;125:861-78. 10.1007/s00401-013-1111-z [DOI] [PubMed] [Google Scholar]
- 68.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci 2016;17:5-21. 10.1038/nrn.2015.1 [DOI] [PubMed] [Google Scholar]
- 69.Morales I, Jimenez JM, Mancilla M, et al. Tau oligomers and fibrils induce activation of microglial cells. J Alzheimers Dis 2013;37:849-56. [DOI] [PubMed] [Google Scholar]
- 70.Kanmert D, Cantlon A, Muratore CR, et al. C-Terminally Truncated Forms of Tau, But Not Full-Length Tau or Its C-Terminal Fragments, Are Released from Neurons Independently of Cell Death. J Neurosci 2015;35:10851-65. 10.1523/JNEUROSCI.0387-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dujardin S, Lecolle K, Caillierez R, et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol Commun 2014;2:14. 10.1186/2051-5960-2-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bolos M, Llorens-Martin M, Jurado-Arjona J, et al. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J Alzheimers Dis 2016;50:77-87. 10.3233/JAD-150704 [DOI] [PubMed] [Google Scholar]
- 73.Holmes BB, Furman JL, Mahan TE, et al. Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci U S A 2014;111:E4376-85. 10.1073/pnas.1411649111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 2015;16:358-72. 10.1038/nrn3880 [DOI] [PubMed] [Google Scholar]
- 75.Dudvarski Stankovic N, Teodorczyk M, Ploen R, et al. Microglia-blood vessel interactions: a double-edged sword in brain pathologies. Acta Neuropathol 2016;131:347-63. 10.1007/s00401-015-1524-y [DOI] [PubMed] [Google Scholar]
- 76.Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007;53:337-51. 10.1016/j.neuron.2007.01.010 [DOI] [PubMed] [Google Scholar]
- 77.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 2012;71:362-81. 10.1097/NEN.0b013e31825018f7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wes PD, Easton A, Corradi J, et al. Tau overexpression impacts a neuroinflammation gene expression network perturbed in Alzheimer's disease. PLoS One 2014;9:e106050. 10.1371/journal.pone.0106050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol 2007;184:69-91. 10.1016/j.jneuroim.2006.11.017 [DOI] [PubMed] [Google Scholar]
- 80.Ferri CP, Prince M, Brayne C, et al. Global prevalence of dementia: a Delphi consensus study. Lancet 2005;366:2112-7. 10.1016/S0140-6736(05)67889-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol 2014;10:217-24. 10.1038/nrneurol.2014.38 [DOI] [PubMed] [Google Scholar]
- 82.Wood LB, Winslow AR, Proctor EA, et al. Identification of neurotoxic cytokines by profiling Alzheimer's disease tissues and neuron culture viability screening. Sci Rep 2015;5:16622. 10.1038/srep16622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Franceschi C, Capri M, Monti D, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev 2007;128:92-105. 10.1016/j.mad.2006.11.016 [DOI] [PubMed] [Google Scholar]
- 84.Hart AD, Wyttenbach A, Perry VH, et al. Age related changes in microglial phenotype vary between CNS regions: grey versus white matter differences. Brain Behav Immun 2012;26:754-65. 10.1016/j.bbi.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer's disease. Curr Pharm Des 2010;16:2766-78. 10.2174/138161210793176572 [DOI] [PubMed] [Google Scholar]
- 86.Wang WY, Tan MS, Yu JT, et al. Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann Transl Med 2015;3:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jiang T, Yu JT, Tan L. Novel disease-modifying therapies for Alzheimer's disease. J Alzheimers Dis 2012;31:475-92. 10.3233/JAD-2012-120640 [DOI] [PubMed] [Google Scholar]
- 88.Weitz TM, Town T. Microglia in Alzheimer's Disease: It's All About Context. Int J Alzheimers Dis 2012;2012:314185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spencer NG, Schilling T, Miralles F, et al. Mechanisms Underlying Interferon-gamma-Induced Priming of Microglial Reactive Oxygen Species Production. PLoS One 2016;11:e0162497. 10.1371/journal.pone.0162497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Andersson U, Tracey KJ. Reflex principles of immunological homeostasis. Annu Rev Immunol 2012;30:313-35. 10.1146/annurev-immunol-020711-075015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hannestad J, Gallezot JD, Schafbauer T, et al. Endotoxin-induced systemic inflammation activates microglia: [(1)(1)C]PBR28 positron emission tomography in nonhuman primates. Neuroimage 2012;63:232-9. 10.1016/j.neuroimage.2012.06.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer's disease. Neuropharmacology 2010;58:561-8. 10.1016/j.neuropharm.2009.11.010 [DOI] [PubMed] [Google Scholar]
- 93.Lim SL, Rodriguez-Ortiz CJ, Kitazawa M. Infection, systemic inflammation, and Alzheimer's disease. Microbes Infect 2015;17:549-56. 10.1016/j.micinf.2015.04.004 [DOI] [PubMed] [Google Scholar]
- 94.Pardon MC, Yanez Lopez M, Yuchun D, et al. Magnetic Resonance Spectroscopy discriminates the response to microglial stimulation of wild type and Alzheimer's disease models. Sci Rep 2016;6:19880. 10.1038/srep19880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang B, Gaiteri C, Bodea LG, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell 2013;153:707-20. 10.1016/j.cell.2013.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013;368:107-16. 10.1056/NEJMoa1211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guerreiro R, Hardy J. TREM2 and neurodegenerative disease. N Engl J Med 2013;369:1569-70. [DOI] [PubMed] [Google Scholar]
- 98.Britschgi M, Rufibach K, Huang SL, et al. Modeling of pathological traits in Alzheimer's disease based on systemic extracellular signaling proteome. Mol Cell Proteomics 2011;10:M111.008862. [DOI] [PMC free article] [PubMed]
- 99.Hu WT, Chen-Plotkin A, Arnold SE, et al. Novel CSF biomarkers for Alzheimer's disease and mild cognitive impairment. Acta Neuropathol 2010;119:669-78. 10.1007/s00401-010-0667-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Knuesel I, Chicha L, Britschgi M, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol 2014;10:643-60. 10.1038/nrneurol.2014.187 [DOI] [PubMed] [Google Scholar]
- 101.Kim J, Castellano JM, Jiang H, et al. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron 2009;64:632-44. 10.1016/j.neuron.2009.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer's disease: an update. Annu Rev Neurosci 2014;37:79-100. 10.1146/annurev-neuro-071013-014300 [DOI] [PubMed] [Google Scholar]
- 103.Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol 2015;14:388-405. 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hickman SE, Kingery ND, Ohsumi TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 2013;16:1896-905. 10.1038/nn.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hsieh CL, Koike M, Spusta SC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem 2009;109:1144-56. 10.1111/j.1471-4159.2009.06042.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tan L, Wang HF, Tan MS, et al. Effect of CLU genetic variants on cerebrospinal fluid and neuroimaging markers in healthy, mild cognitive impairment and Alzheimer's disease cohorts. Sci Rep 2016;6:26027. 10.1038/srep26027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: Current evidence and future directions. Alzheimers Dement 2016;12:719-32. 10.1016/j.jalz.2016.02.010 [DOI] [PubMed] [Google Scholar]
- 108.Derecki NC, Katzmarski N, Kipnis J, et al. Microglia as a critical player in both developmental and late-life CNS pathologies. Acta Neuropathol 2014;128:333-45. 10.1007/s00401-014-1321-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arima Y, Kamimura D, Sabharwal L, et al. Regulation of immune cell infiltration into the CNS by regional neural inputs explained by the gate theory. Mediators Inflamm 2013;2013:898165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maeda T, Kanda T. Blood-brain barrier and blood-nerve barrier in neuroinflammatory diseases. Nihon Rinsho 2013;71:789-94. [PubMed] [Google Scholar]
- 111.Stolp HB, Dziegielewska KM. Review: Role of developmental inflammation and blood-brain barrier dysfunction in neurodevelopmental and neurodegenerative diseases. Neuropathol Appl Neurobiol 2009;35:132-46. 10.1111/j.1365-2990.2008.01005.x [DOI] [PubMed] [Google Scholar]
- 112.Raj DD, Moser J, van der Pol SM, et al. Enhanced microglial pro-inflammatory response to lipopolysaccharide correlates with brain infiltration and blood-brain barrier dysregulation in a mouse model of telomere shortening. Aging Cell 2015;14:1003-13. 10.1111/acel.12370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Steinman L. Inflammatory cytokines at the summits of pathological signal cascades in brain diseases. Sci Signal 2013;6:pe3. 10.1126/scisignal.2003898 [DOI] [PubMed] [Google Scholar]
- 114.Takeda S, Sato N, Ikimura K, et al. Increased blood-brain barrier vulnerability to systemic inflammation in an Alzheimer disease mouse model. Neurobiol Aging 2013;34:2064-70. 10.1016/j.neurobiolaging.2013.02.010 [DOI] [PubMed] [Google Scholar]
- 115.Malik M, Simpson JF, Parikh I, et al. CD33 Alzheimer's risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci 2013;33:13320-5. 10.1523/JNEUROSCI.1224-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, et al. Microglial genes regulating neuroinflammation in the progression of Alzheimer's disease. Curr Opin Neurobiol 2016;36:74-81. 10.1016/j.conb.2015.10.004 [DOI] [PubMed] [Google Scholar]
- 117.Zhang Y, Chen K, Sloan SA, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 2014;34:11929-47. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Moreno B, Jukes JP, Vergara-Irigaray N, et al. Systemic inflammation induces axon injury during brain inflammation. Ann Neurol 2011;70:932-42. 10.1002/ana.22550 [DOI] [PubMed] [Google Scholar]
- 119.Scheltens P, Blennow K, Breteler MM, et al. Alzheimer's disease. Lancet 2016;388:505-17. 10.1016/S0140-6736(15)01124-1 [DOI] [PubMed] [Google Scholar]
- 120.Xu W, Tan L, Wang HF, et al. Meta-analysis of modifiable risk factors for Alzheimer's disease. J Neurol Neurosurg Psychiatry 2015;86:1299-306. [DOI] [PubMed] [Google Scholar]
- 121.Grande G, Vanacore N, Maggiore L, et al. Physical activity reduces the risk of dementia in mild cognitive impairment subjects: a cohort study. J Alzheimers Dis 2014;39:833-9. [DOI] [PubMed] [Google Scholar]
- 122.Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer's disease. Acta Neuropathol 2013;126:461-77. 10.1007/s00401-013-1182-x [DOI] [PubMed] [Google Scholar]
- 123.Vlad SC, Miller DR, Kowall NW, et al. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008;70:1672-7. 10.1212/01.wnl.0000311269.57716.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wong WB, Lin VW, Boudreau D, et al. Statins in the prevention of dementia and Alzheimer's disease: a meta-analysis of observational studies and an assessment of confounding. Pharmacoepidemiol Drug Saf 2013;22:345-58. 10.1002/pds.3381 [DOI] [PubMed] [Google Scholar]
- 125.Marsh SE, Abud EM, Lakatos A, et al. The adaptive immune system restrains Alzheimer's disease pathogenesis by modulating microglial function. Proc Natl Acad Sci U S A 2016;113:E1316-25. 10.1073/pnas.1525466113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vom Berg J, Prokop S, Miller KR, et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat Med 2012;18:1812-9. 10.1038/nm.2965 [DOI] [PubMed] [Google Scholar]
- 127.He J, Liao T, Zhong GX, et al. Alzheimer's Disease-like Early-phase Brain Pathogenesis: Self-curing Amelioration of Neurodegeneration from Pro-inflammatory 'Wounding' to Anti-inflammatory 'Healing'. Curr Alzheimer Res 2017;14:1123-35. 10.2174/1567205014666170417111420 [DOI] [PubMed] [Google Scholar]
- 128.Lopategui Cabezas I, Herrera Batista A, Penton Rol G. The role of glial cells in Alzheimer disease: potential therapeutic implications. Neurologia 2014;29:305-9. 10.1016/j.nrl.2012.10.006 [DOI] [PubMed] [Google Scholar]