

Since the introduction of modern capillary electrophoresis (CE) by Jorgenson and Lukacs in 1981, CE has evolved into a highly mature and versatile separation technique. After a first decade of development studies and instrument commercialization, CE took its place among established analytical techniques and, for instance, became the method of choice for fast high-resolution DNA sequencing in the nineties of the last century. Although with a considerably smaller footprint than liquid and gas chromatography, CE remains to play an essential role in contemporary analytics. For example, with the strong advent of biopharmaceuticals, CE has shown to be particularly useful for routine quality control of therapeutic proteins, such as monoclonal antibodies. Current CE applications range from determination of small inorganic ions to characterization of high-molecular-weight biomolecules, and even particles and intact cells. The research field of CE remains very active, as exhibited by a steady and significant flow of scientific reports on theory, separation modes, new instrumentation, and applications of CE techniques in various areas.
The present review provides a brief cross section of new developments in the broad field of CE, covering the period between September 2015 and September 2017. An initial search on Web of Science, considering keywords related to all modes of CE including their acronyms, yielded about 7000 articles. A first screening in which reviews and irrelevant references were omitted, provided 1200 papers of significance. From these a selection of about 200 was made based on originality, interesting developments and relevance. Notably, studies utilizing electrophoretic principles in microfluidic devices were not included in the present review.
Techniques and Methods
Preconcentration
Injected sample amounts in CE are inherently small. Moreover, when using optical detectors, the optical pathway is limited. Consequently, the concentration sensitivity of CE methods can be poor. Improvement of concentration detection limits by sample preconcentration, either online or offline, remains a topic of research activity.
Electrophoretic Preconcentration
Electrophoretic sample preconcentration in CE often relies on an abrupt and temporary reduction of the migration velocity of analytes. This is clear in commonly applied methods like field-amplified sample stacking (FASS),1 large-volume sample stacking/injection (LVSS/LVSI),2 field-enhanced sample injection (FESI),3,4 sweeping,5 and electrokinetic supercharging (EKS).6 The fundamentals of electrokinetic processes occurring during FASS were studied experimentally and with computer simulation by Sestak and Thormann.7 The authors investigated the effect of injected plug lengths, buffer concentration, sample composition, and linear velocity for the analysis of cationic compounds.
Another study described the use of a free liquid membrane (FLM) to further enhance stacking efficiency of a EKS-CE-UV method.8 The FLM presents a water-immiscible organic solvent interface facilitating the electrically induced transfer of charged analytes, such as paraquat and diquat. The sensitivity gain was almost 2000-fold. Similar improvements in sensitivity were achieved by Cheng et al., who combined LVSI, anion selective exhaustive injection, and sweeping for the online preconcentration of tetrahydrocannabinol and metabolites.9 The resulting CE method allowed direct detection of target analytes in urine.
Multiple isotachophoresis (M-ITP) injections were explored to enhance sensitivity.10 In M-ITP, the ITP sample preconcentration procedure is repeated several times allowing injection of up to 300 times the normal volume. With 6 M-ITP cycles, quantification of the Aβ 1-40 amyloid peptide down to 50 nM was achieved using UV detection.
Chromatographic Preconcentration
Solid-phase extraction (SPE) remains a popular chromatographic preconcentration technique for CE. Zhao et al. demonstrated the effectiveness of offline C5 reversed-phase liquid chromatography (RPLC) prior to top-down capillary zone electrophoresis coupled to electrospray ionization mass spectrometry (CZE-ESI-MS) analysis of a yeast proteome.11 In total, 580 proteoforms and 180 protein groups were identified from 23 proteome fractions analyzed. Another study used C8 SPE cartridges for reduction of sample complexity and preconcentration.12 Melatonin and indole compounds in plant extracts could be detected down to low ppb levels. Rodriguez et al. performed SPE based on synthesized Fe3O4–fullerene–activated carbon magnetic adsorbents for analysis of azo dyes in wastewater in the low mg/L range.13
In online SPE-CE a small trapping column is introduced just before or in the initial part of the separation capillary. Tascon et al. proposed an online SPE-CE-MS method for sensitive alkaloid analysis.14 A micro-C18-column ensured sample cleanup and simultaneous preconcentration, providing limits of detection (LODs) in the 2–77 pg/mL range for algal extracts. Espina-Benitez et al. used a short segment of silica-based monolith with a locally functionalized acrylamide derivative of phenylboronic acid to isolate and preconcentrate diols from 2 μL of complex matrixes (Figure 1).15 Column elution with a small plug of acidic solution allowed FASS of the analytes prior to their CE separation, ensuring LODs in the ng/mL range. This inline coupling was subsequently successfully used for the fully automated analysis of catecholamines (neurotransmitters) in urine samples.
Figure 1.

Schematic illustration of the different steps of cis-diols compounds analysis. A miniaturized boronate affinity monolithic column (μBAMC) at the inlet of an open fused silica capillary is used to preconcentrate and purify cis-diol containing molecules. After their capture under alkaline conditions, catecholamines are eluted in a short acidic plug before their subsequent inline CE separation and UV detection. Reprinted from J. Chromatogr. A, Vol. 1494, Espina-Benitez, M. B.; Randon, J.; Demesmay, C.; Dugas, V., Development and application of a new in-line coupling of a miniaturized boronate affinity monolithic column with capillary zone electrophoresis for the selective enrichment and analysis of cis-diol-containing compounds, pp. 65–76 (ref (15)). Copyright 2017, with permission from Elsevier.
Miscellaneous
An online sample preconcentration method was developed exclusively for catecholamines that were fluorogenically derivatized with naphthalene-2,3-dicarboxaldehyde.16 It takes advantage of diol-borate complexation, through which one negative charge is added to the analytes. The sample was electrokinetically introduced via flow-gated injection. The analytes were selectively focused to a narrow zone by reversible complexation, leading to 100-fold preconcentration for catecholamines in artificial cerebrospinal fluid.
Shimura and Nagai combined immunoaffinity chromatography (IAC) with isoelectric focusing (IEF) in a single capillary.17 IAC was ensured by immobilizing an anti-E-tag antibody at the inlet of the capillary. The remainder of the capillary was coated with neutral polydimethylacrylamide to ensure efficient IEF separations. Fluorescently labeled recombinant Fab with an E-tag spiked at 16 pM to 10 nM in 50% serum was separated and detected with high precision.
An online high-throughput microdialysis-capillary electrophoresis (MD-CE) assay was designed to investigate branched-chain amino acids as possible biomarkers.18 Analytes were sampled using microdialysis, fluorescently labeled in an online reaction, separated using CE and detected using laser-induced fluorescence (LIF) in a sheath flow cuvette. CE separations were performed in less than 30 s, and the temporal resolution of the online MD-CE assay was within 60 s. In a next study, the MD-CE assay was used to monitor in vivo dynamics, achieving a temporal resolution of 22 s for small bioamines.19
Coatings
When conventional bare fused-silica capillaries are used in CE, resolution and peak widths and shapes may be compromised by adverse interactions of the analytes with the inner capillary surface. Furthermore, adsorption of sample matrix components, e.g., proteins, may cause uncontrollable changes of the electroosmotic flow (EOF) and poor migration-time reproducibility. In order to avoid unwanted adsorptions, coating of the capillary wall is a common strategy which remains the subject of research.
Poulsen et al. posed new capillary coating procedures using polyethylene glycol (PEG).20 These include in-capillary surface-initiated atom transfer radical polymerization ensuring covalent binding to the capillary wall and an electrostatic adsorption process. Coating procedures were followed by monitoring adsorption of 2-propylisochinolinium bromide and Sunset Yellow as a positive and negative marker, respectively. Multiple injections of high concentrations of proteins covering a pI range of 3.4–8.4 could be performed without depletion of capillary performance, indicating coating stability of at least 100 days.
A capillary coating procedure allowing regulation of the magnitude and direction of the EOF was proposed by Fu and co-workers.21 This was achieved by coadsorption of polydopamine and polyethylenimine of different molecular weights in variable mass ratios. The polymer chains were stabilized by complexation with Fe3+. The obtained coatings were further characterized by field emission scanning electron microscopy and attenuated total reflection Fourier-transform infrared spectroscopy analysis.
Moreno-Gordaliza et al. used pretreated surface layer protein A from Lactobacillus acidophilus bacteria as a capillary coating,22 which was characterized by contact angle, fluorescence, and atomic force microscopy (AFM) analysis. The new coatings were used for analysis of lipoproteins from human serum with capillary ITP (cITP). The coating could be used for over 100 injections without loss of separation performance with coefficients of variation of 3% for protein migration times over a period of 7 days.
AFM with an adhesive tip was used by Stock et al. to assess topographic and charge-induced features on capillary coatings.23 The charge distribution of different successive multiple ionic polymer layer (SMIL) coatings was assessed with nanometer resolution employing avidin as a single molecule sensor. The acquired surface properties of a four-layer SMIL with poly(acrylamide-co-2-acrylamido-2-methyl-1-propansulfonate) as the terminal layer were related to the observed EOF and CE performance for model proteins and peptides on the same capillary.24
Optimization of capillary coating procedures and their tuning toward specific applications commonly is time-consuming and a trial-and-error process. Monteferrante et al. proposed a method to describe the EOF behavior of a polymer coating as a function of pH, allowing predictive analysis of electroosmosis under different polymeric coating conditions.25 By means of a theoretical argument and numerical simulations involving the linearized Poisson–Boltzmann equation and the Lattice Boltzmann scheme, the experimental curve for the EOF of an acrylamide/methacrylate coating is assessed.
Separation Media
Pseudostationary Phases
Pseudostationary phases (PSP) enable separations not achievable with regular CZE. Micellar electrokinetic chromatography (MEKC) was used for the resolution of insulin and closely related peptides.26 The use of neutral surfactants such as Thesit and Tween20 increased selectivity by reducing adverse interactions with the capillary wall,26 avoiding the use of a capillary coating while using an aqueous background electrolyte (BGE). For separation of different insulins, negatively charged surfactants were required,27 from which perfluorooctanoic acid was found to provide the best resolution.
The new chiral ionic ligand 1-ethyl-3-methyl imidazole L-tartrate ([EMIM][L-Tar]) was introduced for the separation of tryptophan, tyrosine, and phenylalanine enantiomers by chiral-ligand-exchange CE. A comparison with L-tartaric and [EMIM]l-proline indicated the potential of [EMIM][L-Tar] for the enantioseparation of amino acids (AAs).28 Liu et al. synthesized and used the sugar-based surfactant poly(sodium N-alkenyl-α-d-glucopyranoside) with various size and headgroup functionalities for chiral separation of ephedrine alkaloids and β-blockers by MEKC analysis.29 Polymers as compared to monomers showed to strongly enhance separation, while sulfate groups gave less resolution enhancement than phosphate head groups.
Capillary Electrochromatography
Capillary electrochromatography (CEC) offers a dual separation mechanism based on analyte partitioning between a stationary and mobile phase as well as analyte electrophoretic mobility. Over the years, routine CEC has proven difficult, partly due to robustness issues related to bubble formation in frits of microparticulate columns at higher electric fields. These problems may be circumvented by use of open-tubular (OT) or monolithic columns. Sepehrifar et al. used pH-responsive poly(2-dimethylaminoethyl methacrylate)-block-poly(-acrylic acid) as stationary phase in an OT column for efficient CEC analysis of acidic and basic compounds.30 Selectivity could be manipulated via differential contributions from chromatographic and electrophoretic mechanisms by changing the pH or the ionic strength of the BGE.31 The established packing was used to analyze egg white samples containing β-lactoglobulin and ovalbumin resulting in the separation of, respectively, two and eight variants thereof.
Zhao et al. designed glutathione-, somatostatin acetate-, and ovomucoid-functionalized silica-monolithic stationary phases achieving hydrophilic interaction and chiral selectivity in CEC.32 The phases showed EOF tunability by pH control of the mobile phase. Enantioseparation capabilities were enhanced by incorporating gold nanoparticles (NPs) in the glutathione-silica monolithic column. Application to dl-AAs and drug enantiomers was demonstrated.
Stationary phases for CEC based on metal–organic frameworks (MOFs) were introduced by Pan et al. by in situ rapid synthesis of the homochiral MOF [Zn(s-nip)2]n accompanied by zinc oxide nucleating agents.33 Monoamine neurotransmitter enantiomers, nitrophenol isomers, and bisphenols analogues were separated by the new CEC method, showing similar performance as layer-by-layer assembled stationary phases but having the advantage of a significantly reduced preparation and analysis time.
In an attempt to enhance the separation of chiral compounds, Kulsing et al. used peak sharpening effects in CEC by employing molecularly imprinted porous layer OT.34 Enantioseparation required overloading and introduction of an acetonitrile-enriched sharpening zone.
Chiral Media
Enantioseparation can be achieved by CE using chiral selectors in the BGE. CE has the advantages of high peak efficiency and resolution, relatively fast separation, and low sample and reagent consumption. Still, chiral selectors can be expensive and their selection time-consuming. Therefore, the group of Escuder-Gilabert presented a modeling approach to predict enantioresolution for a given selector.35 The model is based on trial runs of selected compounds, correlating mobility to their physicochemical and topological characteristics. Principal component analysis and partial least-squares discriminant analysis models were used to determine the most essential parameters.
Cyclodextrins (CDs) are still the most common chiral selectors used in CE,36−38 but alternative selectors have been studied. Zhang et al. investigated the performance of the spirocyclic chiral ionic liquids (CILs) BMIm+BLHvB– and BMIm+BSMB– for the enantioseparation of five racemic drugs.39 The CILs did not provide chiral separation on their own, but in combination with the CDs, superior enantioseparation was achieved. Molecular dynamics were used to predict interaction configurations and chiral resolution.
Microemulsion electrokinetic chromatography employing sodium dodecyl sulfate (SDS) and potassium sodium tartrate (PST) as a chiral selector was used to separate caffeoylquinic acid (CQA) isomers.40 The PST concentration as well as the pH of the BGE was essential for analyte resolution, which was based on differences in chiral carbon positions. The method was successfully applied to determine individual CQA isomers in honeysuckle and its preparations.
Multidimensional Separations
For the analysis of complex samples, two-dimensional separations are increasingly applied. CE in principle encompasses an interesting separation dimension as it may provide an orthogonal mechanism and favorable separation efficiency at high speed. However, coupling of CE to other separation principles is not straightforward due to voltage handling and the very small volumes involved and therefore demands ingenuity to be established.
Johnson and Bowser described the first coupling of CE to continuous free flow electrophoresis (FFE), circumventing voltage and sampling issues.41 The CE separation capillary was directly inserted into the FFE separation channel (Figure 2). Proof-of-principle measurements involved CE × FFE analysis of fluorescently labeled peptides and bioamines. Taking into to account that these analytes covered only 20–30% of the available separation space, corrected peak capacities of 778 (in 7.6 min) and 377 (in 1.8 min) were obtained for peptides and bioamines, respectively.
Figure 2.

Schematic representation of the CE × FFE-LIF system. (A) CE peaks migrate into the FFE separation channel where a lateral deflection ensures a second dimension separation based on mobility. (B) Visualization of the 2D separation based on CE migration time and the FFE deflection distance. (C) CE × μFFE separation of a fluorescently labeled BSA tryptic digest. Reproduced from Johnson, A. C.; Bowser, M. T. Anal. Chem.2017, 89, 1665–1673 (ref (41)). Copyright 2017 American Chemical Society.
Kohl and co-workers developed a heart-cut two-dimensional CE system (CE-CE) employing a fully insulated mechanical valve with an internal loop of 20 nL, in order to overcome compatibility issues frequently encountered in CE-ESI-MS.42 A portion of interest in the first CE dimension is cut by the loop of the valve and reintroduced to the second CE dimension where interfering compounds are removed, followed by ESI-MS detection. The valve accommodates different CE modes. Capillary isoelectric focusing (cIEF), employing MS-incompatible ampholytes and buffers, was combined with CE-MS method allowing selective distinction of charge variants of a monoclonal antibody (mAb) as well.43
Detection
Mass Spectrometry
The high identification power of MS has driven development of new CE-MS interfaces and optimization/application of existing CE-MS interfaces, achieving better sensitivity and/or ease of use. Wenz et al. reported a collaborative study on the robustness of CE-MS for peptide analysis involving 13 laboratories.44 The equipment used varied in brand, type, and software engaged, while using the same batch of samples, reagents, and capillaries. Relative migration time and peak area reproducibility were below 1.4% relative standard deviation (RSD) and 30% RSD, respectively, indicating that CE-MS performance allows method transfer across multiple laboratories.
CE-MS interfacing remains a field of development. An ideal interface is easy to assemble and use, while achieving efficient analyte ionization and minimize peak dispersion. Gonzalez-Ruiz et al. proposed a low sheath-flow interface operating in the nanospray regime without using nebulizer gas.45 Sensitivity and separation efficiency were somewhat improved as compared to conventional sheath liquid interfacing. Guo et al. posed a new interface based on an integrated metal-coated ESI emitter46 (Figure 3). Employing a 30-μm capillary inner diameter with a similarly sized tapered outer diameter emitter, clogging was avoided. ITP/CZE-MS quantification of peptides using single-reaction monitoring indicated limits of quantification down to 5 amol. A new commercialized system, developed by the lab of Dovichi, was extensively evaluated for proteomics applications over the past few years.47 In this interface, the outlet of the separation capillary is connected to a borosilicate glass spray emitter tip through a tee providing sheath liquid. According to the authors, the ESI voltage generates a steady electroosmotic flow providing a nanoflow of sheath liquid which aids analyte ionization.
Figure 3.

Schematic of the sheathless CITP/CZE-nanoESI-MS setup. After CITP/CZE separation, the CE effluent passes through an integrated metal-coated ESI emitter. An electrically conductive liquid that is in contact with the outer surface of the emitter ensures the electrical connection required for CE prior to single reaction monitoring mass spectrometry (SRM-MS). Reproduced from Guo, X. J.; Fillmore, T. L.; Gao, Y. Q.; Tang, K. Q. Anal. Chem.2016, 88, 4418–4425 (ref (46)). Copyright 2016 American Chemical Society.
With the development of nanospray interfaces for CE-MS, strategies to enhance both ionization efficiency and spray stability have also surfaced. For example, nitrogen gas enriched with an organic dopant in combination with sheathless CE-MS was evaluated for glycopeptide analysis.48 In combination with online preconcentration, this technique yielded higher sensitivity than nano-LC-ESI-MS and sheath-liquid CE-ESI-MS.
Optical Spectroscopy
Fluorescence provides one of the most sensitive means of detection in CE. Two fluorescently labeled mirror image aptamers (Spiegelmers) were developed and used as affinity probes in noncompetitive affinity CE assays with fluorescence detection for the analysis of glucagon and amylin in picomolar concentrations.49 The Spiegelmers specifically bind to the target compounds and offer good stability in biological matricxs as opposed to the conventional aptamers.
Boutonnet et al. evaluated the differences between pulsed and continuous light sources in CE with fluorescence detection studying the compounds 7-hydroxycoumarin, Tamra, and tryptophan.50 Pulsed high energy lasers showed to induce photodegradation of compounds, which could be observed using a dual fluorescence detector setup. ESI-MS was used for structural elucidation of the degradation products. The authors propose the use of continuous LED light sources for the excitation of the analytes.
Gogiashvili et al. developed a cITP-nuclear magnetic resonance (NMR) method employing a microslot probe.51 cITP ensures sample preconcentration, whereas the microslot eliminates spectral broadening due to the magnetic field induced by the separation current, thereby allowing continuous-flow measurements. High-resolution NMR spectra of charged analytes were obtained, but changes in chemical shift were observed at currents above 20 μA. The potential of the microslot probe for hyphenated electrophoretic separations was demonstrated by performing cITP focusing and online 1H NMR detection of a system containing spermine and aniline.
Capacitively Coupled Contactless Conductivity Detection
Capacitively coupled contactless conductivity detection (C4Ds) allows simple and contactless detection in CE, without relying on electrochemical reactions and electrode surfaces (as amperometric and potentiometric detection do). Nyugen et al. developed an in-house built portable CE-C4D system and used it for the separation and detection of 14 rare earth elements within 12 min.52 Achieved LODs were comparable to inductively coupled plasma (ICP) MS detection.
C4Ds were also used to study the fundamental dynamics of CE separation in various modes. Caslavska et al. placed eight C4Ds along a 70 cm capillary to attain temporal insights during CE separation and compared that with theoretical simulations.53 The authors focused their study on the dynamics regarding electroosmosis and hydrodynamic flow, CZE, discontinuous buffer systems, and ITP.
Miscellaneous
Adelantado et al. used an online triple-tube based CE evaporative light scattering detector (ELSD) for the concentration and size determination of silicon dioxide NPs in aqueous solution.54 The coupling thereof is straightforward and bypasses conventional sensitivity discrepancies related to spectroscopic characteristics. ELSD can be used for various analytes provided that the analyte is less volatile than the buffer solution. The method showed to be applicable toward 20–100 nm NPs and resulted in ng/mL LODs.
Cao et al. developed a highly sensitive, in situ real-time imaging of individual metal NPs flowing inside a capillary by using light-sheet scattering microscopy with a supercontinuum55 (Figure 4). The method was applied to measure single plasmonic gold NPs with different sizes or chemical modifications, where separation was achieved in a few minutes based on their different electrophoretic mobility. The fast movement of small NPs with scattering cross-section equivalent to a ∼20 nm gold NP or less was successfully captured. The main issue of Rayleigh scattering background interference from the glass capillary wall was reduced with either a narrow slit or orthogonal polarization detection leading to high S/N imaging of the NPs inside the capillary.
Figure 4.
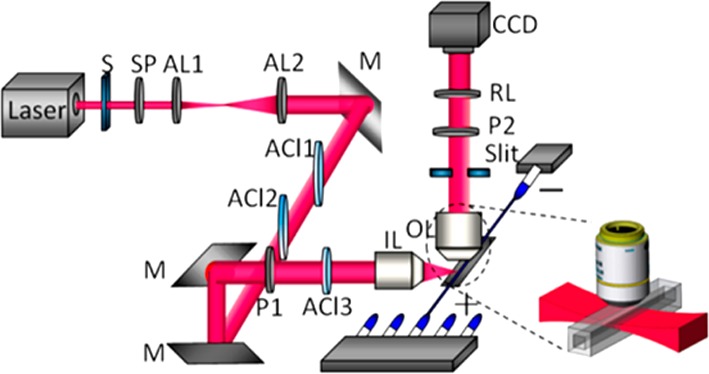
Schematic of the supercontinuum laser light-sheet plasmonic imaging system for CE detection. The combination of the apochromatic (cylindrical) lenses ensure a laterally compressed beam shaped into a planar sheet which forms a detection volume of ∼0.02 nL. Abbreviations: S, shutter; SP, short pass filter; AL1, AL2, ACl1, ACl2, ACl3, apochromatic (cylindrical) lens; M, reflection mirror; P1, P2, polarizer; IL, illumination lens; OL, objective lens; RL, relay lens system. Reproduced from Cao, X.; Feng, J. J.; Pan, Q.; Xiong, B.; He, Y.; Yeung, E. S. Anal. Chem.2017, 89, 2692–2697 (ref (55)). Copyright 2017 American Chemical Society.
Applications
Inorganic Compounds
The analysis of inorganic materials remains a field of interest in which CE has been applied for either quantitative,56 complexation,57 or kinetic analysis.58 Such analyses were predominantly performed using (indirect) UV–vis absorbance detection,59,60 but C4D is increasingly applied. Saiz et al. utilized CE-C4D for the analysis of common cations and compared the performance of the conductivity detection-compatible BGEs (MES/HIS, Lac/His, and Lac/β-Ala) and emphasized that addition of 18-crown-6 and hydroxyisobutyric acid were essential for increasing the detection sensitivity.61 CE-C4D was also used for the determination of bromate in water.62 Utilizing electromembrane extraction as a sample pretreatment, LODs were in the subng/mL range.
Double opposite end injection (DOEI) was shown to allow detection of both metal cations and anions in a single analysis. DOEI-CE-C4D enabled Durc et al. to detect Cl–, Na+, and K+ simultaneously from skin-wipe sweat samples in order to diagnose cystic fybrosis.63 Their approach proved more robust and reliable by monitoring the Cl–/K+ ratio rather than the Cl– concentration only. A similar DOEI-CE-C4D approach was used to monitor four cations and eight anions in saliva of wrestlers that were attempting rapid weight loss.64 Correlations were established between cortisol concentrations and those of several salivary ions. Another option for speeding up analysis is the use of multiple capillaries. Mai et al. developed a portable CE-C4D instrument with three capillary channels employing different BGEs allowing the determination of three different categories of charged analytes65 (Figure 5). The system was used for the concurrent separation of cations and anions in various beverage and food matrixes. Kuban and Bocek developed a dual microelectromembrane extraction approach that enables simultaneous extraction of anions and cations which could subsequently be quantified by CE-C4D.66
Figure 5.

CE-C4D electropherograms for the concurrent separations of inorganic cations, inorganic anions or artificial sweeteners, and organic anions. Channel 1, inorganic cations. Channel 2, option 1, inorganic anions; option 2, artificial sweeteners, aspartame (Asp), cyclamate (Cyc), saccharine (Sac), and acesulfame-K (Ace). Channel 3, organic anions (1) oxalate, (2) formate, (3) tartrate, (4) malate, (5) succinate, (6) citrate, (7) pyruvate, (8) acetate, (9) lactate, and (10) ascorbate. Reprinted from Anal. Chim. Acta, Vol. 911, Mai, T. D., Le, M. D., Sáiz J., Duong, H. A., Koenka, I. J., Pham, H. V., Hauser, P. C., Triple-channel portable capillary electrophoresis instrument with individual background electrolytes for the concurrent separations of anionic and cationic species, pp. 121–128 (ref (65)). Copyright 2016, with permission from Elsevier.
For rare earth elements and actinide analysis, different approaches have been described. Optical detection in combination with either analyte complextion67 or sample stacking68 provided the required sensitivity. However, ICP-optical emission spectroscopy or ICPMS remain the preferred detection techniques, as demonstrated by Bonin et al. and Matczuk and co-workers. They successfully established complexation parameters for tetravalent actinide-diethylenetriaminepentaacetic acid systems,69 respectively, and studied the interaction of quantum dots (QDs) with biologically relevant proteins.70
Nanoparticles
CE has seen significant applied in the still expanding field of NPs, such as QDs and gold NPs, which exhibit high stability, ease of chemical synthesis, and low toxicity. CE was mainly used for obtaining information on the size and surface characteristics of NPs and their interaction with biomolecules. Efficient CE separations of NPs often require addition of stabilizers to the BGE. For example, poly(4-styrenesulfonate) (PSS) was studied as an alternative to SDS for improved separation and size determination of AuNPs.71 The addition of PSS along with a stepwise field gradient significantly improved the resolution for AuNPs with diameters ranging from 5 to 20 nm. Similar observations were made for stabilizers such as Pluronic F-127, citrate, and cetyltrimethylammonium allowing differences in AuNP surface chemistry and size to be revealed.72 In order to establish selective UV detection of titanium dioxide nanoparticles, they were bound to single stranded (ss)DNA followed by coating with PEG,73 whereas detection of zinc oxide NPs was achieved by their interaction with dithiothreitol in phosphate buffer.74 These approaches led to 13–27-fold enhanced UV absorbance signal intensities, respectively.
Qu et al. studied the use of CE for NP size determinations and evaluated the effect of surface chemistry on the electrophoretic mobility of NPs.75 For referencing, surface coated gold NPs in complex matrixes (natural organic matters and fetal bovine serum) were analyzed by CE-ICPMS. Even after addition of surfactants to the BGE, it was not possible to eliminate matrix effects on NP mobility, highlighting the necessity of size calibration using surface coating and matrix-matched standards.
Fichtner et al. studied aqueous dispersions of amorphous silica NPs of various sizes by CE.76 The method allowed the determination of the mean, dispersion width, and skewness of monomodal or multimodal NP size distributions. There was no need for calibration or additional microscopic techniques, under the assumption that the investigated NPs had a constant, size-invariant zeta potential.
CE was also used for the investigation of interactions between proteins and NPs used for biological applications. CE-ICPMS of functionalized Au nanorods interacting with serum proteins revealed metal-specific protein profiles for the differently functionalized AuNPs.77 However, identification of the proteins was not always possible due to the large number of possible candidates.
The development and assessment of new ligands for QDs has developed into a popular field of interest.78 CE with fluorescence detection was extensively applied in order to explore the binding of QDs and polypeptide ligands.78−82 For example, based on meta-affinity driven assembly, the CdSe/ZnS-QDs were functionalized using polyhistidine peptide tags. Longer polyhistidine tags (n = 6) provided optimal self-assembly efficiency.80 New His-peptide ligands were synthesized providing CE-LIF assays for the analysis of the interaction between QDs and functionally important biomolecules.82
Affinity
CE has shown particular useful for the study of (bio)molecular interactions, providing short analysis times, low sample size requirements, high separation efficiencies, and ability to cover a large range of affinities. Li et al. employed affinity CE (ACE) in order to study the binding of sulfated β-CD to uranyl compounds in aqueous solutions.83 ACE was also employed for establishing apparent binding constants of complexes between enantiomers of acyclic nucleoside phosphonates (ANPs) and β-CD in aqueous alkaline medium.84 Estimation of the equilibrium dissociation constants by nonlinear regression and linearized plots showed that the ANP-β-CD complexes are relatively weak. Limitations of ACE for quantification of the supramolecular interactions between the CD cavity and ionic liquids and their effect on the stability of the inclusion complexes were investigated as well.85
The effect of surface oxidation state on the intensity and mode of particle–protein conjugation was quantitatively evaluated by CZE and ACE methods.86 Partial filling (PF) ACE was combined with adsorption energy distribution to determine the heterogeneity of interaction of apoB-100 containing lipoproteins and their antibodies.87 The interaction proved homogeneous and PF-ACE results were in alignment with quartz crystal microbalance experiments.
The use of CE for studying enzymatic activity and inhibition gained attention. The inhibition of human neutrophil elastase was studied using both transverse diffusion of laminar flow profiles (TDLFP) and microscale thermophoresis with LIF detection88 (Figure 6). Two natural pentacyclic triterpenes, ursolic and oleanolic acid, were used to validate the developed CE assay. The method enabled estimation of the IC50 and Ki values of these interactions, which were in agreement with those reported in the literature. In a similar work, cIEF-LIF was compared to CZE for its ability to simultaneously study composition and inhibition of multiple protein kinases.89 The method was successful regardless of structure and charge of the substrate peptides. The use of nanogels to physically constrain an enzyme in a separation capillary while performing electrophoretically mediated microanalysis (EMMA) was proposed for improving sensitivity and separation specificity.90 As immobilization of enzymes is not required, more precise estimation of the Michaelis–Menten constants was achieved. Moreover, it enabled the study of different degrees of stereo specificity in the presence of substrates with different linkages. A comparison between EMMA, pressure-mediated microanalysis, and a spectrophotometric assay was reported for probing the enzyme kinetics of tyrosinase and its inhibition by kojic acid.91
Figure 6.

Inhibition assay of human neutrophil elastase (HNE) using transverse diffusion of laminar flow profiles and CZE-LIF. (A) Online CE assay steps: (a) injection sequence of inhibition buffer (IB), enzyme (E), inhibitor (I), and substrate (S); (b) mixing and incubation by transverse diffusion; (c) separation of product (P), I, S, and E. (B) Examples of electropherograms obtained by CZE-LIF for HNE inhibition by increasing concentrations ursolic acid. Blank assays were conducted by injecting the IB instead of E (0% enzymatic activity) or instead of I (100% enzymatic activity). Reprinted (adapted) from J. Chromatogr. A, Vol. 1431, Syntia, F., Nehmé, R., Claude, B., Morin, P., Human neutrophil elastase inhibition studied by capillary electrophoresis with laser-induced fluorescence detection and microscale thermophoresis, pp. 215–223 (ref (88)). Copyright 2016, with permission from Elsevier.
MS detection has been introduced in ACE in order to assess the interaction of heterogeneous proteins with a target protein.92 This approach allowed multiple parameters to be established in a single run: (i) molecular weight of the separate protein components, (ii) determination of protein–protein dissociation constants, and (iii) determination of the protein complex stoichiometry. Similarly, CE-MS with preincubation was used to estimate the kinetic constants of the degradation of hyaluronic acid by hyaluronidase93 and for the screening of β-secretase inhibitors as potential Alzheimer’s disease therapeutics.94
Nucleic Acids
Kanoatov and Krylov pointed out that an ACE experiment under physiological relevant conditions for the study of DNA–ligand interactions is feasible. With DNA molecules in phosphate buffered saline, they used a pressure-facilitated nonequilibrium CE of equilibrium mixtures approach to attain insights in DNA-ligand kinetics.95 Similarly, Tohala et al. used native separation conditions to study the interaction between homopolymeric sequences and weak DNA binding enantiomers.96
Studies focusing on the CE analysis of microRNA (miRNA) remain scarce. Recent advances in separation performances for long noncoding RNA may alter this.97 Direct separation of miRNA with CE is not always trivial and often requires an additive to the buffer for enhanced performance. ssDNA can be used for such purposes as was shown by Wegman et al., using a hybridization assay with miRNA-specific DNA probes labeled with a fluorophore for LIF detection.98 To separate the miRNA-DNA hybrids from each other and from the probe excess, an ssDNA binding protein as well a different sizes of probes were incorporated in the workflow. The developed approach was sufficiently robust to allow its integration with sample preconcentration by ITP to achieve an LOD below 10 pM
A CE-LIF method for the analysis of the nucleosides adenosine (Ado) and inosine (Ino) in brain samples was developed.99 Interestingly, the fluorescently labeled analytes exhibited about 25-fold fluorescence enhancement upon the formation of inclusion complexes with γ-CD. Ado and Ino were simultaneously quantified in homogenized rat forebrain samples, which were desalted by ultrafiltration in the presence γ-CD, concentrated on-capillary by LVSS to achieve detection limits of 32 and 38 nM for Ado and Ino, respectively.
Although often the identification of nucleic acids is done by polyacrylamide gel electrophoresis, or PAGE, and polymerase chain reaction, or PCR, analysis, the capability of attaining insights in post-transcriptional modifications is limited.100 Recently, CE-MS was used to identify and quantify these modifications.101 The authors could detect two endogenous human circulating miRNAs isolated from B-cell chronic lymphocytic leukemia serum. The CE separation and following MS analysis provided label-free quantitation and revealed 5′-phosphorylation and 3′-uridylation as modifications of miRNAs.
Viruses and Bacteria
Van Tricht et al.102 developed a capillary gel electrophoresis (CGE) method for fast and selective characterization and quantification of viral proteins in influenza vaccines. Dilution of the gel buffer allowed higher separation voltages which led to shorter run times and improved efficiencies. The CGE method allowed analysis of 100 samples in 4 days making it very suitable for quality control purposes. In order to enable characterization of low quantities of adeno-associated virus capsid proteins, Zhang et al. developed a head-column FASS method as an online sample preconcentration technique compatible with CGE103 (Figure 7). The effects of a short water plug, SDS concentration both in sample matrix and in the matrix exchanging solution, as well as the effect of sample injection time were investigated. With LODs in the low-picomolar range, the new method provided 3 orders of magnitude sensitivity enhancement as compared to conventional CGE.
Figure 7.
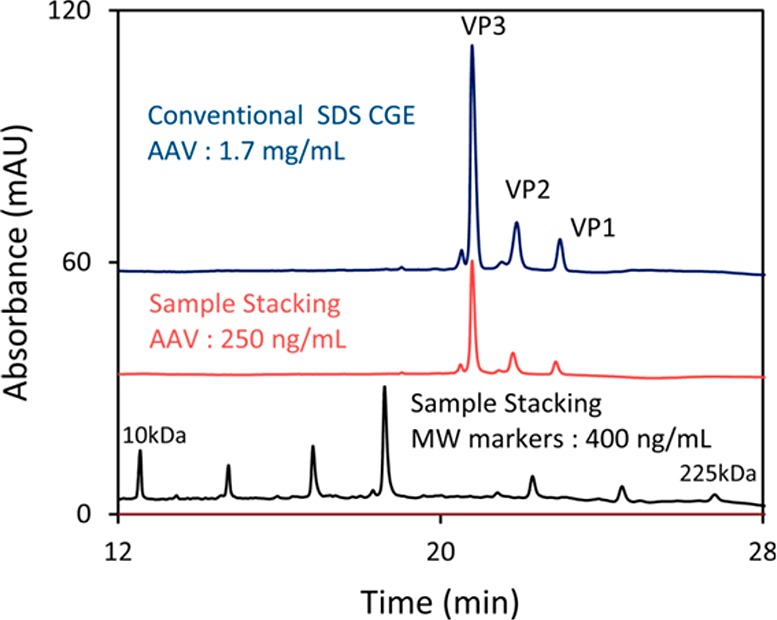
Comparison of sample stacking to conventional CGE. Top trace: conventional CGE analysis of three capsid proteins (i.e., VP3, VP2, and VP1). Middle trace: Sample stacking analysis of the same three capsid proteins. Bottom trace: Sample stacking analysis of SDS-MW Size Standards (10–225 kDa). Indicated concentrations are total protein concentration. Reproduced from Zhang, C. X.; Meagher, M. M. Anal. Chem.2017, 89, 3285–3292 (ref (103)). Copyright 2017 American Chemical Society.
CZE was used for the quantitative analysis of intact adenovirus types Ad26 and Ad35, both in the upstream and downstream processing.104 Because of the complex matrix and the adverse effects of adsorptions in the bare-fused silica capillaries, charged and neutral capillary coatings were tested. Best results were obtained with neutrally coated capillaries in combination with the use of polysorbate-20 in the BGE, low capillary temperatures, and pre- and in-between-run flushing with phosphoric acid. A study by Betonville et al. showed similar results in terms of method requirements for intact virus-like particles of human papillomavirus.105 Moreover, the presence of polysorbate in the sample was indicated to avoid viral particle adsorption to the sample container.
CE has also been evaluated as a tool for the separation and characterization of living bacterial cells. Phung et al. used a dual-stage ITP method for the inline fluorescence in situ hybridization (FISH) and subsequent quantitation of bacteria.106 ITP was recommended as it seems to enhance hybridization kinetics. With an LOD (6.0 × 104 cells/mL) comparable to the CE analysis of a sample processed using an offline FISH protocol, the total analysis time was reduced from 2.5 h to 30 min. With the selection of the appropriate hybridization probe, this approach could be used for specific detection of bacterial cells in aqueous samples.
The characterization of intact phytopathogen bacteria was investigated using cIEF, CZE, and matrix-assisted laser-desorption ionization-time-of-flight (MALDI-TOF).107 In total, 43 strains of the Dickeya and Pectobacterium species were selected among of which some that could not be classified with the traditional methods. In the case of cIEF, the major challenge was the similarity on the pI values of some subspecies. On the other hand, most of these species could be discriminated unambiguously by CZE. Most discriminatory power was obtained with MALDI-TOF-MS as unique mass spectral profiles were obtained for all respective species or subspecies.
Metabolites
One of the major applications of CE relates to the determination of metabolites in various types of biologically relevant samples. MS detection plays an important role since many metabolites cannot be optically detected without prior derivatization, and it provides opportunities for the identification of unknowns. To aid in the latter, a chemoinformatics approach for ranking candidate structures of unidentified peaks was developed.108 The approach uses information about the known metabolites detected in samples containing unidentified peaks and was successfully applied to identify two unknown compounds observed in a CE-MS urinary metabolite profile. Another study focused more on big-data handling in a SPE-CE-MS for identifying biomarkers (in mice) related to Huntington’s disease.109 The workflow ensured significant data reduction prior to multivariate curve resolution asymmetric least-squares analysis.
Cationic metabolite profiling by CE-MS is routinely applied, however, profiling of anions proves more problematic. Yamamoto et al. showed that alkaline ammonia-based buffers (pH > 9) often used for these analyses react with polyimide outer coatings of fused-silica capillaries resulting in frequent capillary fractures and poor long-term performance.110 By making minor adaptations to the BGE, robust high-throughput profiling of anionic metabolites was achieved. In an alternative approach, Gulersonmez et al. show that anions can also be analyzed using a method normally applied for cationic profiling.111 By applying a reversed polarity separation with additional pressure, migration of the anions toward the mass spectrometer was ensured.
A targeted CE-TOF-MS method was established that enables identification of potential biomarkers for hepatocellular carcinoma based on the creatine/betaine ratio.112 In a similar targeted approach, the metabolic changes in the polyamine-pathway produced in colon cancer HT-29 cells by difluoromethylornithine were investigated.113 More global profiles were used to look into metabolic changes related to, for example, type 2 diabetes mellitus evolution114 and the effect of exercising.115 Single-cell CE-MS analysis with multisolvent extraction was used to identify metabolic differences between left and right blastomeres in 8-cell frog embryos.116 To quantify metabolite production between left and right cells, they analyzed 24 different cells in technical duplicate–triplicate measurements. Statistical and multivariate analysis revealed 10 distinct metabolites that were significantly differentially accumulated in the left or right cells. Other fields of CE-MS activity over the last years included pharmaceutical,117 food,118 and plant metabolism.119
Notably, in many metabolomics studies CE-MS was used next to other analytical platforms. CE-MS often is used to target highly polar analytes, like amino acids, organic acids, and carnitines,120,121 although in untargeted approaches also often a significant overlap with other platforms is observed.122
Amino Acids
The sensitivity of a CE-based method for AA analysis and characterization is highly associated with their derivatization, a process that can be laborious, time-consuming, and difficult to standardize.123 Two optimized methods were developed using EMMA123 and TDLFP124 with simultaneous on-capillary derivatization of standard AAs by naphthalene-2,3-dicarboxaldehyde allowing LIF detection. Optimal reactant mixing was achieved by TDLFP, leading to a more generic and robust methodology. However, the optimized EMMA approach proved to be most suitable for human plasma analysis. AAs from Dunaliella salina green algae were analyzed by CE-LIF after labeling with fluorescein isothiocyanate using microwave-assisted derivatization at 80 °C (680 W) minimizing the derivatization time to 150 s.125
A new CE-ESI-MS method was proposed for separation and quantitation of nonstandard AAs.126 After optimization, separation of 27 AAs, including the isomers l-leucine, l-isoleucine, and l-alloisoleucine, was achieved in less than 30 min. The applicability of the method was demonstrated for urine samples from children with vesicoureteral reflux and was proposed as a potentially useful diagnostic tool to inspect these samples.
An alternative approach for detection of low or non-UV absorbing compounds like AAs in CE was developed. It is based on a photochemical reaction of the AAs in the detection window of the separation capillary under strong alkaline conditions, introducing a chromophoric group.127 A systematic study was conducted focusing on the reaction mechanism, the influence of BGE concentration, and the irradiation time of analytes in the detection window. The method was successfully applied to the determination of seven essential AAs.
The use of proteases for generation of AAs for C-terminal sequencing of peptides was reported by Tian et al.128 (Figure 8). The described method involved carboxypeptidase Y (CPY) digestion in combination with rapid online derivatization using OPA/β-ME and optically gated CE with LIF detection. Gaining temporal resolutions of 50 s and the possibility to measure AA release in a time-dependent manner were some of the method’s advantages.
Figure 8.

(A) Serial monitoring of the hydrolysis of the peptide AC-Gln-Arg-Glu-Trp-Phe-Met-Asn-Ser-Tyr by carboxypepsidase Y using optically gated CE with LIF detection. (B) Evolution of the concentrations of released amino acids from the C-terminus of the peptide. Reprinted (adapted) from J. Chromatogr. A, Vol. 1459, Tian, M., Zhang, N., Liu, X., Guo, L., Yanh, L., Sequential online C-terminal sequencing of peptides based on carboxypeptidase Y digestion and optically gated capillary electrophoresis with laser-induced fluorescence detection, pp. 152–159 (ref (128)). Copyright 2016, with permission from Elsevier.
Chiral separation of AAs is an active field of study due to the importance of d-AAs for living organisms. More recent approaches focus on the development of chiral ligands28 and different derivatization agents,129 as well as the coupling of these approaches with MS. Prior et al. developed chiral CE-MS methods for enantioselective analysis of proteinogenic AAs in biological samples.130,131 One method employed β-CD as chiral selector and fluorenylmethyloxycarbonyl chloride as derivatization agent yielding improved AA enantiomer separation and ESI efficiency. In the other method the use of involatile chiral selectors is circumvented by employing (+)-1-(9-fluorenyl)ethyl chloroformate as a chiral AA derivatizing agent and ammonium perfluorooctanoate as a volatile pseudostationary phase for separation of the formed AA diastereomers. d-AA detection in cerebrospinal fluid was shown for both methods. A similar, fully automatized CE-MS method using in-capillary derivatization enabled the successful separation of the diastereomeric derivatives of 14 AAs.132 A different approach was followed in order to distinguish between AAs formed by abiotic versus biotic processes using their chemical distributions.133 AAs were labeled with 5-carboxyfluorescein succinimidyl ester and two separation methods ensured fingerprinting of all 17 relevant AAs.
Peptides
CE-MS of peptides has become quite a routine technique and is applied frequently in untargeted proteomics and peptidomics fields. For example, urine samples were screened for peptides134−137 as suitable biomarkers of disease, revealing specific biomarkers related to heart failure,134 deep vein thrombosis, and pulmonary embolism.135 Related to this, the suitability of mice as models for human aging was investigated by investigating the urinary proteome of the mice.137 Advanced CE-ESI-MS was used for quantitative analysis of diverse proteins in single embryonic cells using untargeted bottom-up proteomics.138,139 The identification of 500–800 protein groups was achieved from single blastomeres isolated from 16-cells of frog embryos, with minimum derivatization steps and label-free quantification for single cells.139
As LC-MS is the predominant proteomics technology, several studies focused on benchmarking CE-MS against LC-MS. For example, a systematic comparison between UPLC-MS/MS and CZE-MS/MS for analysis of an enriched phosphoproteome from the MCF-10A cell line showed that, when the same sample loading amounts (2–200 ng) are used, CZE-MS/MS consistently outperformed UPLC-MS/MS in terms of phosphorylated peptide and total peptide identifications.140 For Xenopus laevis fertilized egg digests, which were prefractionated by RPLC,141 4134 and 5787 proteins could be identified by CE-MS and LC-MS, respectively. The combination of orthogonal separation technologies as RPLC and CE can significantly improve protein coverage, as also shown for the complex proteome of a yeast mitochondrial extract.142
CZE-MS has shown to be very powerful for the characterization of post-translational modifications (PTMs) on the peptide level. A sheathless CZE-ESI-MS/MS method was developed for separation of asparagine deamidation (deaN) and aspartic acid isomerization (isoD).143 CZE could separate the unmodified peptide from modified homologous exhibiting deaN, isoD, or both with a resolution above 1.29. The CE-MS method was successfully applied for the characterization of PTMs on monoclonal antibodies and complex protein mixture. In a similar study, CE-MS with a sheathless interface was used to analyze challenging modifications, such as asparagine deamidation, aspartate isomerization, arginine citrullination, and phosphorylation.144 High resolution was achieved for asparagine, aspartic acid, and isoaspartic acid containing peptides. Applying the CE-MS method for fast and sensitive analysis of intact and enzymatically digested histone H4 revealed a variety of citrullinated proteoforms.
Surfactants often present a source of undesirable interferences during (glyco)proteomics studies. Whereas the surfactants are required to ensure full digestion, their elimination at low pH (when using RapiGest) has shown to lead to loss of sialic acid residues on glycan structures of transferrin and distortion of glycopeptide peaks in general.145 Changing the type of acid and sample desalting helped minimizing desialylation and excellent peak shapes were obtained. CE enabled fast and efficient separation of the sialylated glycopeptides improving detection sensitivity due to decreased ion suppression. Differences in transferrin glycoforms serum from a healthy control and patients with congenital disorders of glycosylation could be observed.
Proteins
CE holds very strong potential for intact protein analysis and methodological improvements remain the subject of research. Double injection capillary zone electrophoresis was used for identification of human chorionic gonadotropin.146 Samples of unknown content were analyzed together with a reference standard and identification was based on the closeness of agreement between the observed migration times of the two peaks. Fu et al. used velocity gap CE to separate groups of reference protein mixtures, obtaining increasing separation efficiency, especially for low-abundance protein species.147
A CE-TOF-MS method was developed for the characterization of intact Mycobacterium tuberculosis (TB) antigens TB10.4 and Ag85B and their chemically produced glycoconjugates, which are glycovaccine candidates against TB.148 A SMIL coating of Polybrene-dextran sulfate-Polybrene was used in combination with acetic acid as BGE in order to prevent protein adsorption and allow the efficient separation of different antigen proteoforms and degradation products. The presence of several closely related degradation products, including truncated, oxidized, deamidated, and conformational variants, were revealed next to the glycoform composition of the neo-glycoproteins.
CE allows protein analysis under near-physiological conditions, allowing detection of the different conformers.149−151 For example, the unfolding of wild-type β2-microglobulin under nondenaturing conditions was investigated with CE-UV149 and CE-ESI-MS.150 In the latter study, several interfaces and mass spectrometers were compared. With CE-ESI-TOF MS proteoforms differing by 1 Da only could be assigned and sheathless interfacing appeared best suited to preserve protein structure integrity. A CE-MS method developed to separate the conformers and dimers of the drug antithrombin employed a neutral poly(vinyl alcohol)-coated capillary.151 The protein conformation was preserved by using a BGE at physiological pH. The developed method allowed the detection of the native, latent, and heterodimer conformers in formulation.
Iwabuchi et al. developed a CGE method for α-synuclein using nonlinear laser wave-mixing detection, which has a quadratic dependence on analyte concentration, allowing small changes in concentration to be monitored.152 Various fluorescently labeled protein oligomers could be separated and detected down to sub-pM concentrations, making the method potentially suitable for Parkinson’s Disease diagnostics. In another method, prostate specific antigen (PSA) was isolated from serum in a way that is compatible for further analysis with CZE.153 The protocol employs an anti-PSA column for isolation. SDS-PAGE followed by Western blotting, circular dichroism, and CZE were used to check for possible protein alterations during the procedure. A CZE method for the characterization of human serum protein components like immunoglobulins and albumins was described by Christians et al.154 As all requirements of the ICH guidelines on validation were met, the developed method could replace conventional CGE methods used to characterize these proteins.
Carbohydrates
CE methods remain of particular interest for identification and quantification of carbohydrates (CHs). CHs are typically UV inactive and require additional consideration or treatment to achieve their detection. These involve indirect UV,155 complexation with a ligand for direct UV analysis,156 or derivatization with, e.g., the fluorescent label APTS for subsequent LIF detection.157
CGE-LIF is commonly used for the analysis of released glycans. For identification purposes, a software tool was developed that correlates the relative migration of a glycan to a glucose size ladder and expresses this in so-called glucose unit (GU) values.158 A database with known structures and correlated GU values aids in identifying structures. GU values strongly depend on temperature, making temperature control essential.159 Interestingly, by using a temperature gradient during the CGE separation, the separation selectivity for branched glycans can be incresed.159,160 This method was demonstrated for mixtures of complex N-glycans (afucosylated, fucosylated, and high mannose oligosaccharides) of biopharmaceutical and biomedical importance. In order to overcome the problem of migration-time shifts, a triple-internal standard approach was proposed.161 Glucose α-1-4-linked oligomers with a degree of polymerization of 2, 3, and 15 were employed. The method provided small errors and showed to be in good agreement with available reference data. Another study looked into determination of activation energies of migration regarding maltooligosaccharides with and without the presence of a monomeric viscosity modifier (ethylene glycol) and a polymeric additive (linear polyacrylamide) in correlation with the GU.162 This study provided an insight in molecular conformation changes of the labeled glycans and possible matrix interaction effects.
Normally, glycans are released from protein in solution. However, within biobanks there are many formalin fixated and paraffin embedded (FFPE) tissue samples of which glycan profiling could be of interest. CGE-LIF was used to identify possible protein N-glycosylation alterations due to FFPE.163 The N-linked sugars of FFPE treated samples were released using PNGase F digestion. No significant changes were revealed in the N-glycome profile upon FFPE, indicating that global N-glycome analysis on the FFPE samples might be feasible.
Hennig et al. performed a longitudinal sampling for biomarker discovery164 (Figure 9). For this complex study, the analysis of the plasma N-glycome was performed by multiplexed CGE-LIF. The results proved a long-term stability of the plasma N-glycome over the examined period of seven years and minor longitudinal changes are more correlated with lifestyle and environmental factors. This work can be considered a step forward for personalized diagnostics.
Figure 9.

(A) CGE-LIF generated human plasma N-glycan fingerprints. Signal intensity in relative fluorescence units [RFU] is plotted over the normalized migration time [MTU″]. The fingerprint is of a native (sialylated) frozen normal control plasma-derived N-glycome, labeled with APTS. (B) Evaluation of the intraindividual and interindividual variability of the plasma N-glycome, by plotting three peaks (high, middle, and low abundance) over the sampling time points (volunteer 3, blue circles; volunteer 5, red squares) and making box plots of the relative peak height over the whole time period per volunteer and total population. Reprinted (adapted) from Biochim. Biophys. Acta Gen. Subj., Vol. 1860, Hennig, R., Cajic, S., Borowiak, M., Hoffmann, M., Kottler, R., Reichl, U., Rapp, E., Toward personalized diagnostics via longitudinal study of the human plasma N-glycome, pp. 1728–1738, DOI: 10.1016/j.bbagen.2016.03.035, under the terms of the Creative Commons Attribution License (CC BY) (ref (164)).
Guan et al. developed a CE method for the precise and accurate determination of glucose levels in blood.165 The first part of the capillary is used as a microreactor for the coupled enzymatic assay of glucose oxidase and horseradish peroxidase. These enzymes are separately injected and govern two sequential reactions, where through peroxide formation a fluorogenic reagent is converted into fluorescein. The latter is subsequently separated via CE analysis from the other reagents and detected with LIF. LODs were down to 10 nM and the method showed potential toward additional peroxide generating systems.
In the work of Bucsella et al., a CE-UV method utilizing dynamic coating for the separation and quantification of structurally very similar nucleotides and nucleotide sugars was developed.166 A total of 11 nucleotides and 6 nucleotide sugars were analyzed. The addition of PEG was indicated to enhance the separation efficiency. The method was tested for Chinese hamster ovarian cell extracts where 3 sugar nucleotides and 7 nucleotides were identified and quantified.
Pharmaceutics
Biopharmaceuticals
CE has emerged as an essential tool for the characterization of biopharmaceuticals, including mAbs, using both intact and middle-up approaches. A useful protocol based on a two-phase-four-step mode was proposed for rapid CZE method development for top-down and middle-up analysis of mAbs.167 This approach focused on the screening of the effect and subsequent optimization of the pH and ionic strength of the BGE, the percentage of organic additive, and viscosity enhancer. The protocol was tested using commercially available mAbs.
The dose determination of a newly developed recombinant subunit envelope protein-based vaccine against all four serotypes produced in Drosophila S2 cells was performed in a comparative study comprising LC and CE methods.168 CZE appeared more suitable as a concentration assay for the tetravalent dengue subunit-based vaccine as separation of all four units was achieved.
Francois et al. described the use of offline CZE-UV/ESI-MS for the middle-up characterization of Fc/2 variants of cetuximab.169 Obtained mass spectral information was cross-validated with CZE-UV/MALDI-MS. In a subsequent publication, a top-down characterization after sample enrichment using CE-UV/MALDI-MS was used for the middle-down characterization of Fc/2 cetuximab variants.170 About 9% sequence coverage of Fc/2 cetuximab fragments was achieved, showing the feasibility of the strategy for middle-down characterization. CZE separation was mainly based on Fc/2 fragments with and without C-terminal truncation.
A sheathless CZE-MS method for middle up analysis of the antibody-drug conjugate (ADC) brentuximab vedotin was developed.171 Native MS was achieved using a nanoESI infusion apparatus, which allowed accurate mass determination with parallel estimation of the average drug to antibody ratio and drug load distribution. In a further step, middle up CZE-ESI-MS/MS analysis was performed after proteolysis with IdeS. This method increased the level of characterization, as complete amino acid sequence identification was achieved alongside glycosylation and drug-loaded-peptides. A similar approach was used for the analysis of the ADC trastuzumab emtansine.172 Here, various analytical techniques including MS, imaging cIEF, and CGE were used for structural characterization and probing protein interactions.
The charge heterogeneity of mAbs is an important quality attribute. An improved method for the characterization of the acidic and basic variants of an IgG1 antibody utilized preparative immobilized pH gradient isoelectric focusing fractionation.173 The fractions were further characterized by CGE and Lys-C peptide mapping via LC-MS/MS. Deamidation, sialylation, glycation, and fragmentation were identified as the main modifications contributing to acidic variants of the mAb, whereas C-terminal lysine, C-terminal proline amidation, and uncyclized N-terminal glutamine were the major species contributing to the basic variants. Another approach for isolation of mAb charge variants was based on FFE.174 Both acidic and basic variants were successfully identified and related back to the cIEF charge profile. SEC, CGE, reduced and intact LC-MS, and LC-MS/MS tryptic peptide mapping were subsequently used to characterize the collected fractions.
In many studies CGE is employed to characterize mAbs. The United States Pharmacopeia (USP) has released a protocol for this method. However, it was shown that the USP method run under nonreducing conditions induced fragmentation for three different mAbs.175 An in-house developed method did not show this extent of fragmentation for the same samples. The findings support the conclusion that molecule-specific methods are still essential to minimize method induced artifacts and address molecule specific behavior.
Incompatibility of the separation medium components is one of the greatest limitations in hyphenation of separation technologies with MS detection. A CE-MS method was recently developed for the analysis of mAbs in SDS-complex matrixes.176 This in-capillary approach incorporates the coinjection of cationic surfactants (CTAB or ADBAC) in the presence of methanol. Successful removal of the SDS was achieved in neutral coated capillaries as well as positively charged coated ones with simultaneous application of reversed polarity. This method allows direct MS analysis of SDS denatured antibodies and protein samples without extensive sample pretreatment. The same authors also presented a heart-cut CZE-CZE-MS setup with an implemented mechanical four-port valve that used a generic ε-aminocaproic acid based BGE in the first dimension and acetic acid in the second dimension177 (Figure 10). Interference-free, highly precise mass data (deviation less than 1 Da) of charge variants of trastuzumab were obtained.
Figure 10.

(A) Complete two-dimensional CE-CE-MS setup. During the separation in the first dimension, the mechanical valve is kept in the loading position (top), where the sample loop is connected to channels S and W. When the desired analyte is located in the sample loop, the valve is switched to the inject position (bottom), transferring the analyte from the first dimension to the second dimension. (B) CZE-UV electropherogram of deglycosylated trastuzumab. Separation was performed with the CE-CE-MS setup. The raw and deconvoluted mass spectra obtained after the second dimension separation of the main variant (M, 10-nL cut), acidic variant A2 (20-nL cut), and acidic variant A3 (20-nL cut) show the presence of deamidated antibody variants. Adapted by permission from Springer, Anal. Bioanal. Chem., Two-dimensional capillary zone electrophoresis–mass spectrometry for the characterization of intact monoclonal antibody charge variants, including deamidation products, Jooß, K., Hühner, J., Kiessig, S., Moritz, B., Neusüß, C., Vol. 409, pp. 6057–6067, Copyright 2017 (ref (177)).
In order to facilitate large scale N-glycosylation analysis, multiplexed CGE-LIF has sparked interest. The technique was used to evaluate the purification of rhEPO glycoforms when using serotonin as affinity ligand for preparative chromatography by monitoring changes in the N-glycan fingerprint.178 An automated sample preparation workflow based on a magnetic bead protocol for N-glycosylation analysis of antibodies was proposed.179 The protocol comprised the endoglycosidase digestion, fluorophore labeling, and cleanup, with CGE-LIF separations of less than 3 min, leading to the entire analysis of the 96-well plate format in a couple of hours. Along the same lines, a multiple injection approach for rapid large scale CE analysis followed by multicomponent optical detection with LIF was proposed.180 As a proof of principle, the protocol was applied for rapid and large scale analysis of major monoclonal antibody (IgG) N-glycans. Nearly 100 samples were introduced in a single capillary, leading to a full analysis time of 4 h rather than 12 h when the conventional approach was applied. For the analysis of even more complex samples, the potential of multidimensional systems were explored. A multiplexing CE mapping method was introduced for the identification of N-glycans of two human IgG samples that gave similar electropherograms, in both CZE and MEKC modes.181 Different orthogonal CE separation mechanisms were combined in parallel applications of CZE, MEKC, and CGE all providing acceptable detection limits, repeatability, and linearity. The combination of CZE and MEKC mechanisms showed optimal orthogonality allowing a larger space for glycan analysis.
A CZE-ESI-MS/MS method was developed for the intact analysis of recombinant human interferon-β1 (rhIFN-β1), a biopharmaceutical with complex glycosylation at a single N-linked site.182 Top-down MS/MS and exoglycosidase digestion were applied in order to elucidate the complex structures. Charged species due to deamidation and sialylation were sufficiently separated by CZE, and high resolution MS and MS/MS along with the enzymatic treatments proved essential for characterization.
Low-Molecular-Weight Drugs
CE in all of its variants is well-established in analysis of small drug analysis. Recent work focused on sensitivity improvement by including sample-stacking approaches183,184 and probing drug chirality.185,186
A fast CZE method for the simultaneous analysis of glibenclamide and its impurities (IA and IB) in pharmaceutical dosage forms was fully developed within a quality by design framework.187 Critical quality attributes were represented by IA peak efficiency, critical resolution between glibenclamide and IB, and analysis time. After optimized conditions were selected, the full separation of the analytes was obtained in less than 2 min. The method was fully validated and was applied to real samples of glibenclamide tablets.
Malá et al. describe the combination of electrophoretic focusing on inverse electromigration dispersion (EMD) gradient with ESI-MS detection.188 The separation of analytes along the electromigrating EMD profile proceeds so that each analyte is at a particular position governed by its pKa and ionic mobility. The focused zones are transported to the capillary end by electromigration, electroosmotic flow, and ESI suction. The system allowed sensitive analyses of trace amounts of weak acids in the pKa range between 6 and 9. The analysis of several sulfonamides at the nanomolar-level in waters was reported.
A new method for screening tyrosinase inhibitors from traditional Chinese medicines utilizing immobilized enzyme reactor technology was described.189 Parallel molecular docking was used to investigate the interaction between the enzyme and inhibitors. Aiming for high-throughput and shorten analysis time, a short-end injection in CE was implemented, leading to a successful method with which four compounds were determined as tyrosinase active inhibitors.
Interactions between the consumed drug and HSA and other serum proteins play a major role in drug distribution and pharmacokinetics. Fluorophore-assisted carbohydrate electrophoresis was used for the evaluation of the affinity and drug–protein interactions between HSA or its equivalent glycated form and the first generation of sulfonylurea antidiabetics.190 The binding constants vary between the different drugs for normal and glycated human serum albumins, with glycated giving lower values.
Polymers
CE finds increasing application for the characterization of complex (bio)polymers. CE under critical conditions has shown to be a suitable approach to separate both natural and synthetic charged polymers.191 Composition distributions heterogeneity of branching could be probed by the obtained separation distributions. The dispersity was quantified, including monitoring of grafting on polymers. Using peak dispersity values, a numerical representation of the mobility and composition distributions was calculated allowing comparisons between samples.
Nitrocelluloses (NCs) and glycosaminoglycans (GAGs) have also been analyzed by CE. Alinat et al. provided a method for the characterization of nonexplosive NCs.192 The CE instrument was used as a viscometer rather than for separation. The viscosity of NC samples was successfully correlated to molecular weight and nitrogen content. A CE-MS method to characterize the GAG heparin was developed by Lin et al.,193 based on a method proposed earlier.194 Reverse polarity CE separation and negative-mode electrospray ionization were optimized using a volatile methanolic ammonium acetate BGE and sheath liquid. An Orbitrap mass spectrometer appeared useful in disaccharide compositional analysis and bottom-up and top-down analysis of low molecular weight heparin.
A CE-UV method was developed for the qualitative and quantitative determination of oligosaccharides.195 Reference compounds consisting of five xylo-, three manno-, and five cello-oligosaccharides were concurrently measured in a highly alkaline solution without derivatization. The method was applied for the determination oligosaccharides from hot-water extracts of a bleached birch and pine kraft pulp in order to study the degradation of hemicelluloses into oligosaccharides as functions of time and temperature.
Binding characteristics of oppositely charged poly(l-lysine) (PLL) and copolymers of acrylamide and 2-acrylamido-2-methyl-1-propanesulfonate (PAMAMPS) were determined by frontal analysis continuous CE at different ionic strengths.196 PAMAMPS charge densities between 15% and 100% were investigated showing that the chain stoichiometry decreases with increasing PAMAMPS charge density, while the charge stoichiometry was in good agreement with a general predicting rule. Modeling of binding constants was used to predict the interaction binding constant between PLL and PAMAMPS of different charge densities at a given ionic strength.
Acknowledgments
Robert L. C. Voeten and Iro K. Ventouri acknowledge the HOSAna Project, which is funded by The Netherlands Organization for Scientific Research (NWO) in the framework of the Programmatic Technology Area PTA-COAST4 of the Fund New Chemical Innovations (Project No. 053.21.117)
Biographies
Robert L. C. Voeten is currently a Ph.D. candidate in the Division of Bioanalytical Chemistry at the Vrije Universiteit Amsterdam (VUA). He focuses on the fundamental and applied aspects of separation methods hyphenated to trapped ion mobility mass spectrometry for intact protein and synthetic polymer characterization. He obtained his Master’s degree at the VUA focussing on the kinetic aspects of a Diels–Alder reaction in microreactors probed by gas chromatography- and direct infusion-mass spectrometry.
Iro K. Ventouri obtained her Master’s degree from the University of Amsterdam in 2017. Her thesis research centered on native protein analysis. Currently she is a Ph.D. candidate in the research group of Prof. Peter Schoenmakers at the Analytical Chemistry group of Van’t Hoff Institute for Molecular Sciences at University of Amsterdam. There she focuses on the optimization and combination of liquid separation (SEC, HDC, FFF) and detection (MALS, MS) for the establishment of nondestructive separation, with the ultimate goal the development of an integrated platform for higher order structural analysis.
Rob Haselberg obtained his Ph.D. degree in 2010 at Utrecht University on the development and application of capillary electrophoresis-mass spectrometry (CE-MS) technologies for the analysis of biopharmaceuticals. Subsequent postdoctoral research at Utrecht University and Vrije Universiteit Amsterdam (VUA) focused on the evaluation of CE-MS for protein characterization as well as setting up affinity CE-MS methods to study protein–protein interactions. In 2013, Haselberg joined the BioMolecular Analysis group of Prof. Govert W. Somsen at VUA, where his research focuses on the characterization of (intact) biomacromolecular compounds using a variety of analytical platforms.
Govert W. Somsen is Professor of Biomolecular Analysis/Analytical Chemistry at Vrije Universiteit Amsterdam. He obtained his Doctorate in Amsterdam and subsequently was Assistant and Associate Professor at the University of Groningen and at Utrecht University in The Netherlands. His current research interests include compositional and conformational characterization of intact biomacromolecules, bioactivity screening of compounds in complex samples, and metabolite analysis with particular attention for compound chirality. His group built an internationally recognized expertise in the hyphenation of capillary electrophoresis and liquid chromatography with mass spectrometry and optical spectroscopy for the detailed profiling of pharmaceutical proteins. Somsen is (co)author of over 150 peer-reviewed papers and is editor of Journal of Chromatography B.
Author Contributions
R.L.C.V. and I.K.V. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Cao K.; Xu Y.; Mu X. N.; Zhang Q.; Wang R. J.; Lv J. J. J. Sep. Sci. 2016, 39, 4243–4250. 10.1002/jssc.201600762. [DOI] [PubMed] [Google Scholar]
- Liu J.; Tian J.; Li J.; Azietaku J. T.; Zhang B. L.; Gao X. M.; Chang Y. X. Electrophoresis 2016, 37, 1632–1639. 10.1002/elps.201500426. [DOI] [PubMed] [Google Scholar]
- Guo L.; Wang Y.; Zheng Y. L.; Huang Z. P.; Cheng Y. Y.; Ye J. N.; Chu Q. C.; Huang D. P. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1014, 70–74. 10.1016/j.jchromb.2016.01.052. [DOI] [PubMed] [Google Scholar]
- Chen Y. L.; Huang Y. C.; Wang C. C. Talanta 2015, 143, 27–34. 10.1016/j.talanta.2015.04.086. [DOI] [PubMed] [Google Scholar]
- Kubalczyk P.; Chwatko G.; Glowacki R. Electrophoresis 2016, 37, 1155–1160. 10.1002/elps.201500411. [DOI] [PubMed] [Google Scholar]
- Thang L. Y.; Breadmore M. C.; See H. H. J. Chromatogr. A 2016, 1461, 185–191. 10.1016/j.chroma.2016.07.067. [DOI] [PubMed] [Google Scholar]
- Sestak J.; Thormann W. J. Chromatogr. A 2017, 1502, 51–61. 10.1016/j.chroma.2017.04.041. [DOI] [PubMed] [Google Scholar]
- Chui M. Q.; Thang L. Y.; See H. H. J. Chromatogr. A 2017, 1481, 145–151. 10.1016/j.chroma.2016.12.042. [DOI] [PubMed] [Google Scholar]
- Cheng H. L.; Tsai Y. H.; Hsu W. L.; Lin Y. H. J. Chromatogr. A 2015, 1426, 226–232. 10.1016/j.chroma.2015.11.032. [DOI] [PubMed] [Google Scholar]
- Mai T. D.; Oukacine F.; Taverna M. J. Chromatogr. A 2016, 1453, 116–123. 10.1016/j.chroma.2016.05.048. [DOI] [PubMed] [Google Scholar]
- Zhao Y. M.; Sun L. L.; Zhu G. J.; Dovichi N. J. J. Proteome Res. 2016, 15, 3679–3685. 10.1021/acs.jproteome.6b00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez F. J. V.; Hernandez I. G.; Cerutti S.; Silva M. F. Microchem. J. 2015, 123, 22–27. 10.1016/j.microc.2015.05.013. [DOI] [Google Scholar]
- Rodriguez J. A.; Ibarra I. S.; Miranda J. M.; Barrado E.; Santos E. M. Anal. Methods 2016, 8, 8466–8473. 10.1039/C6AY02631A. [DOI] [Google Scholar]
- Tascon M.; Gagliardi L. G.; Benavente F. Anal. Chim. Acta 2017, 954, 60–67. 10.1016/j.aca.2016.12.012. [DOI] [PubMed] [Google Scholar]
- Espina-Benitez M. B.; Randon J.; Demesmay C.; Dugas V. J. Chromatogr. A 2017, 1494, 65–76. 10.1016/j.chroma.2017.03.014. [DOI] [PubMed] [Google Scholar]
- Zhang Q. Y.; Gong M. J. J. Chromatogr. A 2016, 1450, 112–120. 10.1016/j.chroma.2016.04.080. [DOI] [PubMed] [Google Scholar]
- Shimura K.; Nagai T. Sci. Rep. 2016, 6, 39221. 10.1038/srep39221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harstad R. K.; Bowser M. T. Anal. Chem. 2016, 88, 8115–8122. 10.1021/acs.analchem.6b01846. [DOI] [PubMed] [Google Scholar]
- Weisenberger M. M.; Bowser M. T. Anal. Chem. 2017, 89, 1009–1014. 10.1021/acs.analchem.6b04516. [DOI] [PubMed] [Google Scholar]
- Poulsen N. N.; Ostergaard J.; Petersen N. J.; Daasbjerg K.; Iruthayaraj J.; Dedinaite A.; Makuska R.; Jensen H. J. Sep. Sci. 2017, 40, 779–788. 10.1002/jssc.201600878. [DOI] [PubMed] [Google Scholar]
- Fu Q. F.; Li X. J.; Zhang Q. H.; Yang F. Q.; Wei W. L.; Xia Z. N. J. Chromatogr. A 2015, 1416, 94–102. 10.1016/j.chroma.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Moreno-Gordaliza E.; Stigter E. C. A.; Lindenburg P. W.; Hankemeier T. Anal. Chim. Acta 2016, 923, 89–100. 10.1016/j.aca.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Leitner M.; Stock L. G.; Traxler L.; Leclercq L.; Bonazza K.; Friedbacher G.; Cottet H.; Stutz H.; Ebner A. Anal. Chim. Acta 2016, 930, 39–48. 10.1016/j.aca.2016.05.002. [DOI] [PubMed] [Google Scholar]
- Stock L. G.; Leitner M.; Traxler L.; Bonazza K.; Leclercq L.; Cottet H.; Friedbacher G.; Ebner A.; Stutz H. Anal. Chim. Acta 2017, 951, 1–15. 10.1016/j.aca.2016.10.030. [DOI] [PubMed] [Google Scholar]
- Monteferrante M.; Sola L.; Cretich M.; Chiari M.; Marconi U. M. B.; Melchionna S. J. Chem. Phys. 2015, 143, 184907. 10.1063/1.4934998. [DOI] [PubMed] [Google Scholar]
- Lamalle C.; Servais A. C.; Demelenne A.; Crommen J.; Fillet M. J. Sep. Sci. 2016, 39, 1189–1194. 10.1002/jssc.201501093. [DOI] [PubMed] [Google Scholar]
- Lamalle C.; Roland D.; Crommen J.; Servais A. C.; Fillet M. Electrophoresis 2015, 36, 2504–2506. 10.1002/elps.201500178. [DOI] [PubMed] [Google Scholar]
- Huang L.; Yu L. S.; Chen Y. T.; Li Y. X. LC GC Eur. 2016, 29, 618–623. [Google Scholar]
- Liu Y. J.; Wu B. L.; Wang P.; Shamsi S. A. Electrophoresis 2016, 37, 913–923. 10.1002/elps.201500434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepehrifar R.; Boysen R. I.; Danylec B.; Yang Y. Z.; Saito K.; Hearn M. T. W. Anal. Chim. Acta 2016, 917, 117–125. 10.1016/j.aca.2016.02.045. [DOI] [PubMed] [Google Scholar]
- Zhang Y. M.; Wang W. T.; Ma X. D.; Jia L. Anal. Biochem. 2016, 512, 103–109. 10.1016/j.ab.2016.08.015. [DOI] [PubMed] [Google Scholar]
- Zhao L. C.; Yang L. M.; Wang Q. Q. J. Chromatogr. A 2016, 1446, 125–133. 10.1016/j.chroma.2016.04.014. [DOI] [PubMed] [Google Scholar]
- Pan C. J.; Wang W. F.; Chen X. G. J. Chromatogr. A 2016, 1427, 125–133. 10.1016/j.chroma.2015.12.020. [DOI] [PubMed] [Google Scholar]
- Kulsing C.; Yang Y. Z.; Chowdhury J. M.; Boysen R. I.; Hearn M. T. W. Electrophoresis 2017, 38, 1179–1187. 10.1002/elps.201600532. [DOI] [PubMed] [Google Scholar]
- Escuder-Gilabert L.; Martin-Biosca Y.; Medina-Hernandez M. J.; Sagrado S. J. Chromatogr. A 2016, 1467, 391–399. 10.1016/j.chroma.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Sanchez-Lopez E.; Marcos A.; Ambrosio E.; Marina M. L.; Crego A. L. J. Chromatogr. A 2016, 1467, 372–382. 10.1016/j.chroma.2016.06.053. [DOI] [PubMed] [Google Scholar]
- Baciu T.; Borrull F.; Calull M.; Aguilar C. Electrophoresis 2016, 37, 2352–2362. 10.1002/elps.201600149. [DOI] [PubMed] [Google Scholar]
- Theurillat R.; Sandbaumhuter F. A.; Bettschart-Wolfensberger R.; Thormann W. Electrophoresis 2016, 37, 1129–1138. 10.1002/elps.201500468. [DOI] [PubMed] [Google Scholar]
- Zhang Y. J.; Du S. J.; Feng Z. J.; Du Y. X.; Yan Z. Anal. Bioanal. Chem. 2016, 408, 2543–2555. 10.1007/s00216-016-9356-8. [DOI] [PubMed] [Google Scholar]
- Tang B.; Guo D.; Li Y. P.; Yang H. Q.; Huang Y. M.; Li H. Anal. Methods 2016, 8, 189–196. 10.1039/C5AY02313K. [DOI] [Google Scholar]
- Johnson A. C.; Bowser M. T. Anal. Chem. 2017, 89, 1665–1673. 10.1021/acs.analchem.6b03768. [DOI] [PubMed] [Google Scholar]
- Kohl F. J.; Montealegre C.; Neususs C. Electrophoresis 2016, 37, 954–958. 10.1002/elps.201500579. [DOI] [PubMed] [Google Scholar]
- Huhner J.; Jooss K.; Neusubb C. Electrophoresis 2017, 38, 914–921. 10.1002/elps.201600457. [DOI] [PubMed] [Google Scholar]
- Wenz C.; Barbas C.; Lopez-Gonzalvez A.; Garcia A.; Benavente F.; Sanz-Nebot V.; Blanc T.; Freckleton G.; Britz-McKibbin P.; Shanmuganathan M.; de l’Escaille F.; Far J.; Haselberg R.; Huang S.; Huhn C.; Pattky M.; Michels D.; Mou S.; Yang F.; Neusuess C.; Tromsdorf N.; Baidoo E. E. K.; Keasling J. D.; Park S. S. J. Sep. Sci. 2015, 38, 3262–3270. 10.1002/jssc.201500551. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Ruiz V.; Codesido S.; Far J.; Rudaz S.; Schappler J. Electrophoresis 2016, 37, 936–946. 10.1002/elps.201500523. [DOI] [PubMed] [Google Scholar]
- Guo X. J.; Fillmore T. L.; Gao Y. Q.; Tang K. Q. Anal. Chem. 2016, 88, 4418–4425. 10.1021/acs.analchem.5b04912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M.; Rock B. M.; Pearson J. T.; Rock D. A. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1011, 24–32. 10.1016/j.jchromb.2015.12.045. [DOI] [PubMed] [Google Scholar]
- Kammeijer G. S. M.; Kohler I.; Jansen B. C.; Hensbergen P. J.; Mayboroda O. A.; Falck D.; Wuhrer M. Anal. Chem. 2016, 88, 5849–5856. 10.1021/acs.analchem.6b00479. [DOI] [PubMed] [Google Scholar]
- Yi L.; Wang X.; Bethge L.; Klussmann S.; Roper M. G. Analyst 2016, 141, 1939–1946. 10.1039/C5AN02468D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutonnet A.; Morin A.; Petit P.; Vicendo P.; Poinsot V.; Couderc F. Anal. Chim. Acta 2016, 912, 146–155. 10.1016/j.aca.2016.01.036. [DOI] [PubMed] [Google Scholar]
- Gogiashvili M.; Telfah A.; Lambert J.; Hergenroder R. Anal. Bioanal. Chem. 2017, 409, 2471–2475. 10.1007/s00216-017-0196-y. [DOI] [PubMed] [Google Scholar]
- Nguyen T. A. H.; Nguyen V. R.; Le D. D.; Nguyen T. T. B.; Cao V. H.; Nguyen T. K. D.; Saiz J.; Hauser P. C.; Mai T. D. J. Chromatogr. A 2016, 1457, 151–158. 10.1016/j.chroma.2016.06.050. [DOI] [PubMed] [Google Scholar]
- Caslavska J.; Koenka I. J.; Hauser P. C.; Thormann W. Electrophoresis 2016, 37, 699–710. 10.1002/elps.201500424. [DOI] [PubMed] [Google Scholar]
- Adelantado C.; Rodriguez-Farinas N.; Martin-Doimeadios R. C. R.; Zougagh M.; Rios A. Anal. Chim. Acta 2016, 923, 82–88. 10.1016/j.aca.2016.03.055. [DOI] [PubMed] [Google Scholar]
- Cao X.; Feng J. J.; Pan Q.; Xiong B.; He Y.; Yeung E. S. Anal. Chem. 2017, 89, 2692–2697. 10.1021/acs.analchem.6b03844. [DOI] [PubMed] [Google Scholar]
- Vortmann-Westhoven B.; Lurenbaum C.; Winter M.; Nowak S. Electrophoresis 2017, 38, 540–546. 10.1002/elps.201600445. [DOI] [PubMed] [Google Scholar]
- Kautenburger R.; Sander J. M.; Hein C. Electrophoresis 2017, 38, 930–937. 10.1002/elps.201600488. [DOI] [PubMed] [Google Scholar]
- Sato Y.; Hoshino H.; Iki N. J. Inorg. Biochem. 2016, 161, 122–127. 10.1016/j.jinorgbio.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Sursyakova V. V.; Burmakina G. V.; Rubaylo A. I. J. Coord. Chem. 2017, 70, 431–440. 10.1080/00958972.2016.1270450. [DOI] [Google Scholar]
- Peng L. Q.; Ye L. H.; Cao J.; Du L. J.; Xu J. J.; Zhang Q. D. Microchim. Acta 2017, 184, 1747–1754. 10.1007/s00604-017-2172-9. [DOI] [Google Scholar]
- Saiz J.; Martin-Alberca C.; Mai T. D.; Garcia-Ruiz C. Electrophoresis 2016, 37, 2896–2902. 10.1002/elps.201600220. [DOI] [PubMed] [Google Scholar]
- Zhang X. L.; Guo L.; Zhang D. X.; Ge X. X.; Ye J. N.; Chu Q. C. Food Anal. Meth. 2016, 9, 393–400. 10.1007/s12161-015-0208-5. [DOI] [Google Scholar]
- Durc P.; Foret F.; Pokojova E.; Homola L.; Skrickova J.; Herout V.; Dastych M.; Vinohradska H.; Kuban P. Anal. Bioanal. Chem. 2017, 409, 3507–3514. 10.1007/s00216-017-0318-6. [DOI] [PubMed] [Google Scholar]
- Mori M.; Ishikawara F.; Tomoda T.; Yamada S.; Okamoto M.; Itabashi H.; Seki Y.; Matsumoto R.; Shoho Y.; Martha L.; Sumino H.; Murakami M. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1012, 178–185. 10.1016/j.jchromb.2016.01.037. [DOI] [PubMed] [Google Scholar]
- Mai T. D.; Le M. D.; Saiz J.; Duong H. A.; Koenka I. J.; Pham H. V.; Hauser P. C. Anal. Chim. Acta 2016, 911, 121–128. 10.1016/j.aca.2016.01.029. [DOI] [PubMed] [Google Scholar]
- Kuban P.; Bocek P. Anal. Chim. Acta 2016, 908, 113–120. 10.1016/j.aca.2016.01.007. [DOI] [PubMed] [Google Scholar]
- Deblonde G. J. P.; Chagnes A.; Cote G.; Vial J.; Rivals I.; Delaunay N. J. Chromatogr. A 2016, 1437, 210–218. 10.1016/j.chroma.2016.01.075. [DOI] [PubMed] [Google Scholar]
- Kratii E.; Nikonorov V.; Nikitina T. Microchem. J. 2017, 130, 198–204. 10.1016/j.microc.2016.09.008. [DOI] [Google Scholar]
- Bonin L.; Aupiais J.; Kerbaa M.; Moisy P.; Topin S.; Siberchicot B. RSC Adv. 2016, 6, 62729–62741. 10.1039/C6RA08121E. [DOI] [Google Scholar]
- Matczuk M.; Legat J.; Timerbaev A. R.; Jarosz M. Analyst 2016, 141, 2574–2580. 10.1039/C6AN00276E. [DOI] [PubMed] [Google Scholar]
- Ciriello R.; Iallorenzi P. T.; Laurita A.; Guerrieri A. Electrophoresis 2017, 38, 922–929. 10.1002/elps.201600478. [DOI] [PubMed] [Google Scholar]
- Pallotta A.; Boudier A.; Leroy P.; Clarot I. J. Chromatogr. A 2016, 1461, 179–184. 10.1016/j.chroma.2016.07.031. [DOI] [PubMed] [Google Scholar]
- Alsudir S.; Lai E. P. C. Anal. Bioanal. Chem. 2017, 409, 1857–1868. 10.1007/s00216-016-0130-8. [DOI] [PubMed] [Google Scholar]
- Alsudir S.; Lai E. P. C. Talanta 2017, 169, 115–122. 10.1016/j.talanta.2017.03.019. [DOI] [PubMed] [Google Scholar]
- Qu H.; Linder S. W.; Mudalige T. K. Anal. Bioanal. Chem. 2017, 409, 979–988. 10.1007/s00216-016-0012-0. [DOI] [PubMed] [Google Scholar]
- Fichtner A.; Jalil A.; Pyell U. Langmuir 2017, 33, 2325–2339. 10.1021/acs.langmuir.6b04543. [DOI] [PubMed] [Google Scholar]
- Matczuk M.; Legat J.; Scaletti F.; Messori L.; Timerbaev A. R.; Jarosz M. J. Chromatogr. A 2017, 1499, 222–225. 10.1016/j.chroma.2017.03.081. [DOI] [PubMed] [Google Scholar]
- Liu F. F.; Wang J. H.; Yang L.; Liu L.; Ding S. M.; Fu M. L.; Deng L. H.; Gao L. Q. Electrophoresis 2016, 37, 2170–2174. 10.1002/elps.201600165. [DOI] [PubMed] [Google Scholar]
- Wang J. H.; Fan J.; Li J. C.; Liu L.; Wang J. P.; Jiang P. J.; Liu X. Q.; Qiu L. J. Sep. Sci. 2017, 40, 933–939. 10.1002/jssc.201600937. [DOI] [PubMed] [Google Scholar]
- Wang J. H.; Li J. Y.; Chen Y.; Teng Y. W.; Wang C. L.; Li J. C.; Liu L.; Dong B. Y.; Qiu L.; Jiang P. J. Electrophoresis 2015, 36, 2419–2424. 10.1002/elps.201500205. [DOI] [PubMed] [Google Scholar]
- Wang J. H.; Li J. Y.; Li J. C.; Qin Y. Q.; Wang C. L.; Qiu L.; Jiang P. J. Electrophoresis 2015, 36, 1523–1528. 10.1002/elps.201500073. [DOI] [PubMed] [Google Scholar]
- Wang J. H.; Zhang C. C.; Liu L.; Kalesh K. A.; Qiu L.; Ding S. M.; Fu M. L.; Gao L. Q.; Jiang P. J. Electrophoresis 2016, 37, 2156–2162. 10.1002/elps.201600164. [DOI] [PubMed] [Google Scholar]
- Li L. N.; Zhang Y. D.; Li X. J.; Shen S. S.; Huang H. X.; Bai Y.; Liu H. W. Electrophoresis 2016, 37, 2567–2573. 10.1002/elps.201600074. [DOI] [PubMed] [Google Scholar]
- Solinova V.; Mikyskova H.; Kaiser M. M.; Janeba Z.; Holy A.; Kasicka V. Electrophoresis 2016, 37, 239–247. 10.1002/elps.201500337. [DOI] [PubMed] [Google Scholar]
- Mofaddel N.; Fourmentin S.; Guillen F.; Landy D.; Gouhier G. Anal. Bioanal. Chem. 2016, 408, 8211–8220. 10.1007/s00216-016-9931-z. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Li P.; Bian W. W.; Yu J. K.; Zhan J. H. Sci. Rep. 2016, 6, 25909. 10.1038/srep25909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Multia E.; Siren H.; Andersson K.; Samuelsson J.; Forssen P.; Fornstedt T.; Oorni K.; Jauhiainen M.; Riekkola M. L. Anal. Biochem. 2017, 518, 25–34. 10.1016/j.ab.2016.10.024. [DOI] [PubMed] [Google Scholar]
- Syntia F.; Nehme R.; Claude B.; Morin P. J. Chromatogr. A 2016, 1431, 215–223. 10.1016/j.chroma.2015.12.079. [DOI] [PubMed] [Google Scholar]
- Xiao L. X.; Liu S. Q.; Lin L. H.; Yao S. Z. Electrophoresis 2016, 37, 2075–2082. 10.1002/elps.201600090. [DOI] [PubMed] [Google Scholar]
- Gattu S.; Crihfield C. L.; Holland L. A. Anal. Chem. 2017, 89, 929–936. 10.1021/acs.analchem.6b04074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. M.; Yang J. L.; Ha W.; Chen J.; Shi Y. P. Anal. Biochem. 2017, 525, 54–59. 10.1016/j.ab.2017.02.020. [DOI] [PubMed] [Google Scholar]
- Dominguez-Vega E.; Haselberg R.; Somsen G. W.; de Jong G. J. Anal. Chem. 2015, 87, 8781–8788. 10.1021/acs.analchem.5b01701. [DOI] [PubMed] [Google Scholar]
- Fayad S.; Nehme R.; Langmajerova M.; Ayela B.; Colas C.; Maunit B.; Jacquinet J. C.; Vibert A.; Lopin-Bon C.; Glatz Z.; Morin P. Anal. Chim. Acta 2017, 951, 140–150. 10.1016/j.aca.2016.11.036. [DOI] [PubMed] [Google Scholar]
- Schejbal J.; Slezackova L.; Reminek R.; Glatz Z. J. Chromatogr. A 2017, 1487, 235–241. 10.1016/j.chroma.2017.01.057. [DOI] [PubMed] [Google Scholar]
- Kanoatov M.; Krylov S. N. Anal. Chem. 2016, 88, 7421–7428. 10.1021/acs.analchem.6b02117. [DOI] [PubMed] [Google Scholar]
- Tohala L.; Oukacine F.; Ravelet C.; Peyrin E. Electrophoresis 2017, 38, 1383–1390. 10.1002/elps.201600516. [DOI] [PubMed] [Google Scholar]
- Li Z. Q.; Liu C. C.; Zhang D. W.; Luo S. P.; Yamaguchi Y. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1011, 114–120. 10.1016/j.jchromb.2015.12.057. [DOI] [PubMed] [Google Scholar]
- Wegman D. W.; Ghasemi F.; Stasheuski A. S.; Khorshidi A.; Yang B. B.; Liu S. K.; Yousef G. M.; Krylov S. N. Anal. Chem. 2016, 88, 2472–2477. 10.1021/acs.analchem.5b04682. [DOI] [PubMed] [Google Scholar]
- Stephen T. K. L.; Guillemette K. L.; Green T. K. Anal. Chem. 2016, 88, 7777–7785. 10.1021/acs.analchem.6b01796. [DOI] [PubMed] [Google Scholar]
- Goedecke S.; Muhlisch J.; Hempel G.; Fruhwald M. C.; Wunsch B. Electrophoresis 2015, 36, 2939–2950. 10.1002/elps.201500242. [DOI] [PubMed] [Google Scholar]
- Khan N.; Mironov G.; Berezovski M. V. Anal. Bioanal. Chem. 2016, 408, 2891–2899. 10.1007/s00216-015-9277-y. [DOI] [PubMed] [Google Scholar]
- van Tricht E.; Geurink L.; Pajic B.; Nijenhuis J.; Backus H.; Germano M.; Somsen G. W.; Sanger-van de Griend C. E. Talanta 2015, 144, 1030–1035. 10.1016/j.talanta.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Zhang C. X.; Meagher M. M. Anal. Chem. 2017, 89, 3285–3292. 10.1021/acs.analchem.6b02933. [DOI] [PubMed] [Google Scholar]
- van Tricht E.; Geurink L.; Backus H.; Germano M.; Somsen G. W.; Sanger-van de Griend C. E. Talanta 2017, 166, 8–14. 10.1016/j.talanta.2017.01.013. [DOI] [PubMed] [Google Scholar]
- Bettonville V.; Nicol J. T. J.; Thelen N.; Thiry M.; Fillet M.; Jacobs N.; Servais A. C. Electrophoresis 2016, 37, 579–586. 10.1002/elps.201500431. [DOI] [PubMed] [Google Scholar]
- Phung S. C.; Cabot J. M.; Macka M.; Powell S. M.; Guijt R. M.; Breadmore M. C. Anal. Chem. 2017, 89, 6513. 10.1021/acs.analchem.7b00598. [DOI] [PubMed] [Google Scholar]
- Salplachta J.; Kubesova A.; Horky J.; Matouskova H.; Tesarova M.; Horka M. Anal. Bioanal. Chem. 2015, 407, 7625–7635. 10.1007/s00216-015-8920-y. [DOI] [PubMed] [Google Scholar]
- Yamamoto H.; Sasaki K. Electrophoresis 2017, 38, 1053–1059. 10.1002/elps.201600328. [DOI] [PubMed] [Google Scholar]
- Pont L.; Benavente F.; Jaumot J.; Tauler R.; Alberch J.; Gines S.; Barbosa J.; Sanz-Nebot V. Electrophoresis 2016, 37, 795–808. 10.1002/elps.201500378. [DOI] [PubMed] [Google Scholar]
- Yamamoto M.; Ly R.; Gill B.; Zhu Y. J.; Moran-Mirabal J.; Britz-McKibbin P. Anal. Chem. 2016, 88, 10710–10719. 10.1021/acs.analchem.6b03269. [DOI] [PubMed] [Google Scholar]
- Gulersonmez M. C.; Lock S.; Hankemeier T.; Ramautar R. Electrophoresis 2016, 37, 1007–1014. 10.1002/elps.201500435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J.; Huang X.; Zhou L. N.; Tan Y. X.; Hu C. X.; Wang X. M.; Niu J. Q.; Wang H. Y.; Lin X. H.; Yin P. Y. Sci. Rep. 2015, 5, 16101. 10.1038/srep16101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez C.; Simo C.; Valdes A.; Campone L.; Piccinelli A. L.; Garcia-Canas V.; Cifuentes A. J. Pharm. Biomed. Anal. 2015, 110, 83–92. 10.1016/j.jpba.2015.03.001. [DOI] [PubMed] [Google Scholar]
- Ciborowski M.; Adamska E.; Rusak M.; Godzien J.; Wilk J.; Citko A.; Bauer W.; Gorska M.; Kretowski A. Electrophoresis 2015, 36, 2286–2293. 10.1002/elps.201500021. [DOI] [PubMed] [Google Scholar]
- Kuehnbaum N. L.; Gillen J. B.; Kormendi A.; Lam K. P.; DiBattista A.; Gibala M. J.; Britz-McKibbin P. Electrophoresis 2015, 36, 2226–2236. 10.1002/elps.201400604. [DOI] [PubMed] [Google Scholar]
- Onjiko R. M.; Morris S. E.; Moody S. A.; Nemes P. Analyst 2016, 141, 3648–3656. 10.1039/C6AN00200E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocato M. Z.; Moreira F. D.; de Albuquerque N. C. P.; de Gaitani C. M.; de Oliveira A. R. M. J. Pharm. Biomed. Anal. 2016, 128, 528–537. 10.1016/j.jpba.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Pena D.; Dudzik D.; Colina-Coca C.; de Ancos B.; Garcia A.; Barbas C.; Sanchez-Moreno C. J. Funct. Foods 2015, 19, 363–375. 10.1016/j.jff.2015.09.033. [DOI] [Google Scholar]
- Dobrecky C. B.; Flor S. A.; Lopez P. G.; Wagner M. L.; Lucangioli S. E. Electrophoresis 2017, 38, 1292–1300. 10.1002/elps.201600533. [DOI] [PubMed] [Google Scholar]
- Andreas N. J.; Hyde M. J.; Gomez-Romero M.; Lopez-Gonzalvez M. A.; Villasenor A.; Wijeyesekera A.; Barbas C.; Modi N.; Holmes E.; Garcia-Perez I. Electrophoresis 2015, 36, 2269–2285. 10.1002/elps.201500011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz S.; Calderon A. A.; Garcia A.; Gallafrio J.; Mestre R. T.; Gonzalez E. G.; de Cabo C. M.; Delgado M. C. M.; Balanza J. A. L.; Simionato A. V. C.; Vaeza N. N.; Barbas C.; Ruperez F. J. Electrophoresis 2015, 36, 2303–2313. 10.1002/elps.201500169. [DOI] [PubMed] [Google Scholar]
- Zhao Y. N.; Zhao J. Y.; Zhao C. X.; Zhou H. N.; Li Y. L.; Zhang J. J.; Li L. L.; Hu C. X.; Li W. Z.; Peng X. J.; Lu X.; Lin F. C.; Xu G. W. Sci. Rep. 2015, 5, 16346. 10.1038/srep16346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cela A.; Madr A.; Glatz Z. J. Chromatogr. A 2017, 1499, 203–210. 10.1016/j.chroma.2017.03.080. [DOI] [PubMed] [Google Scholar]
- Cela A.; Madr A.; Dedova T.; Pelcova M.; Jeseta M.; Zakova J.; Crha I.; Glatz Z. Electrophoresis 2016, 37, 2305–2312. 10.1002/elps.201500587. [DOI] [PubMed] [Google Scholar]
- Nehme R.; Atieh C.; Fayad S.; Claude B.; Chartier A.; Tannoury M.; Elleuch F.; Abdelkafi S.; Pichon C.; Morin P. J. Sep. Sci. 2017, 40, 558–566. 10.1002/jssc.201601005. [DOI] [PubMed] [Google Scholar]
- Rodrigues K. T.; Mekahli D.; Tavares M. F. M.; Van Schepdael A. Electrophoresis 2016, 37, 1039–1047. 10.1002/elps.201500534. [DOI] [PubMed] [Google Scholar]
- Xie X.; Chang F. X.; Li X.; Li M. X.; Zhu Z. W. Talanta 2017, 162, 362–367. 10.1016/j.talanta.2016.10.046. [DOI] [PubMed] [Google Scholar]
- Tian M. M.; Zhang N.; Liu X. X.; Guo L. P.; Yang L. J. Chromatogr. A 2016, 1459, 152–159. 10.1016/j.chroma.2016.07.001. [DOI] [PubMed] [Google Scholar]
- Kuhnreich R.; Holzgrabe U. Chromatographia 2016, 79, 1013–1022. 10.1007/s10337-016-3122-0. [DOI] [Google Scholar]
- Prior A.; Sanchez-Hernandez L.; Sastre-Torano J.; Marina M. L.; de Jong G. J.; Somsen G. W. Electrophoresis 2016, 37, 2410–2419. 10.1002/elps.201600015. [DOI] [PubMed] [Google Scholar]
- Prior A.; Moldovan R. C.; Crommen J.; Servais A. C.; Fillet M.; de Jong G. J.; Somsen G. W. Anal. Chim. Acta 2016, 940, 150–158. 10.1016/j.aca.2016.08.040. [DOI] [PubMed] [Google Scholar]
- Moldovan R. C.; Bodoki E.; Kacso T.; Servais A. C.; Crommen J.; Oprean R.; Fillet M. J. Chromatogr. A 2016, 1467, 400–408. 10.1016/j.chroma.2016.08.035. [DOI] [PubMed] [Google Scholar]
- Creamer J. S.; Mora M. F.; Willis P. A. Anal. Chem. 2017, 89, 1329–1337. 10.1021/acs.analchem.6b04338. [DOI] [PubMed] [Google Scholar]
- Rossing K.; Bosselmann H. S.; Gustafsson F.; Zhang Z. Y.; Gu Y. M.; Kuznetsova T.; Nkuipou-Kenfack E.; Mischak H.; Staessen J. A.; Koeck T.; Schou M. PLoS One 2016, 11, e0157167. 10.1371/journal.pone.0157167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von zur Muhlen C.; Koeck T.; Schiffer E.; Sackmann C.; Zurbig P.; Hilgendorf I.; Reinohl J.; Rivera J.; Zirlik A.; Hehrlein C.; Mischak H.; Bode C.; Peter K. Proteomics: Clin. Appl. 2016, 10, 574–584. 10.1002/prca.201500105. [DOI] [PubMed] [Google Scholar]
- Martin-Pozuelo G.; Gonzalez-Barrio R.; Barbera G. G.; Albalat A.; Garcia-Alonso J.; Mullen W.; Mischak H.; Periago M. J. Int. J. Mol. Sci. 2016, 17, 1789. 10.3390/ijms17111789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkuipou-Kenfack E.; Schanstra J. P.; Bajwa S.; Pejchinovski M.; Vinel C.; Dray C.; Valet P.; Bascands J. L.; Vlahou A.; Koeck T.; Borries M.; Busch H.; Bechtel-Walz W.; Huber T. B.; Rudolph K. L.; Pich A.; Mischak H.; Zurbig P. PLoS One 2017, 12, e0166875. 10.1371/journal.pone.0166875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard-Banek C.; Moody S. A.; Nemes P. Angew. Chem., Int. Ed. 2016, 55, 2454–2458. 10.1002/anie.201510411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard-Banek C.; Reddy S.; Moody S. A.; Nemes P. Mol. Cell. Proteomics 2016, 15, 2756–2768. 10.1074/mcp.M115.057760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig K. R.; Sun L. L.; Zhu G. J.; Dovichi N. J.; Hummon A. B. Anal. Chem. 2015, 87, 9532–9537. 10.1021/acs.analchem.5b02457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X. J.; Sun L. L.; Zhu G. J.; Cox O. F.; Dovichi N. J. Proteomics 2016, 16, 2945–2952. 10.1002/pmic.201600262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M.; Gahoual R.; Enkler L.; Becker H. D.; Chicher J.; Hammann P.; Francois Y. N.; Kuhn L.; Leize-Wagner E. J. Chromatogr. Sci. 2016, 54, 653–663. 10.1093/chromsci/bmw005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahoual R.; Beck A.; Francois Y. N.; Leize-Wagner E. J. Mass Spectrom. 2016, 51, 150–158. 10.1002/jms.3735. [DOI] [PubMed] [Google Scholar]
- Faserl K.; Sarg B.; Maurer V.; Lindner H. H. J. Chromatogr. A 2017, 1498, 215–223. 10.1016/j.chroma.2017.01.086. [DOI] [PubMed] [Google Scholar]
- Barroso A.; Gimenez E.; Benavente F.; Barbosa J.; Sanz-Nebot V. Electrophoresis 2016, 37, 987–997. 10.1002/elps.201500255. [DOI] [PubMed] [Google Scholar]
- Durgaryan A.; Rundlof T.; Laven M.; Amini A. Anal. Methods 2016, 8, 4188–4196. 10.1039/C6AY01078D. [DOI] [Google Scholar]
- Fu X.; Xiao H. T.; Liang S.; Bao J. J.; Li T. X.; Zhang Y. Analyst 2016, 141, 305–310. 10.1039/C5AN01680K. [DOI] [PubMed] [Google Scholar]
- Tengattini S.; Dominguez-Vega E.; Temporini C.; Terreni M.; Somsen G. W. Anal. Bioanal. Chem. 2016, 408, 6123–6132. 10.1007/s00216-016-9723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoletti L.; Bisceglia F.; Colombo R.; Giorgetti S.; Raimondi S.; Mangione P. P.; De Lorenzi E. Electrophoresis 2015, 36, 2465–2472. 10.1002/elps.201500148. [DOI] [PubMed] [Google Scholar]
- Bertoletti L.; Schappler J.; Colombo R.; Rudaz S.; Haselberg R.; Dominguez-Vega E.; Raimondi S.; Somsen G. W.; De Lorenzi E. Anal. Chim. Acta 2016, 945, 102–109. 10.1016/j.aca.2016.10.010. [DOI] [PubMed] [Google Scholar]
- Marie A. L.; Dominguez-Vega E.; Saller F.; Plantier J. L.; Urbain R.; Borgel D.; Tran N. T.; Somsen G. W.; Taverna M. Anal. Chim. Acta 2016, 947, 58–65. 10.1016/j.aca.2016.10.016. [DOI] [PubMed] [Google Scholar]
- Iwabuchi M. F.; Hetu M. M.; Tong W. G. Anal. Biochem. 2016, 500, 51–59. 10.1016/j.ab.2016.01.010. [DOI] [PubMed] [Google Scholar]
- Farina-Gomez N.; Barrabes S.; Gomez-Lopez J. E.; Gonzalez M.; Puerta A.; Navarro-Calderon D.; Albers-Acosta E.; Olivier C.; Diez-Masa J. C.; Peracaula R.; de Frutos M. J. Pharm. Biomed. Anal. 2017, 134, 220–227. 10.1016/j.jpba.2016.11.045. [DOI] [PubMed] [Google Scholar]
- Christians S.; van Treel N. D.; Bieniara G.; Eulig-Wien A.; Hanschmann K. M.; Giess S. Biologicals 2016, 44, 234–241. 10.1016/j.biologicals.2016.04.001. [DOI] [PubMed] [Google Scholar]
- Aid T.; Paist L.; Lopp M.; Kaljurand M.; Vaher M. J. Chromatogr. A 2016, 1447, 141–147. 10.1016/j.chroma.2016.04.027. [DOI] [PubMed] [Google Scholar]
- Yu J. H.; Aboshora W.; Zhang S. Q.; Zhang L. F. Anal. Bioanal. Chem. 2016, 408, 1657–1666. 10.1007/s00216-015-9276-z. [DOI] [PubMed] [Google Scholar]
- Goetz S.; Rejzek M.; Nepogodiev S. A.; Field R. A. Carbohydr. Res. 2016, 433, 97–105. 10.1016/j.carres.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvas G.; Szigeti M.; Guttman A. Electrophoresis 2015, 36, 3094–3096. 10.1002/elps.201500397. [DOI] [PubMed] [Google Scholar]
- Guttman A.; Kerekgyarto M.; Jarvas G. Anal. Chem. 2015, 87, 11630–11634. 10.1021/acs.analchem.5b03727. [DOI] [PubMed] [Google Scholar]
- Szigeti M.; Guttman A. Anal. Chem. 2017, 89, 2201–2204. 10.1021/acs.analchem.7b00016. [DOI] [PubMed] [Google Scholar]
- Jarvas G.; Szigeti M.; Chapman J.; Guttman A. Anal. Chem. 2016, 88, 11364–11367. 10.1021/acs.analchem.6b03596. [DOI] [PubMed] [Google Scholar]
- Kerekgyarto M.; Jarvas G.; Novak L.; Guttman A. Electrophoresis 2016, 37, 573–578. 10.1002/elps.201500394. [DOI] [PubMed] [Google Scholar]
- Donczo B.; Szigeti M.; Ostoros G.; Gacs A.; Tovari J.; Guttman A. Electrophoresis 2016, 37, 2292–2296. 10.1002/elps.201500446. [DOI] [PubMed] [Google Scholar]
- Hennig R.; Cajic S.; Borowiak M.; Hoffmann M.; Kottler R.; Reichl U.; Rapp E. Biochim. Biophys. Acta, Gen. Subj. 2016, 1860, 1728–1738. 10.1016/j.bbagen.2016.03.035. [DOI] [PubMed] [Google Scholar]
- Guan Y. Q.; Zhou G. B. Electrophoresis 2016, 37, 834–840. 10.1002/elps.201500395. [DOI] [PubMed] [Google Scholar]
- Bucsella B.; Fornage A.; Le Denmat C.; Kalman F. Chimia 2016, 70, 732–735. 10.2533/chimia.2016.732. [DOI] [PubMed] [Google Scholar]
- Suba D.; Urbanyi Z.; Salgo A. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1032, 224–229. 10.1016/j.jchromb.2016.07.026. [DOI] [PubMed] [Google Scholar]
- Hamm M.; Wang F.; Rustandi R. R. Electrophoresis 2015, 36, 2687–2694. 10.1002/elps.201500186. [DOI] [PubMed] [Google Scholar]
- Francois Y. N.; Biacchi M.; Said N.; Renard C.; Beck A.; Gahoual R.; Leize-Wagner E. Anal. Chim. Acta 2016, 908, 168–176. 10.1016/j.aca.2015.12.033. [DOI] [PubMed] [Google Scholar]
- Biacchi M.; Said N.; Beck A.; Leize-Wagner E.; Francois Y. N. J. Chromatogr. A 2017, 1498, 120–127. 10.1016/j.chroma.2017.02.064. [DOI] [PubMed] [Google Scholar]
- Said N.; Gahoual R.; Kuhn L.; Beck A.; Francois Y. N.; Leize-Wagner E. Anal. Chim. Acta 2016, 918, 50–59. 10.1016/j.aca.2016.03.006. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Kim M. T.; Zheng L.; Deperalta G.; Jacobson F. Bioconjugate Chem. 2016, 27, 2037–2047. 10.1021/acs.bioconjchem.6b00316. [DOI] [PubMed] [Google Scholar]
- Dada O. O.; Jaya N.; Valliere-Douglass J.; Salas-Solano O. Electrophoresis 2015, 36, 2695–2702. 10.1002/elps.201500219. [DOI] [PubMed] [Google Scholar]
- Hosken B. D.; Li C.; Mullappally B.; Co C.; Zhang B. Y. Anal. Chem. 2016, 88, 5662–5669. 10.1021/acs.analchem.5b03946. [DOI] [PubMed] [Google Scholar]
- Esterman A. L.; Katiyar A.; Krishnamurthy G. J. Pharm. Biomed. Anal. 2016, 128, 447–454. 10.1016/j.jpba.2016.06.006. [DOI] [PubMed] [Google Scholar]
- Sanchez-Hernandez L.; Montealegre C.; Kiessig S.; Moritz B.; Neususs C. Electrophoresis 2017, 38, 1044–1052. 10.1002/elps.201600464. [DOI] [PubMed] [Google Scholar]
- Jooß K.; Hühner J.; Kiessig S.; Moritz B.; Neusüß C. Anal. Bioanal. Chem. 2017, 409, 6057–6067. 10.1007/s00216-017-0542-0. [DOI] [PubMed] [Google Scholar]
- Meininger M.; Stepath M.; Hennig R.; Cajic S.; Rapp E.; Rotering H.; Wolff M. W.; Reichl U. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1012, 193–203. 10.1016/j.jchromb.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Szigeti M.; Lew C.; Roby K.; Guttman A. Jala 2016, 21, 281–286. 10.1177/2211068215608767. [DOI] [PubMed] [Google Scholar]
- Kovacs Z.; Szarka M.; Szigeti M.; Guttman A. J. Pharm. Biomed. Anal. 2016, 128, 367–370. 10.1016/j.jpba.2016.06.002. [DOI] [PubMed] [Google Scholar]
- Feng H. T.; Su M.; Rifai F. N.; Li P. J.; Li S. F. Y. Anal. Chim. Acta 2017, 953, 79–86. 10.1016/j.aca.2016.11.043. [DOI] [PubMed] [Google Scholar]
- Bush D. R.; Zang L.; Belov A. M.; Ivanov A. R.; Kargert B. L. Anal. Chem. 2016, 88, 1138–1146. 10.1021/acs.analchem.5b03218. [DOI] [PubMed] [Google Scholar]
- Wu X. Y.; Xu Z. Q.; Huang Z.; Shao C. Y. Electrophoresis 2016, 37, 2963–2969. 10.1002/elps.201600189. [DOI] [PubMed] [Google Scholar]
- Hernandez-Mesa M.; Airado-Rodriguez D.; Garcia-Campana A. M.; Cruces-Blanco C. Electrophoresis 2015, 36, 2538–2541. 10.1002/elps.201500193. [DOI] [PubMed] [Google Scholar]
- An N.; Wang L. J.; Zhao J. J.; Lv L. L.; Wang N.; Guo H. Z. Anal. Methods 2016, 8, 1127–1134. 10.1039/C5AY02686E. [DOI] [Google Scholar]
- Feng Y.; Wang T. T.; Jiang Z. J.; Chankvetadze B.; Crommen J. Electrophoresis 2015, 36, 1358–1364. 10.1002/elps.201400462. [DOI] [PubMed] [Google Scholar]
- Furlanetto S.; Orlandini S.; Pasquini B.; Caprini C.; Mura P.; Pinzauti S. Anal. Bioanal. Chem. 2015, 407, 7637–7646. 10.1007/s00216-015-8921-x. [DOI] [PubMed] [Google Scholar]
- Mala Z.; Gebauer P.; Bocek P. Anal. Chim. Acta 2016, 935, 249–257. 10.1016/j.aca.2016.06.016. [DOI] [PubMed] [Google Scholar]
- Cheng M. X.; Chen Z. L. Electrophoresis 2017, 38, 486–493. 10.1002/elps.201600367. [DOI] [PubMed] [Google Scholar]
- Michalcova L.; Glatz Z. J. Sep. Sci. 2016, 39, 3631–3637. 10.1002/jssc.201600713. [DOI] [PubMed] [Google Scholar]
- Thevarajah J. J.; Sutton A. T.; Maniego A. R.; Whitty E. G.; Harrisson S.; Cottet H.; Castignolles P.; Gaborieau M. Anal. Chem. 2016, 88, 1674–1681. 10.1021/acs.analchem.5b03672. [DOI] [PubMed] [Google Scholar]
- Alinat E.; Delaunay N.; Archer X.; Gareil P. Carbohydr. Polym. 2015, 128, 99–104. 10.1016/j.carbpol.2015.04.013. [DOI] [PubMed] [Google Scholar]
- Lin L.; Liu X. Y.; Zhang F. M.; Chi L. L.; Amster I. J.; Leach F. E.; Xia Q. W.; Linhardt R. J. Anal. Bioanal. Chem. 2017, 409, 411–420. 10.1007/s00216-016-9662-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X. J.; Lin L.; Liu X. Y.; Zhang F. M.; Chi L. L.; Xia Q. W.; Linhardt R. J. Anal. Chem. 2016, 88, 1937–1943. 10.1021/acs.analchem.5b04405. [DOI] [PubMed] [Google Scholar]
- Hiltunen S.; Siren H.; Heiskanen I.; Backfolk K. Cellulose 2016, 23, 3331–3340. 10.1007/s10570-016-1011-1. [DOI] [Google Scholar]
- Lounis F. M.; Chamieh J.; Leclercq L.; Gonzalez P.; Cottet H. Soft Matter 2016, 12, 9728–9737. 10.1039/C6SM01811D. [DOI] [PubMed] [Google Scholar]