

Abstract
We have explored semicrystalline poly(decamethylene terephthalamide) (PA 10T) based thermosets as single-component high-temperature (>200 °C) shape memory polymers (SMPs). The PA 10T thermosets were prepared from reactive thermoplastic precursors. Reactive phenylethynyl (PE) functionalities were either attached at the chain termini or placed as side groups along the polymer main chain. The shape fixation and recovery performance of the thermoset films were investigated using a rheometer in torsion mode. By controlling the Mn of the reactive oligomers, or the PE concentration of the PE side-group functionalized copolyamides, we were able to design dual-shape memory PA 10T thermosets with a broad recovery temperature range of 227–285 °C. The thermosets based on the 1000 g mol–1 reactive PE precursor and the copolyamide with 15 mol % PE side groups show the highest fixation rate (99%) and recovery rate (≥90%). High temperature triple-shape memory behavior can be achieved as well when we use the melt transition (Tm ≥ 200 °C) and the glass transition (Tg = ∼125 °C) as the two switches. The recovery rate of the two recovery steps are highly dependent on the crystallinity of the thermosets and vary within a wide range of 74%–139% and 40–82% for the two steps, respectively. Reversible shape memory events could also be demonstrated when we perform a forward and backward deformation in a triple shape memory cycle. We also studied the angular recovery velocity as a function of temperature, which provides a thermokinematic picture of the shape recovery process and helps to program for desired shape memory behavior.
Keywords: semiaromatic polyamides, semicrystalline thermosets, high-temperature shape memory, dual-shape memory, triple-shape memory, recovery velocity
1. Introduction
Interest in shape memory polymers (SMPs) has grown rapidly since the 1980s.1−5 Typical applications are heat-shrink tubing, temperature sensors and actuators, biomedical and surgical materials, and aerospace devices.6,7 Thermoresponsive SMPs are the most investigated systems that polymers are able to adopt a temporary shape upon deformation and to revert back to the permanent shape upon exposure to heat.7,8 They are generally composed of a polymer network to maintain the permanent shape and a reversible switch responsible for the shape fixation and recovery.8−11 The glass transition temperature (Tg) and melting temperature (Tm) are the two most important thermal transitions for thermoresponsive SMPs. Polyurethanes, polyesters, and (methyl)acrylate-based polymer networks have been investigated as SMPs based on the glass transition. Cross-linked semicrystalline networks or (multi)block copolymer systems have been developed to design Tm-based SMPs, such as polyolefins, polyethers, and polyesters.6 However, most SMPs exhibit a switching temperature lower than 100 °C, which may not meet the requirements for high temperature aerospace, automotive, or electronic applications.
Recently, several examples of high-temperature (>200 °C) SMPs have been reported.12−21 Vaia and co-workers synthesized an amorphous fluorinated polyimide with a shape recovery temperature of 220 °C.20 This was the first example of a single-component high-temperature SMP. However, this fluorinated polyimide can only be obtained as thin films, which excludes the possibility to produce complex shapes using melt processing methods. Weiss et al. have introduced ionic moieties into poly(ether ether ketone) (PEEK) leading to a Tg-based SMP with a switching temperature close to 200 °C, where the exact switching temperature depends on the metal counterions used.14 In a later publication, this system was further developed into a Tm-based SMP by incorporation of sodium oleate, which displays a higher switching temperature of 230–240 °C due to the melting of sodium oleate.15
In contrast to the dual SMPs mentioned above, triple SMPs featuring two independent temporary shapes were first reported by Lendlein’s group.22 They require two programming steps and show two recovery steps. Triple-shape memory effects can be realized in a polymer material possessing two distinct thermal transitions15,22−24 or one single broad transition such as a broad glass transition.16,25 High temperature triple SMPs based on a single-component thermotropic liquid crystalline poly(esterimide) thermosets have recently been reported by Guan et al. The polymer shows two glass transition temperatures at ∼120 and ∼200 °C, both of which can be used as switches for triple SMPs.21
In previous publications we reported on the synthesis and (thermo)mechanical properties of semiaromatic polyamide thermosets based on poly(decamethylene terephthalamide) (PA 10T).26,27 We showed that this class of polymers can be processed into semicrystalline thermoset films where the degree of crystallinity can be controlled. The melting/crystallization of the crystalline phase and the covalent network can be used as the high-temperature switch and permanent scaffold, respectively, for a dual shape memory effect (SME). More sophisticated triple and one-way reversible SME can be designed with the glass transition as the second switch. To the best of our knowledge, this is the first demonstration of a high-temperature SMP based on a single-component semicrystalline polyamide thermoset.
2. Experimental Section
2.1. Materials
The syntheses of phenylethynyl (PE) end-capped PA 10T oligomers (Scheme 1A) and PE side-group-functionalized copolyamides (Scheme 1B) have been described in detail elsewhere.26,27 In order to obtain a covalently cross-linked polyamide network, the reactive precursors were thermally cured. A standard melt compression technique was used to prepare the thermoset films according to the following procedure: The precursor polymers were ground into a fine powder using a mortar and pestle. The powder was placed between two metal plates lined with Kapton film, and this stack was consolidated in a Joos hot-press using a predetermined temperature program and a 5 kN force. The temperature program was set to heat to 350 °C at 5 °C min–1, hold for 15 min, and cooled to 50 °C at 20 °C min–1. The obtained cured films were used for further characterization.
Scheme 1. Structures of Precursors via (A) Reactive End-Group Approach and (B) Reactive Side-Group Approach.

The film samples prepared from PA 10T reactive oligomers (Mn of 1000 and 3000 g mol–1) are denoted as PE-1K and PE-3K. TPE-5, TPE-10, and TPE-15 represent the film samples prepared from reactive TPE-copolyamides with 5, 10, and 15 mol % of reactive TPE comonomer, respectively. A PA 10T thermoplastic sample (Mn = 7500 g mol–1) was used as the reference in this paper and denoted as Ref. The 1H NMR spectra of the synthesized precursors (Supporting Information, Figure S1) confirms that the concentration of reactive functionalities, either as end-caps or as side groups, is consistent with the feed ratio of the monomers.
2.2. Characterization
1H NMR spectra were recorded on a 400 MHz Bruker WM-400 at 25 °C using trifluoroacetic acid-d as solvent. DSC was conducted on a PerkinElmer Sapphire DSC under a nitrogen atmosphere at a heating/cooling rate of 20 °C min–1. DMTA was performed on a PerkinElmer Diamond DMTA with film samples (0.2–0.3 mm thick) at a heating rate of 2 °C min–1 under a nitrogen atmosphere. Data were collected at a frequency of 1 Hz.
The SME was characterized in a cyclic torsion mode as illustrated in Figure 1. Compared to the traditional extension or bending tests,28 torsion tests involve nonhomogeneous strains and stresses in the cross section of a rectangular bar and enable the SMPs to reach large deformations with moderate strains, which are believed to be more representative of practical applications of SMPs.29−32
Figure 1.
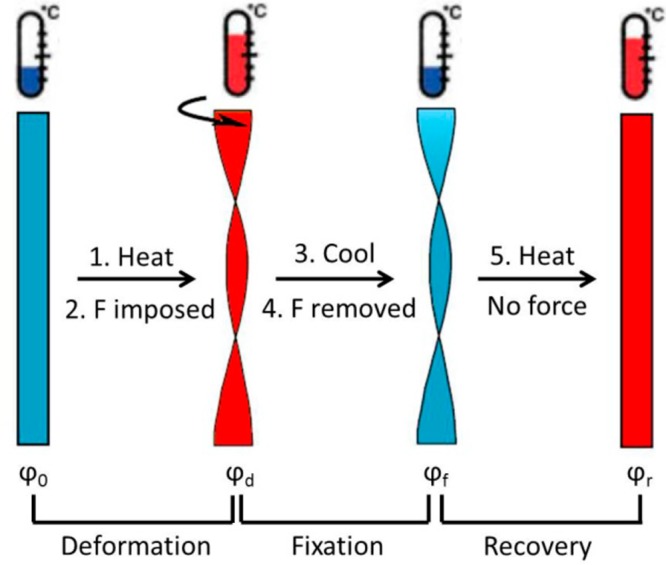
Illustration of a shape deformation, fixation, and recovery cycle of the dual SME in torsion mode.
The shape programming and recovery cycles were performed using a Thermofisher Haake MARS III rheometer equipped with a solid clamp geometry under a N2 atmosphere. Rectangular thin films with a width of ∼2.5 mm and thickness of ∼0.25 mm were loaded into the setup with a length of 15 mm between the clamps. The samples were deformed in a torsion mode at a constant strain rate of 0.1% s–1 equivalent to a rotation speed of 3.4° s–1.
The procedure of one cycle for a dual SME test includes the following steps: (1) heat the sample to the programming temperature (Tprog); (2) rotate the sample to the predetermined angle (ϕd); (3) keep the angle constant, cool to the fixation temperature (Tf) at 10 °C min–1 and stabilize for 10 min; (4) remove the stress; (5) heat the sample to Tprog at 10 °C min–1 in a stress-free condition followed by an isothermal hold at Tprog for 10 min to stabilize. This cycle was repeated multiple times to characterize the reproducibility of the shape memory performance. The rotation angle of the sample was monitored and recorded during the whole test. For triple SME measurements, the sample was consecutively deformed in two steps in one programming cycle. Two different Tprogs were used, one being above Tm and the other between Tg and Tm.
3. Results and Discussion
3.1. Melting and Thermomechanical Properties
In general, either the Tg or the Tm transition of a polymer can be applied to trigger a dual-shape memory event; however, to achieve high-temperature triple-shape memory behavior in a single-component polymer is challenging, as two high-temperature temporary networks with distinct rubbery plateaus are required.15,22−24 One solution to realize this type of structure is to introduce moderate cross-links into a high-Tg semicrystalline polymer. In our previous work, we have explored a semiaromatic polyamide, PA 10T (Tg = ∼125 °C, Tm = ∼315 °C), as the base polymer to prepare semicrystalline polyamide thermosets. Phenylethynyl (PE) shows high cure-temperature of 330–370 °C and thus is able to provide a proper processing window for the PA 10T-based precursors. Therefore, we have synthesized a reactive PE-based end-cap and a reactive PE-based comonomer, which were copolymerized with terephthalic acid and 1,10-diaminodecane to yield curable PE end-capped oligomers and PE side-group functionalized copolymers. The polyamide thermosets were obtained after a subsequent thermal cure at a temperature of 350 °C for 15 min. PE-1K and PE-3K refer to the cured end-capped oligomers with Mn = 1000 and 3000 g mol–1, respectively. TPE-5, TPE-10, and TPE-15 represent the cured side-group functionalized copolyamides with 5, 10, and 15 mol % PE side groups, respectively.
The melting curves and properties of a thermoplastic PA 10T reference polymer (Ref) and the resultant polyamide thermosets are shown in Figure 2 and Table 1. The thermoset samples after cure are semicrystalline showing a Tm of 227–288 °C and ΔHm of 8–33 J g–1. These values are much lower than the Tm and the ΔHm of the reference polymer (Ref) (318 °C and 82 J g–1), which means the crystallizability of the polymer chains in the thermosets is strongly suppressed.
Figure 2.
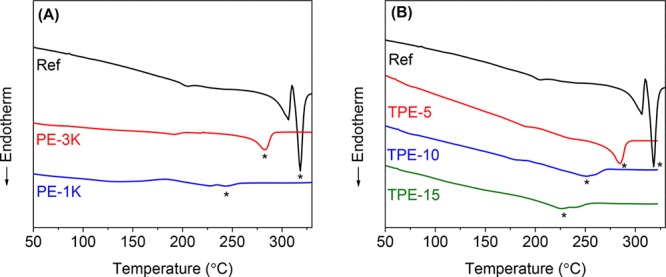
DSC heating scans (first heat) of (A) reference polymer (Ref), PE-3K, and PE-1K and (B) reference polymer (Ref), TPE-5, TPE-10, and TPE-15. The asterisk refers to melting peaks (N2 atmosphere and heating rate of 20 °C min–1).
Table 1. Thermal Properties of the Thermoplastic Reference Polymer (Ref) and the Cured Thermoset Samples.
| sample | Tma (°C) | ΔHm (J g–1) | Tgb (°C) | cross-linking densityc (kmol m–3) |
|---|---|---|---|---|
| Ref | 318 | 82 | 127 | |
| PE-3K | 283 | 33 | 127 | 0.68 |
| PE-1K | 244 | 8 | 129 | 0.72 |
| TPE-5 | 285 | 33 | 123 | 0.94 |
| TPE-10 | 250 | 27 | 123 | 1.79 |
| TPE-15 | 227 | 20 | 125 | 2.34 |
Tm refers to the max of the melting peak as observed in DSC experiments.
Tg refers to the max of E″ as observed in DMTA experiments.
Cross-linking density (υ) was
calculated using 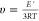 , where E′
is the storage modulus of cured films at 350 °C (T = 623 K) and R the universal gas constant (8.314
J K–1 mol–1).
, where E′
is the storage modulus of cured films at 350 °C (T = 623 K) and R the universal gas constant (8.314
J K–1 mol–1).
Figure 3 shows the thermomechanical behavior of the reference polymer (Ref) and the thermosets. The Ref film exhibits a Tg at 127 °C, but this film fails at 297 °C because it has reached the melting point (Tm). In contrast to the reference polymer (Ref), the thermoset samples show two plateau regions (Tg – Tm and >Tm) in the DMTA profiles. The second plateau of the thermosets is stable up to ∼400 °C, which confirms the presence of a network structure. The Tgs of both thermoset samples remain virtually unchanged (123–129 °C) when compared to that of the reference polymer (Ref).
Figure 3.

DMTA of (A) reference polymer (Ref), PE-1K, and PE-3K films and (B) reference polymer (Ref), TPE-5, TPE-10, and TPE-15 films. Heating rate 2 °C min–1 under N2 atmosphere and a frequency of 1 Hz. The insets show E″ at the glass transition temperature.
The molecular structure of the resultant semicrystalline thermosets is depicted in Figure 4. PE groups combine at the cure temperature resulting in cross-links with multiple after-cure chemical structures depending on cure temperature, time, and PE concentration. We attempted to investigate the cure chemistry using Raman and FTIR spectroscopy.33,34 However, Raman spectroscopy failed due to a strong fluorescence background, and FTIR cannot detect the acetylene bond of PE because of IR insensitivity. Hence, direct evidence is not available to confirm the chemical change of the PE functionalities. The DMTA results of the cured samples show a rubbery plateau above Tm, and this plateau is stable up to ∼400 °C, which confirms that cross-linking has taken place during cure. The two-plateau thermal behavior enables triple-shape memory properties by taking both Tg and Tm transitions as the switches. The Tm transition can be solely used as a switch to design a high-temperature (>200 °C) dual-shape memory polymer.
Figure 4.
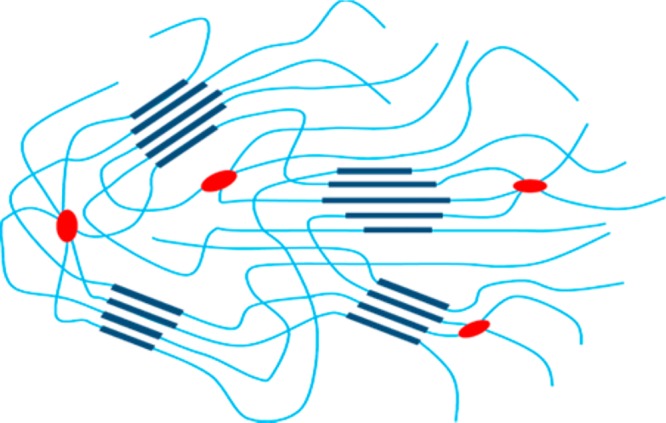
Molecular representation of the semicrystalline PA 10T thermosets. Crystalline polymer (dark blue) is embedded in an amorphous matrix (light blue), and the red dots represent covalent cross-link points.
3.2. Dual-Shape Memory Behavior
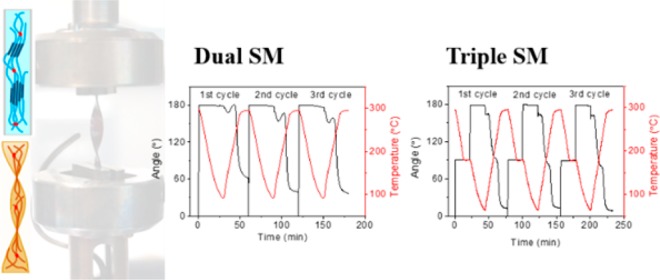
Because of the semicrystalline nature of the synthesized polyamide thermosets, their melting and crystallization processes can be used as the thermoresponsive switch for dual-shape memory behavior. Three consecutive deformation, fixation, and recovery cycles in a torsion mode were conducted for each sample to test the shape memory performance over multiple cycles.
Shape fixation rate (Rf) and shape recovery rate (Rr) are the most important parameters to characterize the shape memory performance.35Rf describes how accurately the temporary shape can be fixed, and Rr quantifies the ability of the polymer to memorize its permanent shape. When performing the measurements in torsion mode, Rf and Rr can be calculated using eqs 1 and 2.
| 1 |
| 2 |
where φd, φf, and φr denote the rotational angle after deformation, at the fixed temporary shape at Tf, and after recovery, respectively. The instantaneous recovery velocity Vr can be calculated as the time derivative of the angle as shown in eq 3.
| 3 |
Plotting Vr against temperature reveals the temperature range corresponding to the shape recovery process.21 This provides a clear thermokinematic view of the shape recovery and helps to program for desired SME.
The thermoplastic reference film (ref) cannot be deformed above the melting temperature because of the absence of a rubbery plateau, thus the glass transition was used as the switch for the SME. As shown in Figure 5A, the sample was deformed at 220 °C, which lies between Tg (127 °C) and Tm (318 °C), and the shape was fixed at 100 °C where the sample is in a glassy state. Therefore, the crystalline domains act as the scaffold, and the deformation of the polymer takes place in the amorphous state.
Figure 5.

Three consecutive dual-shape memory cycles for (A) reference polymer (Ref), (B) PE-3K, and (C) PE-1K. Shape recovery velocity as a function of temperature in the second cycle for (D) reference polymer (Ref), (E) PE-3K, and (F) PE-1K. Cooling/heating rate 10 °C min–1 and N2 atmosphere.
In contrast to the reference polymer (Ref), PE-3K and PE-1K thermoset films are stable up to ∼400 °C in DMTA experiments and exhibit two plateau regions (Tg – Tm and >Tm), as shown in Figure 3. This allows for deforming the film samples in the second plateau region (>Tm). Figures 5B and 5C show that PE-3K and PE-1K were deformed at 315 and 275 °C, respectively, which is about 30 °C higher than their Tms (283 and 244 °C). At this temperature the covalent network acts as the scaffold, and the temporary shape is fixed by crystallization and vitrification at the fixation step. At the recovery step, the angle shows a slight decrease when the sample passes through the glass transition, which is associated with the release of the stress trapped in the mobile amorphous region. A subsequent slow increase in the angle is observed, which can be explained by thermal expansion.36 When the sample starts melting, the angle recovers rapidly due to the fast release of the trapped stress.
The shape-memory performance of the first few cycles is usually not representative, as can be seen in Figure 5B,C. This is generally attributed to residual strain from the processing history of the sample.13,35Table 2 shows the fixation and recovery results of the first three cycles of the reference thermoplastic polymer (Ref), the PE-3K, and PE-1K thermoset films. The PE-3K film shows a very low Rr of 36% in the first cycle compared to the following cycles (Rr = 72% and 76%), whereas the Rr of the PE-1K film shows a medium change over cycles (Rr = 81%, 90%, and 88%). Such significant difference can originate from the different cross-linking densities of these two samples. PE-3K has a lower cross-linking density and therefore longer polymer chains between cross-links, which is more likely to store residual strain. The recovery of both samples cannot reach 100% because the irreversible chain-segment orientation and the relaxation effect in the polymer network partly dissipate the stored entropy.20
Table 2. Dual-Shape Fixation and Recovery Results of the Reference Thermoplastic Polymer (Ref), PE-3K, and PE-1K Thermoset Films.
| cycle 1 |
cycle 2 |
cycle 3 |
||||||
|---|---|---|---|---|---|---|---|---|
| sample | Tproga (°C) | Trb (°C) | Rf (%) | Rr (%) | Rf (%) | Rr (%) | Rf (%) | Rr (%) |
| Ref | 220 | 210 | 89 | 86 | 89 | 82 | 89 | 84 |
| PE-3K | 315 | 285 | 93 | 36 | 94 | 72 | 94 | 76 |
| PE-1K | 275 | 144, 240 | 99 | 81 | 98 | 90 | 95 | 88 |
Tprog refers to the programming temperature.
Tr refers to the temperature at the maximum recovery velocity.
The shape recovery velocity of the second cycle is plotted as a function of temperature as shown in Figure 5D–F. The reference polymer (Ref) shows a low velocity (<20° min–1) through the recovery process because the shape recovery is triggered by the activation of the amorphous chain segments, which are strongly restricted by the crystalline domains. A low recovery velocity is generally observed in Tg-based shape-memory polymers.8 In contrast, PE-3K shows a quick recovery in the temperature range of 260–310 °C and reaches a maximum Vr of ∼60° min–1 at 285 °C, which is close to its Tm (283 °C). The shape recovery is triggered by melting of the crystalline domains, which is a relatively fast process.
It is worthy to note that the PE-1K film was deformed at 275 °C in one step; however, this polymer shows two distinct recovery steps with a maximum recovery velocity of 20–30° min–1 at around 144 and 240 °C, respectively. The first recovery takes place when the sample passes through the glass transition and reaches an Rr of ∼42% at 200 °C. The second recovery is triggered by the melting of the crystalline domains. These two recovery steps indicate that the crystalline domains in PE-1K are not able to form a penetrating scaffold throughout the sample and thus cannot completely lock the amorphous chains. This allows the sample to partially recover at the Tg – Tm range. These results demonstrate that the degree of crystallinity, which is reflected in the ΔHm values in Table 1, strongly affects the shape memory behavior of our semicrystalline PAs.
The dual-shape memory behavior of the TPE-based thermosets were investigated using the same method. Table 3 shows the shape fixation and recovery results for these samples. All samples reveal good shape fixation with Rf values of 93–99%. The TPE-15 samples with the highest cross-linking density shown in Table 1 exhibit excellent recovery efficiency (Rr > 90%), whereas the other samples show Rr values of 62–66% in the first cycle and 78–82% in the second and third cycles.
Table 3. Dual-Shape Fixation and Recovery Results of the TPE Thermoset Films.
| cycle 1 |
cycle 2 |
cycle 3 |
||||||
|---|---|---|---|---|---|---|---|---|
| sample | Tproga (°C) | Trb (°C) | Rf (%) | Rr (%) | Rf (%) | Rr (%) | Rf (%) | Rr (%) |
| TPE-5 | 315 | 277 | 93 | 62 | 93 | 82 | 94 | 82 |
| TPE-10 | 295 | 242 | 98 | 66 | 96 | 78 | 96 | 79 |
| TPE-15 | 275 | 140, 227 | 99 | 93 | 99 | 93 | 97 | 96 |
Tprog refers to the programming temperature.
Tr refers to the temperature at the maximum recovery velocity.
Figure 6 shows the dual-shape memory cycles and the recovery velocity of the TPE thermoset films. Similar to the PE-3K sample, TPE-5 and TPE-10 exhibit one recovery step in the temperature ranges of 260–300 and 230–270 °C, respectively. Interestingly, the TPE-15 film also displays two recovery steps, which is similar to that of the PE-1K film. The two recovery steps originate from the low degree of crystallinity in TPE-15 and PE-1K, where the amorphous chain segments cannot be completely fixed when the temperature is above Tg. TPE-5, TPE-10, and PE-3K, on the other hand, show higher degrees of crystallinity, and this prevents the Tg-induced shape recovery.
Figure 6.

Three consecutive dual-shape memory cycles for the TPE series (A) TPE-5, (B) TPE-10, and (C) TPE-15. Shape recovery velocity as a function of temperature in the second cycle for the TPE series (D) TPE-5, (E) TPE-10, and (F) TPE-15. Cooling and heating rate 10 °C min–1 and N2 atmosphere.
The difference between these samples clearly indicates that the shape memory behavior strongly depends on the degree of crystallinity of the semicrystalline thermosets. By changing the concentration of the PE side groups in the reactive copolyamides, the cross-linking density of the thermosets can be controlled, consequently leading to adjustable crystallinity and shape memory behavior.
3.3. Triple-Shape Memory Behavior
Based on our study above, two reversible processes, the glass transition and the melting process, can both act as the switches to trigger shape recovery. Unlike the traditional high-temperature triple-SMP composed of multiple components, we were able to design a single-component triple-SMP using both switches to achieve two distinct recovery processes.
Figure 7A shows the three consecutive triple-shape memory cycles for PE-3K. The film sample was first heated up to 315 °C, which is above the Tm (283 °C) and rotated by 90° from the original shape A (φA) to a temporary shape B (φB). The sample was cooled to 200 °C, a temperature between Tm and Tg, to fix the shape B. A second rotation of 90° was then applied to reach the temporary shape C (φC). The sample was subsequently cooled to 60 °C, which is below the Tg of 127 °C, to fix the shape C. The external stress was then removed, leading to the final fixed temporary shape (φf). A shape fixation rate (Rf) of ∼90% was calculated using eq 4.
| 4 |
Figure 7.

Three consecutive triple-shape memory cycles for the PE series (A) PE-3K and (B) PE-1K. Shape recovery velocity as a function of temperature in the second cycle for the PE series (C) PE-3K and (D) PE-1K. Cooling and heating rate 10 °C min–1 and N2 atmosphere.
In the subsequent recovery step, the sample was heated up to 315 °C in a stress-free state to monitor the recovery of the rotational angle. The shape recovery was accomplished in two distinct steps, as shown in Figure 7A. This means the thermoset can memorize two temporary shapes in one single shape memory cycle. The shape recovery rate Rr(C→B) and Rr(B→A) were calculated based on eqs 5 and 6, respectively.21
| 5 |
| 6 |
where φA, φB, and φC denote the rotational angle of shape A (φA = 0°), shape B (φB = 90°), and shape C (φC = 180°); φB/rec and φA/rec are the rotational angles of the first and second recovered shapes, respectively. Figure 7B shows the triple-shape memory cycles of PE-1K. This polymer exhibits a lower Tm (244 °C) compared to PE-3K (283 °C); therefore, lower programming temperatures (275 and 150 °C) were used.
PE-3K shows moderate Rr(C→B) values of 67–80% in the first recovery step and Rr(B→A) values of 60–73% in the second recovery step, as listed in Table 4. Compared to PE-3K, PE-1K shows much higher Rr(C→B) values of 128–138% and lower Rr(B→A) values of 40–49%. The first recovery is triggered by the glass transition and is due to the release of stress in the amorphous phase. The major recovery takes place in the first step, indicating that the crystalline phase cannot completely lock the stress in the amorphous phase. The stress, which is supposed to release in the second recovery step, is actually partially released in the first recovery step. This, together with the stress induced by the second deformation, drives the sample to reach a high recovery rate (>100%) in the first step. This result is consistent with the result we obtained from the dual-shape memory experiments and originates from the low degree of crystallinity of the PE-1K thermoset.
Table 4. Triple-Shape Fixation and Recovery Results of the PE- and TPE-Series Thermoset Films.
| cycle 1 |
cycle 2 |
cycle 3 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sample | Tprog1a (°C) | Tprog2a (°C) | Tr1b (°C) | Tr2b (°C) | Rf (%) | Rr(C–B) (%) | Rr(B–A) (%) | Rf (%) | Rr(C–B) (%) | Rr(B–A) (%) | Rf (%) | Rr(C–B) (%) | Rr(B–A) (%) |
| PE-3K | 315 | 200 | 137 | 289 | 91 | 67 | 60 | 93 | 80 | 66 | 89 | 79 | 73 |
| PE-1K | 275 | 150 | 144 | 240 | 97 | 128 | 49 | 97 | 135 | 44 | 93 | 139 | 40 |
| TPE-5 | 315 | 200 | 132 | 285 | 88 | 64 | 69 | 86 | 74 | 79 | 86 | 79 | 79 |
| TPE-10 | 295 | 175 | 138 | 250 | 92 | 79 | 91 | 92 | 89 | 80 | 91 | 91 | 82 |
| TPE-15 | 275 | 150 | 133 | 236 | 93 | 97 | 84 | 92 | 123 | 55 | 93 | 136 | 45 |
Tprog1 and Tprog2 refer to the first and second programming temperature, respectively.
Tr1 and Tr2 refer to the temperature at the first and second maximum recovery velocity, respectively.
Figures 7C and 7D show the shape recovery velocity of PE-3K and PE-1K samples as a function of temperature. The first recovery step of PE-3K shows a lower Vr (13° min–1) than the second step (28° min–1) because of the limited mobility of the amorphous chain segments restricted by the crystalline domains. In contrast, the PE-1K sample shows much higher Vr (54° min–1) in the first step, which is due to its low degree of crystallinity.
The triple-shape memory cycles and recovery velocity of the TPE thermosets are shown in Figure 8. The TPE-5 and TPE-10 films show moderate Rr(C→B) and Rr(B→A) (64–91%) and higher Vr values in the second recovery step, which is similar to the behavior of PE-3K, whereas the TPE-15 sample, similar to PE-1K, exhibits high Rr(C→B) (97–136%) and Vr values (38° min–1) in the first recovery step, because this polymer has the lowest degree of crystallinity.
Figure 8.

Three consecutive triple-shape memory cycles for (A) TPE-5, (B) TPE-10, and (C) TPE-15. Angular velocity of shape recovery as a function of temperature in the second cycle for (D) TPE-5, (E) TPE-10, and (F) TPE-15. Cooling and heating rate 10 °C min–1 and N2 atmosphere.
The temperatures at the first maximum recovery velocity (Tr1) of all samples are within a narrow range of 132–144 °C because the Tg of all samples is around 125 °C. However, the temperatures of the second maximum recovery velocity (Tr2), which depend on the Tm of the samples, display a broad variation between 236 and 289 °C. Therefore, the triple-shape memory behavior of the thermosets is highly tunable over a wide temperature regime.
Besides deforming the sample in one direction only, more interestingly, we can involve both forward and backward deformations in a triple-shape memory cycle; therefore, the corresponding temporary shapes would recover in a reverse direction. To demonstrate this, the TPE-10 thermoset sample, which shows a high Rf (>90%), Rr(C–B) (∼90%), and Rr(B–A) (∼80%), was selected as a representative example.
The results of test I are shown in Figure 9A,B. The sample was programmed forward and backward by 180° in two steps and reached a final temporary shape, which is identical to the original shape. The subsequent heating resulted in an increase in angle in the first recovery step achieving a maximum recovery velocity at 142 °C and an Rr(C–B) of 79–91% at 213 °C. By increasing the temperature further, the sample recovered in the reverse direction reaching a maximum recovery velocity at 243 °C and an Rr(B–A) of 81–87% (Table 5). These results demonstrate that our semicrystalline PA thermosets show one-way reversible shape memory behavior. The reversible shape changes in this experiment occurred without application of any external force. The shape changes were driven by internal stress of the oppositely strained networks formed in the first and second programming steps.
Figure 9.

Three consecutive triple-shape memory cycles for TPE-10 and shape recovery velocity as a function of temperature in the second cycle. (A) and (B) deformation of 180° and −180° in two steps, respectively; (C) and (D) deformation of 180° and −360° in two steps, respectively.
Table 5. Triple-Shape Fixation and Recovery Results of TPE-10 Deformed in Two Opposite Directions.
| test | cycle no. | Δφd1a (deg) | Δφd2a (deg) | Rf (%) | Rr(C–B) (%) | Rr(B–A) (%) | Tmaxb (°C) |
|---|---|---|---|---|---|---|---|
| I | 1 | 180 | –180 | 97 | 91 | 81 | 213 |
| 2 | 180 | –180 | 98 | 85 | 81 | 211 | |
| 3 | 180 | –180 | 99 | 79 | 87 | 209 | |
| II | 1 | 180 | –360 | 90 | 86 | 54 | 218 |
| 2 | 180 | –360 | 89 | 83 | 59 | 213 | |
| 3 | 180 | –360 | 91 | 82 | 61 | 212 |
Δφd1 and Δφd2 refer to the deformation angles in the first and second deformation steps.
Tmax refers to the temperature at the maximum angle.
We can adjust the second programming step from Δφd2 = −180° to Δφd2 = −360° in test II, which results in two opposite temporary shapes, as shown in Figure 9C,D. The sample shows a Rf of 89–91%, Rr(C–B) of 82–86%, and Rr(B–A) of 54–61% (Table 5). The maximum recovery velocity occurs at 142 and 244 °C in the first and second recovery steps, respectively, which is consistent with the results of Test I. Our results clearly demonstrate that high-temperature SME with tunable recovery directions and amplitudes can be designed based on our single-component semicrystalline polyamide thermosets.
4. Conclusions
We have investigated two novel series semicrystalline PA thermosets and demonstrated that they can be as single-component high-temperature (>200 °C) shape memory polymers (SMPs). Two molecular design approaches based on reactive phenylethynyl (PE) functionalities have been explored: cross-linked semicrystalline PA films were prepared by curing reactive thermoplastic PA 10T oligomers, and films were prepared by curing reactive thermoplastic side-group functionalized copolyamides. Compared to the thermoplastic PA 10T reference polymer, the PE-3K, TPE-5, and TPE-10 thermoset films show high-temperature dual-shape memory behavior (>200 °C) when Tm is used as the switching temperature. The densely cross-linked PE-1K and TPE-15 films show the highest fixation rate (99%) and recovery rate (≥90%) and two distinct recovery steps because the degree of crystallinity is too low and cannot provide a scaffold that locks the shape at temperatures between Tg and Tm. Triple-shape memory behavior can be demonstrated when the Tg (∼125 °C) is used as the second switching temperature. The recovery rate of the two recovery steps is highly dependent on the degree of crystallinity of the thermosets and vary within a broad range of 74%–139% and 40–82% for the first and second step, respectively. One-way reversible shape memory events can also be designed when we perform forward and backward deformation in a triple shape memory cycle. We also studied the recovery velocity as a function of temperature to elaborate the thermokinematics of the shape recovery process. The use of phenylethynyl reactive functionalities allows us to control the degree of crystallinity in the final polyamide thermosets and in turn tune their shape memory behavior in terms of recovery temperature, velocity, and efficiency. We believe that the design rules presented herein will help in designing new shape memory polymers based on well-known semicrystalline polyamide chemistries.
Acknowledgments
This research forms part of the research program of the Dutch Polymer Institute (DPI), Project #743. The authors thank the DPI for financial support. The authors thank Prof. S. Sheiko, Dr. J. Bijleveld, Dr. R. Bose, and Dr. R. Rulkens for fruitful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.8b03658.
Figure S1 (PDF)
Author Present Address
T.J.D.: Department of Applied Physical Sciences, University of North Carolina at Chapel Hill, 1113 Murray Hall, 121 South Road, Chapel Hill, NC 27599-3050.
The authors declare no competing financial interest.
Supplementary Material
References
- Xie T. Recent advances in polymer shape memory. Polymer 2011, 52 (22), 4985–5000. 10.1016/j.polymer.2011.08.003. [DOI] [Google Scholar]
- Hu J.; Zhu Y.; Huang H.; Lu J. Recent advances in shape–memory polymers: Structure, mechanism, functionality, modeling and applications. Prog. Polym. Sci. 2012, 37 (12), 1720–1763. 10.1016/j.progpolymsci.2012.06.001. [DOI] [Google Scholar]
- Rousseau I. A. Challenges of shape memory polymers: A review of the progress toward overcoming SMP’s limitations. Polym. Eng. Sci. 2008, 48 (11), 2075–2089. 10.1002/pen.21213. [DOI] [Google Scholar]
- Behl M.; Razzaq M. Y.; Lendlein A. Multifunctional Shape-Memory Polymers. Adv. Mater. 2010, 22 (31), 3388–3410. 10.1002/adma.200904447. [DOI] [PubMed] [Google Scholar]
- Pilate F.; Toncheva A.; Dubois P.; Raquez J.-M. Shape-memory polymers for multiple applications in the materials world. Eur. Polym. J. 2016, 80, 268–294. 10.1016/j.eurpolymj.2016.05.004. [DOI] [Google Scholar]
- Hager M. D.; Bode S.; Weber C.; Schubert U. S. Shape memory polymers: Past, present and future developments. Prog. Polym. Sci. 2015, 49-50, 3–33. 10.1016/j.progpolymsci.2015.04.002. [DOI] [Google Scholar]
- Leng J.; Lan X.; Liu Y.; Du S. Shape-memory polymers and their composites: stimulus methods and applications. Prog. Mater. Sci. 2011, 56 (7), 1077–1135. 10.1016/j.pmatsci.2011.03.001. [DOI] [Google Scholar]
- Zhao Q.; Qi H. J.; Xie T. Recent progress in shape memory polymer: New behavior, enabling materials, and mechanistic understanding. Prog. Polym. Sci. 2015, 49-50, 79–120. 10.1016/j.progpolymsci.2015.04.001. [DOI] [Google Scholar]
- Wu X.; Huang W. M.; Zhao Y.; Ding Z.; Tang C.; Zhang J. Mechanisms of the shape memory effect in polymeric materials. Polymers 2013, 5 (4), 1169–1202. 10.3390/polym5041169. [DOI] [Google Scholar]
- Meng H.; Li G. A review of stimuli-responsive shape memory polymer composites. Polymer 2013, 54 (9), 2199–2221. 10.1016/j.polymer.2013.02.023. [DOI] [Google Scholar]
- Lewis C. L.; Dell E. M. A review of shape memory polymers bearing reversible binding groups. J. Polym. Sci., Part B: Polym. Phys. 2016, 54 (14), 1340–1364. 10.1002/polb.23994. [DOI] [Google Scholar]
- Xiao X.; Kong D.; Qiu X.; Zhang W.; Liu Y.; Zhang S.; Zhang F.; Hu Y.; Leng J. Shape memory polymers with high and low temperature resistant properties. Sci. Rep. 2015, 5, 14137. 10.1038/srep14137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X.; Kong D.; Qiu X.; Zhang W.; Zhang F.; Liu L.; Liu Y.; Zhang S.; Hu Y.; Leng J. Shape-memory polymers with adjustable high glass transition temperatures. Macromolecules 2015, 48 (11), 3582–3589. 10.1021/acs.macromol.5b00654. [DOI] [Google Scholar]
- Shi Y.; Weiss R. Sulfonated Poly (ether ether ketone) Ionomers and Their High Temperature Shape Memory Behavior. Macromolecules 2014, 47 (5), 1732–1740. 10.1021/ma500119k. [DOI] [Google Scholar]
- Shi Y.; Yoonessi M.; Weiss R. High temperature shape memory polymers. Macromolecules 2013, 46 (10), 4160–4167. 10.1021/ma302670p. [DOI] [Google Scholar]
- Yang Z.; Chen Y.; Wang Q.; Wang T. High performance multiple-shape memory behaviors of Poly (benzoxazole-co-imide) s. Polymer 2016, 88, 19–28. 10.1016/j.polymer.2016.02.001. [DOI] [Google Scholar]
- Xiao X.; Qiu X.; Kong D.; Zhang W.; Liu Y.; Leng J. Optically transparent high temperature shape memory polymers. Soft Matter 2016, 12 (11), 2894–2900. 10.1039/C5SM02703A. [DOI] [PubMed] [Google Scholar]
- Bai Y.; Mao L.; Liu Y. High temperature shape memory polyimide ionomer. J. Appl. Polym. Sci. 2016, 133 (30), 43630–43637. 10.1002/app.43630. [DOI] [Google Scholar]
- Wang Q.; Bai Y.; Chen Y.; Ju J.; Zheng F.; Wang T. High performance shape memory polyimides based on π–π interactions. J. Mater. Chem. A 2015, 3 (1), 352–359. 10.1039/C4TA05058D. [DOI] [Google Scholar]
- Koerner H.; Strong R. J.; Smith M. L.; Wang D. H.; Tan L.-S.; Lee K. M.; White T. J.; Vaia R. A. Polymer design for high temperature shape memory: Low crosslink density polyimides. Polymer 2013, 54 (1), 391–402. 10.1016/j.polymer.2012.11.007. [DOI] [Google Scholar]
- Guan Q.; Picken S. J.; Sheiko S. S.; Dingemans T. J. High-Temperature Shape Memory Behavior of Novel All-Aromatic (AB)n-Multiblock Copoly(ester imide)s. Macromolecules 2017, 50 (10), 3903–3910. 10.1021/acs.macromol.7b00569. [DOI] [Google Scholar]
- Bellin I.; Kelch S.; Langer R.; Lendlein A. Polymeric triple-shape materials. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (48), 18043–18047. 10.1073/pnas.0608586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl M.; Lendlein A. Triple-shape polymers. J. Mater. Chem. 2010, 20 (17), 3335–3345. 10.1039/b922992b. [DOI] [Google Scholar]
- Xie T.; Xiao X.; Cheng Y. T. Revealing triple-shape memory effect by polymer bilayers. Macromol. Rapid Commun. 2009, 30 (21), 1823–1827. 10.1002/marc.200900409. [DOI] [PubMed] [Google Scholar]
- Xie T. Tunable polymer multi-shape memory effect. Nature 2010, 464 (7286), 267–270. 10.1038/nature08863. [DOI] [PubMed] [Google Scholar]
- Li M.; Dingemans T. J. Synthesis and characterization of semi-crystalline poly (decamethylene terephthalamide) thermosets. Polymer 2017, 108, 372–382. 10.1016/j.polymer.2016.12.004. [DOI] [Google Scholar]
- Li M.; Bijleveld J.; Dingemans T. J. Synthesis and Properties of Semi-crystalline Poly(decamethylene terephthalamide) Thermosets from Reactive Side-group Copolyamides. Eur. Polym. J. 2018, 98, 273–284. 10.1016/j.eurpolymj.2017.11.019. [DOI] [Google Scholar]
- Wagermaier W.; Kratz K.; Heuchel M.; Lendlein A.. Characterization methods for shape-memory polymers. In Shape-Memory Polymers; Springer: 2009; pp 97–145. [Google Scholar]
- Diani J.; Fredy C.; Gilormini P.; Merckel Y.; Régnier G.; Rousseau I. A torsion test for the study of the large deformation recovery of shape memory polymers. Polym. Test. 2011, 30 (3), 335–341. 10.1016/j.polymertesting.2011.01.008. [DOI] [Google Scholar]
- Baghani M. Analytical study on torsion of shape-memory-polymer prismatic bars with rectangular cross-sections. Int. J. Eng. Sci. 2014, 76, 1–11. 10.1016/j.ijengsci.2013.11.016. [DOI] [Google Scholar]
- Baghani M.; Naghdabadi R.; Arghavani J.; Sohrabpour S. A constitutive model for shape memory polymers with application to torsion of prismatic bars. J. Intell. Mater. Syst. Struct. 2012, 23 (2), 107–116. 10.1177/1045389X11431745. [DOI] [Google Scholar]
- Diani J.; Gilormini P.; Frédy C.; Rousseau I. Predicting thermal shape memory of crosslinked polymer networks from linear viscoelasticity. Int. J. Solids Struct. 2012, 49 (5), 793–799. 10.1016/j.ijsolstr.2011.11.019. [DOI] [Google Scholar]
- Roberts C. C.; Apple T. M.; Wnek G. E. Curing chemistry of phenylethynyl-terminated imide oligomers: Synthesis of 13C-labeled oligomers and solid-state NMR studies. J. Polym. Sci., Part A: Polym. Chem. 2000, 38 (19), 3486–3497. . [DOI] [Google Scholar]
- Iqbal M.; Norder B.; Mendes E.; Dingemans T. J. All-aromatic liquid crystalline thermosets with high glass transition temperatures. J. Polym. Sci., Part A: Polym. Chem. 2009, 47 (5), 1368–1380. 10.1002/pola.23245. [DOI] [Google Scholar]
- Lendlein A.; Kelch S. Shape-memory polymers. Angew. Chem., Int. Ed. 2002, 41 (12), 2034–2057. . [DOI] [PubMed] [Google Scholar]
- Véchambre C.; Buléon A.; Chaunier L.; Gauthier C.; Lourdin D. Understanding the mechanisms involved in shape memory starch: macromolecular orientation, stress recovery and molecular mobility. Macromolecules 2011, 44 (23), 9384–9389. 10.1021/ma202019v. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.