

Abstract
As a hot topic of epigenetic studies, histone deacetylases (HDACs) are related to lots of diseases, especially cancer. Further researches indicated that different HDAC isoforms played various roles in a wide range of tumor types. Herein a novel series of HDAC inhibitors with isatin-based caps and o-phenylenediamine-based zinc binding groups have been designed and synthesized through scaffold hopping strategy. Among these compounds, the most potent compound 9n exhibited similar if not better HDAC inhibition and antiproliferative activities against multiple tumor cell lines compared with the positive control entinostat (MS-275). Additionally, compared with MS-275 (IC50 values for HDAC1, 2 and 3 were 0.163, 0.396 and 0.605 μM, respectively), compound 9n with IC50 values of 0.032, 0.256 and 0.311 μM for HDAC1, 2 and 3 respectively, showed a moderate HDAC1 selectivity.
Keywords: Anticancer, Epigenetics, HDAC inhibitors, Isatin
1. Introduction
The onset and progression of cancer, traditionally seen as a genetic disease, are revealed to be associated with epigenetic changes along with genetic abnormalities in recent years.1 Histone deacetylases (HDACs) as novel targets for the discovery of anticancer agents are a series of enzymes that regulate histone deacetylation, which may be the best understood type of epigenetic modifications.2 HDACs can enrich the histone tails with positive charges via removing an acetyl group from the ε-amino group of lysine residue, forcing the compaction of the chromatin containing electronegative DNA. The packaged chromatin prevents transcription factors from DNA promoters, thereby repressing the expression of genes, including various tumor-suppressor genes.3 In addition, a mass of non-histone proteins with diverse biological functions are also substrates of HDACs, such as transcription factors (p53), nuclear factors (NF-κB), α-tubulin, heat-shock protein 90 (Hsp90), estrogen receptor (ERα), DNA repair protein Ku70 and so on.4 18 mammalian HDACs have been identified and grouped into classes I–IV based on their homology to the yeast (Saccharomyces cerevisiae) HDACs.5 Class I HDACs comprising HDAC1, 2, 3 and 8 are homologous to the yeast transcriptional regulator reduced potassium dependency-3 (RPD3). Class II HDACs which are structurally close to yeast histone deacetylase1 (Had1) are divided into two subfamilies. HDAC4, 5, 7 and 9 compose the class IIa HDACs, while HDAC6 and 10 constitute the class IIb HDACs. Class III HDACs consisting of seven isoforms (SIRT1-7) are most related to the yeast silent information regulator (sir 2) family. HDAC11, the lastly discovered isoform, is the only member of class IV HDAC which shares the homology with class I and II HDACs. The classical HDACs (class I, II, and IV) are Zn2+ dependent enzymes, whereas class III HDACs needs the co-factor NAD+ for their activity.3
Higher expression levels of class I HDACs were observed in tumor cells than in normal cells. HDAC1 has been implicated in the initiation and progression of various types of tumor. For instance, high level of HDAC1 is associated with rapid tumor cell proliferation and genomic instability in prostate cancer.6 Moreover, the knockdown of HDAC1 in leukemia cells K562, HL-60, and U937 significantly inhibited cell cycle progression and cell proliferation.7 HDAC2 overexpression is an independent marker of poor prognosis in patients with oral, prostate, ovarian, endometrial or gastric cancer.8 HDAC3 always forms a stable complex with the nuclear receptor co-repressor (N-CoR) and the silencing mediator of retinoic and thyroid receptors (SMRT), which are relevant to a wide range of biological processes, such as cell differentiation, proliferation and apoptosis.9–11 Therefore, the discovery of novel class I-selective HDAC inhibitors (HDACIs) as antineoplastic agents are of interest for clinical use.
In recent decades, more than 20 chemical entities have been studied as HDACIs in different clinical trials. Among them, five compounds were successfully accepted by U.S.FDA or Chinese FDA (CFDA) as novel anti-cancer agents (Fig. 1). Vorinostat (SAHA) and romidepsin (FK228) were approved by U.S.FDA for the chemotherapeutics against refractory cutaneous T-cell lymphoma (CTCL). Belinostat (PXD-101) and panobinostat (LBH-589) were permitted by U.S.FDA to remedy for relapsed or refractory peripheral T-cell lymphoma (PTCL) and multiple myeloma, respectively. Unlike the above four pan-HDACIs, chidamide (CS055/HBI-8000) which obtained the approval from CFDA as a monotherapy for relapsed and refractory peripheral T-cell lymphoma (PTCL) is an isoform-selective HDACI.12 Chidamide, as well as its benzamide derivatives entinostat (MS-275) and CI-994 (Fig. 2), shows more potent inhibition against class I HDACs (HDAC1, 2, and 3) than other classes. The pharmacophore of most HDACIs comprises three units: zinc binding group (ZBG) binding the Zn2+ at the bottom of the active site; cap group (CAP) specifically interacting with the amino acid residues around the entrance of the active site; a linker that connects the ZBG and CAP occupying the 11 Å channel of the active site (Fig. 1).13,14
Fig. 1.
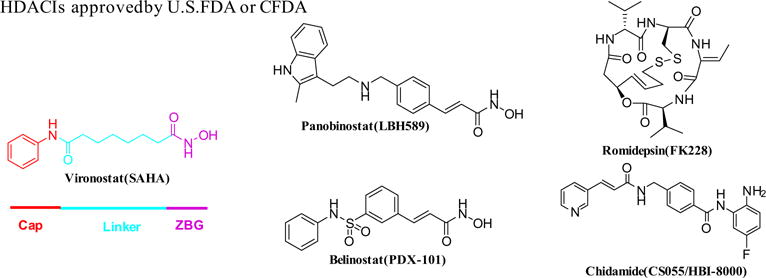
Pharmacophore model and chemical structures of representative HDACIs.
Fig. 2.
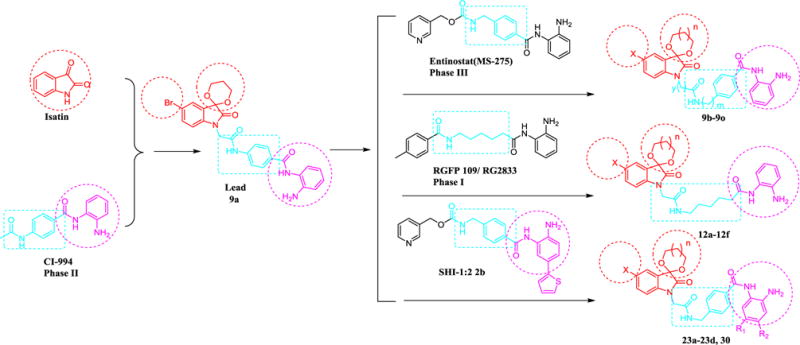
The design of HDACIs through scaffold splicing and hopping strategy.
As shown in Fig. 2, indoline-2, 3-dione also called isatin is an endogenous molecule with a wide range of biological activities, including antiviral activity, antitumor and antiangiogenic activities, antibacterial, antitubercular, antifungal, anticonvulsant and anxyolytic activities.15 For example, indirubin as a derivative of isatin is the active ingredient of Danggui Longhui Wan used to treat chronic myelocytic leukemia (CML).16 In our pioneering work, a series of hydroxamic acid derivatives containing isatin-based caps have been synthesized and evaluated as potential HDACIs. Meanwhile, a benzamide-based molecule 9a has been obtained by splicing isatin to the structure of the clinical HDACI, CI-994.17Although compound 9a and the positive control MS-275 exhibited similar HDACs inhibition, 9a exhibited inferior in vitro antiproliferative activity. Herein, structural modification of the lead compound 9a was carried out through scaffold hopping strategy (Fig. 2) to find more potent HDACIs.
2. Results and discussion
2.1. Chemistry
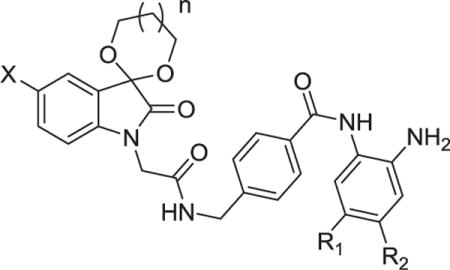
The synthesis of compounds 9a–9o was shown in Scheme 1. Reaction of starting materials 1a–1c, hydroxylamide hydrochloride and chloral hydrate led to compounds 2a–2c, respectively. Sandmeyer reaction was used for the synthesis of compounds 3a–3c under the condition of concentrated sulfuric acid at 80 °C. Compounds 4a–4f obtained from 3a–3c by ketalization reaction in the presence of ethane-1, 2-diol or propane-1, 3-diol were then reacted with methyl 2-bromoacetate or methyl 3-bromo-propanoate to get compounds 5a–5h. The hydrolysis of 5a–5h was implemented to produce intermediates 6a–6h, which further underwent condensation reaction with methyl-4-aminobenzoate or methyl 4-(aminomethyl) benzoate to afford 7a–7o. Compounds 8a–8o were hydrolytic products of 7a–7o, respectively. Finally, target compounds 9a–9o were obtained by condensation of o-phenylenediamine and compounds 8a–8o in the presence of 2-(1H-benzotriazole-1-yl)-1, 1, 3, 3-tetramethyluronium tetrafluo-roborate (TBTU).
Scheme 1.
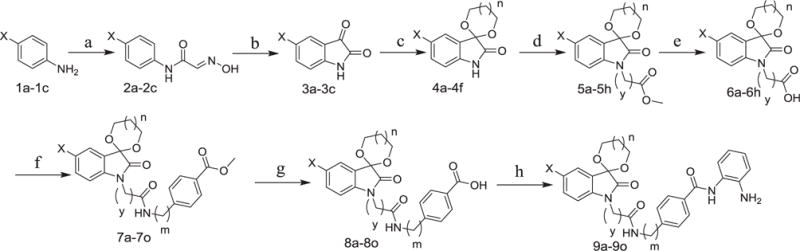
(1) X = Br, Cl or F; n = 0 or 1. (2) Reagents and conditions: (a) Cl3CH(OH) 2, NH2OH, Na2SO4, 60–85 °C; (b) concentrated H2SO4, 80 °C; (c) ethane-1, 2-diol or propane-1, 3-diol, TsOH, toluene, 140 °C; (d) BrCH2COOCH3 or BrCH2CH2COOCH3, TBAB, K2CO3, KI, acetone, 60 °C; (e) KOH, H2O, THF/CH3OH, r.t., then acidified to pH 1 with HCl; (f) methyl-4-aminobenzoate or methyl 4-(aminomethyl) benzoate, TBTU, Et3N, THF/CH2Cl2, r.t.; (g) NaOH, H2O, THF/CH3OH, reflux; then acidified to pH 1 with HCl; (h) o-phenylenediamine, TBTU, Et3N, THF/CH2Cl2, r.t.
Condensation of 6a–6f with methyl 6-aminohexanoate hydrochloride, followed by hydrolysis and condensation with o-phenylenediamine led to target compounds 12a–12f (Scheme 2).
Scheme 2.
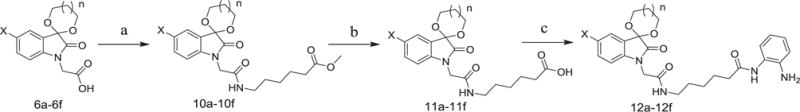
(1) X = Br, Cl or F; n = 0 or 1. (2) Reagents and conditions: (a) Methyl 6-aminohexanoate hydrochloride, TBTU, Et3N, THF/CH2Cl2, r.t.; (b) NaOH, H2O, THF/CH3OH, reflux; then acidified to pH 1 with HCl; (c) o-phenylenediamine, TBTU, Et3N, THF/CH2Cl2, r.t.
Unlike Schemes 1 and 2, the synthetic route affording desired compounds with an aromatic R1 was illustrated in Scheme 3. Firstly, the amino group of 4-bromo-2-nitroaniline (compound 13) was protected by di-tert-butyl dicarbonate to get compound 14, and then thienyl or phenyl group was introduced through Suzuki coupling to get 15a or 15b. Compounds 16a and 16b derived from the reduction of 15a and 15b were reacted with 19 to get 20a and 20b, respectively. At last, the deprotected compounds 21a and 21b were coupled with intermediates 6b and 6e to give 22a–22d, which were treated with hydrogen chloride in ethyl acetate to remove the Boc group to yield the desired compounds 23a–23d.
Scheme 3.
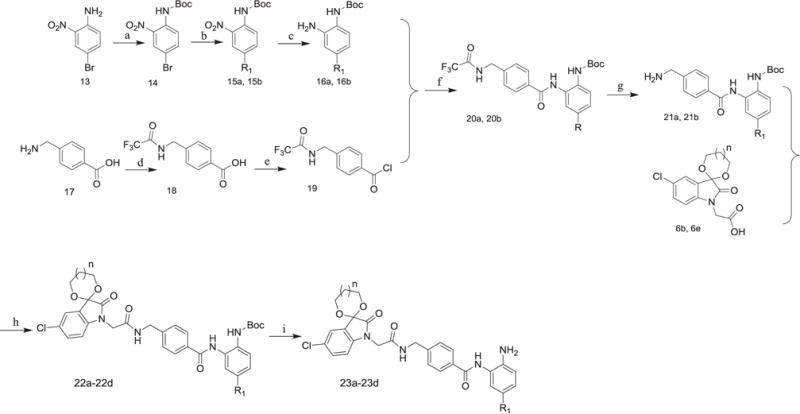
(1) R1 = thienyl or phenyl. (2) Reagents and conditions: (a) (Boc)2O, Et3N, DMAP, CH2Cl2, r.t.; (b) thiophene-2-boronic acid or phenylboronic acid, Pd[P(pH3)4], Na2CO3, 1, 4-dioxane, H2O, 90 °C; (c) Pd/C, H2, CH3OH/EtOAc, r.t.; (d) (CF3CO)2O, 0 °C; (e) SOCl2, THF; reflux; (f) pyridine, r.t.; (g) K2CO3, H2O, THF/CH3OH, 70 °C; (h) o-phenylenediamine, TBTU, Et3N, THF/CH2Cl2, r.t.; (i) EtOAc/HCl, r.t.
The preparation of the compound 30 with fluoro-substituent was illustrated in Scheme 4. Boc protection of the starting material 24 followed by reduction led to 26. Reaction of 26 with 19 led to 27, which was transformed to the target compound 30 using similar methods described in Scheme 3.
Scheme 4.
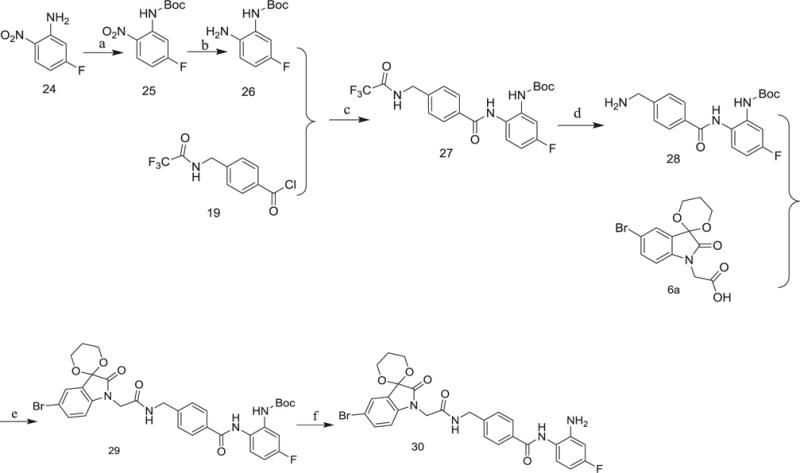
Reagents and conditions: (a) (Boc)2O, Et3N, DMAP, CH2Cl2, r.t.; (b) Pd/C, H2, CH3OH/EtOAc, r.t.; (c) pyridine, r.t.; (d) K2CO3, H2O, THF/CH3OH, 70 °C; (e) o-phenylenediamine, TBTU, Et3N, THF/CH2Cl2, r.t.; (f) EtOAc/HCl, r.t.
2.2. HeLa nuclear extract inhibitory assay
HeLa nuclear extract was used as the enzyme source to evaluate the HDACs inhibitory activities of our target compounds with MS-275 as the positive control. Firstly, the X group of the isatin-based cap in 9a was replaced by different halogens to get analogs 9b and 9c. Then compounds 9e–9f were synthesized with different sizes of ketal spiro ring at the 3-postion of the isatin-based caps. In the following work, the length of the linker was extended by replacing the methyl-4-aminobenzoate with methyl 4-(aminomethyl) ben-zoate according to the structure of MS-275 so as to gain compounds 9g–9l. Meanwhile, compounds 9m, 9n and 9o with different linkers relative to compounds 9d, 9e and 9h respectively were synthesized to evaluate the effect of linker length on HDAC inhibition.
Results in Table 1 showed that compounds 9a–9c with six-membered spirocycle had better HDAC inhibitory activities than their five-membered spirocycle analogs 9d–9f, respectively. The same structure-activity relationship could also be observed between 9g–9i (n = 1) and 9j–9l (n = 0), though not that robust. Among compounds 9a–9c (m = 0), bromo- or chloro-substituted compounds (9a, 9b) exhibited better HDAC inhibition than fluoro-substituted compound 9c. The same structure-activity relationship was also observed among 9d–9f (m = 0), but not among 9g–9i (m = 1) or 9j–9l (m = 1). Compounds 9m and 9n with the ethidine linker in the y position displayed more potent activities than their counterparts 9d and 9e with the methylene linker, respectively. However, compound 9o with the ethidine linker in the y position exhibited similar potency to its analogue 9h with the methylene linker. Above structure-activity relationships indicated that cap part and the linker part could simultaneously affect the HDAC inhibitory potency of these benzamide-based HDACIs.
Table 1.
HDAC Inhibitory Activity of Compounds 9a–9o, and MS-275.
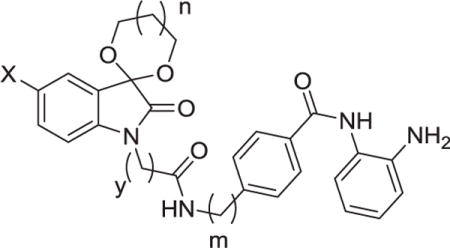
| |||||
|---|---|---|---|---|---|
| Compd | X | n | m | y | IC50 of HeLa nuclear extract (μM)a |
| 9a | −Br | 1 | 0 | 1 | 1.44 ± 0.42 |
| 9b | −Cl | 1 | 0 | 1 | 1.08 ± 0.08 |
| 9c | −F | 1 | 0 | 1 | 3.75 ± 0.65 |
| 9d | −Br | 0 | 0 | 1 | 2.66 ± 0.45 |
| 9e | −Cl | 0 | 0 | 1 | 2.17 ± 0.18 |
| 9f | −F | 0 | 0 | 1 | >6.00 |
| 9g | −Br | 1 | 1 | 1 | 2.00 ± 0.13 |
| 9h | −Cl | 1 | 1 | 1 | 1.25 ± 0.08 |
| 9i | −F | 1 | 1 | 1 | 1.56 ± 0.72 |
| 9j | −Br | 0 | 1 | 1 | 2.14 ± 0.73 |
| 9k | −Cl | 0 | 1 | 1 | 1.93 ± 0.34 |
| 9l | −F | 0 | 1 | 1 | 1.84 ± 0.62 |
| 9m | Br | 1 | 0 | 2 | 0.51 ± 0.03 |
| 9n | Cl | 1 | 0 | 2 | 0.43 ± 0.19 |
| 9o | Cl | 1 | 1 | 2 | 1.03 ± 0.25 |
| MS-275 | 1.12 ± 0.21 | ||||
Results expressed as the mean ± standard deviation of at least three separate determinations.
Then, compounds 12a–12f with the similar aliphatic linker as RG2833 were synthesized.18 Results in Table 2 showed that the aliphatic linker, however, may decrease the HDAC inhibitory activities of the target compounds. This should be associated with the loss of the π-π stacking interaction between aliphatic linker of 12a–12f and the two parallel phenylalanine residues composing the hydrophobic channel of HDACs active site.19,20
Table 2.
HDAC Inhibitory Activity of Compounds 12a–12f, and MS-275.
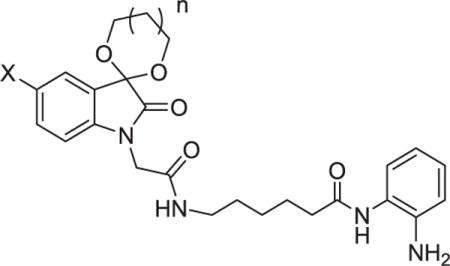
| |||
|---|---|---|---|
| Compd | X | n | IC50 of HeLa nuclear extract (mM)a |
| 12a | −Br | 1 | 8.42 ± 1.59 |
| 12b | −Cl | 1 | 8.30 ± 0.87 |
| 12c | −F | 1 | 6.50 ± 0.09 |
| 12d | −Br | 0 | 7.53 ± 1.32 |
| 12e | −Cl | 0 | 9.70 ± 2.83 |
| 12f | −F | 0 | 27.18 ± 0.28 |
| MS-275 | 1.12 ± 0.21 | ||
Results expressed as the mean ± standard deviation of at least three separate determinations.
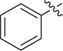
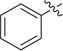
Besides a hydrophobic 11 Å channel, class I HDACs active site also contains a 14 Å internal cavity at the bottom of the active site.21,22 The 14 Å cavity as an escaping route for acetate following deacetylation could be used for the design of HDAC1/2 selective inhibitors.23,24 Thereby, we designed and synthesized several 5-substituted-o-phenylenediamines 23a–23d, which should be HDAC1/2 selective over HDAC3. Moreover, one 4-fluorine-o-phenylenediamine analogue 30 was also synthesized. As shown in Table 3, compounds 23a–23d and 30 exhibited significantly decreased inhibitory activities toward HeLa nuclear extract.
Table 3.
HDAC Inhibitory Activity of Compounds 23a–23d, 30, and MS-275.
| |||||
|---|---|---|---|---|---|
| Compd | X | n | R1 | R2 | IC50 of HeLa nuclear extract (μM)a |
| 23a | Cl | 1 |
|
H | >100.0 |
| 23b | Cl | 0 |
|
H | >100.0 |
| 23c | Cl | 1 |
|
H | >100.0 |
| 23d | Cl | 0 |
|
H | >100.0 |
| 30 | Br | 1 | H | F | >100.0 |
| MS-275 | 1.12 ± 0.21 | ||||
Results expressed as the mean ± standard deviation of at least three separate determinations.
2.3. In vitro antiproliferative assay
Based on the results of HDACs inhibitory assay, several compounds were selected to test their antitumor activities against five cancer cell lines, including K562 (chronic myelogenous leukemia cell), Molt-4 (human T lymphoblast; acute lymphoblastic leukemia cell), HEL (human erythrocyte leukemia cell), PC-3 (human prostate cancer cell) and SK-N-BE(2) (human neuroblastoma) (Table 4). Generally, the antiproliferative activities of these compounds were consistent with their HDACs inhibitory activities. Compounds 9m and 9n with the most potent HDACs inhibitory potency also exhibited the most potent antiproliferative activities, which were comparable to that of MS-275.
Table 4.
In vitro antiproliferative activity for selected HDAC inhibitors.
| Compd | IC50 (μM)a
|
||||
|---|---|---|---|---|---|
| K562 | Molt-4 | HEL | PC-3 | SK-N-BE(2) | |
| 9a | NDb | ND | 2.27 | ND | ND |
| 9d | 1.75 | 1.24 | 0.69 | 1.27 | 0.93 |
| 9h | 2.83 | 2.46 | 1.15 | 11.35 | 1.92 |
| 9m | 1.54 | 0.75 | 0.53 | 4.82 | 0.96 |
| 9n | 1.48 | 0.70 | 0.58 | 3.23 | 1.01 |
| 23a | 5.28 | 5.13 | 2.75 | 10.19 | 8.98 |
| MS-275 | 1.32 | 0.65 | 0.48 | 4.89 | 0.33 |
Assays were performed in replicate (n ≥ 2), the SD values are <20% of the mean.
Not determined.
2.4. HDAC class I and class IIa Whole-cell assay
Whole-cell HDAC inhibition assay in HEL cells was used to evaluate the HDACs class selectivity of five representative compounds. Results in Table 5 showed that except 9a and 23a, which was not active for both class I and class IIa HDACs, all tested o-phenylene-diamine-based HDACIs exhibited selectivity for class I HDACs over class IIa.
Table 5.
HDAC class I and IIa cellular activity for selected HDAC inhibitors.a
| Compd | HDAC class I cellular IC50 (μM)b |
HDAC class IIa cellular IC50 (μM)c |
|---|---|---|
| 9a | >2.00 | >2.00 |
| 9d | 0.61 | >2.00 |
| 9h | 0.93 | >2.00 |
| 9m | 0.37 | >2.00 |
| 9n | 0.51 | >2.00 |
| 23a | >2.00 | >2.00 |
| MS-275 | 0.43 | >2.00 |
Assays were performed in replicate (n ≥ 2), the SD values are <20% of the mean.
Boc-Lys (acetyl)-AMC substrate.
Boc-Lys (triflouroacetyl)-AMC substrate.
2.5. In vitro HDACs isoform selectivity fluorescence assay
The compounds were tested for their inhibition against HDAC1, 2 and 3 to further explore their HDAC isoform selective profiles (Table 6). MS-275 was used as the positive control. As expected, 5-substituted-o-phenylenediamine 23a exhibited dramatic HDAC1/2 selectivity over HDAC3. It is worth noting that compared with MS-275, compound 9n exhibited moderate HDAC1 selectivity over HDAC2 and HDAC3.
Table 6.
HDACs inhibitory activity and isoform selectivity for selected HDAC inhibitors.
| Compd | IC50 of HDAC1 (μM)a | IC50 of HDAC2 (μM)a | IC50 of HDAC3 (μM)a | HDAC2/HDAC1 isoform selectivity | HDAC3/HDAC1 isoform selectivity |
|---|---|---|---|---|---|
| 9d | 0.046 | 0.128 | 0.667 | 2.8 | 14.5 |
| 9h | 0.137 | 0.223 | 0.096 | 1.6 | 0.7 |
| 9m | 0.012 | 0.026 | 0.019 | 2.2 | 1.6 |
| 9n | 0.032 | 0.256 | 0.311 | 8.0 | 9.7 |
| 23a | 0.023 | 0.179 | 151.8 | 7.8 | 6600 |
| MS-275 | 0.163 | 0.396 | 0.605 | 2.3 | 3.7 |
Assays were performed in replicate (n ≥ 2), the SD values are <20% of the mean.
2.6. Docking Study
To explore the binding mode of these compounds in HDACs, compounds 9h and 9n were docked into the active site of HDAC1 (PDB code 5ICN). Results in Fig. 3 showed that 9h and 9n shared a similar binding mode. For both compounds, the isatin-based caps were solvent-exposed (Fig. 3a) and the o-phenylenediamine-based zinc binding groups chelated the Zn2+ (Fig. 3b). However, compared with 9h, the phenyl group in the linker part of 9n could form more favorable p-p stacking interaction with the two parallel phe-nyl groups of Phe 150 and Phe 205 in HDAC1 (Fig. 3b). This could rationalize the better HDAC1 inhibitory activity of 9n.
Fig. 3.
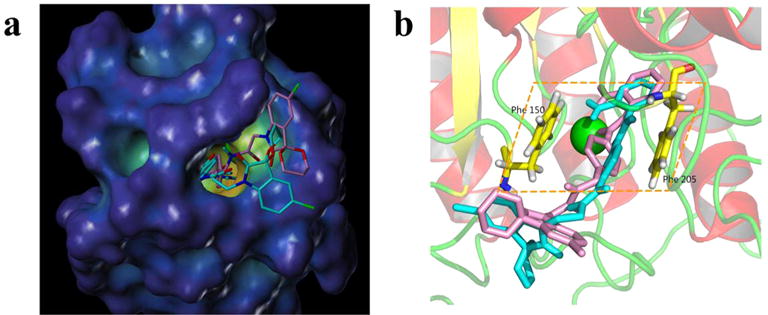
Proposed binding modes of compounds 9h (pink) and 9n (blue) in the active site of HDAC1 (derived by modification of PDB code 5ICN using Tripos SYBYL-X 1.3). The zinc ion is shown as a green sphere.
3. Conclusions
A novel series of compounds with isatin-based caps and o-phenylenediamine-based zinc binding groups as HDACIs were designed and synthesized based on the previous work in our laboratory.17All synthesized target compounds were evaluated for their HDAC inhibition using HeLa nuclear extract. Given their better HDACs inhibitory potency, compounds 9d, 9h, 9m, 9n and 23a were chosen for the antiproliferative assays, and compounds 9m and 9n showed promising antiproliferative effects which were consistent with their potent HDAC inhibitory potency. More importantly, compared with the MS-275, compound 9n presented moderate HDAC1 selectivity over HDAC2 and HDAC3, which could be used as a lead compound to find more selective HDAC1 inhibitors.
4. Experimental section
4.1. Chemistry
4.1.1. Materials and methods
All materials, reagents and solvents used without further purification unless otherwise stated were purchased from commercial sources. All reactions were monitored by TLC with 0.25 mm silica gel plates (60 GF-254). UV light, iodine stain and ninhydrin were used to visualize the spots. 1H and 13C were recorded on a Bruker DRX spectrometer at 300/400 MHz with tetramethylsilane (TMS) as an internal reference, δ in parts per million (ppm) and J in Hertz. High-resolution mass spectra were obtained from Shandong Analysis and Test Centre in Ji’nan, China. ESI-MS spectra were recorded on an API 4000 spectrometer. Melting points were determined on an electrothermal melting point apparatus without correcting. Silica gel was used for column chromatography purification. The purity of the tested compounds was assessed by an Agilent 1200 HPLC instrument. An ODS HYPERSIL column (4.6 mm 250 mm, 5 mm) was used with 30% water/70% methanol (method A for compounds 9a–9d, 9n–9o, 12a–12f, 23a–23d and 30) or 35% water/65% methanol (method B for compounds 9e–9m) over 18 min at a flow rate of 1.0 mL/min with detection at 254 nm. All final compounds were determined to be at least 95% pure by HPLC analysis.
(E)-N-(4-Bromophenyl)-2-(hydroxyimino) acetamide (2a), 5-bromoindoline-2, 3-dione (3a), 5-bromospiro [indoline-3, 2′-[1,3] dioxan]-2-one (4a), methyl 2-(5-bromo-2-oxospiro [indoline-3, 2′-[1,3]dioxan]-1-yl) acetate (5a), 2-(5-bromo-2-oxospiro[indo-line-3,2′-[1,3]dioxan]-1-yl)acetic acid (6a), methyl 4-(2-(5-bromo-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)ben-zoate (7a), 4-(2-(5-bromo-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)benzoic acid (8a), N-(2-aminophenyl)-4-(2-(5-bromo-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)ben-zamide (9a), methyl 6-(2-(5-bromo-2-oxospiro[indoline-3,2′-[1,3]-dioxan]-1-yl)acetamido)hexanoate (10a) and their analogues 2b–2c, 3b–3c,4b–4f, 5b–5h, 6b–6h, 7b–7o and 10b–10f in relevant steps were synthesized as described previously.17
4.1.2. General procedure for the preparation of compounds 8b and its analogue 8c–8o
4.1.2.1. 4-(2-(5-Chloro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)ac-etamido)benzoic acid (8b)
To a solution of compound 7b (0.29 g, 0.67 mmol) in 6 mL THF and 3 mL CH3OH was added 3 M aqueous NaOH (0.7 mL). Keep the reaction for 2 h at room temperature (r.t.). Then 1 M aqueous HCl was added to the mixture gradually until the solution became acidic. After filtration, compound 8b was obtained as white solid (0.20 g, 72% yield). Mp: 226–228 °C, ESI-MS m/z: 417.8 [M+H]+.
4.1.2.2. 4-(2-(5-Fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)ac-etamido)benzoic acid (8c)
White solid, 91% yield. Mp: >250 °C, ESI-MS m/z: 401.1 [M+H]+.
4.1.2.3. 4-(2-(5-Bromo-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl) acetamido)benzoic acid (8d)
White solid, 92% yield. Mp: >250 °C, ESI-MS m/z: 447.0 [M+H]+.
4.1.2.4. 4-(2-(5-Chloro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl) acetamido)benzoic acid (8e)
White solid, 78% yield. Mp: >250 °C, 1H NMR (400 MHz, DMSO-d6) δ 12.77 (s, 1H), 10.70 (s, 1H), 7.90 (d, J = 8.8 Hz, 2H), 7.68 (d, J = 8.8 Hz, 2H), 7.52 (d, J = 2.1 Hz, 1H), 7.48 (dd, J = 8.4, 2.2 Hz, 1H), 7.10 (d, J = 8.4 Hz, 1H), 4.56 (s, 2H), 4.37–4.32 (m, 4H).
4.1.2.5. 4-(2-(5-Fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl) acetamido)benzoic acid (8f)
White solid, 86% yield. Mp: >250 °C, 1H NMR (400 MHz, DMSO-d6) δ 12.76 (s, 1H), 10.71 (s, 1H), 7.91 (d, J = 8.7 Hz, 2H), 7.69 (d, J = 8.7 Hz, 2H), 7.37 (dd, J = 7.6, 2.6 Hz, 1H), 7.28 (td, J = 9.2, 2.7 Hz, 1H), 7.08 (dd, J = 8.6, 4.0 Hz, 1H), 4.56 (s, 2H), 4.38–4.31 (m, 4H).
4.1.2.6. 4-((2-(5-Bromo-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl) acetamido)methyl)benzoic acid (8g)
White solid, 88% yield. Mp: >250 °C, ESI-MS m/z: 475.1 [M+H]+.
4.1.2.7. 4-((2-(5-Chloro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl) acetamido)methyl)benzoic acid (8h)
White solid, 91% yield. Mp: 228–230 °C, 1H NMR (400 MHz, DMSO-d6) δ 12.88 (s, 1H), 8.85 (t, J = 5.9 Hz, 1H), 7.91 (d, J = 8.3 Hz, 2H), 7.45 (dd, J = 8.4, 2.2 Hz, 1H), 7.42–7.39 (m, 2H), 7.38 (s, 1H), 6.96 (d, J = 8.4 Hz, 1H), 4.72 (td, J = 11.5, 2.4 Hz, 2H), 4.39 (s, 2H), 4.37 (s, 2H), 3.98–3.91 (m, 2H), 2.26–2.12 (m, 1H), 1.74–1.66 (m, 1H).
4.1.2.8. 4-((2-(5-Fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl) acetamido)methyl)benzoic acid (8i)
White solid, 90% yield. Mp: >250 °C, ESI-MS m/z: 415.1 [M+H]+.
4.1.2.9. 4-((2-(5-Bromo-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl) acetamido)methyl)benzoic acid (8j)
White solid, 71% yield. Mp: >250 °C, ESI-MS m/z: 461.0 [M+H]+.
4.1.2.10. 4-((2-(5-Chloro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)methyl)benzoic acid (8k)
White solid, 93% yield. Mp: >250 °C, 1H NMR (400 MHz, DMSO-d6) δ 12.88 (s, 1H), 8.85 (t, J = 5.9 Hz, 1H), 7.91 (d, J = 8.3 Hz, 2H), 7.52–7.45 (m, 2H), 7.38 (d, J = 8.3 Hz, 2H), 6.99 (dd, J = 7.6, 1.3 Hz, 1H), 4.38 (s, 2H), 4.37 (s, 2H), 4.36–4.28 (m, 4H).
4.1.2.11. 4-((2-(5-Fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl) acetamido)methyl)benzoic acid (8l)
White solid, 59% yield. Mp: 230–232 °C, ESI-MS m/z: 401.1 [M+H]+.
4.1.2.12. 4-(3-(5-Bromo-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl) propanamido)benzoic acid (8m)
White solid, 86% yield. Mp: >250 °C, 1H NMHR (400 MHz, DMSO-d6) δ 12.52 (s, 1H), 10.32 (s, 1H), 7.92–7.85 (m, 2H), 7.65 (d, J = 8.7 Hz, 2H), 7.61 (dd, J = 8.4, 2.1 Hz, 1H), 7.51 (d, J = 2.0 Hz, 1H), 7.14 (t, J = 5.7 Hz, 1H), 4.70 (td, J = 11.4, 2.2 Hz, 2H), 3.97–3.88 (m, 4H), 2.69 (t, J = 7.0 Hz, 2H), 2.23–2.10 (m, 1H), 1.72–1.63 (m, 1H).
4.1.2.13. 4-(3-(5-Chloro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl) propanamido)benzoic acid (8n)
White solid, 95% yield. Mp: >250 °C, ESI-MS m/z: 431.1 [M+H]+.
4.1.2.14. 4-((3-(5-Chloro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl) propanamido)methyl)benzoic acid (8o)
White solid, 98% yield. Mp: >250 °C, ESI-MS m/z: 445.1 [M+H]+.
4.1.3. General procedure for the preparation of compounds 9b and its analogue 9c–9o
4.1.3.1. N-(2-Aminophenyl)-4-(2-(5-chloro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)benzamide (9b)
To a solution of 8b (0.15 g, 0.36 mmol) in dry THF and CH2Cl2 was added TBTU (0.14 g, 0.43 mmol), followed by Et3N (0.2 mL, 1.44 mmol). After 40 min in room temperature, o-phenylenediamine (0.05 g, 0.43 mmol) was added. The mixture was stirred at the same temperature overnight. Then the solvent was evaporated under vacuum. The residue was dissolved in EtOAc and washed with brine (3×20 mL). Organic phase was combined and dried by MgSO4 overnight. The EtOAc was removed under low pressure to give a crude material that was purified via silica-gel column chromatography (CH2Cl2/CH3OH = 100:1), yielding the desired compound 9b as a reddish white solid (0.25 g, 66% yield). Mp: >250 °C, 1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H), 9.60 (s, 1H), 7.98 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 8.7 Hz, 2H), 7.52–7.39 (m, 2H), 7.16 (d, J = 7.5 Hz, 1H), 7.09 (d, J = 8.2 Hz, 1H), 7.02–6.92 (m, 1H), 6.78 (dd, J = 7.9, 0.9 Hz, 1H), 6.65–6.55 (m, 1H), 4.89 (s, 2H), 4.74 (t, J = 10.5 Hz, 2H), 4.59 (s, 2H), 3.99–3.95 (m, 2H), 2.27–2.13 (m, 1H), 1.73–1.69 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 171.34, 165.66, 165.10, 143.61, 141.80, 141.77, 131.15, 129.82, 129.30, 128.72, 127.41, 127.15, 126.87, 124.45, 123.91, 118.70, 116.74, 116.62, 111.78, 93.44, 61.17, 42.79, 25.21. HRMS (AP-ESI) m/z calcd for C26H23ClN4O5 [M+H]+ 507.1430, found 507.1563. HPLC tR = 8.37 min, 97.5%.
4.1.3.2. N-(2-Aminophenyl)-4-(2-(5-fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)benzamide (9c)
White solid, 51% yield. Mp: >250 °C, 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 9.59 (s, 1H), 7.97 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 8.7 Hz, 2H), 7.30 (dd, J = 7.7, 2.6 Hz, 1H), 7.25 (td, J = 9.2, 2.7 Hz, 1H), 7.16 (d, J = 7.5 Hz, 1H), 7.07 (dd, J = 8.6, 4.1 Hz, 1H), 6.96 (t, J = 7.6 Hz, 1H), 6.78 (d, J = 8.7 Hz, 1H), 6.59 (t, J = 7.9 Hz, 1H), 4.88 (s, 2H), 4.75 (dd, J = 11.5, 9.3 Hz, 2H), 4.58 (s, 2H), 3.97 (dd, J = 11.4, 2.9 Hz, 2H), 2.27–2.13 (m, 1H), 1.72–1.69 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 171.57, 165.77, 165.02, 160.19, 157.81, 143.62, 141.82, 139.05, 129.80, 129.30, 128.47, 128.39, 127.15, 126.87, 123.90, 118.69, 117.73, 117.50, 116.73, 116.61, 112.35, 112.10, 111.27, 111.20, 93.54, 61.09, 42.80, 25.21. HRMS (AP-ESI) m/z calcd for C26H23FN4O5 [M +H]+ 491.1725, found 491.1851. HPLC tR = 5.64 min, 95.4%.
4.1.3.3. N-(2-Aminophenyl)-4-(2-(5-bromo-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)benzamide (9d)
White solid, 60% yield. Mp: >250 °C,1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H), 9.59 (s, 1H), 7.95 (t, J = 12.3 Hz, 2H), 7.69 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 6.8 Hz, 2H), 7.15 (d, J = 7.6 Hz, 1H), 7.07 (d, J = 9.0 Hz, 1H), 6.96 (t, J = 7.4 Hz, 1H), 6.78 (d, J = 7.8 Hz, 1H), 6.59 (t, J = 7.4 Hz, 1H), 4.89 (s, 2H), 4.57 (s, 2H), 4.39–4.30 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 172.84, 165.60, 165.11, 143.70, 143.60, 141.77, 134.68, 129.86, 129.28, 127.88, 127.13, 126.85, 126.81, 123.93, 118.73, 116.74, 116.62, 115.27, 112.57, 101.52, 66.39, 46.18, 43.09. HRMS (AP-ESI) m/z calcd for C25H21BrN4O5 [M+H]+ 537.0768, found 537.0769. HPLC tR = 6.50 min, 95.3%.
4.1.3.4. N-(2-Aminophenyl)-4-(2-(5-chloro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)benzamide (9e)
White solid, 55% yield. Mp: >250 °C,1H NMR (400 MHz, DMSO-d6) δ 10.65 (s, 1H), 9.58 (s, 1H), 7.97 (d, J = 8.7 Hz, 2H), 7.69 (d, J = 8.7 Hz, 2H), 7.53 (d, J = 2.1 Hz, 1H), 7.50 (dd, J = 8.4, 2.2 Hz, 1H), 7.16 (d, J = 7.0 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.00–6.93 (m, 1H), 6.78 (dd, J = 8.0, 1.2 Hz, 1H), 6.59 (td, J = 7.8, 1.2 Hz, 1H), 4.90 (s, 2H), 4.58 (s, 2H), 4.39–4.30 (m, 4H).13C NMR (100 MHz, DMSO-d6) δ 172.96, 165.63, 165.10, 143.61, 143.26, 141.77, 131.87, 129.85, 129.29, 127.70, 127.14, 126.86, 126.47, 125.18, 123.91, 118.72, 116.74, 116.61, 112.10, 101.56, 66.38, 43.11. HRMS (AP-ESI) m/z calcd for C25H21ClN4O5 [M+H]+ 493.1273, found 493.1410. HPLC tR = 8.36 -min, 97.7%.
4.1.3.5. N-(2-Aminophenyl)-4-(2-(5-fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)benzamide (9f)
White solid, 66% yield. Mp: >250 °C,1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 8.25 (t, J = 5.4 Hz, 1H), 7.26 (dd, J = 7.7, 2.7 Hz, 1H), 7.23–7.14 (m, 2H), 6.93–6.83 (m, 2H), 6.71 (dd, J = 8.0, 1.2 Hz, 1H), 6.53 (td, J = 7.7, 1.4 Hz, 1H), 4.81 (s, 2H), 4.73 (td, J = 11.5, 2.4 Hz, 2H), 4.26 (s, 2H), 3.99–3.88 (m, 2H), 3.09 (q, 6.6 Hz, 2H), 2.32 (t, J = 7.4 Hz, 2H), 2.25–2.10 (m, 1H), 1.72–1.65 (m, 1H), 1.65–1.55 (m, 2H), 1.52–1.41 (m, 2H), 1.39–1.29 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.95, 171.55, 166.13, 142.28, 140.55, 126.19, 125.76, 124.09, 118.35, 118.11, 116.69, 116.40, 112.88, 112.64, 111.41, 111.33, 101.67, 66.23, 42.39, 39.12, 36.16, 29.26, 26.57, 25.47. HRMS (AP-ESI) m/z calcd for C25H21FN4O5 [M+H]+ 477.1569, found 477.1717. HPLC tR = 11.55 min, 95.2%.
4.1.3.6. N-(2-Aminophenyl)-4-((2-(5-bromo-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)methyl)benzamide (9g)
White solid, 34% yield. Mp: 219–221 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 1H), 8.88 (t, J = 5.7 Hz, 1H), 7.95 (d, J = 8.0 Hz, 2H), 7.59 (dd, J = 8.4, 1.9 Hz, 1H), 7.52 (d, J = 1.9 Hz, 1H), 7.40 (d, J = 8.0 Hz, 2H), 7.17 (d, J = 7.7 Hz, 1H), 6.97 (t, J = 7.6 Hz, 1H), 6.92 (d, J = 8.4 Hz, 1H), 6.78 (d, J = 7.8 Hz, 1H), 6.60 (t, J = 7.4 Hz, 1H), 4.86 (s, 2H), 4.72 (t, J = 10.6 Hz, 2H), 4.39 (d, J = 7.0 Hz, 4H), 3.94 (dd, J = 11.1, 2.9 Hz, 2H), 2.27–2.09 (m, 1H), 1.69 (d, J = 13.4 Hz, 1H).13C NMR (100 MHz, DMSO-d6) δ 171.09, 166.58, 165.54, 143.61, 143.00, 142.23, 133.82, 129.08, 128.33, 127.45, 127.17, 127.11, 126.66, 123.80, 116.66, 114.88, 112.13, 93.36, 61.13, 42.52, 42.13, 25.21. HRMS (AP-ESI) m/z calcd for C27H25BrN4O5 [M+H]+ 565.1081, found 565.1086. HPLC tR = 11.94 min, 98.3%.
4.1.3.7. N-(2-Aminophenyl)-4-((2-(5-chloro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)methyl)benzamide (9h)
White solid, 31% yield. Mp: 203–205 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 1H), 8.87 (t, J = 5.8 Hz, 1H), 7.96 (d, J = 8.0 Hz, 2H), 7.46 (dd, J = 8.4, 2.1 Hz, 1H), 7.44–7.41 (m, 2H), 7.40 (s, 1H), 7.18 (d, J = 7.6 Hz, 1H), 6.98 (t, J = 7.2 Hz, 2H), 6.79 (d, J = 7.3 Hz, 1H), 6.61 (t, J = 7.4 Hz, 1H), 4.90 (s, 2H), 4.73 (t, J = 10.5 Hz, 2H), 4.41 (s, 2H), 4.39 (s, 2H), 3.97–3.94 (m, 2H), 2.27–2.12 (m, 1H), 1.72–1.69 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 171.19, 166.61, 165.55, 143.62, 143.00, 141.79, 133.72, 131.05, 128.75, 128.33, 127.45, 127.33, 127.18, 126.96, 124.41, 123.78, 116.72, 116.59, 111.64, 93.40, 61.11, 42.52, 42.15, 25.20. HRMS (AP-ESI) m/z calcd for C27H25ClN4O5 [M+H]+ 521.1586, found 521.1702. HPLC tR = 10.54 min, 98.1%.
4.1.3.8. N-(2-Aminophenyl)-4-((2-(5-fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)methyl)benzamide (9i)
White solid, 39% yield. Mp: 198–200 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.64 (s, 1H), 8.87 (t, J = 5.7 Hz, 1H), 7.95 (d, J = 7.9 Hz, 2H), 7.40 (d, J = 8.0 Hz, 2H), 7.28 (dd, J = 7.6, 2.3 Hz, 1H), 7.25–7.20 (m, 1H), 7.17 (d, J = 7.6 Hz, 1H), 7.01–6.91 (m, 2H), 6.79 (d, J = 7.7 Hz, 1H), 6.60 (t, J = 7.4 Hz, 1H), 4.90 (s, 2H), 4.74 (dd, J = 11.2, 9.7 Hz, 2H), 4.43–4.36 (m, 4H), 3.95 (dd, J = 10.8, 3.0 Hz, 2H), 2.25–2.11 (m, 1H), 1.71–1.68 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 171.41, 166.71, 165.54, 160.15, 157.77, 143.63, 143.03, 139.05, 133.72, 128.32, 127.45, 127.18, 126.95, 123.78, 117.63, 117.40, 116.72, 116.59, 112.32, 112.07, 111.12, 111.04, 93.52, 61.03, 42.51, 42.18, 25.21. HRMS (AP-ESI) m/z calcd for C27H25FN4O5 [M+H]+ 505.1882, found 505.1986. HPLC tR = 6.89 min, 97.2%.
4.1.3.9. N-(2-Aminophenyl)-4-((2-(5-bromo-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)methyl)benzamide (9j)
White solid, 18% yield. Mp: 231–233 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.86 (t, J = 5.8 Hz, 1H), 7.95 (d, J = 8.1 Hz, 2H), 7.65–7.58 (m, 2H), 7.39 (d, J = 8.2 Hz, 2H), 7.17 (d, J = 7.5 Hz, 1H), 7.01–6.92 (m, 2H), 6.78 (d, J = 9.1 Hz, 1H), 6.60 (t, J = 7.5 Hz, 1H), 4.91 (s, 2H), 4.39 (s, 2H), 4.37 (s, 2H), 4.39–4.31 (m, 4H).13C NMR (100 MHz, DMSO-d6) δ 172.68, 166.53, 165.57, 143.72, 143.57, 142.98, 134.56, 133.77, 128.32, 127.82, 127.46, 127.13, 126.90, 123.85, 116.75, 116.62, 115.18, 112.41, 101.51, 66.35, 42.55, 42.47. HRMS (AP-ESI) m/z calcd for C26H23BrN4O5 [M+H]+ 551.0928, found 551.0926. HPLC tR = 7.93 min, 95.8%.
4.1.3.10. N-(2-Aminophenyl)-4-((2-(5-chloro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)methyl)benzamide (9k)
White solid, 41% yield. Mp: >250 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.85 (t, J = 5.9 Hz, 1H), 7.95 (d, J = 8.1 Hz, 2H), 7.53–7.46 (m, 2H), 7.39 (d, J = 8.2 Hz, 2H), 7.17 (d, J = 7.3 Hz, 1H), 7.03–6.93 (m, 2H), 6.79 (dd, J = 8.0, 1.3 Hz, 1H), 6.60 (td, J = 7.7, 1.2 Hz, 1H), 4.89 (s, 2H), 4.39 (s, 2H), 4.37 (s, 2H), 4.36–4.28 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 172.80, 166.56, 165.56, 143.60, 143.27, 142.99, 133.75, 131.70, 128.32, 127.62, 127.46, 127.16, 126.94, 126.56, 125.12, 123.81, 116.73, 116.61, 111.94, 101.55, 66.34, 42.53, 42.48. HRMS (AP-ESI) m/z calcd for C26H23-ClN4O5 [M+H]+ 507.1430, found 507.1538. HPLC tR = 7.17 min, 98.3%.
4.1.3.11. N-(2-Aminophenyl)-4-((2-(5-fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)methyl)benzamide (9l)
White solid, 50% yield. Mp: 230–232 °C, 1H NMR (300 MHz, DMSO-d6) δ 9.62 (s, 1H), 8.84 (t, J = 5.9 Hz, 1H), 7.95 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 8.3 Hz, 2H), 7.34 (dd, J = 7.7, 2.7 Hz, 1H), 7.31–7.23 (m, 1H), 7.20–7.14 (m, 1H), 6.97 (ddd, J = 9.5, 6.1, 1.7 Hz, 2H), 6.78 (dd, J = 8.0, 1.3 Hz, 1H), 6.60 (td, J = 7.7, 1.3 Hz, 1H), 4.88 (s, 2H), 4.42–4.36(m, 4H), 4.35–4.30 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 173.04, 166.66, 165.55, 160.34, 157.96, 143.61, 143.02, 140.55, 133.74, 128.32, 128.06, 127.76, 127.46, 127.17, 126.94, 123.80, 118.36, 118.12, 116.72, 116.60, 112.94, 112.69, 111.46, 111.39, 66.26, 42.49. HRMS (AP-ESI) m/z calcd for C26H23FN4O5 [M+H]+ 491.1725, found 491.1836. HPLC tR = 9.09 min, 95.2%.
4.1.3.12. N-(2-Aminophenyl)-4-(3-(5-bromo-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)propanamido)benzamide (9m)
White solid, 44% yield. Mp: 218–220 °C, 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 9.57 (s, 1H), 7.95 (d, J = 8.6 Hz, 2H), 7.67 (d, J = 8.7 Hz, 2H), 7.62 (dd, J = 8.4, 2.0 Hz, 1H), 7.52 (d, J = 2.0 Hz, 1H), 7.19–7.13 (m, 2H), 7.00–6.94 (m, 1H), 6.79 (dd, J = 7.9, 0.9 Hz, 1H), 6.63–6.57 (m, 1H), 4.89 (s, 2H), 4.72 (dd, J = 11.5, 9.4 Hz, 2H), 3.98–3.89 (m, 4H), 2.70 (t, J = 7.0 Hz, 2H), 2.25–2.11 (m, 1H), 1.69 (d, J = 13.4 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 170.98, 169.46, 165.17, 143.58, 142.14, 141.58, 134.07, 129.54, 129.32, 129.14, 127.25, 127.12, 126.85, 123.96, 118.78, 116.76, 116.62, 114.98, 112.18, 93.20, 61.07, 36.16, 34.72, 25.19. HRMS (AP-ESI) m/z calcd for C27H25BrN4O5 [M+H]+ 565.1081, found 565.1078. HPLC tR = 18.11 min, 97.5%.
4.1.3.13. N-(2-Aminophenyl)-4-(3-(5-chloro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)propanamido)benzamide (9n)
White solid, 25% yield. Mp: 220–222 °C, 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 9.55 (s, 1H), 7.94 (d, J = 8.7 Hz, 2H), 7.66 (d, J = 8.7 Hz, 2H), 7.49 (dd, J = 8.4, 2.2 Hz, 1H), 7.41 (d, J = 2.2 Hz, 1H), 7.20 (d, J = 8.4 Hz, 1H), 7.16 (d, J = 6.9 Hz, 1H), 7.02–6.92 (m, 1H), 6.78 (dd, J = 8.0, 1.2 Hz, 1H), 6.60 (td, J = 7.7, 1.3 Hz, 1H), 4.89 (s, 2H), 4.72 (td, J = 11.5, 2.4 Hz, 2H), 4.03–3.87 (m, 4H), 3.32 (s, 2H), 2.70 (t, J = 7.0 Hz, 2H), 2.24–2.10 (m, 1H), 1.74–1.63 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 171.08, 169.46, 165.18, 143.52, 142.14, 141.16, 131.20, 129.55, 129.13, 129.02, 127.39, 127.10, 126.84, 124.55, 124.00, 118.79, 116.80, 116.65, 111.68, 93.26, 61.06, 36.18, 34.73, 25.19. HRMS (AP-ESI) m/z calcd for C27 H25ClN4O5 [M+H]+ 521.1586, found 521.1579. HPLC tR = 10.44 min, 95.8%.
4.1.3.14. N-(2-Aminophenyl)-4-((3-(5′-chloro-2′-oxospiro[[1,3]diox-ane-2,3′-indolin]-1′-yl)propanamido)methyl)benzamide (9o)
White solid, 37% yield. Mp: 171–173 °C,1H NMR (400 MHz, DMSO-d6) δ 9.61 (s, 1H), 8.56 (t, J = 5.9 Hz, 1H), 7.88 (d, J = 8.1 Hz, 2H), 7.44 (dd, J = 8.3, 2.2 Hz, 1H), 7.41 (d, J = 2.0 Hz, 1H), 7.29 (d, J = 8.2 Hz, 2H), 7.16 (d, J = 7.3 Hz, 1H), 7.11 (d, J = 8.3 Hz, 1H), 7.01–6.93 (m, 1H), 6.78 (dd, J = 8.0, 1.3 Hz, 1H), 6.65–6.56 (m, 1H), 4.89 (s, 2H), 4.74 (td, J = 11.5, 2.5 Hz, 2H), 4.32 (d, J = 5.8 Hz, 2H), 3.98–3.91 (m, 2H), 3.88 (t, J = 7.0 Hz, 2H), 2.52 (t, J = 8.7 Hz, 2H), 2.25–2.11 (m, 1H), 1.75–1.66 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 171.04, 170.16, 165.54, 143.55, 143.21, 141.17, 133.58, 131.15, 128.98, 128.23, 127.37, 127.12, 126.91, 124.50, 123.87, 116.76, 116.63, 111.72, 93.27, 61.08, 42.35, 36.49, 33.66, 25.22. HRMS (AP-ESI) m/z calcd for C28H27ClN4O5 [M+H]+ 535.1743, found 535.1742. HPLC tR = 8.14 min, 97.3%.
4.1.4. General procedure for the preparation of compounds 11a and its analogue 11a–11f
4.1.4.1. 6-(2-(5-Bromo-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)ac-etamido)hexanoic acid (11a)
To a solution of compound 10a (0.20 g, 0.43 mmol) in 6 mL THF and 3 mL CH3OH was added 3 M aqueous NaOH (0.42 mL). Keep the reaction for 2 h at room temperature. Then 1 M aqueous HCl was added to the mixture gradually until the solution became acidic. After filtration, the compound 11a was obtained as white solid (0.18 g, 93% yield). ESI-MS m/z: 455.1 [M+H]+.
4.1.4.2. 6-(2-(5-Chloro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)ac-etamido)hexanoic acid (11b)
White solid, 89% yield. Mp: 143–145 °C, ESI-MS m/z: 411.1 [M+H]+.
4.1.4.3. 6-(2-(5-Fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)ac-etamido)hexanoic acid (11c)
White solid, 93% yield. Mp: 144–146 °C, ESI-MS m/z: 395.2 [M+H]+.
4.1.4.4. 6-(2-(5-Bromo-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl) acetamido)hexanoic acid (11d)
White solid, 74% yield. Mp: 165–167 °C, ESI-MS m/z: 441.1 [M+H]+.
4.1.4.5. 6-(2-(5-Chloro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl) acetamido)hexanoic acid (11e)
White solid, 98% yield. Mp: 170–172 °C, 1H NMR (400 MHz, DMSO-d6) δ 11.98 (s, 1H), 8.22 (t, J = 5.5 Hz, 1H), 7.50–7.44 (m, 2H), 6.92 (dd, J = 8.0, 0.6 Hz, 1H), 4.38–4.28 (m, 4H), 4.25 (s, 2H), 3.05 (q, J = 6.7 Hz, 2H), 2.19 (t, J = 7.4 Hz, 2H), 1.54–1.45 (m, 2H), 1.45–1.37 (m, 2H), 1.32–1.22 (m, 2H).
4.1.4.6. 6-(2-(5-Fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl) acetamido)hexanoic acid (11f)
White solid, 79% yield. Mp: 133–135 °C, 1H NMR (300 MHz, DMSO-d6) δ 11.98 (s, 1H), 8.24 (t, J = 5.4 Hz, 1H), 7.32 (dd, J = 7.7, 2.7 Hz, 1H), 7.30–7.20 (m, 1H), 6.90 (dd, J = 8.6, 4.1 Hz, 1H), 4.41–4.26 (m, 4H), 4.24 (s, 2H), 3.06 (q, 6.6 Hz, 2H), 2.20 (t, J = 7.3 Hz, 2H), 1.59–1.46 (m, 2H), 1.45–1.36 (m, 2H), 1.33–1.19 (m, 2H).
4.1.5. General procedure for the preparation of compounds 12a and its analogue 12b–12f
4.1.5.1. N-(2-Aminophenyl)-6-(2-(5-bromo-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)hexanamide (12a)
To a solution of 11a (0.20 g, 0.44 mmol) in dry THF and CH2Cl2 was added TBTU (0.17 g, 0.53 mmol), followed by Et3N (0.25 mL, 1.76 mmol). After 40 min in room temperature, o-phenylenediamine (0.06 g, 0.53 mmol) was added. The mixture was stirred at the same temperature overnight. Then the solvent was evaporated under vacuum. The residue was dissolved in EtOAc and washed with brine (3×20 mL). Organic phase was combined and dried by MgSO4 overnight. The EtOAc was removed under vacuum to give a crude material that was purified via silica-gel column chromatography (CH2Cl2/CH3OH = 100:1), yielding the desired compound 12a as white solid (0.25 g, 54% yield). Mp: 139–140 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.08 (s, 1H), 8.24 (t, J = 5.0 Hz, 1H), 7.58–7.48 (m, 2H), 7.16 (d, J = 7.7 Hz, 1H), 6.93–6.80 (m, 2H), 6.71 (d, J = 7.9 Hz, 1H), 6.54 (t, J = 7.5 Hz, 1H), 4.80 (s, 2H), 4.71 (t, J = 10.6 Hz, 2H), 4.26 (s, 2H), 3.92 (d, J = 2.8 Hz, 2H), 3.09 (dd, J = 12.2, 6.3 Hz, 2H), 2.31 (t, J = 7.3 Hz, 2H), 2.25–2.10 (m, 1H),1.68 (d, J = 13.4 Hz, 1H), 1.60 (dt, J = 14.7, 7.4 Hz, 2H), 1.51–1.41 (m, 2H), 1.38–1.28 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.54, 171.02, 166.04, 142.34, 142.25, 133.90, 129.06, 127.13, 126.15, 125.75, 124.08, 116.66, 116.38, 114.80, 112.06, 93.35,61.11, 60.21, 42.02, 39.15, 36.17, 29.25, 26.58, 25.47, 25.21. HRMS (AP-ESI) m/z calcd for C25H29BrN4O5 [M+H]+ 545.1394, found 545.1395. HPLC tR = 8.54 min, 96.6%.
4.1.5.2. N-(2-Aminophenyl)-6-(2-(5-chloro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)hexanamide (12b)
White solid, 53% yield. Mp: 123–125 °C, 1H 1H NMR (400 MHz, DMSO-d6) δ 9.10 (s, 1H), 8.26 (t, J = 5.4 Hz, 1H), 7.45–7.39 (m, 2H), 7.16 (d, J = 7.2 Hz, 1H), 6.89 (dd, J = 7.8, 3.7 Hz, 2H), 6.71 (d, J = 7.2 Hz, 1H), 6.54 (t, J = 7.5 Hz, 1H), 4.72 (t, J = 10.5 Hz, 2H), 4.27 (s, 2H), 3.95–3.92 (m, 2H), 3.09 (q, J = 6.4 Hz, 2H), 2.32 (t, J = 7.4 Hz, 2H), 2.25–2.11 (m, 1H), 1.70–1.67 (m, 1H), 1.65–1.55 (m, 2H), 1.50–1.43 (m, 2H), 1.37–1.29 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.54, 171.11, 166.07, 142.35, 141.81, 131.04, 128.70, 127.25, 126.16, 125.77, 124.41, 124.04, 116.65, 116.37, 111.58, 93.39, 61.08, 42.02, 39.13, 36.16, 29.26, 26.57, 25.48, 25.20. HRMS (AP-ESI) m/z calcd for C25H29ClN4O5 [M+H]+ 501.1899, found 501.2034. HPLC tR = 7.71 min, 98.5%.
4.1.5.3. N-(2-Aminophenyl)-6-(2-(5-fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)hexanamide (12c)
White solid, 42% yield. Mp: 129–131 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 8.25 (t, J = 5.4 Hz, 1H), 7.26 (dd, J = 7.7, 2.7 Hz, 1H), 7.23–7.14 (m, 2H), 6.93–6.83 (m, 2H), 6.71 (dd, J = 8.0, 1.2 Hz, 1H), 6.53 (td, J = 7.7, 1.4 Hz, 1H), 4.81 (s, 2H), 4.73 (td, J = 11.5, 2.4 Hz, 2H), 4.26 (s, 2H), 3.99–3.88 (m, 2H), 3.09 (q, 6.6 Hz, 2H), 2.32 (t, J = 7.4 Hz, 2H), 2.25–2.10 (m, 1H), 1.72–1.65 (m, 1H), 1.65–1.55 (m, 2H), 1.52–1.41 (m, 2H), 1.39–1.29 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.54, 171.33, 166.17, 142.36, 139.08, 128.46, 128.39, 126.16, 125.77, 124.05, 117.61, 117.37, 116.65, 116.37, 112.25, 112.00, 111.05, 110.97, 61.01, 42.06, 39.12, 36.16, 29.27, 26.58, 25.48, 25.21. HRMS (AP-ESI) m/z calcd for C25H29FN4-O [M+H]+ 485.2195, found 485.2249. HPLC tR = 5.46 min, 96.6%.
4.1.5.4. N-(2-Aminophenyl)-6-(2-(5-bromo-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)hexanamide (12d)
White solid, 31% yield. Mp: 158–160 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 8.25 (s, 1H), 7.58 (d, J = 7.8 Hz, 2H), 7.16 (d, J = 7.6 Hz, 1H), 6.88 (t, J = 9.2 Hz, 2H), 6.71 (d, J = 7.7 Hz, 1H), 6.53 (t, J = 7.3 Hz, 1H), 4.81 (s, 2H), 4.37–4.28 (m, 4H), 4.25 (s, 2H), 3.13–3.02 (m, 2H), 2.31 (t, J = 7.2 Hz, 2H), 1.63–1.56 (m, 2H), 1.47–1.42 (m, 2H), 1.37–1.28 (m, 2H).13C NMR (100 MHz, DMSO-d6) δ 172.59, 171.55, 166.00, 143.73, 142.34, 134.56, 127.76, 126.83, 126.15, 125.76, 124.08, 116.67, 116.38, 115.10, 112.36, 101.50, 66.32, 42.36, 39.15, 36.17, 29.24, 26.57, 25.47. HRMS (AP-ESI) m/z calcd for C24H27BrN4O5 [M+H]+ 531.1238, found 531.1656. HPLC tR = 6.00 min, 95.9%.
4.1.5.5. N-(2-Aminophenyl)-6-(2-(5-chloro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)hexanamide (12e)
White solid, 69% yield. Mp: 170–172 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.10 (s, 1H), 8.25 (t, J = 5.2 Hz, 1H), 7.48 (s, 1H), 7.47–7.42 (m, 1H), 7.16 (d, J = 7.8 Hz, 1H), 6.93–6.87 (m, 2H), 6.71 (d, J = 7.9 Hz, 1H), 6.55 (t, 7.4 Hz, 1H), 4.84 (s, 2H), 4.38–4.27 (m, 4H), 4.25 (s, 2H), 3.08 (dd, J = 12.5, 6.4 Hz, 2H), 2.31 (t, J = 7.3 Hz, 2H), 1.65–1.54 (m, 2H), 1.49–1.42 (m, 2H), 1.36–1.28 (m, 2H).13C NMR (100 MHz, DMSO-d6) δ 172.71, 171.54, 166.02, 143.28, 142.33, 131.71, 127.54, 126.48, 126.16, 125.77, 125.06, 124.05, 116.67, 116.38, 111.89, 101.54, 66.31, 42.36, 39.19, 36.15, 29.25, 26.57, 25.54. HRMS (AP-ESI) m/z calcd for C24H27ClN4O5 [M+H]+ 487.1743, found 487.1842. HPLC tR = 5.53 min, 95.3%.
4.1.5.6. N-(2-Aminophenyl)-6-(2-(5-fluoro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)hexanamide (12f)
White solid, 46% yield. Mp: 183–185 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.73 (s, 1H), 8.87 (t, J = 5.4 Hz, 1H), 7.98 (d, J = 7.8 Hz, 2H), 7.63–7.50 (m, 3H), 7.45–7.37 (m, 4H), 7.37–7.32 (m, 2H), 7.31–7.22 (m, 2H), 6.98 (dd, J = 8.4, 3.8 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H), 5.13 (s, 2H), 4.45–4.37 (m, 4H), 4.37–4.25 (m, 4H).
13C NMR (100 MHz, DMSO-d6) δ 173.05, 166.67, 165.72, 160.34, 157.96, 143.20, 143.09, 140.65, 140.53, 133.70, 129.27, 128.64, 128.38, 127.47, 126.50, 125.98, 125.27, 125.17, 124.09, 118.36, 118.13, 117.02, 112.96, 112.71, 111.47, 111.39, 101.68, 66.31, 42.52. HRMS (AP-ESI) m/z calcd for C24H27FN4O5 [M+H]+ 471.2083, found 471.2183. HPLC tR = 4.26 min, 97.6%.
4.1.6. tert- Butyl (4-Bromo-2-nitrophenyl) carbamate (14)
4-Bromo-2-nitroaniline (13) (1.0 g, 4.61 mmol) was dissolved in dry CH2Cl2 (15 mL), and then Et3N (1.92 mL, 13.82 mmol) was added followed by (Boc)2O (1.51 g, 6.91 mmol) and DMAP (0.06 g, 0.50 mmol) solubilized in dry CH2Cl2. After 1 h in room temperature, the solution was washed with water, dried by Na2SO4 and concentrated under low pressure. The residue was washed with petroleum ether (PE) and EtOAc to afford the product 14 as a yellow solid (1.80 g, 55% yield).1H NMR (400 MHz, DMSO-d6) δ 8.43 (d, J = 2.2 Hz, 1H), 8.11 (dd, J = 8.8, 2.5 Hz, 1H), 7.64 (d, J = 8.3 Hz, 1H), 1.35 (s, 9H).
4.1.7. tert-Butyl (2-nitro-4-(thiophen-2-yl) phenyl) carbamate (15a) and tert-butyl (3-nitro-[1, 1′-biphenyl]-4-yl) carbamate (15b)
4.1.7.1. tert-Butyl (2-nitro-4-(thiophen-2-yl) phenyl) carbamate (15a)
Compound 14 (0.40 g, 1.26 mmol) and 2-thienylboronic acid were dissolved in degassed 1, 4-dioxane, and then Pd[P (pH3)4] was added quickly. Degassed 3 M aqueous Na2CO3 (1.25 mL) was added into the solution, which was degassed for 10 min by sparging with argon gas. The reaction mixture was heated at 90 °C for 5 h under argon atmosphere. The solvent was removed with rotary evaporator. EtOAc was added. The organic layer was washed with brine, dried over anhydrous MgSO4 and purified by chromatography on a silica gel column (PE/EtOAc = 90:1) to obtain compound 15a as a pure yellow powder (0.35 g, 87% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.17 (d, J = 2.4 Hz, 1H), 7.94 (dd, J = 8.5, 2.3 Hz, 1H), 7.69 (d, J = 8.5 Hz, 1H), 7.63–7.58 (m, 2H), 7.16 (dd, J = 5.2, 3.5 Hz, 1H), 1.45 (s, 9H).
4.1.7.2. tert-Butyl (3-Nitro-[1, 1′-biphenyl]-4-yl) carbamate (15b)
Yellow pure solid, 25% yield. ESI-MS m/z: 337.6 [M+Na]+.
4.1.8. tert-Butyl (2-Amino-4-(thiophen-2-yl)phenyl)carbamate (16a) and tert-butyl (3-amino-[1,1′-biphenyl]-4-yl)carbamate (16b)
4.1.8.1. tert-Butyl (2-amino-4-(thiophen-2-yl)phenyl)carbamate (16a)
Compound 15a (0.53 g, 1.66 mmol) was solubilized in methanol and EtOAc (6 + 16 mL) and stirred at room temperature for 20 h with Pd/C (0.05 g, 10% w/w) under hydrogen atmosphere. The catalyst was filtered off, and the solvent was removed and washed with PE to give compound 16a as a white solid (0.47 g, 98% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.34 (s, 1H), 7.44–7.42 (m, 1H), 7.29 (dd, J = 3.5, 1.2 Hz, 1H), 7.26 (d, J = 8.5 Hz, 1H), 7.11 (dd, J = 5.3, 3.5 Hz, 1H), 6.97 (d, J = 2.0 Hz, 1H), 6.78 (dd, J = 8.5, 2.2 Hz, 1H), 5.04 (s, 2H), 1.45 (s, 9H).
4.1.8.2. tert-Butyl (3-amino-[1,1′-biphenyl]-4-yl)carbamate (16b)
White solid, 90% yield. Mp: 171–173 °C, ESI-MS m/z: 307.4 [M +Na]+.
4.1.9. 4-((2,2,2-Trifluoroacetamido)methyl)benzoic acid (18)
Trifluoroacetic anhydride (4 mL) was dropwise added to compound 17 (1.00 g, 6.62 mmol) under the ice baths. The suspension was stirred at room temperature for 2 h, and quenched by adding ice water. Compound 18 was collected via filtration and dried under vacuum to get a white solid (1.50 g, 93% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.96 (s, 1H), 10.09 (t, J = 5.9 Hz, 1H), 7.93 (d, J = 8.3 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 4.47 (d, J = 6.0 Hz, 2H).
4.1.10. tert-Butyl (4-(Thiophen-2-yl)-2-(4-((2,2,2-trifluoroacetamido) methyl) benzamido) phenyl) carbamate (20a) and tert-butyl (3-(4-((2,2,2-trifluoroacetamido)methyl) benzamido)-[1,1′-biphenyl]-4-yl) carbamate (20b)
4.1.10.1. tert-Butyl (4-(Thiophen-2-yl)-2-(4-((2,2,2-trifluoroac-etamido) methyl) benzamido) phenyl) carbamate (20a)
To a solution of compound 18 (0.5 g, 2.02 mmol) in anhydrous THF (7 mL) was added SOCl2 (0.74 mL, 10.11 mmol) slowly under ice baths. The mixture was heated at reflux for 3 h, and concentrated under vacuum to get crude material 19 as yellow oil. The residue solubi-lized in pyridine (3 mL) was added to the solution of the compound 16a (0.43 g, 1.50 mmol) in pyridine (3 mL) slowly under ice baths. The mixture was stirred overnight at room temperature. Pyridine was removed. The residue was extracted with CH2Cl2 (3×20 mL) and washed with 1 M HCl (3×20 mL), saturated NaHCO3 (3×20 mL) and brine (3×20 mL). The under solvent was evaporated vacuum to give the crude product which was recrystallized by EtOAc. Finally, the desired compound 20a was achieved as a white solid (0.55 g, 71% yield).
4.1.10.2. tert-Butyl (3-(4-((2,2,2-Trifluoroacetamido)methyl) benza-mido)-[1,1′-biphenyl]-4-yl) carbamate (20b)
White solid, 52% yield. 1H NMR (300 MHz, DMSO-d6) δ 10.12 (t, J = 6.0 Hz, 1H), 9.96 (s, 1H), 8.80 (s, 1H), 8.00 (d, J = 8.3 Hz, 2H), 7.88 (d, J = 2.1 Hz, 1H), 7.71–7.62 (m, 3H), 7.53 (dd, J = 8.5, 2.2 Hz, 1H), 7.50–7.43 (m, 4H), 7.36 (t, J = 7.3 Hz, 1H), 4.50 (d, J = 5.9 Hz, 2H), 1.47 (s, 9H).
4.1.11. tert-Butyl (2-(4-(aminomethyl)benzamido)-4-(thiophen-2-yl) phenyl)carbamate (21a) and tert-butyl (3-(4-(aminomethyl) benzamido)-[1,1′-biphenyl]-4-yl)carbamate (21b)
4.1.11.1. tert-Butyl (2-(4-(aminomethyl)benzamido)-4-(thiophen-2-yl)phenyl)carbamate (21a)
To a solution of compound 20a (0.37 g, 0.72 mmol) in THF (4 mL), methanol (3 mL) and water (2 mL) was added K2CO3 (0.40 g, 2.88 mmol). The mixture was kept at reflux for 6 h, and then cooled to room temperature. The solution was concentrated and dissolved in CH2Cl2 (30 mL) again. After washing with water, the product 21a precipitated from the solvent and filtered to obtain a white solid (0.30 g, 100% yield).
4.1.11.2. tert-Butyl (3-(4-(aminomethyl)benzamido)-[1,1′-biphenyl]-4-yl)carbamate (21b)
White solid, 88% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 8.1 Hz, 2H), 7.58–7.48 (m, 3H), 7.45 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 8.1 Hz, 1H), 6.94–6.86 (m, 1H), 4.24 (d, J = 6.3 Hz, 1H), 3.79 (s, 2H), 1.45 (s, 9H).
4.1.12. General procedure of preparation of 22a and its analogues 22b–22d
4.1.12.1. tert-Butyl (2-(4-((2-(5-Chloro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)methyl)benzamido)-4-(thiophen-2-yl) phenyl)carbamate (22a)
Compound 6b (0.19 g, 0.65 mmol) was dissolved in anhydrous THF and CH2Cl2, then TBTU (0.25 g, 0.77 mmol) was added followed by Et3N (0.33 mL, 2.36 mmol). After 40 min, another reactant, 22a (0.25 g, 0.59 mmol) was added. The mixture solution was stirred at room temperature overnight. Solvent was concentrated and dissolved in EtOAc. The organic layer was washed with saturated NaHCO3 (3×20 mL) and brine (3×20 mL), dried by MgSO4, and the solution was evaporated in vacuum. The crude product was purified by silica chromatography column (CH2Cl2/CH3OH = 100:1) to get compound 22a as a white solid (0.41 g, 89% yield). Mp: 176–178 °C, 1H NMR (300 MHz, DMSO-d6) δ 9.95 (s, 1H), 8.93 (t, J = 5.9 Hz, 1H), 8.77 (s, 1H), 7.97 (d, J = 8.3 Hz, 2H), 7.84 (d, J = 2.1 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.56–7.51 (m, 2H), 7.48–7.41 (m, 5H), 7.14 (dd, J = 5.1, 3.6 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 4.73 (td, J = 11.3, 2.1 Hz, 2H), 4.42 (s, 2H), 4.41 (s, 2H), 3.99–3.90 (m, 2H), 2.28–2.10 (m, 1H), 1.74–1.65 (m, 1H), 1.46 (s, 9H).
4.1.12.2. tert-Butyl (2-(4-((2-(5-chloro-2-oxospiro[indoline-3,2′-[1,3]-dioxolan]-1-yl)acetamido)methyl)benzamido)-4-(thiophen-2-yl) phenyl)carbamate (22b)
White solid, 89% yield. Mp: 174–176 °C, 1H NMR (300 MHz, DMSO-d6) δ 10.01 (s, 1H), 8.99 (t, J = 5.9 Hz, 1H), 8.80 (s, 1H), 7.98 (d, J = 8.3 Hz, 2H), 7.83 (d, J = 2.1 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.54 (dd, J = 3.6, 1.5 Hz, 2H), 7.51–7.45 (m, 4H), 7.43 (s, 1H), 7.13 (dd, J = 5.1, 3.6 Hz, 1H), 7.02 (dd, J = 7.7, 1.1 Hz, 1H), 4.41 (s, 2H), 4.39 (s, 2H), 4.37–4.30 (m, 4H), 1.46 (s, 9H).
4.1.12.3. tert-Butyl (3-(4-((2-(5-chloro-2-oxospiro[indoline-3,2′-[1,3]-dioxan]-1-yl)acetamido)methyl)benzamido)-[1,1′-biphenyl]-4-yl)car-bamate (22c)
White solid, 60% yield. Mp: 141–143 °C, 1H NMR (300 MHz, DMSO-d6) δ 9.91 (s, 1H), 8.88 (t, J = 5.9 Hz, 1H), 8.78 (s, 1H), 7.96 (d, J = 8.3 Hz, 2H), 7.87 (d, J = 2.1 Hz, 1H), 7.65 (d, J = 8.3 Hz, 3H), 7.55–7.41 (m, 7H), 7.40–7.33 (m, 1H), 6.98 (d, J = 8.3 Hz, 1H), 4.79–4.67 (m, 2H), 4.41 (s, 4H), 4.00–3.90 (m, 2H), 2.26–2.10 (m, 1H), 1.75–1.65 (m, 1H), 1.47 (s, 9H).
4.1.12.4. tert-Butyl (3-(4-((2-(5-Chloro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)methyl)benzamido)-[1,1′-biphenyl]-4-yl)carbamate (22d)
White solid, 62% yield. Mp: 183–185 °C, 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.04 (s, 1H), 8.84 (s, 1H), 8.00 (dd, J = 8.2, 3.9 Hz, 2H), 7.88 (d, J = 1.3 Hz, 1H), 7.66 (dd, J = 8.2, 3.2 Hz, 3H), 7.52 (dd, J = 8.5, 2.2 Hz, 1H), 7.51–7.43 (m, 6H), 7.36 (t, J = 7.3 Hz, 1H), 7.02 (d, J = 7.9 Hz, 1H), 4.44–4.38 (m, 4H), 4.38–4.30 (m, 4H), 1.47 (s, 9H).
4.1.13. General procedure for the preparation of 23a and its analogues 23b–23d
4.1.13.1. N-(2-Amino-5-(thiophen-2-yl)phenyl)-4-((2-(5-chloro-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)methyl)benza-mide (23a)
Compound 22a (0.40 g, 0.57 mmol) was solubilized in the solution of EtOAc (4 mL) saturated by anhydrous HCl gas. After 5 h, the precipitate was collected by filter as a yellow solid. The crude product was washed with saturated NaHCO3, extracted with EtOAc, and purified by chromatography on a silica gel column (CH2Cl2/CH3OH = 80:1) to afford compound 23a as a white solid (0.11 g, 32% yield). Mp: 208–210 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.73 (s, 1H), 8.88 (t, J = 5.8 Hz, 1H), 7.99 (d, J = 8.1 Hz, 2H), 7.49 (d, J = 1.6 Hz, 1H), 7.46 (dd, J = 8.4, 2.2 Hz, 1H), 7.44–7.43 (m, 2H), 7.41 (s, 1H), 7.36 (dd, J = 5.0, 0.7 Hz, 1H), 7.31 (dd, J = 8.3, 2.1 Hz, 1H), 7.25 (d, J = 2.7 Hz, 1H), 7.06 (dd, J = 5.0, 3.7 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.83 (d, J = 8.4 Hz, 1H), 5.16 (s, 2H), 4.73 (dd, J = 11.5, 9.4 Hz, 2H), 4.41 (s, 2H), 4.40 (s,2H), 3.96 (dd, J = 11.4, 2.9 Hz, 2H), 2.27–2.13 (m, 1H), 1.72–1.69 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 171.19, 166.62, 165.72, 144.71, 143.51, 143.12, 141.80, 133.59, 131.05, 128.75, 128.69, 128.40, 127.45, 127.33, 124.42, 123.85, 123.70, 122.72, 121.42, 116.79, 111.64, 93.41, 61.11, 42.53, 42.16, 25.21. HRMS (AP-ESI) m/z calcd for C31H27ClN4O5S [M+H]+ 603.1463, found 603.1598. HPLC tR = 16.93 min, 95.5%.
4.1.13.2. N-(2-Amino-5-(thiophen-2-yl)phenyl)-4-((2-(5-chloro-2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)methyl)benza-mide (23b)
White solid, 47% yield. Mp: 198–200 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 8.90 (t, J = 5.9 Hz, 1H), 7.99 (d, J = 8.1 Hz, 2H), 7.53 (d, J = 1.7 Hz, 1H), 7.52–7.47 (m, 2H), 7.43 (s, 1H), 7.39 (dd, J = 6.5, 1.1 Hz, 2H), 7.36 (dd, J = 8.3, 2.0 Hz, 1H), 7.29 (d, J = 3.3 Hz, 1H), 7.07 (dd, J = 5.0, 3.7 Hz, 1H), 7.01 (d, J = 8.4 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H), 4.40 (s, 2H), 4.39(s, 2H), 4.37–4.27 (m, 4H).13C NMR (100 MHz, DMSO-d6) δ 172.80, 166.59, 165.81, 144.36, 143.27, 143.23, 133.44, 131.71, 128.76, 128.44, 127.62, 127.47, 126.55, 125.13, 124.36, 124.14, 121.96, 117.92, 111.95, 101.55, 66.34, 42.53, 42.48. HRMS (AP-ESI) m/z calcd for C30H25ClN4O5S [M+H]+ 589.1307, found 589.1436. HPLC tR = 11.31 min, 95.4%.
4.1.13.3. N-(4-Amino-[1,1′-biphenyl]-3-yl)-4-((2-(5-chloro-2-oxos-piro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)methyl)benzamide (23c)
White solid, 65% yield. Mp: 218–220 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.73 (s, 1H), 8.88 (t, J = 5.9 Hz, 1H), 7.99 (d, J = 8.1 Hz, 2H), 7.56 (d, J = 7.5 Hz, 2H), 7.54 (d, J = 1.5 Hz, 1H), 7.46 (dd, J = 8.4, 2.2 Hz, 1H), 7.44–7.36 (m, 5H), 7.34 (dd, J = 8.3, 2.1 Hz, 1H), 7.24 (t, J = 7.3 Hz, 1H), 6.97 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H), 5.11 (s, 2H), 4.73 (t, J = 10.5 Hz, 2H), 4.41 (s, 2H), 4.39(s, 2H), 3.97–3.93 (m, 2H), 2.19 (m, 1H), 1.71–1.68 (m, 1H).13C NMR (100 MHz, DMSO-d6) δ 171.20, 166.62, 165.71, 143.27, 143.07, 141.80, 140.66, 133.70, 131.05, 129.27, 128.75, 128.60, 128.38, 127.47, 127.33, 126.50, 125.97, 125.28, 125.17, 124.42, 124.05, 116.99, 111.64, 93.41, 61.11, 42.53, 42.16, 25.21. HRMS (AP-ESI) m/z calcd for C33H29ClN4O5 [M+H]+ 597.1899, found 597.1966. HPLC tR = 19.26 min, 95.1%.
4.1.13.4. N-(4-Amino-[1,1′-biphenyl]-3-yl)-4-((2-(5-chloro-2-oxos-piro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetamido)methyl)benzamide (23d)
White solid, 98% yield. Mp: 191–192 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.74 (s, 1H), 8.89 (t, J = 5.9 Hz, 1H), 7.99 (d, J = 8.2 Hz, 2H), 7.55 (m, 3H), 7.51–7.47 (m, 2H), 7.43–7.37 (m, 4H), 7.34 (dd, J = 8.3, 2.2 Hz, 1H), 7.24 (t, J = 7.3 Hz, 1H), 7.00 (d, J = 8.6 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H), 5.11 (s, 2H), 4.40 (s, 2H), 4.38 (s, 2H), 4.37–4.28 (m, 4H).13C NMR (100 MHz, DMSO-d6) δ 172.80, 166.58, 165.73, 143.27, 143.07, 140.67, 133.72, 131.71, 129.27, 128.60, 128.38, 127.62, 127.47, 126.56, 126.49, 125.98, 125.27, 125.13, 124.07, 116.99, 111.94, 101.55, 66.34, 42.54, 42.48. HRMS (AP-ESI) m/z calcd for C32H27ClN4O5 [M+H]+ 583.1743, found 583.1787. HPLC tR = 12.89 min, 97.3%.
4.1.14. tert-Butyl (5-fluoro-2-(4-((2,2,2-trifluoroacetamido)methyl) benzamido)phenyl)carbamate (27)
Using the synthetic method for compounds 20a, compounds 19 and 26 gave the compound 27 as a white solid, 57% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H), 9.81 (s, 1H), 8.80 (s, 1H), 7.95 (d, J = 8.2 Hz, 2H), 7.53 (dd, J = 11.1, 2.9 Hz, 1H), 7.45 (dd, J = 12.4, 7.3 Hz, 3H), 6.97 (td, J = 8.5, 3.0 Hz, 1H), 4.48 (s, 2H), 1.45 (s, 9H).
4.1.15. tert-Butyl (2-(4-(aminomethyl)benzamido)-5-fluorophenyl)carbamate (28)
Using the synthetic method for compounds 21a, compound 27 gave the compound 28 as a white solid, 98% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 8.1 Hz, 2H), 7.58–7.48 (m, 3H), 7.45 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 8.1 Hz, 1H), 6.94–6.86 (m, 1H), 4.24 (d, J = 6.3 Hz, 1H), 3.79 (s, 2H), 1.45 (s, 9H).
4.1.16. tert-Butyl (2-(4-((2-(5-bromo-2-oxospiro[indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)methyl)benzamido)-5-fluorophenyl)carbamate (29)
Using the synthetic method for compounds 22a, compounds 6a and 28 gave the compound 29 as a white solid, 46% yield. 1H NMR (400 MHz, DMSO-d6) δ 9.78 (s, 1H), 8.88 (t, J = 5.9 Hz, 1H), 8.77 (s, 1H), 7.94 (d, J = 8.1 Hz, 2H), 7.58 (dd, J = 8.4, 2.0 Hz, 1H), 7.52 (td, J = 6.0, 3.0 Hz, 2H), 7.49–7.41 (m, 3H), 7.00–6.91 (m, 2H), 4.72 (dd, J = 11.6, 9.3 Hz, 2H), 4.39 (d, J = 4.1 Hz, 4H), 3.95 (dd, J = 8.1, 6.4 Hz, 2H), 2.25–2.15 (m, 1H), 1.71–1.67 (m, 1H), 1.45 (s, 9H).
4.1.17. N-(2-Amino-4-fluorophenyl)-4-((2-(5-bromo-2-oxospiro [indoline-3,2′-[1,3]dioxan]-1-yl)acetamido)methyl)benzamide (30)
Using the synthetic method for compounds 23a, compound 29 gave the compound 30 as a white solid, 98% yield. Mp: 237–239 °C, 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 1H), 8.88 (t, J = 5.7 Hz, 1H), 7.95 (d, J = 8.1 Hz, 2H), 7.59 (dd, J = 8.4, 1.9 Hz, 1H), 7.53 (d, J = 1.9 Hz, 1H), 7.39 (d, J = 8.1 Hz, 2H), 7.11 (dd, J = 8.3, 6.6 Hz, 1H), 6.92 (d, J = 8.4 Hz, 1H), 6.54 (dd, J = 11.2, 2.8 Hz, 1H), 6.36 (td, J = 8.5, 2.7 Hz, 1H), 5.24 (s, 2H), 4.72 (dd, J = 11.5, 9.6 Hz, 2H), 4.39 (d, J = 8.5 Hz, 4H), 3.94 (dd, J = 11.3, 2.9 Hz, 2H), 2.25–2.11 (m, 1H), 1.69 (d, J = 13.4 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 171.08, 166.59, 165.82, 143.03, 142.22, 133.91, 133.60, 129.07, 128.35, 127.41, 127.11, 119.74, 114.88, 112.13, 102.60, 102.38, 102.04, 101.78, 93.34, 61.21, 42.52, 42.11, 25.20. HRMS (AP-ESI) m/z calcd for C27H24BrFN4O5 [M+H]+ 583.0987, found 583.0983. HPLC tR = 8.81 min, 97.2%.
4.2. Biological materials and methods
4.2.1. In vitro HDAC inhibition fluorescence assay
In vitro HDAC inhibition assays were conducted as previously described.25 In brief, 10 lL of enzyme solution (HeLa nuclear extract, HDAC1, 2 or 3) was mixed with various concentrations of 50 μL tested compounds (0.039, 0.39, 1.56, 6.25, 25, 100 μM). The mixture was incubated at 37 °C for 1 h, then fluorogenic substrate Boc-Lys (acetyl)-AMC (40 μL) was added, and the suspension was incubated at 37 °C for another 1 h. The reaction was ended by addition of 100 μL of developer containing trypsin and TSA. After incubation under the same situation for 20 min, fluorescence intensity was monitored through a microplate reader at excitation and emission wavelengths of 390 and 460 nm, respectively. The inhibition ratios were calculated from the fluorescence intensity readings of tested wells relative to those of control wells, and the IC50 values were calculated using a regression analysis of the concentration/inhibition data.
4.2.2. In vitro antiproliferative assay
Cell antiproliferative assay was determined by MTT [(3-[4, 5-dimethyl-2-thiazolyl]-2, 5-diphenyl-2H-tetrazolium bromide)] method as previously described.25 All cell lines were cultured in RPMI 1640 medium containing 10% FBS at 37 °C in a 5% CO2 humidified incubator. Cells were plated into 96-well plates (5000/well), and permitted to grow for at least 4 h. After addition of different concentrations of compounds, cells were maintained for 48 h. A solution of 0.5% MTT was added to each well. After incubation for another 4 h, the formazan formed from MTT was extracted by adding 200 μL of DMSO for 15 min. Then the optical density values were measured using a UV–vis spectrophotometer at 570 nm.
4.2.3. Whole-cell HDAC inhibition assay
The whole-cell HDAC inhibition assay was based on assays published by Tessier P. et al. 26, Hildmann C. et al.27 and Marek L. et al.28 with minor modifications. Different subtypes of HDACs have intrinsic differences in substrate recognition. The lysine analogs Boc-Lys (acetyl)-AMC and Boc-Lys (triflouroacetyl)-AMC could permeate into cells and function as cellular class I and class IIa HDAC substrates, respectively. After deacetylation of the lysine residue, the product could be hydrolyzed by trypsin to release 7-amino-4-methylcoumarin (AMC), a well-studied fluorophore detectable by standard fluorescence measurements.
HEL cells were seeded into 96-well black tissue culture plates at a density of 8×103 cells/well in a total volume of 81 μL of culture medium and cultivated for 24 h under standard cell culture conditions. The volume of 9 μL of tested compounds with various concentrations was added to each well. After 3 h, 10 μL of 3 mM Boc-Lys (Ac)-AMC or Boc-Lys-(ε-trifluormethylacetyl)-AMC was added to reach a final concentration of 300 μM. Cells were placed with the substrates for 3 h in 37 °C incubator with 5% CO2. The reaction was stopped by adding 100 μL/well lysis/developer buffer mix (50 mM Tris-HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1% Nonidet-P40, 2.0 mg/mL trypsin, 10 μM TSA). After final incubation for 3 h, fluorescence was measured at excitation of 360 nm and emission of 470 nm.
4.2.4. Molecular docking studies
Compounds were docked into the active site of HDAC1 (PDB entry: 5ICN) using Tripos SYBYL-X 1.3. Before docking process, the protein structure retrieved from PDB site was treated by deleting water molecules, FF99 charges. A 100-step minimization process was performed to further optimize the protein structure. The molecular structures were generated with Sybyl/Sketch module and optimized using Powell’s method with the Tripos force field with convergence criterion setting at 0.005 kcal/(Åmol) and assigned charges with the Gasteiger-Hückel method. Molecular docking was carried out via the Sybyl/Surflex-Dock module. Other docking parameters were kept to the default values.
Supplementary Material
Acknowledgments
This work was supported by National Natural Science Foundation of China (Grant Nos. 21302111, 81373282), National High-Tech R&D Program of China (863 Program) (Grant No. 2014AA020523), Young Scholars Program of Shandong University (YSPSDU, Grant NO. 2016WLJH33), Major Project of Science and Technology of Shandong Province. (2015ZDJS04001). National Cancer Institute of the National Institute of Health (Award no. R01CA163452).
A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2017.03.036.
Footnotes
Conflict of interest
The authors confirm that this article content has no conflict of interest.
References
- 1.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther. 2009;8(6):1409–1420. doi: 10.1158/1535-7163.MCT-08-0860. [DOI] [PubMed] [Google Scholar]
- 3.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370(Pt 3):737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spange S, Wagner T, Heinzel T, et al. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41(1):185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 5.Witt O, Deubzer HE, Milde T, et al. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277(1):8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 6.Burdelski C, Ruge OM, Melling N, et al. HDAC1 overexpression independently predicts biochemical recurrence and is associated with rapid tumor cell proliferation and genomic instability in prostate cancer. Exp Mol Pathol. 2015;98(3):419–426. doi: 10.1016/j.yexmp.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 7.Huang Y, Chen J, Lu C, et al. HDAC1 and Klf4 interplay critically regulates human myeloid leukemia cell proliferation. Cell Death Dis. 2014;5(10):e1491. doi: 10.1038/cddis.2014.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krämer OH. HDAC2: a critical factor in health and disease. Trends Pharmacol Sci. 2009;30(12):647–655. doi: 10.1016/j.tips.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Mariadason JM. HDACs and HDAC inhibitors in colon cancer. Epigenetics. 2008;3(1):28–37. doi: 10.4161/epi.3.1.5736. [DOI] [PubMed] [Google Scholar]
- 10.Gupta M, Han JJ, Stenson M, et al. Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: implications for therapy. Leukemia. 2012;26(6):1356–1364. doi: 10.1038/leu.2011.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moreno DA, Scrideli CA, Cortez MAA, et al. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br J Haematol. 2010;150(6):665–673. doi: 10.1111/j.1365-2141.2010.08301.x. [DOI] [PubMed] [Google Scholar]
- 12.Gao S, Li X, Zang J, et al. Preclinical and clinical studies of chidamide (CS055/HBI-8000), an orally available subtype-selective HDAC inhibitor for cancer therapy. Anti-Cancer Agents Med Chem. 2016 doi: 10.2174/1871520616666160901150427. [DOI] [PubMed] [Google Scholar]
- 13.Bertrand P. Inside HDAC with HDAC inhibitors. Eur J Med Chem. 2010;45(6):2095–2116. doi: 10.1016/j.ejmech.2010.02.030. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Inks ES, Li X, et al. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J Med Chem. 2014;57(8):3324–3341. doi: 10.1021/jm401877m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medvedev A, Buneeva O, Glover V. Biological targets for isatin and its analogues: implications for therapy. Biologics. 2007;1(2):151–162. [PMC free article] [PubMed] [Google Scholar]
- 16.Hoessel R, Leclerc S, Endicott JA, et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat Cell Biol. 1999;1(1):60–67. doi: 10.1038/9035. [DOI] [PubMed] [Google Scholar]
- 17.Jin K, Li S, Li X, et al. Design, synthesis and preliminary biological evaluation of indoline-2, 3-dione derivatives as novel HDAC inhibitors. Bioorg Med Chem. 2015;23(15):4728–4736. doi: 10.1016/j.bmc.2015.05.048. [DOI] [PubMed] [Google Scholar]
- 18.Chutake YK, Lam CC, Costello WN, et al. Reversal of epigenetic promoter silencing in Friedreich ataxia by a class I histone deacetylase inhibitor. Nucleic Acids Res. 2016:gkw107. doi: 10.1093/nar/gkw107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou J, Xie H, Liu Z, et al. Structure-function analysis of the conserved tyrosine and diverse p-stacking among Class I histone deacetylases: a QM (DFT)/MM MD study. J Chem Inf Model. 2014;54(11):3162–3171. doi: 10.1021/ci500513n. [DOI] [PubMed] [Google Scholar]
- 20.Gong K, Xie J, Yi H, et al. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem J. 2012;443(3):735–746. doi: 10.1042/BJ20111685. [DOI] [PubMed] [Google Scholar]
- 21.Witter DJ, Harrington P, Wilson KJ, et al. Optimization of biaryl selective HDAC1&2 inhibitors (SHI-1: 2) Bioorg Med Chem Lett. 2008;18(2):726–731. doi: 10.1016/j.bmcl.2007.11.047. [DOI] [PubMed] [Google Scholar]
- 22.Wambua MK, Nalawansha DA, Negmeldin AT, et al. Mutagenesis studies of the 14 Å internal cavity of histone deacetylase 1: insights toward the acetate-escape hypothesis and selective inhibitor design. J Med Chem. 2014;57(3):642–650. doi: 10.1021/jm401837e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moradei OM, Mallais TC, Frechette S, et al. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J Med Chem. 2007;50(23):5543–5546. doi: 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]
- 24.Stolfa DA, Stefanachi A, Gajer JM, et al. Design, synthesis, and biological evaluation of 2-aminobenzanilide derivatives as potent and selective HDAC inhibitors. ChemMedChem. 2012;7(7):1256–1266. doi: 10.1002/cmdc.201200193. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Inks ES, Li X, et al. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J Med Chem. 2014;57(8):3324–3341. doi: 10.1021/jm401877m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tessier P, Smil DV, Wahhab A, et al. Diphenylmethylene hydroxamic acids as selective class IIa histone deacetylase inhibitors. Bioorg Med Chem Lett. 2009;19(19):5684–5688. doi: 10.1016/j.bmcl.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Hildmann C, Wegener D, Riester D, et al. Substrate and inhibitor specificity of class 1 and class 2 histone deacetylases. J Biotechnol. 2006;124(1):258–270. doi: 10.1016/j.jbiotec.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 28.Marek L, Hamacher A, Hansen FK, et al. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J Med Chem. 2013;56(2):427–436. doi: 10.1021/jm301254q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.