

Abstract
Microtubules are heavily regulated dynamic polymers of αβ-tubulin that are required for proper chromosome segregation and organization of the cytoplasm. Polymerases in the XMAP215 family use arrayed TOG domains to promote faster microtubule elongation. Regulatory factors in the cytoplasmic linker associated protein (CLASP) family that reduce catastrophe and/or increase rescue also contain arrayed TOGs, but how CLASP TOGs contribute to activity is poorly understood. Here, using Saccharomyces cerevisiae Stu1 as a model CLASP, we report structural, biochemical, and reconstitution studies that clarify functional properties of CLASP TOGs. The two TOGs in Stu1 have very different tubulin-binding properties: TOG2 binds to both unpolymerized and polymerized tubulin, and TOG1 binds very weakly to either. The structure of Stu1-TOG2 reveals a CLASP-specific residue that likely confers distinctive tubulin-binding properties. The isolated TOG2 domain strongly suppresses microtubule catastrophe and increases microtubule rescue in vitro, contradicting the expectation that regulatory activity requires an array of TOGs. Single point mutations on the tubulin-binding surface of TOG2 ablate its anti-catastrophe and rescue activity in vitro, and Stu1 function in cells. Revealing that an isolated CLASP TOG can regulate polymerization dynamics without being part of an array provides insight into the mechanism of CLASPs and diversifies the understanding of TOG function.
INTRODUCTION
Microtubules organize the cytoplasm, serve as tracks for motor-based transport, and form the mitotic spindle that is essential for faithful chromosome segregation. Microtubules are hollow, cylindrical polymers that assemble from αβ-tubulin heterodimers and that exhibit dynamic instability, the apparently random switching between phases of growing and shrinking. Dynamic instability facilitates rapid reorganization of microtubule networks, and is heavily regulated in cells: polymerases promote faster growth, depolymerases promote shrinking and/or catastrophe (the switch from growing to shrinking), and rescue factors promote rescue (the switch from shrinking to growing; reviewed in Akhmanova and Steinmetz, 2015). A large number of these microtubule regulatory factors have been identified, but in many cases the molecular origin of their activity remains unclear.
Cytoplasmic linker associated proteins (CLASPs; Pasqualone and Huffaker, 1994; Akhmanova et al., 2001; Bratman and Chang, 2007) comprise a family of microtubule rescue factors. During interphase, CLASP activity is important for stabilizing microtubules and promoting rescue (Mimori-Kiyosue et al., 2005; Drabek et al., 2006). During mitosis, CLASPs localize to kinetochores, where they regulate microtubule dynamics, and at the midzone, where their activity is important for stabilizing antiparallel microtubule overlaps (Inoue et al., 2004; Pereira et al., 2006; Bratman and Chang, 2007). Loss of CLASP activity leads to diminished microtubule stability, collapse of the spindle, and other aberrations (Maiato et al., 2003, 2005; Inoue et al., 2004; Pereira et al., 2006; Bratman and Chang, 2007; Ortiz et al., 2009). In vitro, CLASPs from yeast (Cls1p, Schizosaccharomyces pombe) and Drosophila (mast/Orbit) have been shown to decrease the frequency of microtubule catastrophe and/or increase the frequency of microtubule rescue (Al-Bassam et al., 2010; Moriwaki and Goshima, 2016).
CLASPs contain multiple domains—tubulin-interacting TOG domains, a basic region, and a dimerization element—that collectively determine activity (Al-Bassam et al., 2010; Patel et al., 2012; Leano et al., 2013; Funk et al., 2014). How the domain composition and organization of CLASPs determine activity remains largely unknown, but multiple studies indicate that the tubulin-binding activity of CLASP TOGs is essential (Al-Bassam et al., 2010; Patel et al., 2012; Leano et al., 2013; Funk et al., 2014).
Microtubule polymerases in the Stu2/XMAP215 family also contain arrayed TOG domains (reviewed in Slep, 2009; Al-Bassam and Chang, 2011), and share a common overall domain organization with CLASPs. These better understood Stu2/XMAP215 proteins provide a paradigm for understanding the role of TOG domains. Microtubule polymerase activity requires at least two TOGs (Widlund et al., 2011), each of which binds tightly to curved conformation, unpolymerized tubulin but weakly, if at all, to straight conformation tubulin in the microtubule (Ayaz et al., 2012, 2014); polymerase activity also requires that the TOGs be coupled to a basic region that provides affinity for the negatively charged tubulin C-termini that decorate the microtubule surface (Widlund et al., 2011). The shared domain organization between polymerases and CLASPs might indicate that CLASP activity also requires that multiple TOGs work together (Al-Bassam et al., 2010; Leano et al., 2013). However, recent structural studies of CLASP TOGs revealed a distinctive domain structure that may underlie different tubulin-binding properties (Leano et al., 2013; Maki et al., 2015).
In the present study, we used a combination of biochemical, structural, and reconstitution studies to gain insight into the role of TOG domains in CLASP function. Using the TOG1 and TOG2 domains from Stu1 (Pasqualone and Huffaker, 1994), the Saccharomyces cerevisiae CLASP, we found that only Stu1-TOG2 displays appreciable tubulin-binding activity (1.68 μM affinity), and that Stu1-TOG2 can also bind to microtubules (12 μM affinity). A crystal structure of Stu1-TOG2 revealed a conserved CLASP-specific residue positioned to interact with β-tubulin, likely conferring distinctive tubulin-binding properties to CLASP-family TOGs. In reconstitution assays the isolated Stu1-TOG2 domain potently suppressed microtubule catastrophe and stimulated microtubule rescue. Observing these activities from an isolated TOG domain contradicts the expectation that an array of TOGs would be required to recapitulate the catastrophe-suppressing and rescue-promoting activity of CLASPs. Stu1-TOG2 had little, if any, effect on microtubule growing or shrinking rates, so the observed changes in catastrophe and rescue frequencies are not an indirect consequence stemming from altered kinetics of microtubule growing or shrinking. Stu1-TOG2 also did not detectably increase the number of stabilizing cap sites as detected by Bim1-GFP binding, which suggests that it did not act by reducing GTPase activity in the microtubule lattice. We speculate that Stu1-TOG2 binding to the microtubule tip directly influences the switching propensities there.
Our results demonstrate that a single CLASP-family TOG can suppress catastrophe and promote rescue, without needing to be part of a linked array. These unexpected properties of Stu1-TOG2 provide new insight into the molecular origin of CLASP activity, diversify the understanding of TOG function, and may have implications for other TOG-containing regulatory factors.
RESULTS
Tubulin- and microtubule-binding properties of the Stu1 TOGs
Polymerase-family TOGs like Stu2-TOG1 and Stu2-TOG2 bind tightly to the curved conformation of αβ-tubulin but do not bind appreciably to the straight conformation (Ayaz et al., 2012, 2014). The conformational preference of CLASP-family TOGs has not yet been defined. We used analytical ultracentrifugation and microtubule cosedimentation assays to begin investigating the tubulin- and microtubule-binding properties of the Stu1 TOG domains. The constructs we used for Stu1-TOG1 and for Stu1-TOG2 (Figure 1, A and B; purity of these and other constructs is shown in Supplemental Figure S1) each include segments of the sequence that links TOG1 to TOG2 in the intact protein because expressing more minimal fragments of those domains yielded insoluble or otherwise poorly behaved protein (unpublished data).
FIGURE 1:

Tubulin and microtubule-binding activity of Stu1 TOGs. (A) Domain organization of Stu1 based on our studies and on Funk et al. (2014). Numbers indicate the amino acid boundaries of the different domains. Only the TOG1 and TOG2 domains are studied in this article. (B) Schematics of the expression constructs used for Stu1-TOG1 and Stu1-TOG2. For each TOG, it was necessary to include elements from the linker sequence to obtain soluble, well-behaved protein. The TOG1-TOG2 construct we used encompasses residues 1–567 (is not pictured). (C) Tubulin binding analyzed by sedimentation velocity analytical ultracentrifugation. c(s) distributions (signal population as a function of s) for TOG only (red), tubulin only (black), and a TOG:tubulin mix (green) are shown in each panel. Stu1-TOG1 binds weakly, if at all, to tubulin (top). Stu1-TOG2 binds tubulin and forms a faster-sedimenting complex (middle). For comparison, the tubulin interactions of Stu2-TOG2 are illustrated (bottom; these data were originally presented in Ayaz et al., 2014; the y-axis signal is higher because these data were collected using a shorter wavelength to monitor the sedimentation). These c(s) distributions are taken from a single trial; two more trials gave consistent results (unpublished data). Figure 2 shows results from titrations. (D) Microtubule binding analyzed by cosedimentation. S, supernatant; P, pellet. A fraction of Stu1-TOG2 cosediments with microtubules, indicating some binding; no cosedimentation is observed for Stu1-TOG1 or for Stu2-TOG1, which is shown to provide a comparison with polymerase TOGs. The white * is to draw attention to the increased tubulin in the supernatant fraction with Stu2-TOG1, which actually depolymerizes the stabilized microtubules because of its preference for curved αβ-tubulin. Experiment was repeated three times (unpublished data).
Stu1-TOG1 did not bind appreciably to αβ-tubulin: there was little, if any, change in the sedimentation behavior of αβ-tubulin when it was mixed with TOG1 (Figure 1C; 0.6 μM αβ-tubulin and 3 μM Stu1-TOG1). By contrast, Stu1-TOG2 formed a complex with unpolymerized αβ-tubulin: the TOG2+αβ-tubulin mix showed a clear shift to larger sedimentation coefficients compared with TOG2 or αβ-tubulin alone (Figure 1C; 0.6 μM αβ-tubulin and 3 μM Stu1-TOG2). We showed previously that polymerase TOGs (Stu2-TOG1 and Stu2-TOG2) each bind with comparable affinity to unpolymerized tubulin (Ayaz et al., 2014); a representative experiment from Ayaz et al. (2014) showing this interaction is reproduced in Figure 1C (0.3 μM αβ-tubulin and 7 μM Stu2-TOG2). The lack of tubulin binding we observed for Stu1-TOG1 is consistent with a previous study that used copurification and gel filtration to examine tubulin binding by the isolated Stu1 TOGs (Funk et al., 2014).
Stu1-TOG1 and Stu1-TOG2 also differed in their microtubule-binding properties (Figure 1D; experiments used 3 μM αβ-tubulin and 9 µM of the candidate interaction partner). Stu1-TOG1 did not appreciably cosediment with preformed microtubules, indicating that it binds weakly, if at all, to the microtubule lattice (see also Figure 2). On the other hand, some Stu1-TOG2 cosedimented with preformed microtubules, indicating that Stu1-TOG2 can bind the microtubule lattice in addition to being able to bind unpolymerized tubulin. This ability of Stu1-TOG2 to interact with both unpolymerized tubulin and with microtubules contrasts with polymerase-family TOGs like Stu2-TOG1 and Stu2-TOG2. Indeed, those polymerase TOGs do not bind microtubules and actually induce microtubule depolymerization because they so strongly prefer the curved conformation of tubulin (Figure 1D, starred lane; depolymerization induced by a polymerase TOG domain has been described in Ayaz et al., 2012; Geyer et al., 2015). Thus, Stu1-TOG2 has different conformation selectivity compared with polymerase TOGs: while Stu1-TOG2 can bind to unpolymerized tubulin (presumably in its curved conformation), it can also bind to microtubules, where the tubulin is straight. Not observing microtubule depolymerization in cosedimentation assays using Stu1-TOG2 suggests that Stu1-TOG2 does not prefer curved tubulin as strongly as the polymerase TOGs, and/or that Stu1-TOG2 binds less tightly to curved tubulin.
FIGURE 2:

Binding affinities of Stu1 TOGs to unpolymerized tubulin and the microtubule (MT) lattice. (A) Binding isotherms of TOG:tubulin interaction for TOG1 (left) and TOG2 (right) plotting s-fast against [TOG]. No change in s-fast was observed with increasing concentrations of Stu1-TOG1, indicating very weak or no binding (N = 1 titration). Dose-dependent increases in s-fast were observed for TOG2 (N = 2 titrations; data from both are shown). The low concentration region of the titrations is consistent with a one-site binding reaction, but the titrations reproducibly fail to saturate at high concentrations. To extract the apparent binding constant from the low concentration region, we excluded data where s-fast exceeded 7.2 S (shaded area; 7.2 S is the theoretical maximum S for a 1:1 complex of Stu1-TOG and tubulin; see Materials and Methods and see Supplemental Figure S2 for a complementary binding assay). This yielded an apparent KD = 1.68 [1.49, 1.88] µM. Tubulin concentration in all samples is 0.6 µM. TOG1 concentrations used were 0.19, 0.75, 3, 12, 24 µM and TOG2 concentrations for both titrations were 0.25, 1, 4, 16, 64 µM and 0.25, 1, 4, 16, 64 µM. (B) Representative images from a TIRF microscopy–based microtubule-binding assay using ReAsH-labeled yeast MTs (left column, red) and Alexa-488–labeled Stu1-TOG2 (middle column, green); merged images are shown in the right column. (C) Quantification of fluorescence intensity. Intensity values from 4-pixel-wide linescans perpendicular to MTs are plotted as the solid curves. These curves were fitted with a Gaussian (to quantify the peak height and intensity; colored region) plus a line (to model uneven background intensity; dotted line). Intensity of MTs shown in red; TOG2 in green. (D) Results of the binding titrations for Stu1-TOG constructs on MT lattice. We performed a normalization (see Materials and Methods) to account for day-to-day variations in labeling stoichiometry or laser intensity. n = 15 scans per concentration for each of three independent titrations for Stu1-TOG2 (red; different symbols for the three titrations); n = 15 scans per concentration for a single Stu1-TOG1 titration (black). The fitted dissociation constants are 40 ± 20 μM and 12 ± 3 μM for Stu1-TOG1 and Stu1-TOG2, respectively. Error bars represent SEM. Concentration of TOGs for all titrations were 0.5, 1, 2, 5, 10, 15, 20 µM. Two additional concentrations of TOG2 at 35 and 38 µM were also included. (E) Summary of measured tubulin- and microtubule-binding affinities for Stu1-TOG1 and Stu1-TOG2.
We used sedimentation velocity analytical ultracentrifugation and fluorescence microscopy assays to obtain more quantitative insight into the tubulin- and microtubule-binding affinities of the Stu1-TOG1 (Figure 2). Adding Stu1-TOG1 at concentrations as high as 24 μM did not change the sedimentation coefficient of the reaction boundary (Figure 2A; 0.6 μM αβ-tubulin was used), indicating that the TOG1 domain interacted very weakly, if at all, with unpolymerized tubulin. By contrast, adding Stu1-TOG2 resulted in the formation of a faster sedimenting species (Figure 2A), reflecting a binding interaction between Stu1-TOG2 and αβ-tubulin. We fitted this titration after excluding the high-concentration data points that failed to show saturation (see Materials and Methods for details about the fitting), obtaining a dissociation constant of 1.68 μM for the Stu1-TOG2:αβ-tubulin interaction. We also used microscale thermophoresis to measure the binding affinity in an independent way, obtaining an affinity of 2.6 μM (Supplemental Figure S2).
We used total internal reflection fluorescence (TIRF) microscopy to quantify microtubule-binding affinity. Adding Alexa-488–labeled Stu1-TOG1 or Stu1-TOG2 to ReAsH-labeled yeast microtubules attached to a coverslip yielded dose-dependent increases in TOG fluorescence along the microtubules (Figure 2, B and C). Quantifying the dose dependence of this behavior yielded microtubule-binding affinities for Stu1-TOG1 and Stu1-TOG2 of 40 and 12 μM, respectively (Figure 2D). The binding affinities for αβ-tubulin versus microtubules are summarized in Figure 2E.
The binding experiments reveal marked differences between the Stu1-TOG domains. Whereas Stu1-TOG1 binds very weakly to unpolymerized tubulin (estimated >300 μM affinity) or microtubules (∼40 μM affinity), Stu1-TOG2 binds with moderate affinity to both (∼2 μM affinity for tubulin and ∼12 μM affinity for microtubules). Thus, compared with the polymerase TOGs from Stu2 (Ayaz et al., 2014), the CLASP TOG Stu1-TOG2 binds ∼10-fold less tightly to unpolymerized tubulin, without a strong preference for unpolymerized over polymerized tubulin.
Structure of the TOG2 domain from Stu1
Different conformation selectivity for CLASP-family TOGs might result because in these TOGs the structural arrangement of tubulin-binding residues differs from that in polymerase-family TOGs (Slep and Vale, 2007; Ayaz et al., 2012, 2014; Leano et al., 2013; Maki et al., 2015, and next section).To obtain insight into the structural features that may underlie the tubulin-binding properties of Stu1-TOG2, we determined the structure of Stu1-TOG2—1.89 Å resolution using x-ray crystallography. We were unable to crystallize Stu1-TOG1. The structure of Stu1-TOG2 was phased using a tantalum bromide cluster after attempts at molecular replacement only yielded weak solutions that were difficult to advance (unpublished data). Data collection and phasing statistics for native and derivative crystals are summarized in Table 1. The experimental phases were of high quality, and the model was autobuilt using HKL3000 (Minor et al., 2006) followed by cycles of manual rebuilding and refinement. The refined structure has Rwork/Rfree values of 17.3%/21.2% and good geometry (98.5% of residues in the most favored regions of a Ramachandran plot, MolProbity [Chen et al., 2010] score 1.19 (99th percentile; Table 2).
TABLE 1:
Data collection and phasing.
| Anomalous | Native | |
|---|---|---|
| Space group | P21 | P21 |
| Cell constants (Å or °) |
|
|
| Wavelength (Å) | 1.25478 | 0.97926 |
| Resolution range (Å) |
|
|
| Unique reflections | 26,628 (1009) | 23,899 (1116) |
| Multiplicity | 6.2 (2.5) | 4.5 (3.6) |
| Data completeness (%) | 95.9 (71.6) | 98.6 (95.3) |
| Rmerge (%) | 11.8 (139) | 7.4 (56.8) |
| Rpim (%) | 4.8 (89) | 4.0 (33) |
| CC1/2 (last resolution shell) | 0.143 | 0.812 |
| I/σ(I) | 13.8 (0.5) | 16.7 (2.4) |
| FOMa | 0.2902 | n/a |
| Anomalous signal | 8.4% | n/a |
Values in parentheses refer to the highest resolution shell.
aFigure of merit for phasing is before density modification.
TABLE 2:
Refinement statistics.
| Resolution range (Å) | 40.95−1.89 (1.97−1.89) |
| No. of reflections Rwork/Rfree | 23,025/1152 (2211/116) |
| Data completeness (%) | 95.06 (76) |
| Atoms (non-H protein/solvent) | 2197/161 |
| Rwork (%) | 17.3 (21.4) |
| Rfree (%) | 21.2 (29.3) |
| rmsd bond lengths (Å) | 0.007 |
| rmsd bond angles (°) | 0.810 |
| Mean B-value (Å2; non-H protein/solvent) | 25.7/31.1 |
| Ramachandran plot (%) (favored/additional/disallowed) | 98.5/1.5/0 |
| Maximum likelihood coordinate error (Å) | 0.20 |
| Missing residues, protein | 1–27, 47–63, 243–251 |
| MolProbity Clashscore | 4.02 (98th percentile) |
| MolProbity overall score | 1.19 (99th percentile) |
The overall structure of Stu1-TOG2 (Figure 3) shows the characteristic, paddle-like arrangement of helical hairpins that has been observed in numerous other polymerase- and CLASP-family TOGs (Al-Bassam et al., 2007; Slep and Vale, 2007; Ayaz et al., 2012, 2014; Leano et al., 2013; Fox et al., 2014; Howard et al., 2015; Byrnes and Slep, 2017). Previous structures of other CLASP-family TOGs revealed an “arched” configuration of the long and narrow tubulin-binding interface (Leano et al., 2013; Maki et al., 2015), which in polymerase-family TOGs typically adopts a flatter configuration (Supplemental Figure S3). The spatial arrangement of presumptive tubulin-binding residues in these CLASP-family TOGs is distinct from that observed in polymerase TOGs (Ayaz et al., 2012, 2014). The structure of Stu1-TOG2 (Figure 3, A and B) does not share the arched arrangement previously observed for vertebrate CLASP-family TOGs; instead, Stu1-TOG2 presents a flatter tubulin-binding interface (Supplemental Figure S3). Stu1-TOG2 shows comparable structural similarity to a CLASP-family TOG2 domain (2.6 Å Cα rms coordinate deviation to hCLASP1-TOG2 [Leano et al., 2013], the TOG2 domain from the human CLASP1 isoform) as it does to a polymerase TOG (2.5 Å Cα rms coordinate deviation to Stu2-TOG2 [Ayaz et al., 2014]; Figure 3B, inset). By contrast, vertebrate CLASP-family TOG2 domains show larger structural differences when compared with a polymerase TOG (3.5 Å Cα rms coordinate deviation to Stu2-TOG2).
FIGURE 3:

Crystal structure of Stu1-TOG2. (A) Cartoon representation of the TOG2 structure (orange; the linker sequences are in cyan). Inset shows the construct crystallized (repeated from Figure 1). (B) Rigid body superpositions of Stu1-TOG1 (orange; docked linker, cyan) onto a polymerase TOG (left, Stu2-TOG2, slate) or a CLASP TOG (right, CLASP1-TOG2, gray; docked linker, magenta). Conserved Arg and Trp residues implicated in tubulin binding are shown as spheres and colored to match their respective TOG. The different length brackets illustrate the difference in positioning of the conserved tryptophan. Cα rms coordinate deviation values for the superpositions are presented in the inset table. (C) A distinctive tubulin-binding interface for Stu1-TOG2. Cartoon representation of the Stu2-TOG2:tubulin complex (left; PDB 4U3J), with a region of interest boxed. Close-up view of the TOG:tubulin interface in the region of interest for Stu2 (middle) and Stu1 (right). In Stu1-TOG2 an Arg fills the space normally taken by a tryptophan; this Arg likely contacts tubulin directly, conferring distinctive tubulin-binding properties. (D) Multiple-sequence alignment of CLASP-family (top) and polymerase-family (bottom) TOG domains. W339 (Stu1 numbering) is highly conserved in both CLASP and polymerase TOGs; R386 is highly conserved in CLASPs but in polymerases that position is an asparagine.
One notable feature of the Stu1-TOG2 structure is an additional α-helix positioned along one face of the domain. This helix, colored cyan in Figure 3, forms part of the linker sequence that connects TOG1 to TOG2 in the intact protein. Analogous “linker docking” was also observed in structures of vertebrate CLASP-family TOGs (Leano et al., 2013; Maki et al., 2015), but in those cases the docking occurs in a different place on the TOG (Figure 3B, magenta) and uses a distinct set of linker residues that are not conserved in fungal CLASPs like Stu1 (unpublished data). The presence of this interaction with the linker probably explains why we were unable to purify well-behaved constructs of Stu1-TOG2 that lacked elements from the preceding linker. That both vertebrate and fungal CLASP-family TOGs show docked linkers may indicate that keeping TOG1 and TOG2 close to each other is important for some aspect of CLASP function.
A perplexing feature of previous vertebrate CLASP-family TOG structures has been that a conserved tryptophan residue implicated in tubulin binding appears to be positioned differently than it is in polymerase-family TOGs like Stu2-TOG1 and Stu2-TOG2 (Leano et al., 2013; Maki et al., 2015; see also Figure 3 and Supplemental Figure S3); based on this structural difference it has been speculated that CLASP-family TOGs prefer to interact with a hypercurved conformation of tubulin (Leano et al., 2013). In our structure of Stu1-TOG2, we observe a similar “repositioning” of the presumptive tubulin-contacting tryptophan (Figure 3, B and C). However, we noticed that R386, which is highly conserved in the CLASP family (Figure 3D), packs against this tryptophan and mostly fills the space that the tryptophan normally occupies in the polymerase TOGs (Figure 3C). Previous structures of vertebrate CLASP-family TOG2 domains (Leano et al., 2013; Maki et al., 2015) show the same positioning of this CLASP-family–specific arginine (unpublished data). Thus, it appears that in CLASPs the conserved tryptophan may not directly contact tubulin. Instead, the CLASP-family–specific arginine probably contacts tubulin, thereby conferring distinctive tubulin-binding properties via a more polar interaction surface. We test the functional importance of this residue in a later section.
The TOG1-TOG2 fragment of Stu1 adopts a compact arrangement
The docking of the linker we observed in the Stu1-TOG2 structure effectively shortens the connection between TOG1 and TOG2, and implies that in Stu1 the two domains should be nearer each other than if the linker were disordered, as is thought to be in Stu2. We used limited proteolysis and small-angle x-ray scattering (SAXS) to test this prediction.
The TOG1-TOG2 fragments of Stu1 (CLASP family) and Stu2 (polymerase family) are of similar overall size (567 and 560 residues, respectively), and each contains two TOG domains that are linked by a comparably sized linker (62 and 75 residues, respectively). To probe the accessibility of sequences in the linkers relative to those in the folded TOG domains, we used limited chymotrypsin proteolysis on the TOG1-TOG2 fragments of Stu1 (CLASP family) and Stu2 (polymerase family; 5 µM TOG1-TOG2; Figure 4A). We observed marked differences in the pattern of proteolysis on Stu1 compared with Stu2 (Figure 4A). We observed appreciable cleavage of Stu1-TOG1-TOG2 only into two fragments at the highest levels of chymotrypsin (1:10 chymotrypsin:protein), and the weak intensity of the main proteolytic fragments suggested that the folded domains and the linker sequence were being proteolyzed at comparable rates. Thus, to a first approximation, residues in Stu1-TOG1-TOG2 are uniformly resistant to chymotrypsin treatment and show high protease resistance characteristic of folded domains. Stu2-TOG1-TOG2 behaved quite differently (Figure 4A). Even at the lowest amount of chymotrypsin tested (1:10,000 chymotrypsin:protein), a substantial amount of Stu2-TOG1-TOG2 was cleaved into two fragments, one slightly larger than the other. These proteolytic fragments were much more resistant to proteolysis because they were only appreciably degraded at ∼100-fold higher amounts of protease. Thus, these proteolysis experiments reveal differences in the behavior of the TOG1-TOG2 linker in Stu1 and in Stu2: in Stu1 the linker is approximately as protease resistant as the folded TOG domains, whereas in Stu2 it was much more protease sensitive, consistent with a greater degree of flexibility/disorder (Figure 4A).
FIGURE 4:

Solution analysis of TOG1-TOG2 and TOG2 fragments. (A) Limited chymotrypsin proteolysis of TOG1-TOG2 fragments from Stu1 (left) and Stu2 (right). Stu1-TOG1-TOG2 is less protease sensitive than Stu2-TOG1-TOG2, consistent with a more compact structure (less flexible linker sequence). (B) Left, SAXS scattering intensity profiles for TOG1-TOG2 fragments from Stu1 (black) and Stu2 (gray). Curves are offset for clarity. Inset shows Guinier plots of the data and associated fit (red line). Right, normalized pair-distribution distributions (P(r)) for TOG1-TOG2 fragments of Stu1 (black) and Stu2 (gray). (C) Left, SAXS scattering intensity profile for the Stu1-TOG2 fragment. Inset shows a Guinier plot of the intensity data and associated fit (red line). Right, pair-distribution distributions (P(r)) for TOG2. (D) Crystal structure of Stu1-TOG2 (cartoon) fitted into a three-dimensional (3D) volume reconstructed from the SAXS data for TOG2 (see C). Two orthogonal views of the fitted are shown. (E) Two alternative dockings of TOG2 into the 3D volume reconstructed from the SAXS data for TOG2, with models for disordered parts of the structure represented as a ribbon. Left, same docking as in D showing that in this orientation the disordered N terminus fills an empty region at the bottom of the reconstructed volume; right, alternative docking in which the TOG domain is rotated ∼180° around an axis perpendicular to the page; in this orientation the disordered N terminus fills an empty region at the top right of the reconstructed volume.
We used SAXS to more directly examine the TOG:TOG separation in the TOG1-TOG2 fragments from Stu1 and Stu2 (Figure 4B). In these solution experiments the angular dependence of scattered intensity provides information about the shape of the scattering object. We recorded SAXS intensity profiles for the TOG1-TOG2 fragments of Stu1 and Stu2 (Figure 4B). The low angle scattering provides information about the compactness of the mass distribution in the object, which can be described by a “radius of gyration” (Rg) derived from the slope of a Guinier plot. Despite similar molecular weights (67 and 64 kDa for TOG1-TOG2 fragments from Stu1 and Stu2, respectively), the TOG1-TOG2 fragments of Stu1 and Stu2 yielded different Rg values: 34.7 versus 43.5 Å, indicating a substantial difference in overall compactness (Figure 4B). We also analyzed the scattering profiles in terms of the pair-distribution function, which provides a complementary way to characterize the shape of the scatting object using both low- and high-angle scattering data. Consistent with the Guinier analysis, P(r) analysis also indicated that the TOG1-TOG2 fragment of Stu1 is more compact than that of Stu2: the maximum interatomic distance in Stu1-TOG1-TOG2 was 136 Å, whereas for Stu2 it was 175 Å (Figure 4B). In favorable circumstances where there is one dominant solution conformation, shape reconstruction from SAXS data can provide more detailed insight into the underlying structure. However, and probably as a result of conformational flexibility, the SAXS data for Stu1-TOG1-TOG2 or Stu2-TOG1-TOG2 were not able to be well described by a single, fixed arrangement of TOG1 and TOG2 domains (unpublished data). Thus, while the SAXS data support a closer arrangement of TOG domains in Stu1 compared with Stu2, the data do not allow us to provide a unique model for how the domains are arranged.
We also used SAXS to gain insight into the solution structure of the linker-TOG2 construct that we crystallized (Figure 4C). The scattering profiles we obtained were consistent with the smaller size of this fragment (Rg = 24.4 Å, Dmax = 80 Å; Figure 4C). The extrapolated intensity at zero scattering angle (I0) indicates that the linker-TOG2 was monomeric in solution during the measurement. In this case, ab initio shape reconstructions based on the measured scattering data (see Materials and Methods) yielded a molecular envelope that closely matched our crystal structure (Figure 4D). Two orientations of our structure fitted comparably into this envelope. Figure 4E shows representative examples of these two orientations. In each of the two orientations, the disordered segments fitted into “bumps” in the calculated SAXS envelope that were not well filled by the ordered parts from the crystal structure. That the disordered segments fill the parts of the SAXS envelope not occupied by the structured portion of the domain supports the model fitting into the SAXS envelope and provides evidence that the linker docking we observed is not a crystallization artifact.
Together, the data in this section indicate that the TOGs in Stu1-TOG1-TOG2 adopt a compact arrangement, likely because a segment of the linking sequence docks onto the side of the TOG2 domain as observed in the crystal structure (Figure 3, A and B). This compact arrangement contrasts with the more extended arrangement of Stu2-TOG1-TOG2, wherein the linker sequence is not thought to form an integral part of one of either TOG domain. The functional importance of these differences is not yet clear.
The isolated Stu1-TOG2 domain suppresses catastrophe and stimulates rescue
The ability of TOG2 to bind the microtubule lattice led us to speculate that TOG2 binding might affect microtubule stability and/or dynamics. We therefore used time-lapse differential interference contrast (DIC) microscopy to determine whether the presence of substoichiometric amounts of Stu1-TOG2 affected any of the parameters of microtubule dynamics: growing rate, shrinking rate, catastrophe frequency, or rescue frequency (Figure 5). We used 0.8 μM yeast αβ-tubulin and 200 nM Stu1-TOG2 for these experiments. Figure 5A shows representative kymographs from the control experiment: dynamic microtubules growing from GTPγS-stabilized seeds without any Stu1-TOG2 added. These kymographs are typical for wild-type yeast αβ-tubulin, showing slow growth, frequent catastrophe, very fast shrinking, and no rescues. Figure 5B shows representative kymographs from microtubules growing in the presence of 200 nM Stu1-TOG2. Stu1-TOG2 caused striking changes in microtubule dynamics: growth phases were substantially longer, and there were a number of rescue events. Microtubule growing and shrinking rates were largely unchanged by the addition of Stu1-TOG2 (∼5% and ∼7% difference, respectively; p > 0.05; Figure 5C). By contrast, there were substantial changes in the frequencies of catastrophe (inverse of microtubule lifetime) and rescue (Figure 5C). Median microtubule lifetimes increased from 506 s in the control to 1580 s in the presence of Stu1-TOG2, corresponding to a fourfold decrease in catastrophe frequency (Figure 5C). We also observed an appreciable rescue frequency in the presence of Stu1-TOG2 (∼19 min−1; 37 rescues out of 82 catastrophes recorded); no rescues were observed in control measurements where Stu1-TOG2 was not present (217 catastrophes observed; Figure 5C). Similar experiments using the Stu1-TOG1 domain (200 nM) behaved identically to control reactions without any TOG (Supplemental Figure S4). Thus, the isolated Stu1-TOG2 domain is sufficient to stimulate rescue and suppress catastrophe, and these effects on microtubule transition frequencies occur without substantial changes in the rates of microtubule growing or shrinking.
FIGURE 5:

Anti-catastrophe and rescue activity of Stu1-TOG2. (A) Representative kymographs of yeast αβ-tubulin (0.8 µM) growing from GTPγS-stabilized seeds (pseudocolored green). Microtubule dynamics were imaged using time-lapse DIC microscopy and show slow growth, rapid shrinking, frequent catastrophe, and no rescues. See C for quantification of the dynamics. (B) As above, but in the presence of Stu1-TOG2 (0.2 µM). The presence of Stu1-TOG2 leads to longer growth phases (reduced catastrophe) and elevated rescue (rescues are marked with *). (C) Quantification of the microtubule dynamics from A (in gray) and B (in red). From left to right: growing rates do not change substantially (control: 17.9 ± 0.2 µm/h, n = 245; +TOG2: 18.8 ± 0.3 µm/h, n = 126); shrinking rates are also little changed (control: 170 ± 8 µm/min, n = 11; +TOG2: 183 ± 12 µm/min, n = 18); catastrophe frequency decreases fourfold in the presence of TOG2 (control: 0.098 {0.094, 0.100} min−1, n = 57, 160 catastrophes; +TOG2: 0.025 {0.024, 0.027} min−1, n = 44, 38); rescue frequency increases in the presence of TOG2 (control: no rescues; +TOG2: 19 {18, 20} min−1, n = 21, 16 rescues). Error bars on the scatter plots for growing and shrinking rates indicate overall mean and SD. Values reported for growth and transition frequencies are weighted average over two independent experiments. n gives the total number of observed events for growing and shrinking rates, and the number in each of two trials for the transition frequencies. For the transition frequencies, averages from each separate experiment are given in braces to provide a measure of experimental variation. Values reported for shrinking rates are from a single experiment. Errors reported for growth and shrinking rates are SEM.
Mutations on the tubulin-binding interface of Stu1-TOG2 abolish anti-catastrophe and rescue activity in vitro and Stu1 function in cells
Polymerase-family TOG domains contain conserved residues at the “top” and “bottom” of their tubulin-binding surface that mediate their conformation-selective, high-affinity interactions with curved αβ-tubulin. CLASP-family TOGs contain the same conserved residues, but as mentioned previously they are positioned somewhat differently in the structure (Figure 3 and Supplemental Figure S3). To determine whether both of these conserved residues (W339 and R525 in Stu1-TOG2) are important for the catastrophe-suppressing and rescue-promoting activities of TOG2, we measured the effects of singly (W339A or R525A) or doubly (W339A and R525A) mutated TOG2 on microtubule polymerization dynamics (Figure 6A; 200 nM TOG domain was used). The single or double mutant TOG2 domains failed to suppress catastrophe or to promote rescue: reactions containing W339A, R525A, or W339A and R252A all showed a high frequency of catastrophe, and a complete lack of rescues, similar to what we observed in “no TOG2” control reactions (Figure 6A). The mutant Stu1-TOG2 domains also are impaired for tubulin and microtubule binding (Figure 6B). Circular dichroism spectroscopy confirmed that the mutants were stably folded at 30°C (Supplemental Figure S5; Supplemental Figure S6 shows that the double mutant is also impaired for microtubule binding), so the lack of anti-catastrophe and rescue effects does not result from a mutation-induced defect in folding or stability.
FIGURE 6:

Consequences of mutating the tubulin-binding interface of Stu1-TOG2. (A) W339A (blue) or R525A (green) mutations on the tubulin-binding interface abolish anti-catastrophe (left) and rescue (right) activity comparably to the double mutant W339A, R525A (purple). R386A (pink), a mutation of the CLASP-specific residue identified from the structure, also abolishes anti-catastrophe and rescue-promoting activity. Assays were performed using yeast αβ-tubulin (0.8 µM) with mutant TOG2 (0.2 µM). Catastrophe events for a single trial per mutant are summarized as a survival plot with average frequency (in min−1) and number of measured catastrophes as follows: W339A, 0.118, 153; R525A, 0.0775, 94; W339A, R525A, 0.082, 98; R386A, 0.103, 60. Data with (red) and without (gray) TOG2 are duplicated from Figure 5. (B) W339A (top) and R525A (middle) substantially weaken interactions with unpolymerized αβ-tubulin as detected by sedimentation velocity analytical ultracentrifugation; the double mutant W339A, R252A (bottom) and the CLASP-specific mutant R386A essentially abolishes interactions with tubulin. c(s) distributions are shown for tubulin only (black) and tubulin+mutant (green). (C) Results of a genetic rescue assay in which endogenous Stu1 can be degraded in an auxin-dependent manner, leading to a loss in cell viability. While plasmid-based expression of nondegradable wild-type, full-length Stu1 rescues the inducible growth defect, W339A, R529A, W339A, R529A (double mutant), and R386A point mutations in the full-length protein do not. STU1-AID cells expressing various covering alleles (as listed) were serially diluted and spotted on plates with either DMSO or auxin.
We also investigated the consequences of mutating the CLASP-family–specific residue R386 (Figure 3D). Stu1-TOG2(R386A) failed to suppress catastrophe, and did not enhance rescue (Figure 6A). Stu1-TOG2(R386A) also substantially eliminated the tubulin-binding affinity of Stu1-TOG2 (Figure 6B). Together, these data demonstrate that the CLASP-family–specific residue we identified in the structure is required for the anti-catastrophe, rescue-promoting, and tubulin-binding activities of Stu1-TOG2.
In summary, specific interactions between the isolated Stu1-TOG2 domain and αβ-tubulin underlie the anti-catastrophe and rescue-promoting activities we observed. The CLASP-family–specific residue we identified at the “top” of the presumptive Stu1-TOG2:tubulin interface is essential for these activities. For the polymerase TOGs Stu2-TOG1 and Stu2-TOG2 to bind curved αβ-tubulin, the “top” and “bottom” of the TOG (corresponding to W339 and R525 in Stu1-TOG2) must simultaneously contact αβ-tubulin (Ayaz et al., 2012, 2014). Single mutations in Stu1-TOG2 (W339A or R525A or R386A) abolish anti-catastrophe and rescue activity, so it seems plausible that Stu1-TOG2 exerts its effects on microtubule dynamics by recognizing a particular conformation of αβ-tubulin (see Discussion).
Are the catastrophe-suppressing and rescue-promoting activities of the isolated Stu1-TOG2 domain relevant to the essential function of Stu1 in cells? We addressed this question using a genetic rescue assay (Figure 6C). We prepared cells in which endogenous Stu1 protein could be conditionally depleted (Nishimura et al., 2009): we incorporated a stu1-AID allele at the endogenous STU1 locus, and a transgene to express the TIR1 F-box protein at the HIS3 locus; addition of the plant hormone auxin to these cells results in rapid degradation of Stu1-AID protein and concomitant loss of cell viability. Wild-type Stu1 (nondegradable because no AID), expressed from a centromeric plasmid under control of its endogenous promoter, fully rescues the growth defect caused by depletion of endogenous Stu1-AID (Figure 6C). In contrast, covering alleles with W339A or R525A mutations in the full-length STU1 gene failed to compensate for the loss of endogenous Stu1-AID. A covering allele with the R386A mutation (the CLASP-family–specific residue we implicated in tubulin binding; Figures 3 and 6A) also could not compensate for the loss of endogenous Stu1-AID. Thus, mutations that abolish the tubulin-binding, anti-catastrophe, and rescue-promoting activities of isolated TOG2 in vitro also significantly impair the function of Stu1 in cells.
The results from point mutants in the rescue assay (Figure 6C), and data from others (Funk et al., 2014), all indicate that an intact TOG2 domain is necessary for Stu1 function. Other regions of the protein, for example the dimerization domain or the basic region that mediates binding to the microtubule lattice, have also been shown to make important contributions to the physiological functions of Stu1 (Funk et al., 2014). As a first step toward understanding how other domains of Stu1 might influence the activities of the TOG2 domain, we measured the tubulin- and microtubule-binding affinities of Stu1-TOG1-TOG2, and how it affected microtubule dynamics (Supplemental Figure S7). Even though Stu1-TOG1 by itself does not bind appreciably to αβ-tubulin or microtubules (Figure 2), its presence in Stu1-TOG1-TOG2 modestly increased αβ-tubulin– and microtubule-binding affinity compared with Stu1-TOG2 (Supplemental Figure S7, B and C). In contrast to Stu1-TOG2, the presence of Stu1-TOG1-TOG2 (200 nM) caused a decrease in the rate of microtubule elongation (Supplemental Figure 7A). Thus, it seems that when in the same polypeptide (Stu1-TOG1-TOG2), the presence of the TOG1 domain can influence the activities of TOG2. We do not yet understand the mechanistic origin of the different behaviors of Stu1-TOG1-TOG2.
The TOG2 domain does not detectably increase the end-binding comet
We speculated that Stu1-TOG2 might influence dynamics by affecting the size of the microtubule’s stabilizing cap. End-binding (EB) proteins mark the stabilizing cap region near the growing microtubule end (Bieling et al., 2007; Zanic et al., 2009; Maurer et al., 2012; Duellberg et al., 2016), so we used the yeast EB protein Bim1 (Schwartz et al., 1997) to test whether Stu1-TOG2 influenced the size of the stabilizing cap or the number of EBs therein (Figure 7). We measured Bim1-GFP fluorescence intensity profiles along control microtubules (no Stu1-TOG2 present; Figure 7A) and along microtubules with Stu1-TOG2 (200 nM) present in the assay (Figure 7B). To obtain a higher signal-to-noise measure of the cap, we aligned and averaged multiple profiles together. Then we fitted a Gaussian distribution with different baselines for the lattice and for the background (see Materials and Methods) to quantify the total peak intensity and width of the Bim1-GFP “comet” (Figure 7, C and D). The measured width of the caps (1σ; 0.20 and 0.23 μm for samples without and with TOG2, respectively; Figure 7, C and D) was comparable to the point spread function of our microscope (0.198 µm, also 1σ), which limits our ability to detect small changes in comet length. However, the integrated cap intensity (219 and 194 A.U. for samples without and with TOG2, respectively), which is related to the number of Bim1-GFP proteins bound in the cap region, did not differ significantly (Figure 7, C and D). Thus, while we cannot draw definitive conclusions about Stu1-TOG2–induced changes in the length of the cap, the intensity measurements indicate that the presence of Stu1-TOG2 does not increase the number of high-affinity EB-binding sites in the cap. Consequently, the anti-catastrophe and rescue-promoting effects of Stu1-TOG2 probably do not result from an increase in the size of the microtubule’s stabilizing cap.
FIGURE 7:

TOG2 does not influence the stabilizing cap. (A) Representative still images and associated fluorescence intensity line scans characterizing the comet of Bim1-GFP on the end of growing control microtubules with no Stu1-TOG2 added (microtubules are unlabeled in this experiment, and the direction of growth is left to right). Bim1 is the yeast EB protein. (B) As in A, but with 200 nM Stu1-TOG2 (unlabeled) added to the assay. (C) Aligned and averaged comet profile (n = 18 comets) for Bim1-GFP comets on control microtubules growing without any Stu1-TOG2 present. Solid line shows fit by a Gaussian with different baseline intensity behind the cap (on the microtubule lattice) and in front of the cap (actual fluorescence background; the peak intensity integrates to 219 ± 8 A.U. and the fitted peak width, σ, is 0.20 ± 0.01 µm). (D) As in C, but for the reactions including 200 nM Stu1-TOG2 (n = 30 comets; the peak intensity integrates to 194 ± 6 A.U. and the fitted peak width, σ, 0.23 ± 0.01 µm). Neither peak intensity nor peak width are significantly different between the control and the +Stu1-TOG2 reactions by analysis of variance (for peak intensity: F (1, 37) = 1.667, p = 0.3223; for peak width: F (1, 37) = 1.082, p = 0.9218).
DISCUSSION
Multiple families of microtubule regulatory factors contain arrays of TOG domains. Recent studies of microtubule polymerases have demonstrated that polymerase activity requires at least two linked TOG domains, establishing a paradigm that regulatory function requires arrayed TOGs (Widlund et al., 2011; Ayaz et al., 2014). We began this study with the expectation that the anti-catastrophe and rescue-promoting activity of CLASP-family proteins would also require linked TOGs. However, our experiments revealed that the isolated Stu1-TOG2 domain displays anti-catastrophe and rescue-promoting activity, without the need for a second, linked TOG partner. Whether structurally distinct vertebrate CLASP-family TOGs like hCLASP1-TOG2 possess similar activity remains to be determined. Nevertheless, by demonstrating that an isolated TOG domain can display the same kinds of regulatory activities attributed to the full-length protein from whence the TOG came, the results reported here expand our understanding of what TOG domains can do and how they may do it. In particular, that an isolated CLASP-family TOG can suppress catastrophe and promote rescue indicates that “tethering” of an unpolymerized tubulin to the lattice is not strictly required for these activities.
Not requiring a linked array of TOGs to obtain activities promoted by the intact protein may have implications for other TOG-containing regulatory factors. Indeed, recent structures of TOG3, TOG4, and TOG5 from XMAP215-family microtubule polymerases (Howard et al., 2015; Byrnes and Slep, 2017), and of TOG domains from cilium-localized regulatory factors (Das et al., 2015; Rezabkova et al., 2016), have revealed a range of TOG structures, variable conformation specificity for tubulin binding, and a diversity of linker dockings or other appendages. Understanding how these “alternative” TOGs contribute to different regulatory functions, and whether they need a tethered TOG partner to achieve activity, are interesting questions for future work.
Our data exclude several potential mechanisms for how the isolated Stu1-TOG2 domain suppresses catastrophe and promotes rescue. Less catastrophe and more rescue might in principle be achieved by increasing the rate of microtubule elongation. However, Stu1-TOG2 did not significantly change microtubule elongation rates, so Stu1-TOG2 probably does not exert its effects by changing the conformation of unpolymerized αβ-tubulin or by changing the structure/configuration of the growing microtubule end. Less catastrophe and more rescue could in principle also be achieved by reducing the rate of microtubule shrinking, for example by stabilizing the GDP microtubule lattice. But we observed that the presence of Stu1-TOG2 did not alter microtubule shrinking rates, so it seems unlikely that Stu1-TOG2 exerts its effects by stabilizing the GDP microtubule lattice. Finally, less catastrophe could be achieved by increasing the size of the microtubule’s stabilizing cap. Yet, we measured comparable amounts of Bim1-GFP, the yeast EB protein that marks the stabilizing cap, bound at the growing end in the presence and absence of Stu1-TOG2. Thus, the anti-catastrophe and rescue activity of the isolated Stu1-TOG2 domain cannot be explained by effects on unpolymerized αβ-tubulin, on the configuration of the growing microtubule end, by stabilization of the GDP lattice, or by an increase in the size of the stabilizing cap. Some other as yet unidentified molecular mechanism must be operating.
Whatever the mechanism by which Stu1-TOG2 acts, the genetic rescue assays we performed indicate that the essential function of Stu1 is lost when Stu1-TOG2 interactions with tubulin are abrogated. A similar result was obtained for Cls1, the S. pombe orthologue (Al-Bassam et al., 2010). Given that the isolated Stu1-TOG2 domain is sufficient to suppress catastrophe and promote rescue in vitro, it could be that the other domains of Stu1 serve to recruit more TOG2 to the microtubule lattice (e.g., the basic region) or to amplify the activity of individual TOG2s (e.g., via dimerization). Other regions of CLASPs can dictate specific localization patterns (e.g., an EB-interacting SxIP motif [Honnappa et al., 2009; Patel et al., 2012] or kinetochore localization mediated by Stu1-TOG1 [Funk et al., 2014]). It is also possible that more intimate interdomain cooperation operates to modulate the activity of Stu1-TOG2. Indeed, we find it interesting that both yeast and vertebrate CLASP TOGs feature “docked” linkers that force a more compact arrangement of the linked TOGs. Presumably this conserved feature of CLASP TOGs reflects some important but as yet undetermined constraint on their function.
Suppression of catastrophe and promotion of rescue could in principle represent two distinct molecular activities of Stu1-TOG2. However, given that none of the Stu1-TOG2 mutants we tested separated anti-catastrophe activity from rescue-promoting activity, it seems simpler to consider the possibility that Stu1-TOG2 achieves both effects using a common molecular mechanism. But what is the mechanism? The selective effects on transition frequencies (catastrophe and rescue) without changes in growing or shrinking rates suggest that the isolated Stu1-TOG2 domain acts directly on αβ-tubulin subunits at (or very near) the microtubule end. Stu1-TOG2 affects catastrophe and rescue at low concentrations where its interactions with unpolymerized αβ-tubulin or with the microtubule lattice are far from saturation. One speculative explanation for the different concentration-dependence Stu1-TOG2 binding and activity is that Stu1-TOG2 influences transition frequencies because it binds more tightly to some intermediate state of αβ-tubulin at the microtubule end that is important for catastrophe and rescue but not rate limiting for steady-state growing or shrinking. The same regions required for individual polymerase TOGs to bind curved tubulin are important for the tubulin-binding and anti-catastrophe and rescue-promoting activities of Stu1-TOG2, so this intermediate state might be a partially curved conformation of αβ-tubulin at the microtubule end. This conformation-selective model is appealing because it resonates with the known conformation selectivity of polymerase TOGs like Stu2-TOG1 and Stu2-TOG2. However, our data do not exclude other kinds of models, for example those in which Stu1-TOG2 does not bind more tightly to an intermediate conformation of αβ-tubulin, in which the TOG domain itself undergoes conformational rearrangements on the microtubule, or something else. Resolving this ambiguity will require knowledge about the optimal tubulin conformation for Stu1-TOG2 binding.
MATERIALS AND METHODS
Protein expression and purification
Yeast αβ-tubulin was overexpressed in S. cerevisiae and purified as previously described (Johnson et al., 2011; Geyer et al., 2015). Tubulin aliquots were stored in 10 mM PIPES, pH 6.9, 1 mM MgCl2, 1 mM EGTA (ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid) containing 20 or 50 µM GTP depending on the application. STU1-TOG1 (11-276) and Stu1-TOG2 (305-567) were subcloned into a modified pET28a vector containing N-terminal polyhistidine and SUMO tags (gift from Xuewu Zhang, UT Southwestern), overexpressed in Escherichia coli strain BL21(DE3), and purified using Ni-affinity chromatography. The SUMO tag was cleaved by Ulp protease and removed by either cation exchange chromatography (Stu1-TOG2) or a second Ni-affinity chromatography (Stu1-TOG1). Point mutants in Stu1-TOG2 were prepared using site-directed mutagenesis (Quikchange; Stratagene) and expressed and purified like the wild-type protein. The integrity of all expression constructs was confirmed by DNA sequencing. All proteins were concentrated and dialyzed into RB100 (25 mM Tris, pH 7.5, 100 mM NaCl, 1 mM MgCl2, 1 mM EGTA) for storage. For AUC samples, an additional 20 µM GTP was included in the dialysis buffer.
Crystallization and tantalum bromide soak/phasing
Crystals of STU1-TOG2 were initially obtained using sparse-matrix crystallization screening, performed using a Phenix DT Drop Setter, mixing protein (at ∼10 mg/ml) and precipitant 1:1 for sitting-drop vapor diffusion at 20°C. After optimization, the best crystals were obtained with manual setups using 18% PEG3350, 0.1 M HEPES, pH 7.5, as the precipitant. Crystals were cryoprotected by quick serial transfer through 18% PEG3350, 0.1 M HEPES, pH 7.5, containing 10% glycerol and then 20% glycerol and flash-frozen in liquid nitrogen. To obtain a heavy-atom derivative for experimental phasing, crystals were soaked with tantalum bromide (Jena Bioscience; ∼0.2–0.5 mg) added directly to the 2 μl sitting drop for 1 h before flash-freezing.
Diffraction data were collected by remote control data collection using Argonne National Laboratory APS Beamline 19ID, using wavelengths of 0.97926 and 1.25478 Å for native and derivative data sets, respectively. Diffraction data were processed using HKL3000 (Minor et al., 2006). Crystals adopt space group P21 with one molecule in the asymmetric unit and 42% solvent content. Native and tantalum bromide–soaked crystals diffracted isotropically to 1.89 and 1.80 Å resolution, respectively. The tantalum bromide data set showed a strong anomalous signal of 8.4%. SAD phasing, solvent flattening, and model building were performed using the automated routines in HKL3000 (Minor et al., 2006). Manual rebuilding and refinement against the slightly better diffracting native data set was performed using COOT (Emsley et al., 2010) and PHENIX (Adams et al., 2010), respectively.
Analytical ultracentrifugation
Samples for analytical ultracentrifugation (Stu1-TOG1, Stu1-TOG2 and mutants thereof, yeast αβ-tubulin) were dialyzed into RB100 (25 mM Tris, pH 7.5, 1 mM MgCl2, 1 mM EGTA, 100 mM NaCl) containing 20 μM GTP. Samples were mixed and incubated at 4°C for at least 1 h before the experiment. Analytical ultracentrifugation experiments was performed in an Optima XL-1 centrifuge using an An50-Ti rotor (Beckman Coulter) as previously described (Ayaz et al., 2012, 2014; Geyer et al., 2015), at 20°C after the centrifugation rotor and cells had equilibrated at that temperature for at least 2.5 h. Sedimentation was monitored by absorbance at 280 nm. Buffer viscosities, buffer densities, and protein partial-specific volumes were calculated using SEDNTERP (Laue et al., 1992). SEDFIT (Schuck, 2000) was used to generate c(s) distributions. For binding titrations, c(s) distributions from SEDFIT were imported into GUSSI (Brautigam, 2015) and integrated to generate an isotherm file for SEDPHAT (Schuck, 2010). SEDPHAT was used to fit the binding isotherms for TOG2 and TOG1-TOG2 to a single-site binding model (A + B heteroassociation) with a fixed sAB (maximum sedimentation coefficient allowed) corresponding to a theoretical maximum for a 1:1 tubulin:TOG2/TOG1-TOG2 complex with an assumed frictional ratio of 1.3.
Microtubule cosedimentation assays
Microtubule cosedimentation assays were carried out as previously described (Ayaz et al., 2012; Geyer et al., 2015) using 3 µM wild-type yeast αβ-tubulin in 1× G-PEM assembly buffer (100 mM PIPES, pH 6.9, 20% glycerol, 2 mM MgSO4, 1 mM EGTA). Tubulin samples containing 5 mM GTP were polymerized for 30 min at 30°C before a candidate binding partner was added to a final concentration of 9 µM. Mixed samples were incubated for an additional 30 min at 30°C before pelleted by ultracentrifugation at 60,000 rpm (∼150,000 × g) at 30°C for 30 min in a prewarmed TLA-100 rotor (Beckman Coulter). Afterward, supernatant was carefully removed, and the pellet was resuspended in an equal volume of assembly buffer, and samples were analyzed by SDS–PAGE.
SAXS: data collection, analysis, modeling
Purified protein samples were exchanged into RB100 containing 5% glycerol using a Superdex 200 10/300 column. The samples for SAXS were at a concentration between 1 and 4 mg/ml. Measurements were taken at room temperature using the SAXS instrument at the 12ID-B Beamline of the Advanced Photon Source, Argonne National Laboratory. The energy of x-rays was 14 keV. Data were collected using a Pilatus 2 M detector (DECTRIS) at a sample-to-detector distance of ∼2 m, covering ∼0.004–0.85 A−1 for the scattering vector Q. Immediately before data collection, samples were centrifuged at 15,000 g for 5 min, and the top 80% of the solution was used for the SAXS measurement. This process helps eliminate any aggregates. Samples were measured in a quartz capillary flow cell with a 1.5 mm diameter under a flow rate of 10 µl/s to mitigate potential radiation damage. Successive frames (30–40) with an exposure of 1 s were recorded for each sample, and the resulting data averaged to improve signal and reduce noise. Three concentrations of each sample were measured (“neat,” twofold, and fourfold diluted). Each sample measurement was preceded by the measurement of its matching buffer solution. In addition to being used for background subtraction, the buffer measurements also served as a check on beam properties and the cleanliness of the sample cell between switching of samples.
Scattering profiles (intensity I vs. scattering vector Q) were reduced and SAXS data were merged using beamline software. After buffer background subtraction, to eliminate the possible structure factor, the SAXS data were extrapolated against concentration to obtain the final zero-concentration SAXS data set for further data analysis. Pair distance distribution functions, P(r), were calculated with GNOM (Svergun, 1992).
Low-resolution molecular shape reconstructions of STU1-TOG2 from the experimental scattering data were performed with DAMMIN (Svergun, 1999) or DAMMIF (Franke and Svergun, 2009) with similar results. The scattering profiles were used up to a Q of 0.49 Å−1 for the reconstruction. Multiple calculations were performed, and the resulting bead models were averaged by DAMAVER (Volkov and Svergun, 2003) to generate a final model representing the most probable shape for the protein. Fitting of the crystal structure of STU1-TOG2 into the SAXS envelope was carried out using Chimera (Pettersen et al., 2004).
Limited chymotrypsin proteolysis
TOG1-TOG2 fragments from STU1 or STU2 (5 µM final concentration) were incubated with different amounts of chymotrypsin for 90 min at room temperature. Chymotrypsin was serially diluted (10-fold steps) from a stock concentration of 1 mg/ml. The reaction was quenched by adding SDS–PAGE loading buffer and boiling for 10 min. Samples were analyzed by SDS–PAGE.
Time-lapse DIC microscopy
Flow chambers were prepared as described previously (Gell et al., 2010; Geyer et al., 2015; Piedra et al., 2016) with the exception that GTPγS-stabilized “seed” yeast microtubules were used instead of axonemes to template microtubule growth. To make seeds, 3 µM wild-type yeast αβ-tubulin in 1× G-PEM was allowed to polymerize overnight at 30°C with 2 mM GTPγS. The resulting GTPγS-stabilized microtubules were sheared by vigorous pipetting before being introduced into the flow chamber. Chambers were preincubated for 10 min with an anti-His-tag antibody (Millipore) diluted to 5 µg/ml in BRB80, 80 mM PIPES, pH 6.9, 1 mM MgCl2, 1 mM EGTA (Millipore). Chambers were then blocked with 1% F-127 pluronic in BRB80 for 5 min, and washed with 1× PEM (100 mM PIPES, pH 6.9, 1 mM EGTA, 1 mM MgSO4) containing 1 mM GTP. Then sheared seeds (3 µl) were flowed in and incubated for 10 min. Yeast αβ-tubulin aliquots were taken from −80°C, rapidly thawed, and passed through a 0.1 µm centrifugal filter at 4°C to remove aggregates. The concentration of αβ-tubulin was measured by UV absorbance using an extinction coefficient of 115,000 M−1cm−1. Samples containing αβ-tubulin and accessory protein (STU1 TOGs or mutants) in 1× PEM + 0.1 mg/ml bovine serum albumin (BSA) + 1 mM GTP + 10% RB100 were flowed into the prepared chamber containing seeds and the chamber immediately sealed with VALAP. Control experiments were carried out in the identical buffer and included 10% RB100 (without any accessory protein) to ensure matching buffer conditions.
Microtubule (MT) dynamics were imaged by DIC using an Olympus IX81 microscope with a PlanS Apo N 100×/1.49 NA objective lens and DIC prisms. Illumination at 550 nm was obtained by inserting a bandpass filter of 550/100 nm (Olympus) in the light path. Temperature was maintained at 30°C using a WeatherStation temperature controller with enclosure fitted to the microscope’s body. Micro-Manager 2.0 (Edelstein et al., 2010) was used to control the microscope and a Photometrics Prime95B camera used to record the reactions. MT dynamics were recorded by taking an averaged set of 10 images of 100 ms exposure every 5 s for 37.5 min. Averaging was carried out using the FrameCombiner plug-in in Micro-Manager 2.0. At the end of each movie, an averaged set of 200 out-of-focus background images was taken for background correction. The intensity of each image was normalized to its mean before the entire stack was divided by the background. To correct for temporal illumination variation during data acquisition, a second flat-field correction was performed using a flat-field image created by applying a broad 20 pixel Gaussian filter to the original image. To improve contrast, a 2-pixel variance filter was applied. Kymographs were generated using ImageJ Reslice and MT length over time measured manually. Rates of MT elongation were calculated using MT length and timestamps of images. Lifetimes of each growing phase right before a catastrophe were also calculated and survival plots generated using GraphPad Prism. Catastrophe frequency was calculated as the number of observed catastrophes divided by the sum total of lifetimes. Rescues were noted along with respective shrinking distances to calculate the number of rescues per micrometer of shrinking, then converted to frequency using the shrinking rate (see below).
To measure shrinking rates, chambers were prepared as described above and incubated for 10 min to allow for growth before images were acquired continuously (streaming), obtaining images every 20 ms. We used Micro-Manager’s “On-the-fly-data-processing” plug-in to average 50 images together to form a single frame. Data processing was carried out as described above.
TIRF microscopy (MT lattice binding)
Yeast tubulin with a C-terminal WDCCPGCCK tag (Griffin et al., 2000) was expressed and purified as described above for wild-type yeast tubulin (also described in Geyer et al., 2015). Aliquots (3 µM) were thawed and labeled with ReAsH-EDT2 reagent (Toronto Research Chemicals) by incubating with fivefold molar excess reagent and 2 mM TCEP for 90 min at room temperature. Excess free dye was then removed using Zeba Spin desalting columns (Thermo Fisher), and GTPγS-stabilized microtubules were prepared as above. Stu1-TOG constructs with a KCK tag were purified as described above and stored at −80°C. Aliquots were thawed on each day and labeled with Alexa-488 (similar to the ReAsH labeling protocol above). Preparation of flow chambers was carried out as described above for DIC except GTPγS-stabilized microtubules were not sheared before being flowed in. After microtubule incubation, 20 µl RB100 + 10 µM epothilone (to keep microtubules stable) containing various concentrations of fluorescent Stu1-TOG was flowed in.
Interactions of fluorescent Stu1 TOG constructs with MTs were imaged by total internal reflection fluorescence microscopy using an Olympus IX81 microscope with a TIRF ApoN 100×/1.49 objective lens, a 491 and 561 nm 50 mW solid-state laser, and a Photometrics Prime95B camera. Reactions were done at room temperature and the microscope was controlled as described above. Images were taken with 5 ms exposure for the 561 nm channel and with 20 ms exposure for the 491 nm channel. Bleed through between channels was negligible. The fluorescence intensity from the MT and the background fluorescence were measured by using the PlotProfile function (4-pixel-wide measurement lines oriented perpendicularly to the MTs) in ImageJ (Schneider et al., 2012). These linescans were fitted to a custom function (a sum of a sloped line, representing the background fluorescence and a normal distribution with predetermined sigma from point spread function (PSF) measurements, representing the fluorescence from the MT):
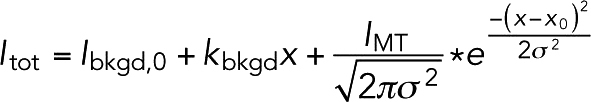 |
where Ibkgd,0 is the background intensity at x = 0 and kbkgd is the slope of background intensity as a function of x. IMT is the fluorescence intensity from the MT, σ = 0.2 µm is the spread of the PSF, and x0 is the location of the MT. The background intensity at x0 was used as the background intensity fluorescence for all following calculations. The fluorescence intensity of MT is proportional (ϕ gives the proportionality constant) to the laser power (LP), the fluorophore labeling efficiency (LE), and the fractional saturation of TOG binding to MTs (f):
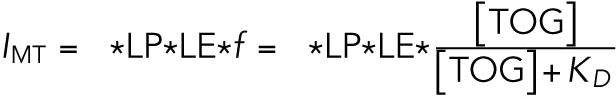 |
The fluorescence intensity of the background is proportional to the LP, the fluorophore LE, and the concentration of the protein:
The quotient of above two intensities does not depend on the LP or the LE (which vary from experiment to experiment). Multiplying the quotient by the concentration of TOG, we obtain a value that is proportional to the fractional saturation:
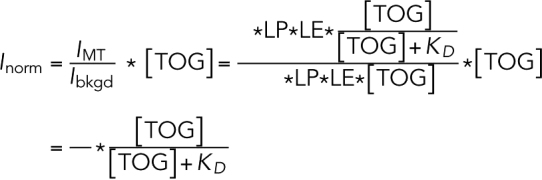 |
Finally, we normalize the green channel (TOG) intensity by the red channel (MT) intensity to account for the presence of some microtubule bundling (based on the distribution of microtubule fluorescence intensities, roughly 69% of the microtubules measured were singles, with ∼29% as two-wide bundles). All measured intensity ratios of all TOG constructs were fitted simultaneously with a binding isotherm with a global maximum intensity ratio:
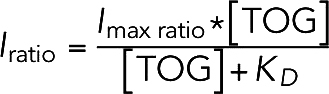 |
Comet analysis using Bim1-GFP
Preparation of flow chambers was carried out as described above. Samples of wild-type αβ-tubulin along with 20 nM Bim1-GFP, with or without 0.2 µM STU1-TOG2, in imaging buffer (1× PEM + 0.1 mg/ml BSA + antifade reagents [glucose, glucose oxidase, catalase]; Gell et al., 2010) were flowed into the chamber. Interactions of Bim1-GFP with MTs were imaged by total internal reflection fluorescence microscopy using an Olympus IX81 microscope with a TIRF ApoN 100×/1.49 objective lens, a 491 nm 50 mW solid-state laser, and a Photometrics Prime95B camera. Reactions were temperature controlled at 30°C and the microscope was controlled as described above. Images of MTs were taken every 5 s for 15 min. Bim1-GFP fluorescence intensity along microtubules and extending beyond their growing ends was obtained using the PlotProfile function in ImageJ (Schneider et al., 2012). These linescans were aligned using a custom-made function in MATLAB (see below). In this function, the linescans are initially aligned by aligning the maximum intensity points from the linescans. Then, the linescans were allowed to move one by one minimizing the average squared distance from the averaged linescan. This process was iterated until all linescans were stably aligned.
The function used for linescan alignment in MATLAB is inspired by Maurer et al. (2014), and includes terms for constant background fluorescence intensity, a Gaussian-shaped Bim1-GFP comet, and a soft roll-off to capture the boundary between the Bim1-GFP fluorescence and the background:
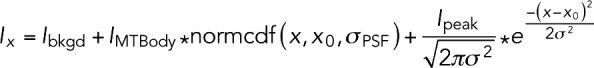 |
where Ix is the fluorescence intensity at position x, x0 is the position of the center of the comet, σPSF represents the PSF of our microscope, and σ is the comet width; Ibkgd, IMTBody, and Ipeak are the intensities of the background, body of the microtubule lattice, and peak, respectively; and normcdf is the cumulative normal distribution (used to model the falloff in fluorescence intensity at the microtubule end).
Genetic rescue assay
We used an auxin-inducible degron system (Nishimura et al., 2009) to create a genetic rescue assay. The S. cerevisiae strains used are described in Supplemental Table S1. STU1-3HA-IAA7 was generated using PCR-based methods described in Longtine et al. (1998) and Miller et al. (2016) and integrated at the endogenous STU1 locus. pGPD1-TIR1 integration plasmid for integration at the His3 locus was a gift from Matthew Miller and Sue Biggins (Fred Hutchinson Cancer Research Institute). Cells from the resulting strain express 1) Stu1 fused to an auxin-responsive protein (IAA7) from endogenous STU1 locus, and 2) TIR1, a protein required for auxin-induced degradation (Nishimura et al., 2009).
Covering alleles were generated as follows. The STU1 gene with upstream and downstream regulatory sequences was amplified using PCR and subcloned into pRS315, a LEU2 centromeric plasmid (gift from Christine Weirich, UT Southwestern). To avoid introducing undesired mutations in the rescue plasmid, point mutations in Stu1-TOG2 were introduced by QuikChange mutagenesis using a separate plasmid containing the coding sequence for residues 1–700 of STU1. After mutations were confirmed by sequencing, the MscI/PstI fragment was liberated for subcloning into the similarly digested pRS315 rescue plasmid containing full-length STU1. The STU1-AID strain was grown overnight in YPD medium, made competent following Frozen EZ-Yeast transformation protocol (Zymogen), transformed with the covering alleles and plated on Leu(–) selective media.
For the spotting assay, the strains transformed with covering alleles were grown overnight in Leu(–) selective media and cells diluted to OD600 = ∼1 from which serial 1:10 dilutions were made and spotted onto YPD plates with dimethyl sulfoxide (DMSO) or 0.4 mM auxin (indole-3-acetic acid; Sigma; dissolved in DMSO). Plates were incubated at 30°C for 2 d.
Analysis of sequence conservation
Sequences for CLASP- and polymerase-family proteins from S. cerevisiae (Stu1 and Stu2), S. pombe (Cls1 and Alp14), Drosophia melanogaster (mast/Orbit and minispindles), Arabidopsis thaliana (CLASP and MOR1), Xenopus laevis (XCLASP1A and XMAP215), and Homo sapiens (CLASP1 and ch-TOG) were obtained from the National Center for Biotechnology Information (NCBI) and aligned using T-coffee (Notredame et al., 2000). Portions of the multiple-sequence alignment in the region of Stu1-TOG2(W339) and Stu1-TOG2(R386) are shown in Figure 3.
Microscale thermophoresis
Protein stocks including labeled tubulin and unlabeled TOG constructs were purified and labeled as above. Buffer condition was identical to AUC: RB100 + 20 μM GTP. A 15-point, 1:1 dilution series of TOG2 with 120 μM as its highest concentration was prepared in 10 μl aliquots. A 16th sample was prepared with no TOG2. To each tube, 10 μl of 80 nM tubulin that had been supplemented to 0.1% (vol/vol) Tween-20 was added and mixed, giving a highest [TOG2] of 60 μM (for TOG1-TOG2, the highest concentration is 32.5 µM), a constant [tubulin] of 40 nM (50 nM for TOG1-TOG2 titrations), and a final concentration of Tween-20 of 0.05%. Samples were incubated in the dark for at least 30 min before loading them into premium-coated capillaries and placing them in the instrument (a NanoTemper NT.115 BLUE/RED device). The blue filter was selected, and three replicate titrations performed using 80% MST, 70% LED, a pre-IR phase of 5 s, an IR-on phase of 30 s, and a post-IR phase of 5 s (95% LED for TOG1-TOG2 titration). The data were analyzed using the 1:1 binding model in the program PALMIST (Scheuermann et al., 2016).
Circular dichroism
Purified proteins (STU1-TOG2, wild type and point mutants) were dialyzed into 10 mM Na phosphate, pH 7.5, 50 mM NaCl and diluted to ∼0.4 mg/ml using the same buffer. Circular dichroism (CD) spectroscopy was performed on a Jasco J-815 CD spectrometer. Samples (300 µl) were prepared and placed in a 0.1 cm CD cuvette, degassed, and spectra collected from 190 to 250 nm. The CD as a function of temperature was monitored at 208 nm and 221 nm. Temperature was varied from 25°C to 95°C at a ramp rate of 1°C/min. Melt data were fitted to the equation
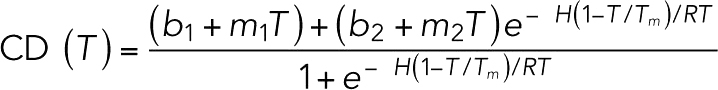 |
where b1 and m1 are, respectively, intercept and slope parameters for the left portion of the curve, and b2 and m2 are, respectively, intercept and slope parameters for the right side of the curve, Tm is the melting temperature, and R is the universal gas constant.
Supplementary Material
Acknowledgments
We thank X. Zhang for the gift of the ppSUMO expression plasmid and M. Miller (S. Biggins lab) and C. Weirich (J. Erzberger lab) for gifts of plasmids and advice on the genetic rescue assay. We also thank the entire lab for helpful discussions and for providing critical comments and E. Bonventre for assistance with cloning and mutagenesis. L.M.R. is the Thomas O. Hicks Scholar in Medical Research. This work was supported by grants to L.M.R. from the National Institutes of Health (NIH; R01GM098543), the Robert A. Welch Foundation (I-1908), and the National Science Foundation (MCB-1615938). T.K. is supported by NIH T32 GM-008297. Results shown in this report are derived from work performed at SBC Beamline 19-ID and at Beamline 12-ID-B at the Advanced Photon Source, Argonne National Laboratory. Advanced Photon Source is a U.S. Department of Energy (DOE) Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Some molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institute of General Medical Sciences P41-GM103311). Atomic coordinates and diffraction data have been deposited in the Protein Data Bank, accession code 6COK.
Abbreviations used:
- AID
auxin-inducible degron
- APS
advanced photon source
- A.U.
arbitrary units
- AUC
analytical ultracentrifugation
- BSA
bovine serum albumin
- CD
circular dichroism
- CLASP
cytoplasmic linker associated protein
- DIC
differential interference contrast
- DOE
Department of Energy
- EB
end-binding
- EGTA
ethylene glycol-bis(β-aminoethyl ether)- N, N, N’, N’-tetraacetic acid
- FOM
figure of merit
- GFP
green fluorescent protein
- GTP
guanosine 5’-triphosphate
- GTPγS
guanosine 5’- O-[gamma-thio]triphosphate
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- MST
microscale thermophoresis
- MT
microtubule
- NCBI
National Center for Biotechnology Information
- NIH
National Institutes of Health
- PEG
polyethylene glycol
- PEM
PIPES, EGTA, magnesium
- PIPES
piperazine- N, N’-bis(2-ethanesulfonic acid)
- PSF
point spread function
- SAD
single-wavelength anomalous dispersion
- SAXS
small-angle x-ray scattering
- Stu1
suppressor of tubulin 1
- Stu2
suppressor of tubulin 2
- SUMO
small ubiquitin-like modifier
- TIRF
total internal reflection fluorescence
- TOG
tumor overexpressed gene
- VALAP
Vaseline lanolin paraffin
- XMAP215
Xenopus microtubule assembly protein 215 kDa.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-12-0748) on April 5, 2018.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr , 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmanova A, Hoogenraad CC, Drabek K, Stepanova T, Dortland B, Verkerk T, Vermeulen W, Burgering BM, De Zeeuw CI, Grosveld F, et al. (2001). Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell , 923–935. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Steinmetz MO. (2015). Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol , 711–726. [DOI] [PubMed] [Google Scholar]
- Al-Bassam J, Chang F. (2011). Regulation of microtubule dynamics by TOG-domain proteins XMAP215/Dis1 and CLASP. Trends Cell Biol , 604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Bassam J, Kim H, Brouhard G, van Oijen A, Harrison SC, Chang F. (2010). CLASP promotes microtubule rescue by recruiting tubulin dimers to the microtubule. Dev Cell , 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Bassam J, Larsen NA, Hyman AA, Harrison SC. (2007). Crystal structure of a TOG domain: conserved features of XMAP215/Dis1-family TOG domains and implications for tubulin binding. Structure , 355–362. [DOI] [PubMed] [Google Scholar]
- Ayaz P, Munyoki S, Geyer EA, Piedra FA, Vu ES, Bromberg R, Otwinowski Z, Grishin NV, Brautigam CA, Rice LM. (2014). A tethered delivery mechanism explains the catalytic action of a microtubule polymerase. Elife , e03069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayaz P, Ye X, Huddleston P, Brautigam CA, Rice LM. (2012). A TOG:αβ-tubulin complex structure reveals conformation-based mechanisms for a microtubule polymerase. Science , 857–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieling P, Laan L, Schek H, Munteanu EL, Sandblad L, Dogterom M, Brunner D, Surrey T. (2007). Reconstitution of a microtubule plus-end tracking system in vitro. Nature , 1100–1105. [DOI] [PubMed] [Google Scholar]
- Bratman SV, Chang F. (2007). Stabilization of overlapping microtubules by fission yeast CLASP. Dev Cell , 812–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam CA. (2015). Calculations and publication-quality illustrations for analytical ultracentrifugation data. Methods Enzymol , 109–133. [DOI] [PubMed] [Google Scholar]
- Byrnes AE, Slep KC. (2017). TOG-tubulin binding specificity promotes microtubule dynamics and mitotic spindle formation. J Cell Biol , 1641–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr , 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Dickinson DJ, Wood CC, Goldstein B, Slep KC. (2015). Crescerin uses a TOG domain array to regulate microtubules in the primary cilium. Mol Biol Cell , 4248–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabek K, van Ham M, Stepanova T, Draegestein K, van Horssen R, Sayas CL, Akhmanova A, Ten Hagen T, Smits R, Fodde R, et al. (2006). Role of CLASP2 in microtubule stabilization and the regulation of persistent motility. Curr Biol , 2259–2264. [DOI] [PubMed] [Google Scholar]
- Duellberg C, Cade NI, Holmes D, Surrey T. (2016). The size of the EB cap determines instantaneous microtubule stability. Elife , 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. (2010). Computer control of microscopes using µManager. Curr Protoc Mol Biol , 14.20-1–14.20-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr , 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JC, Howard AE, Currie JD, Rogers SL, Slep KC. (2014). The XMAP215 family drives microtubule polymerization using a structurally diverse TOG array. Mol Biol Cell , 2375–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Svergun DI. (2009). DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr , 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk C, Schmeiser V, Ortiz J, Lechner J. (2014). A TOGL domain specifically targets yeast CLASP to kinetochores to stabilize kinetochore microtubules. J Cell Biol , 555–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gell C, Bormuth V, Brouhard GJ, Cohen DN, Diez S, Friel CT, Helenius J, Nitzsche B, Petzold H, Ribbe J, et al. (2010). Microtubule dynamics reconstituted in vitro and imaged by single-molecule fluorescence microscopy. Methods Cell Biol , 221–245. [DOI] [PubMed] [Google Scholar]
- Geyer EA, Burns A, Lalonde BA, Ye X, Piedra FA, Huffaker TC, Rice LM. (2015). A mutation uncouples the tubulin conformational and GTPase cycles, revealing allosteric control of microtubule dynamics. Elife , 3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin BA, Adams SR, Jones J, Tsien RY. (2000). Fluorescent labeling of recombinant proteins in living cells with FlAsH. Methods Enzymol , 565–578. [DOI] [PubMed] [Google Scholar]
- Honnappa S, Gouveia SM, Weisbrich A, Damberger FF, Bhavesh NS, Jawhari H, Grigoriev I, van Rijssel FJ, Buey RM, Lawera A, et al. (2009). An EB1-binding motif acts as a microtubule tip localization signal. Cell , 366–376. [DOI] [PubMed] [Google Scholar]
- Howard AE, Fox JC, Slep KC. (2015). Drosophila melanogaster mini spindles TOG3 utilizes unique structural elements to promote domain stability and maintain a TOG1- and TOG2-like tubulin-binding surface. J Biol Chem , 10149–10162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue YH, Savoian MS, Suzuki T, Máthé E, Yamamoto MT, Glover DM. (2004). Mutations in orbit/mast reveal that the central spindle is comprised of two microtubule populations, those that initiate cleavage and those that propagate furrow ingression. J Cell Biol , 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson V, Ayaz P, Huddleston P, Rice LM. (2011). Design, overexpression, and purification of polymerization-blocked yeast αβ-tubulin mutants. Biochemistry , 8636–8644. [DOI] [PubMed] [Google Scholar]
- Laue TM, Shah BD, Ridgeway RM, Pelletier SL. (1992). Analytical Ultracentrifugation in Biochemistry and Polymer Science, ed. Harding SE, Rowe AJ, and Horton JC, Cambridge, UK: The Royal Society of Chemistry, 90–125. [Google Scholar]
- Leano JB, Rogers SL, Slep KC. (2013). A cryptic TOG domain with a distinct architecture underlies CLASP-dependent bipolar spindle formation. Structure , 939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast , 953–961. [DOI] [PubMed] [Google Scholar]
- Maiato H, Fairley EAL, Rieder CL, Swedlow JR, Sunkel CE, Earnshaw WC. (2003). Human CLASP1 is an outer kinetochore component that regulates spindle microtubule dynamics. Cell , 891–904. [DOI] [PubMed] [Google Scholar]
- Maiato H, Khodjakov A, Rieder CL. (2005). Drosophila CLASP is required for the incorporation of microtubule subunits into fluxing kinetochore fibres. Nat Cell Biol , 42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki T, Grimaldi AD, Fuchigami S, Kaverina I, Hayashi I. (2015). CLASP2 has two distinct TOG domains that contribute differently to microtubule dynamics. J Mol Biol , 2379–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer SP, Cade NI, Bohner G, Gustafsson N, Boutant E, Surrey T. (2014). EB1 accelerates two conformational transitions important for microtubule maturation and dynamics. Curr Biol , 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer SP, Fourniol FJ, Bohner G, Moores CA, Surrey T. (2012). EBs recognize a nucleotide-dependent structural cap at growing microtubule ends. Cell , 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MP, Asbury CL, Biggins S. (2016). A TOG protein confers tension sensitivity to kinetochore-microtubule attachments. Cell , 1428–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori-Kiyosue Y, Grigoriev I, Lansbergen G, Sasaki H, Matsui C, Severin F, Galjart N, Grosveld F, Vorobjev I, Tsukita S, et al. (2005). CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J Cell Biol , 141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. (2006). HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr , 859–866. [DOI] [PubMed] [Google Scholar]
- Moriwaki T, Goshima G. (2016). Five factors can reconstitute all three phases of microtubule polymerization dynamics. J Cell Biol , 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods , 917–922. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. (2000). T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol , 205–217. [DOI] [PubMed] [Google Scholar]
- Ortiz J, Funk C, Schäfer A, Lechner J. (2009). Stu1 inversely regulates kinetochore capture and spindle stability. Genes Dev , 2778–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualone D, Huffaker TC. (1994). STU1, a suppressor of a beta-tubulin mutation, encodes a novel and essential component of the yeast mitotic spindle. J Cell Biol , 1973–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel K, Nogales E, Heald R. (2012). Multiple domains of human CLASP contribute to microtubule dynamics and organization in vitro and in Xenopus egg extracts. Cytoskeleton (Hoboken) , 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira AL, Pereira AJ, Maia AR, Drabek K, Sayas CL, Hergert PJ, Lince-Faria M, Matos I, Duque C, Stepanova T, et al. (2006). Mammalian CLASP1 and CLASP2 cooperate to ensure mitotic fidelity by regulating spindle and kinetochore function. Mol Biol Cell , 4526–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem , 1605–1612. [DOI] [PubMed] [Google Scholar]
- Piedra FA, Kim T, Garza ES, Geyer EA, Burns A, Ye X, Rice LM. (2016). GDP-to-GTP exchange on the microtubule end can contribute to the frequency of catastrophe. Mol Biol Cell , 3515–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezabkova L, Kraatz SHW, Akhmanova A, Steinmetz MO, Kammerer RA. (2016). Biophysical and structural characterization of the centriolar Protein Cep104 interaction network. J Biol Chem , 18496–18504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann TH, Padrick SB, Gardner KH, Brautigam CA. (2016). On the acquisition and analysis of microscale thermophoresis data. Anal Biochem , 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. (2012). NIH image to ImageJ: 25 years of image analysis. Nat Methods , 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. (2000). Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J , 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. (2010). Sedimentation patterns of rapidly reversible protein interactions. Biophys J , 2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz K, Richards K, Botstein D. (1997). BIM1 encodes a microtubule-binding protein in yeast. Mol Biol Cell , 2677–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slep KC. (2009). The role of TOG domains in microtubule plus end dynamics. Biochem Soc Trans , 1002–1006. [DOI] [PubMed] [Google Scholar]
- Slep KC, Vale RD. (2007). Structural basis of microtubule plus end tracking by XMAP215, CLIP-170, and EB1. Mol Cell , 976–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI. (1992). Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr , 495–503. [Google Scholar]
- Svergun DI. (1999). Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J , 2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. (2003). Uniqueness of ab-initio shape determination in small-angle X-ray scattering. J Appl Crystallogr , 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlund PO, Stear JH, Pozniakovsky A, Zanic M, Reber S, Brouhard GJ, Hyman AA, Howard J. (2011). XMAP215 polymerase activity is built by combining multiple tubulin-binding TOG domains and a basic lattice-binding region. Proc Natl Acad Sci USA , 2741–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanic M, Stear JH, Hyman AA, Howard J. (2009). EB1 recognizes the nucleotide state of tubulin in the microtubule lattice. PLoS One , e7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.