

Abstract
Previously, we reported the discovery of a series of N-hydroxycinnamamide-based HDAC inhibitors, among which compound 11y exhibited high HDAC1/3 selectivity. In this current study, structural derivatization of 11y led to a new series of benzamide based HDAC inhibitors. Most of the compounds exhibited high HDACs inhibitory potency. Compound 11a (with 4-methoxybenzoyl as N-substituent in the cap and 4-(aminomethyl) benzoyl as the linker group) exhibited selectivity against HDAC1 to some extent, and showed potent antiproliferative activity against several tumor cell lines. In vivo studies revealed that compound 11a displayed potent oral antitumor activity in both hematological tumor cell U937 xenograft model and solid tumor cell HCT116 xenograft model with no obvious toxicity. Further modification of benzamide 3, 11a and 19 afforded new thienyl and phenyl compounds (50a, 50b, 63a, 63b and 63c) with dramatic HDAC1 and HDAC2 dual selectivity, and the fluorine containing compound 56, with moderate HDAC3 selectivity.
Keywords: HDACs, HDACIs, Benzamides, Anti-tumor drugs, In vivo oral activity
1. Introduction
It is widely considered that the initiation and progression of cancer are associated with genetic abnormalities, as well as epigenetic alterations that do not change the nucleotide sequence of DNA [1]. Histone deacetylases (HDACs) are one of the most studied epigenetic modulators, modifying the acetylation status of chromatin histones and non-histone proteins [2]. In detail, HDACs remove acetyl groups from lysine residues, resulting in a “closed” chromatin configuration, which blocks the access of the transcription machinery to DNA, and suppresses gene expression including tumor suppressor genes [3].
Currently a total of 18 HDACs have been identified in humans, which can be divided into 4 classes according to their homology. Class I includes HDACs 1, 2, 3, and 8, and they are homologous to yeast Rpd3; class IIa (HDACs 4, 5, 7 and 9) and class IIb (HDAC6 and 10) are structurally related to yeast Hda1; Class IV has only one member, HDAC11. These HDAC isoforms are all Zn2+-dependent enzymes, which bear a highly conserved catalytic site. The class III HDACs (comprising SIRT 1-7) are related to the yeast protein Sir2 and their activities are depended on NAD+ concentration [4,5]. The exact physiological role of these HDAC subtypes in cells is far from being completely elucidated, however, the division of labor among them has been studied using knockout and transgenic mice and RNAi techniques, in which, class I and class II are the most well studied [6,7]. The knockout of HDAC1 in mice caused cell proliferation defects and developmental retardation, and this unique function cannot be compensated by HDAC2 and HDAC3 [8,9]. HDAC2 is involved in cardiac morphogenesis until the perinatal period of mice [10]. HDAC3 has been reported to function in proliferative mitosis and the regulation of INF expression [11]. In normal human tissues, HDAC8 is associated with the regulation of the contractile capacity [12]. Unlike class I HDACs, Class IIa HDACs (HDAC4, 5, 7 and 9) display tissue-specific expression and are involved in a variety of signal transduction pathways [13]. The well-known class IIb HDAC6 controls the status of tubulin and hsp90 acetylation [14]. All these indicates each HDAC enzyme has its unique function in normal cells, therefore, most pan-HDAC inhibitors (HDACIs), which inhibit a broad spectrum of HDACs, exhibited significant toxicity. For example, panobinostat (LBH589) caused serious thrombocytopenia, atrial fibrillation, eye swelling, neutropenia, and anemia [15]. Thus, increasing efforts are being directed toward developing selective HDACIs for a single class or a single isoform, which are likely to have fewer adverse reactions and a better tolerance. A few Class I selective HDACIs such as MS-275 and MGCD0103 (HDAC1, 2, and 3-selective) are currently in various stages of clinical trial for cancer therapeutics [16], and Chidamide recently has been approved by China food and drug administration (CFDA).
In our previous study, we designed and synthesized N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral anti-leukemia activity [17]. Efforts to improve the selectivity of the dually reported the discovery of a new series of benzamides based HDACIs. The structure-activity relationship (SAR) studies, isotype-selectivity, and anticancer activities of benzamide HDACIs were particularly discussed. One representative compound 11a exhibited selectivity against HDAC1, and showed potent antiproliferative activity against solid and hematological tumor cell lines both in vitro and in vivo. Further modified thienyl and phenyl compounds all exhibited dramatic HDAC1/2 dual selectivity.
1.1. Chemistry
The synthesis of compound 3 was described in Scheme 1. The synthesis of the starting material 1 was described in our previous study [18]. Deprotection of methyl group of 1 in the presence of KOH aqueous solution yielded 2, which was then coupled with o-phenylenediamine in anhydrous THF to yield the final product 3.
Scheme 1.
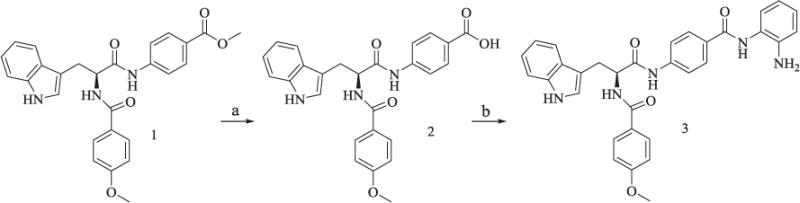
Synthesis of Compound 3.
Reagents and conditions: (a) CH3OH/H2O, KOH, reflux, 85% yield; (b) TBTU, Et3N, anhydrous THF, 30–33% yield.
The synthesis of compounds 11a-11g was described in Scheme 2. Methyl ester protection of 5 gave rise to 6, which was then reacted with Boc-protected L-Trp by TBTU-mediated amide formation to afford 7. N-deprotection of 7 followed by condensation with different carboxylic acids gave 9a-9g, which were deprotected in the presence of KOH aqueous solution to obtained 10a-10h. 10a-10h were then subjected to hydrolysis reaction and amide formation reaction as 2 in Scheme 1 to yield the final products 11a-11g, respectively.
Scheme 2.
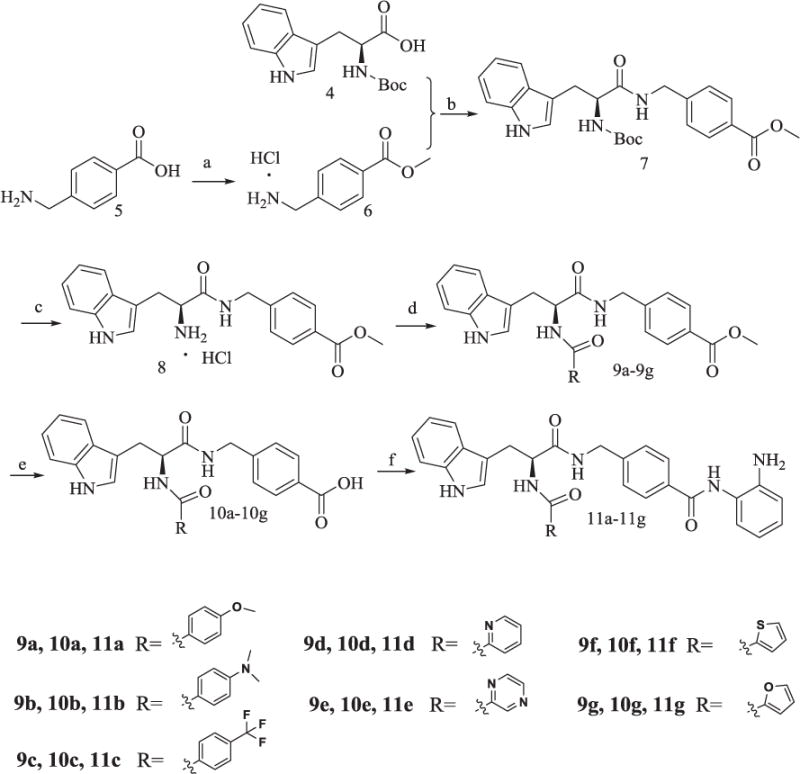
Synthesis of Compounds 11a-11g.
Reagents and conditions: (a) CH3COCl, CH3OH, 95% yield; (b) TBTU, Et3N, anhydrous CH2Cl2, 60–65% yield; (c) AcOEt/HCl, 80% yield; (d) ROOH, TBTU, Et3N, anhydrous CH2Cl2, 60–65% yield; (e) CH3OH/H2O, KOH, reflux, 80% yield; (f) TBTU, Et3N, anhydrous THF, 30–33% yield.
Scheme 3 was similar to that of Scheme 2. 12 was reacted with 4-methoxybenzoic acid via condensation to get 13. 15 was subjected to methyl ester protection to afford 16. 14, the hydrolysis products of 13, was then connected with 16 to get 17 which was subjected to methyl deprotection and amide formation reaction as 3 in Scheme 1 to yield 19.
Scheme 3.
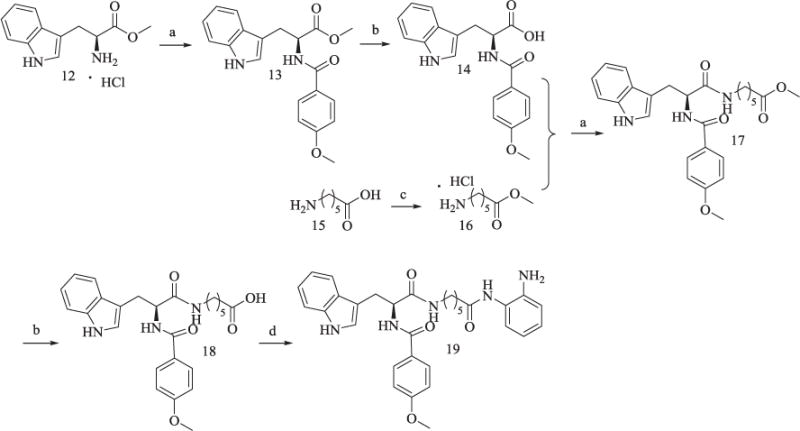
Synthesis of Compound 19.
Reagents and conditions: (a) TBTU, Et3N, anhydrous CH2Cl2, 60–65% yield; (b) CH3OH/H2O, KOH, reflux, 80% yield; (c) CH3COCl, CH3OH, 95% yield; (d) TBTU, Et3N, anhydrous THF, 30–33% yield.
Compound 20 of Scheme 4 was reported as an intermediate in our previous study [17], and it was transformed into compound 22 by the same reaction in Scheme 1.
Scheme 4.
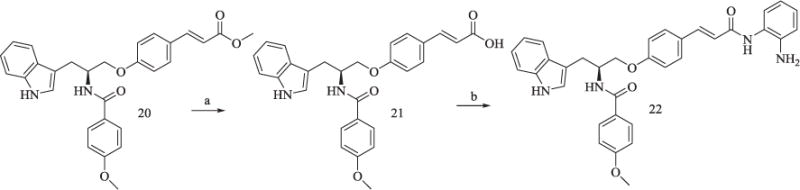
Synthesis of Compound 22.
Reagents and conditions: (a) CH3OH/H2O, KOH, reflux, 90% yield; (b) TBTU, Et3N, anhydrous THF, 30–33% yield.
Methyl ester protection of 24a and 24b gave 25a and 25b, which were connected with 23 through Mitsunobu reaction to give 26a and 26b (Scheme 5). Intermediate 27a and 27b, the N-deprotected product of 26a and 26b, were reacted with 4-methoxybenzoic acid to afford 28a and 28b, which were further subjected to methyl de-protection and condensation to yield the final products 30a and 30b, respectively.
Scheme 5.
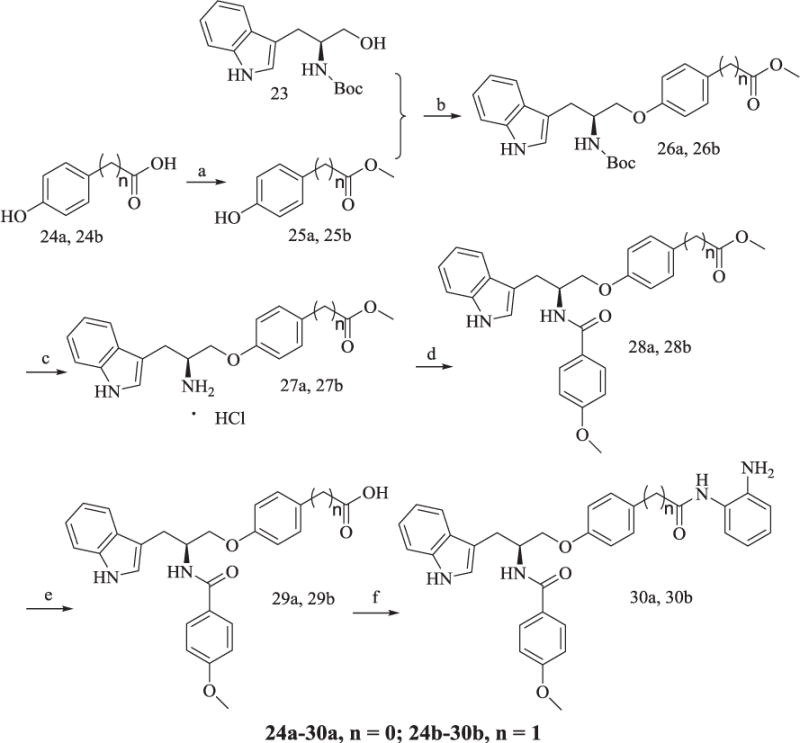
Synthesis of Compounds 30a, 30b.
Reagents and conditions: (a) CH3COCl, CH3OH, 95% yield; (b) PPh3, DEAD, anhydrous THF, 57% yield; (c) AcOEt/HCl, 80% yield; (d) TBTU, Et3N, anhydrous CH2Cl2, 60–65% yield; (e) CH3OH/H2O, KOH, reflux, 80% yield; (f) TBTU, Et3N, anhydrous THF, 30–33% yield.
Compound 37 was synthesized following the procedures described in Scheme 6. Condensation between intermediate 14 and N,O-dimethylhydroxylamine hydrochloride yielded 31, which was subsequently converted to aldehyde 32 by LiAlH4 at −40 °C. 32 was reacted with 6 in the presence of NaBH3CN to afford 33. Boc-protection, methyl deprotection, amide formation reaction and N-deprotection of 33 yielded the target product 37.
Scheme 6.
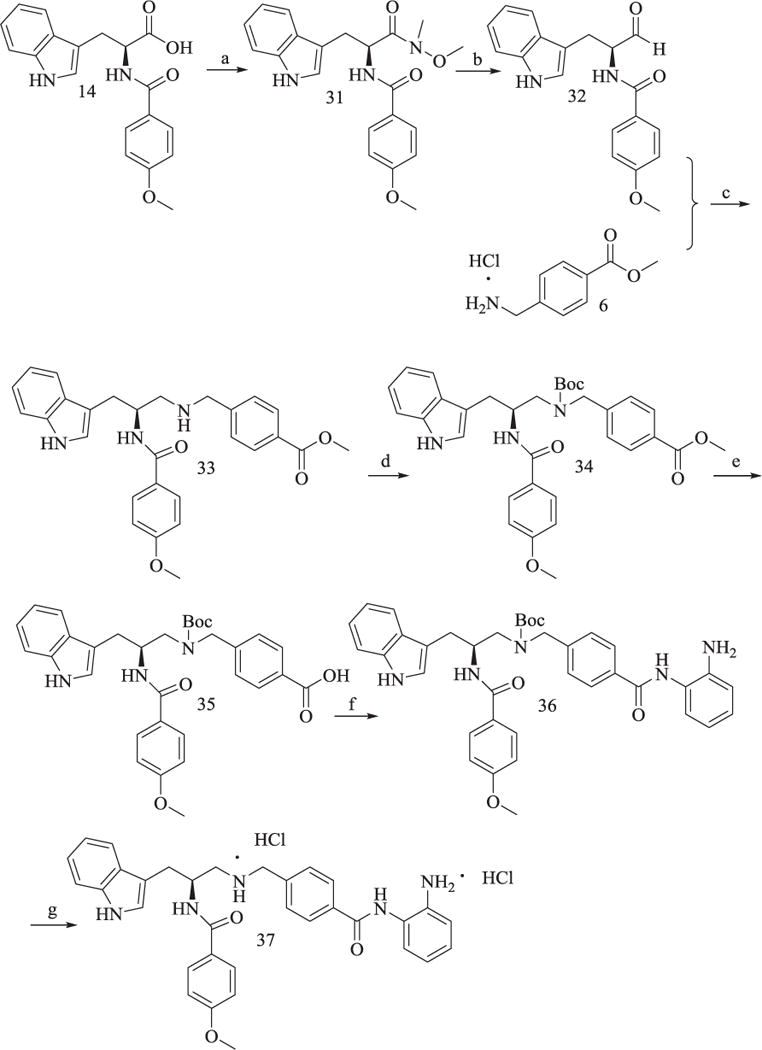
Synthesis of Compound 37.
Reagents and conditions: (a) TBTU, Et3N, anhydrous CH2Cl2, 60–65% yield; (b) LiAlH4, 2 h, 70% yield; (c) NaBH3CN, AcOH, Et3N, CH3OH, 53% yield; (d) (Boc)2O, Et3N, CH2Cl2, 90% yield; (e) CH3OH/H2O, KOH, reflux, 80% yield; (f) TBTU, Et3N, anhydrous THF, 48% yield; (g) AcOEt/HCl, 65% yield.
The reaction for 43a and 43b was described in Scheme 7. Boc-protection of 38 gave 39, which was coupled with thiophene-2-boronic acid or phenylboronic acid using Suzuki reaction in the presence of Pd(OAc)2 to afford 40a or 40b. Catalytic hydrogenation of 40a and 40b gave 41a and 41b, which were subjected to amide formation reaction and N-deprotection to obtain 43a and 43b, respectively.
Scheme 7.
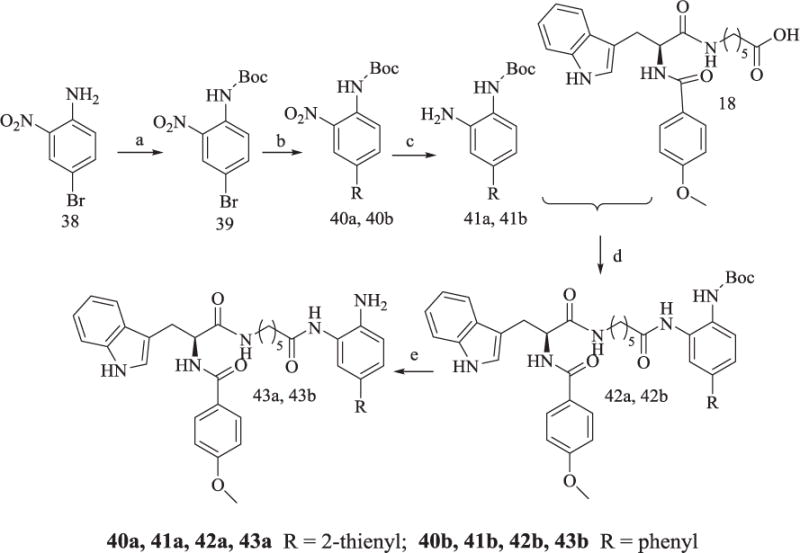
Synthesis of Compounds 43a and 43b.
Reagents and conditions: (a) (Boc)2O, Et3N, CH2Cl2, DMAP, 90% yield; (b) Thiophene-2-boronic acid or phenylboronic acid, Pd(OAc)2, PPh3, Na2CO3, H2O, DMSO, 48% yield; (c) Pd/C, H2, CH3OH, 85% yield; (d) TBTU, Et3N, anhydrous THF, 60–65% yield; (e) CF3COOH, 68% yield.
Emmanuel H. Demont et al. described the synthesis of 46 [19], briefly, double Boc-protection of 44 gave 45, then one Boc-protecting group was removed in the presence of trifluoroacetic acid to afford mono-Boc protected product 46. Reduction of 46 gave 47, which was subjected to a similar reaction of 41 in Scheme 7 to yield 49 (Scheme 8).
Scheme 8.
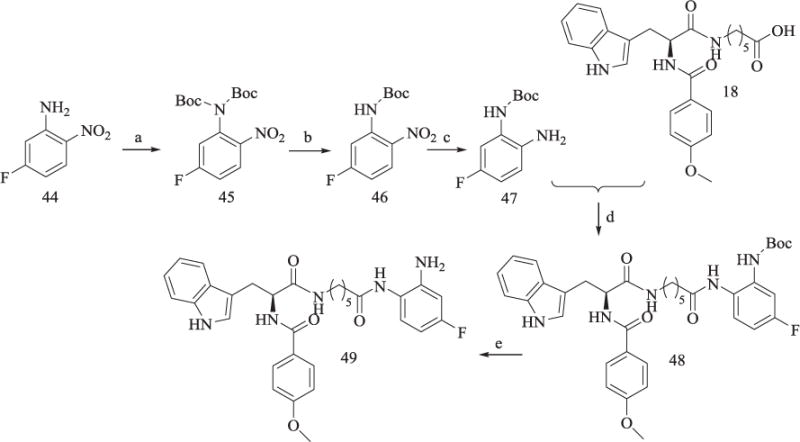
Synthesis of Compound 49.
Reagents and conditions: (a) (Boc)2O, Et3N, CH2Cl2, DMAP, 90% yield; (b) CF3COOH, CH2Cl2, 78% yield; (c) Pd/C, H2, ethyl alcohol, 85% yield; (d) TBTU, Et3N, anhydrous CH2Cl2, 60–65% yield; (e) CF3COOH, 65% yield.
6 and 50 were treated with trifluoroacetic anhydride to yield trifluoroacetyl protected 51a and 51b, respectively. 51a and 51b were converted to acyl chloride 52a and 52b. 52a was subsequently reacted with 41a to afford product 53a, while 52b was reacted with 41a and 41b to achieve 53b and 53c, respectively (Scheme 9). Trifluoroacetyl deprotection of 53a-53c gave 54a-54c, which were subjected to a similar reaction of 41 in Scheme 7 to yield 56a-56c. The synthesis of 60a and 60b in Scheme 10 was similar to the reaction of 52a converting to 56a, and this route would not be described in this section.
Scheme 9.
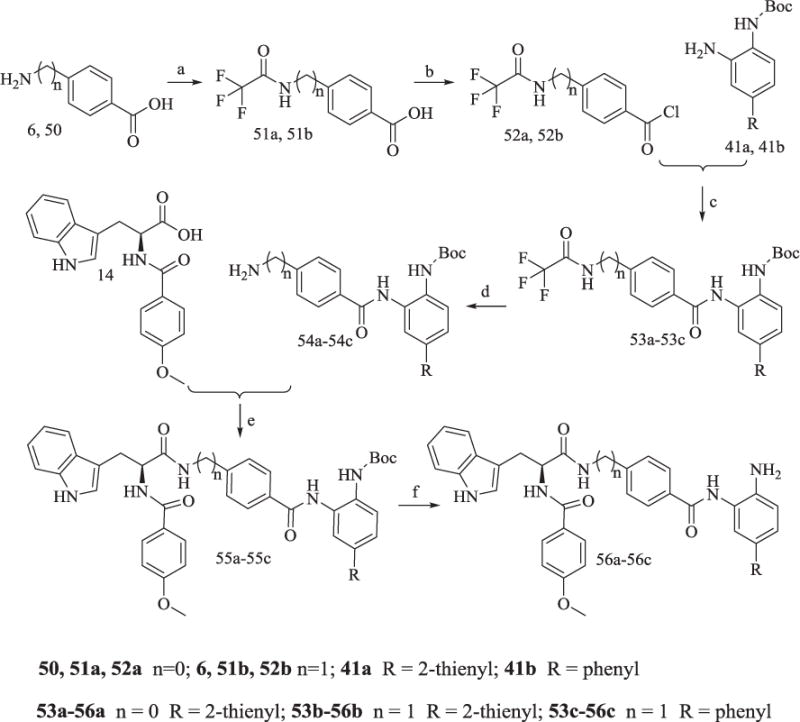
Synthesis of Compounds 56a-56c.
Reagents and conditions: (a) (CF3CO)2O, 95% yield; (b) SOCl2, reflex, 83% yield; (c) pyridine, 78% yield; (d) K2CO3, CH3OH/H2O, 67% yield; (e) TBTU, Et3N, anhydrous CH2Cl2, 60–65% yield; (f) CF3COOH, 65% yield.
Scheme 10.
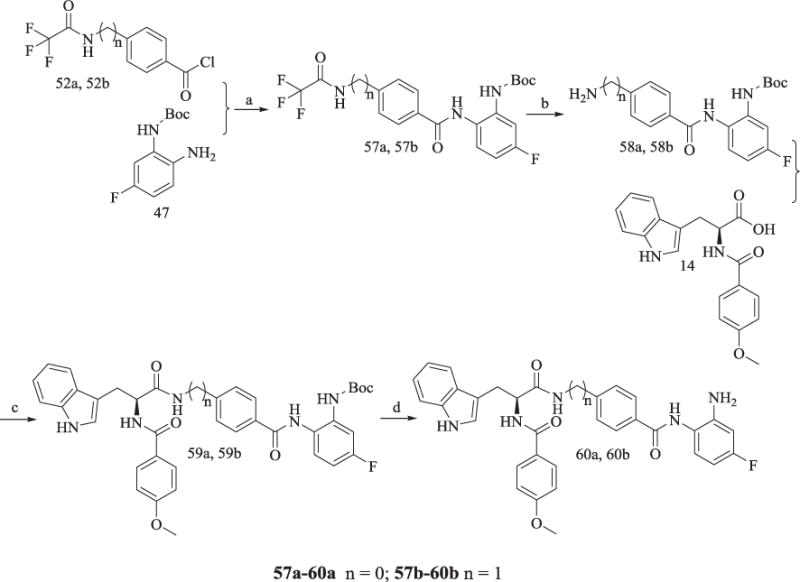
Synthesis of Compounds 60a and 60b.
Reagents and conditions: (a) pyridine, 78% yield; (b) K2CO3, CH3OH/H2O, 67% yield; (c) TBTU, Et3N, anhydrous CH2Cl2, 60–65% yield; (d) CF3COOH, 65% yield.
2. Results and discussion
Our previous study described the discovery of several N-hydroxycinnamamide-based HDACIs with HDAC 1/3 inhibitory activity [17]. However, due to its strong chelating ability with zinc ions, hydroxamates unavoidably exhibited inhibitory activity against other HDAC subtypes including HDAC 2, 8, 6 and so on. Therefore, we attempted to replace hydroxamic acid with other zinc-binding groups to obtain HDACIs with better isoform selectivity. To the best of our knowledge, most of the sub-class I selective inhibitors currently in research are benzamide HDACIs, such as MS-275, MGCD0103, CI994 and RG2833. Therefore, we kept the cap group (or the cap and the linker groups) of the representative HDAC1/3 selective inhibitor 11y unchanged, and introduced the linker and zinc binding group (ZBG) of aforementioned sub-class I selective inhibitors to obtain a novel series of hybrid compounds, 3, 11a, 19, 22, 30a, 30b, 37 (Fig. 1). In detail, 3 was designed by combining cap group of 11y with linker and ZBG group of CI994. Using the same strategy, 11a and 19 were derived by combining the cap group of 11y with the linker and the ZBG group of MS275 and RG2833, respectively. Keeping both the cap group and the linker of 11y unchanged while replacing hydroxamate with the benzamide as ZBG led to 22. Moreover, 30a, 30b and 37 were achieved by further modification of the linker group of 22 and 11a (Fig. 1).
Fig. 1.
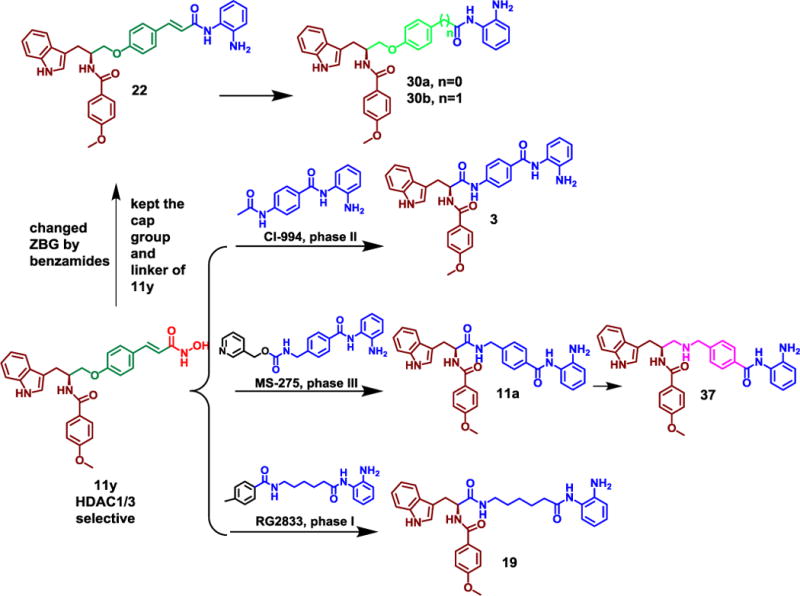
Design of benzamides bearing HDACIs.
HDAC class I whole cell assay and antiproliferative assay based on the human leukemia cell line HL60 were conducted to efficiently screen the newly designed compounds (Table 1). Benzamides 3, 11a, 19, 22, 30a, 30b and 37 with different linker groups exhibited diverse class I HDAC inhibitory activities. To be specific, 3, 11a and 30a, bearing linker structure of N-(p-tolyl)acetamide, N-(4-methylbenzyl)acetamide and 1-ethoxy-4-methylbenzene, respectively, displayed submicromolar IC50 values (0.67–0.85 μM), comparable with that of MS275 (0.62 μM). 19 with N-hexylacetamide, 22 with (E)-1-ethoxy-4-(prop-1-en-1-yl)benzene and 37 with N-(4-methylbenzyl)ethanamine as linker group showed low micromolar IC50 values (1.23–2.95 μM), while 30b having one carbon longer than 30a in the linker chain length displayed no inhibitory activity up to 10 μM. These results indicated that changes in the linkers had obvious effects on the inhibitory activities of benzamide based HDACs inhibitors. The antiproliferative activity of all the compounds was consistent with their cellular enzymatic activity, 30b showed no anti-proliferative activity at the concentration up to 20 μM. The other benzamides displayed low micromolar IC50 values, among which, the most potent compounds 3, 11a and 30a also exhibited comparable antiproliferative activities to MS275.
Table 1.
HDAC class I cellular activity and in vitro antiproliferative activity for synthesized HDAC inhibitors 3, 11a, 19, 22, 30a, 30b and 37.a
| Cpd No. | Structure | IC50 of HDAC cellular class I (μM)b | IC50 of HL60 cell (μM) |
|---|---|---|---|
| 3 |
|
0.85 | 2.14 |
| 11a |
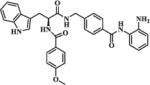
|
0.67 | 1.84 |
| 19 |
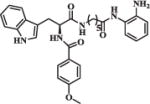
|
1.23 | 7.94 |
| 30a |
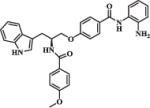
|
0.82 | 1.93 |
| 30b |
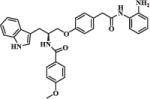
|
>10 | >20 |
| 22 |
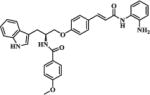
|
1.75 | 5.97 |
| 37 |
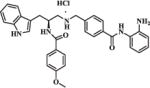
|
2.95 | 8.04 |
| MS275 |
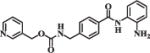
|
0.62 | 1.52 |
Assays were performed in HL60 cells; Values are average of at least two determinations, the SD values are < 20% of the mean.
Boc-Lys (acetyl)-AMC substrate.
Due to its promising enzymatic and antiproliferative activity, further structural modification was focused on the cap of 11a to find more potent derivatives. Analogs 11b-11g were designed and synthesized by replacing the 4-methoxybenzoyl of 11a with various para substituted benzoic acids or heterocyclic rings. Among these analogs, 11f (with 2-thiophenecarboxyl), 11g (with 2-furancarboxyl), and their parent compound 11a displayed higher enzymatic inhibitory and antiproliferative activity than the other compounds (Table 2).
Table 2.
HDAC class I cellular activity and in vitro antiproliferative activity for synthesized HDAC inhibitors 11a-11g.a
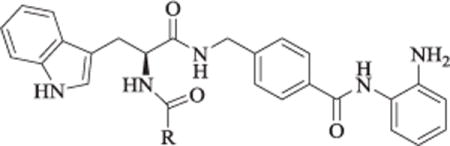
| |||
|---|---|---|---|
|
| |||
| Cpd No. | Structure | IC50 of HDAC cellular class I (μM)b | IC50 of HL60 cell (μM) |
| 11a |
|
0.67 | 1.84 |
| 11b |
|
1.51 | 2.38 |
| 11c |
|
1.98 | 3.09 |
| 11d |
|
1.61 | 6.09 |
| 11e |
|
1.02 | 5.85 |
| 11f |
|
0.78 | 1.74 |
| 11g |
|
0.91 | 2.40 |
Assays were performed in HL60 cells; Values are average of at least two determinations, the SD values are < 20% of the mean.
Boc-Lys (acetyl)-AMC substrate.
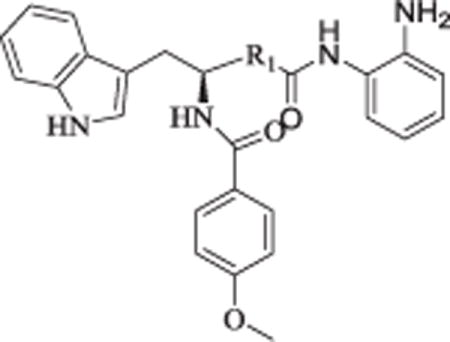
In order to explore the HDAC isoform selective profile, compounds 3, 11a, 19, 22, 30a, 30b, and 37, with different linkers, were evaluated in enzyme inhibitory assays against HDAC1, HDAC2 and HDAC3. Results presented in Table 3 revealed that compounds 3, 11a and 30a exhibited moderate HDAC1 selectivity; 19 displayed moderate HDAC1/3 dual selectivity; 37 showed some preference to HDAC1/2; 30b was not active in cellular assays and also showed no inhibition against HDAC1/2/3 up to 20 μM; 22 showed almost no selectivity among HDAC1/2/3. Considering its moderate HDAC1 selectivity (Table 3) and promising cellular enzyme activity and antiproliferative activity (Table 1), 11a was chosen for further evaluation.
Table 3.
In Vitro inhibition of HDACs isoforms of representative Compounds.a
| ||||
|---|---|---|---|---|
|
| ||||
| Cpd No. | R1 | IC50 (nM)
|
||
| HDAC1 | HDAC2 | HDAC3 | ||
| 3 |
|
49.6 | 441 | 227 |
| 11a |
|
20.6 | 157 | 138 |
| 19 |
|
58.6 | 296 | 42.9 |
| 30a |
|
14.7 | 111 | 90.0 |
| 30b |
|
>20000 | >20000 | >20000 |
| 22 |
|
284 | 217 | 261 |
| 37 |
|
29.9 | 21.2 | 223 |
Assays were performed in replicate (n ≥ 2); The SD values are < 20% of the mean.
The MTT assay results in Table 4 indicated that 11a exhibited similar in vitro antiproliferative activities against several hematological and solid tumor cell lines to MS275. 11a and MS275 displayed low micromolar or submicromolar IC50 values against HEL, K562, U937, U266 and HCT116 cell lines, while showed poor antiproliferative activity against ES-2.
Table 4.
In Vitro antiproliferative Activity of 11a and MS275.a
| Cpd | IC50 (μM)a
|
|||||
|---|---|---|---|---|---|---|
| HEL | K562 | U937 | U266 | HCT116 | ES-2 | |
| 11a | 3.02 | 0.98 | 1.10 | 2.23 | 4.23 | >20 |
| MS275 | 1.29 | 1.68 | 0.77 | 6.10 | 2.07 | >20 |
Assays were performed in replicate (n ≥ 2); The SD values are < 20% of the mean.
Encouraged by its promising in vitro activity, compound 11a was further progressed to in vivo experiments. Firstly, we established a hematological tumor xenograft model, using MS275 as the positive control, to investigate if 11a was active in vivo. 5 × 106 U937 cells were subcutaneously implanted in the right flanks of male nude mice (BALB/c-nu). Once tumor size reached about 100 mm3, mice were randomized into seven per group and were treated with compound 11a (100 mg/kg/day) or MS275 (50 mg/kg/day) by oral gavage for 13 days. Tumor growth inhibition (TGI) and relative increment ratio (T/C) were calculated at the end of experiment to reveal the antitumor effects via tumor weight and tumor volume, respectively. Meanwhile, tumor volume and mice body weight were also measured during the treatments (Fig. 2, Table 5). Although compound 11a displayed potent in vivo oral antitumor activity with TGI value of 51% and T/C value of 49%, it was a little less potent than the positive control MS275 (TGI = 60%, T/C = 33%). However, we could see from Fig. 2d and Table 5 that during treatment, the mice group administrated with MS275 demonstrated obvious body weight loss compared with the control group, which indicated that MS275 had obvious toxicity in the dose of 50 mg/kg/day. This toxicity didn’t appear in the mice treated with 11a in the dose of 100 mg/kg/day. In fact, at the beginning of the in vivo study, mice were treated with MS275 at the same dosage as 11a (100 mg/kg/day). Three days later, serious body weight loss was observed unexpectedly, and after six days, two of the six mice died. Therefore, we had to setup a new in vivo experiment and decreased the dose of MS275 to 50 mg/kg/day. In conclusion, compound 11a exhibited potent in vivo oral antitumor activity in the U937 xenograft model without obvious side effects compared with MS275.
Fig. 2.
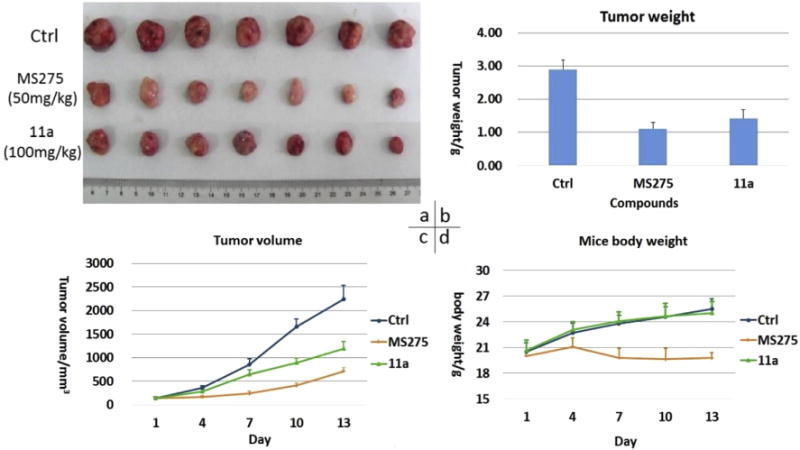
Antitumor activity comparison of 11a and MS275 against U937 human tumor xenografts implanted in mice. (a) Picture of dissected U937 tumor tissues; (b) Tumor weight in different mice group; (c) Mean tumor volume during mice treatment; (d) Mice body weight change after administration.
Table 5.
Data of in vivo study with U937 xenograft model.
| Cpd No. | Does (mg/kg) |
Tumor growth inhibition (TGI %) | Relative increment ratio (T/C %) | Mice body weight (g) |
|---|---|---|---|---|
| 11a | 100 | 51 | 49 | 25.04 ± 1.31 |
| MS275 | 50 | 60 | 32 | 19.77 ± 0.61 |
| Ctrl | / | / | / | 25.39 ± 1.18 |
Another subcutaneous colon cancer cell HCT116 xenograft model was also established to test if 11a was effective against solid tumor in vivo. Mice were treated with 11a (100 mg/kg), SAHA (100 mg/kg) or MS275 (50 mg/kg) by oral gavage for 16 days. Results in Fig. 3 and Table 6 showed that significant tumor growth delays were observed relative to the untreated group for all tested compounds. Compound 11a (TGI = 49%, T/C = 59%) was much more effective than SAHA (TGI = 32%, T/C 74%), while was less potent than positive control MS275 (TGI = 62%, T/C = 47%). Again, compared with the control group, serious body weight loss was observed in MS275 group but not in 11a group and SAHA group (Table 6). Taken together, our benzamide 11a was orally active not only in the hematological tumor cell U937 xenograft model but also in the solid tumor HCT116 xenograft model. Though less potent, 11a possessed much better safety profiles than the clinical compound MS275.
Fig. 3.
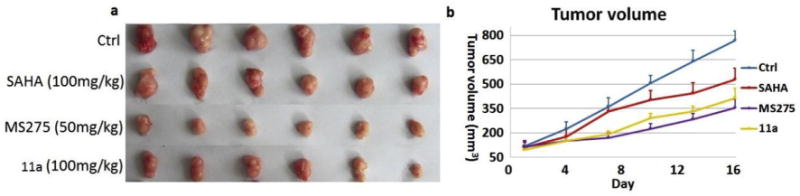
Antitumor activity comparison of 11a, SAHA and MS275 against HCT116 human tumor xenografts implanted in mice. (a) Picture of dissected HCT116 tumor tissues; (b) Mean tumor volume during mice treatment.
Table 6.
Data of in vivo study with HCT116 xenograft model.
| Cpd No. | Does (mg/kg) |
Tumor growth inhibition (TGI %) | Relative increment ratio (T/C %) | Mice body weight (g) |
|---|---|---|---|---|
| 11a | 100 | 49 | 59 | 21.59 ± 1.05 |
| SAHA | 100 | 32 | 73 | 21.23 ± 0.63 |
| MS275 | 50 | 62 | 46 | 18.87 ± 0.80 |
| Ctrl | / | / | / | 19.91 ± 1.26 |
Previous reports demonstrated that a 14 Å internal cavity immediately below the zinc binding group could be used to design HDAC1/2 selective HDACs inhibitors effectively [20]. Another report showed the introduction of fluorine in the para position of acid amide could decrease the activity against HDAC1 to afford a HDAC3 selective inhibitor [21]. Therefore, compounds 43a, 43b, 49, 56a-56c, 60a and 60b were designed based on 3, 11a or 19, and these new compounds were expected to display different selective profiles (Fig. 4).
Fig. 4.
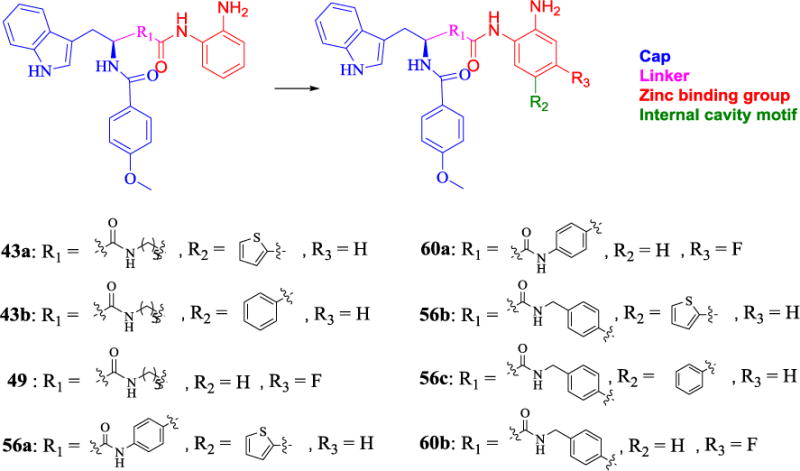
Design of the new series compounds.
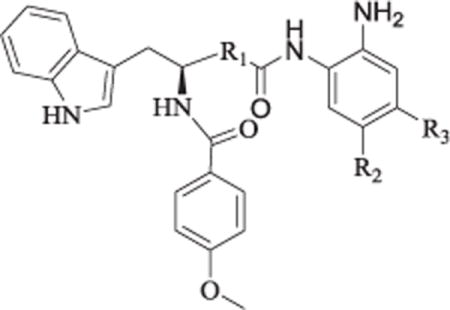
All of the newly designed compounds were evaluated in enzyme inhibitory assays against HDAC1, HDAC2 and HDAC3 along with 19, 3 and 11a (Table 7). Results revealed that regardless of the linker, para aromatic substituent of compounds 19, 3 and 11a could obviously decrease their inhibitory activity against HDAC3, which was consistant with previous reports [20]. Among compounds 49, 60a and 60b with fluorine in the para position of acid amide, only 49 displayed moderate HDAC3 selectivity, which indicated the para fluorine plus the appropriate linker, such as the linear aliphatic liner in 49, co-determined the selective profile of HDAC inhibitor. To further ascertain the selectivity of our compounds across the broader family of HDAC isoforms, we next profiled the representative 43a with para aromatic substituent, 49 with fluorine against HDAC8 (class I), HDAC4 (class IIa), and HDAC6 (class IIb). 43a and 49 displayed almost no activity (>100 μM) against HDAC8, HDAC4 and HDAC6 (see Table 8).
Table 7.
In Vitro inhibition of HDACs isoforms of representative Compounds.a
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Cpd No. | R1 | R2 | R3 | IC50 (nM)
|
||
| HDAC1 | HDAC2 | HDAC3 | ||||
| 19 |
|
H | H | 58.7 | 296 | 42.9 |
| 43a |
|
|
H | 48.0 | 338 | 3569 |
| 43b |
|
|
H | 55.5 | 490 | 5626 |
| 49 |
|
H | F | 511 | 909 | 111 |
| 3 |
|
H | H | 49.7 | 441 | 227 |
| 56a |
|
|
H | 36.5 | 314 | 62540 |
| 60a |
|
H | F | 91.7 | 121 | 167 |
| 11a |
|
H | H | 20.6 | 157 | 138 |
| 56b |
|
|
H | 58.8 | 320 | 8007 |
| 56c |
|
|
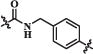
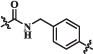
H | 58.4 | 311 | 30850 |
| 60b |
|
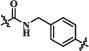
H | F | 171 | 179 | 243 |
Assays were performed in replicate (n ≥ 2); The SD values are < 20% of the mean.
Table 8.
In Vitro inhibition of HDACs isoforms of representative compounds 43a and 49.a
| Cpd No. | Class I (μΜ)
|
Class IIa (μΜ)
|
Class IIb (μΜ)
|
|||
|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC4 | HDAC6 | |
| 43a | 0.048 | 0.337 | 3.57 | >100 | >100 | >100 |
| 49 | 0.511 | 0.908 | 0.110 | >100 | >100 | >100 |
| 11r[17] | 0.012 | 0.50 | 0.004 | 2.0 | 5.7 | 0.31 |
Assays were performed in replicate (n ≥ 2); The SD values are < 20% of the mean.
Several HDAC1/2 selective compounds 43a, 43b, 56b and 56c were tested in antiproliferative assays (Table 9). Results showed all these HDAC1/2 selective compounds displayed much weaker antiproliferative activities than their parent compounds 19 and 11a. In order to explain this result, we conducted western blot assay to determine the levels of acetylated histone H3, acetylated histone H4 and several apoptosis-related proteins including procaspase-3, cleaved caspase-3 and cleaved PARP (Poly ADP ribose polymerase) in tumor cells treated with 11a, 56c and positive control MS275, respectively. Result in Fig. 5 displayed that compound 11a could markedly increase the level of acetylated histone H3 and acetylated histone H4, which was much superior to 56c at the same concentration. In addition, compound 11a and MS275 dramatically reduced the level of procaspase-3 and increased the level of cleaved caspased-3 and cleaved PARP, which indicated the induction of apoptosis, however, the levels of procaspase-3, cleaved caspased-3 and cleaved PARP could be hardly affected by 56c. The above western blot results of 11a, 56c and MS275 were consistent with their antiproliferative activities.
Table 9.
In Vitro Antiproliferative Activity of representative and MS275.a
| Cpd | IC50 (μΜ)a
|
|||||
|---|---|---|---|---|---|---|
| HEL | K562 | U937 | U266 | HCT116 | ES-2 | |
| 10 | 15.4 | 8.51 | 13.20 | 12.7 | 10.6 | >20 |
| 43a | 16.6 | 16.0 | >20 | >20 | >20 | >20 |
| 43b | 25.2 | 15.7 | >20 | >20 | >20 | >20 |
| 11a | 3.02 | 0.98 | 1.10 | 2.23 | 4.23 | >20 |
| 56b | >20 | 13.5 | >20 | >20 | >20 | >20 |
| 56c | >20 | 14.0 | >20 | >20 | >20 | >20 |
| MS275 | 1.29 | 1.68 | 0.77 | 6.10 | 2.07 | >20 |
Assays were performed in replicate (n ≥ 2); The SD values are < 20% of the mean.
Fig. 5.
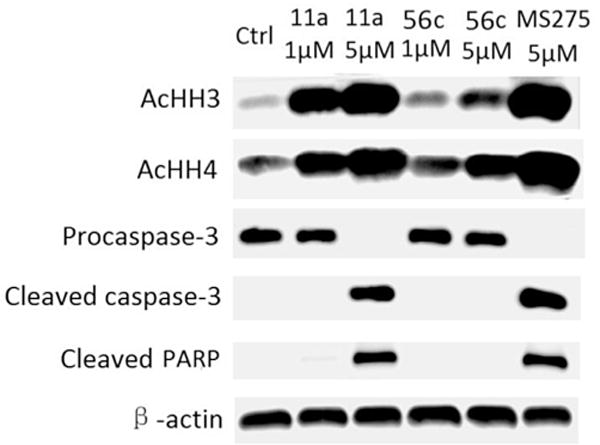
Western blot analysis of acetylated histone H3 (AcHH3), acetylated histone H4 (AcHH4), procaspase-3, cleaved caspase-3 and cleaved PARP in HL60 cell lines after 48 h treatment with compounds using MS275 as positive control. β-actin was used as a loading control.
3. Conclusion
A new series of benzamide-based HDACIs were designed and synthesized, and their structure-activity relationship (SAR), isotype-selectivity, and anticancer activities were described. Most of the compounds exhibited high HDAC inhibitory potency. Any changes in the linker fragment and the N-substituent in the cap all had influence in HDAC inhibitory activity and antiproliferative potency of benzamide HDACIs. 11a, with 4-methoxybenzoyl as N-substituent in the cap and 4-(aminomethyl) benzoyl as linker group, exhibited the best activity and exhibited some HDAC1 selective profile. In addition, 11a also showed potent antiproliferative activity against multiple solid or hematological tumor cell lines. In vivo studies revealed that compound 11a displayed potent oral antitumor activity in the U937 and HCT116 xenograft models. Although it was a little less potent than the positive control MS275, 11a did have a much better tolerance with almost no toxicity in mice.
The newly designed thienyl and phenyl compounds (43a, 43b, 56a, 56b and 56c) based on 19, 3 and 11a exhibited dramatic HDAC1/2 dual selectivity. One representative compound 56c showed low nanomolar IC50 values against HDAC1 (58.4 nM), submicromolar IC50 values against HDAC2 (311 nM) and micromolar IC50 values against HDAC3 (30850 nM). Compounds 49, 60a and 60b with fluorine in the para position of acid amide were also synthesized, among which, 49 displayed moderate HDAC3 selectivity. Disappointingly, our HDAC1/2 selective compounds exhibited poor in vitro antiproliferative activity, which was consistent with the western blot results that 56c displayed much less effects on acetylated histone H3, acetylated histone H4, procaspase-3, cleaved caspase-3 and cleaved PRAP than its parent compound 11a. Considering that it was reported that thienyl based HDAC1/2 inhibitors exhibited better antiproliferative activity than their parent HDAC1/2/3 inhibitors [20], we supposed that the poor cellular potency of 56c should be due to its poor transmembrane permeability rather than its lack of HDAC3 inhibition. Actually, our preliminary results showed that the intracellular fluid concentration of 56c was more than 10 times lower than 11a (data not shown). Besides, we also detected the solubility of 56c by testing lip-water partition coefficient (LogP), however, we could hardly detect 56c in water phase (compound concentration in n-octyl alcohol was 5 mM) due to its poor water solubility, which could be partially responsible for its poor transmembrane permeability. Further study focused on the improvement of compounds’ water solubility and transmembrane permeability is underway in our lab.
3.1. Experiment section
Materials and Methods
High-resolution mass spectrometry was performed by Shandong Analysis and Test Center in Ji’nan, China. ESI-MS spectra were recorded on an API 4000 spectrometer. The [M+H]+ data of HRMS or ESI-MS consisted with the calculated molecular mass of compounds. 1H NMR and 13C NMR spectra were obtained on a Bruker DRX spectrometer at 400 MHz with TMS as an internal standard, δ in parts per million and J in hertz. 1H NMR and 13C NMR peaks consisted with the structures of compounds. All reactions were monitored by TLC using 0.25 mm silica gel plates (60 GF-254). UV light and ferric chloride were used to visualize the spots. Silica gel was used for column chromatography purification. Flash chromatography was accomplished using the automated CombiFlash Rf system from Teledyne ISCO and was done using silica gel of 200–300 mesh. Melting points were determined on an electrothermal melting point apparatus. All tested compounds are >95% pure by HPLC analysis, performed on an Agilent 1100 HPLC instrument using an ODS HYPERSIL column (5 μm,4.6 mm × 250 mm).
3.1.1. General procedure for the preparation of 6 and 16
3.1.1.1. Methyl 4-(aminomethyl)benzoate hydrochloride (6)
Compound 5 (1.5 g, 10 mmol) was dissolved in 200 mL MeOH, then acetyl chloride (2.4 g, 30 mmol) was added dropwise at 0 °C; the mixed solution was refluxed at 75 °C for 5 h. The solvent was evaporated under vacuum, the product was washed by diethyl ether to give compound 6, a white solid powder (1.7 g, 93%).
3.1.1.2. Methyl 6-aminohexanoate hydrochloride (16)
Using the synthetic method for 5, compound 15 gave 16 as a white solid, 94% yield.
3.1.2. General procedure for the preparation of 25a and 25b
3.1.2.1. Methyl 4-hydroxybenzoate (25a)
24a (2.76 g, 20 mmol) was dissolved in 100 mL MeOH, then acetyl chloride (4.7 g, 60 mmol) was added dropwise at 0 °C; the mixed solution was refluxed at 75 °C for 5 h. The solvent was evaporated under vacuum, the product dissolved with EtOAc, washed by saturated NaHCO3 solution (2 × 100 mL), 1 M aqueous citric acid (2 × 100 mL) and brine (2 × 100 mL), dried over MgSO4 overnight and evaporated under vacuum to obtained product compound 25a, a white solid (7.98 g, 87.5%).1H NMR (400 MHz, DMSO-d6) δ 10.33 (s, 1H), 7.84–7.79 (m, 2H), 6.87–6.82 (m, 2H), 3.78 (s, 3H). ESI-MS m/z: 153.2 [M+H]+.
3.1.2.2. Methyl 2-(4-hydroxyphenyl)acetate (25b)
Using the synthetic method for 25a, compound 24b gave 25b as a white solid, 81% yield.
3.1.3. General procedure for the preparation of 26a and 26b
3.1.3.1. Methyl(S)-4-(2-((tert-butoxycarbonyl)amino)-3-(1H-indol-3-yl)propoxy)benzoate (26a)
Compound 23 (2.90 g, 10 mmol), 25a (1.82 g, 12 mmol), and PPh3 (3.95 g, 15 mmol) were dissolved in anhydrous THF, then DEAD (2.61 g, 15 mmol) was added dropwise at 0 °C, the mixture was stirred for 4 h, then THF was evaporated. The crude material was purified via flash chromatography to afford the compound 26a, a white solid (2.52 g, 60%). ESI-MS m/z: 425.4 [M+H]+.
3.1.3.2. Methyl(S)-2-(4-(2-((tert-butoxycarbonyl)amino)-3-(1H-indol-3-yl)propoxy)phenyl) acetate (26b)
Using the synthetic method for 26a, compound 25b gave 26b as a white solid, 55% yield. ESI-MS m/z: 439.6 [M+H]+.
3.1.4. General procedure for the preparation of 7, 9a-9g, 13, 17, 28a, 28b, 31, 39a, 39b
3.1.4.1. Methyl(S)-4-((2-((tert-butoxycarbonyl)amino)-3-(1H-indol-3-yl)propanamido) methyl) benzoate (7)
To a solution of 4 (2.4 g, 8 mmol) in anhydrous CH2Cl2, was added 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU, 2.8 g, 8.8 mmol), followed by Et3N (0.9 g, 8.8 mmol). After 30 min, compound 6 (1.6 g, 8.8 mmol) was added followed by Et3N (0.9 g, 8.8 mmol). 4 h later, the solution of CH2Cl2 was washed with 1 M HCl (2 × 30 mL), saturated Na2CO3 (2 × 30 mL) and brine (2 × 30 mL), dried over MgSO4 overnight, and the solvent was evaporated under vacuum to afford colorless oil. The crude product was purified by recrystallization with EtOAc and petroleum ether. The desired compound 7 was white solid, 55% yield. ESI-MS m/z: 452.2 [M+H]+.
3.1.4.2. Methyl (S)-4-((3-(1H-indol-3-yl)-2-(4 methoxybenzamido) propanamido)methyl) benzoate (9a)
Using the synthetic method for 7, compound 4-methoxybenzoic acid and 8 gave 9a as a white solid, 53% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 8.68 (t, J = 6.0 Hz, 1H), 8.41 (d, J = 7.9 Hz, 1H), 7.91–7.80 (m, 4H), 7.69 (d, J = 7.9 Hz, 1H), 7.32 (dd, J = 8.2, 2.2 Hz, 3H), 7.21 (d, J = 2.2 Hz, 1H), 7.08–7.04 (m, 1H), 7.00–6.96 (m, 3H), 4.79–4.74 (m, 1H), 4.44–4.32 (m, 2H), 3.84 (s, 3H), 3.80 (s, 3H), 3.30–3.23 (m, 1H), 3.21–3.13 (m, 1H). ESI-MS m/z: 486.5 [M+H]+.
3.1.4.3. Methyl (S)-4-((2-(4-(dimethylamino)benzamido)-3-(1H-indol-3yl)propanamido) methyl)benzoate (9b)
Using the synthetic method for 7, compound 4-(dimethylamino)benzoic acid and 8 gave 9b as a white solid, 55% yield.
3.1.4.4. Methyl (S)-4-((3-(1H-indol-3-yl)-2-(4-(trifluoromethyl)benzamido)propanamido) methyl)benzoate (9c)
Using the synthetic method for 7, compound 4-(trifluoromethyl)benzoic acid and 8 gave 9c as a white solid, 52% yield. ESI-MS m/z: 524.5 [M+H]+.
3.1.4.5. Methyl (S)-4-((3-(1H-indol-3-yl)-2-(picolinamido)propanamido)methyl)benzoate (9d)
Using the synthetic method for 7, compound picolinic acid and 8 gave 9d as a white solid, 58% yield.
3.1.4.6. Methyl (S)-4-((3-(1H-indol-3-yl)-2-(pyrazine-2-carboxamido)propanamido)methyl) benzoate (9e)
Using the synthetic method for 7, compound pyrazine-2-carboxylic acid and 8 gave 9e as a white solid, 54% yield.
3.1.4.7. Methyl(S)-4-((3-(1H-indol-3-yl)-2-(thiophene-2-carboxamido)propanamido)methyl)benzoate (9f)
Using the synthetic method for 7, compound thiophene-2-carboxylic acid and 8 gave 9f as a white solid, 54% yield.
3.1.4.8. Methyl (S)-4-((2-(furan-2-carboxamido)-3-(1H-indol-3-yl) propanamido)methyl) benzoate (9g)
Using the synthetic method for 7, compound furan-2-carboxylic acid and 8 gave 9g as a white solid, 56% yield.
3.1.4.9. Methyl (4-methoxybenzoyl)-L-tryptophanate (13)
Using the synthetic method for 7, compound 4-methoxybenzoic acid and 12 gave 13 as a white solid, 52% yield.
3.1.4.10. Methyl (S)-6-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)hexanoate (17)
Using the synthetic method for 7, compound 14 and 16 gave 17 as a white solid, 52% yield.
3.1.4.11. Methyl (S)-4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propoxy)benzoate (28a)
Using the synthetic method for 7, compound 4-methoxybenzoic acid and 27a gave 28a as a white solid, 52% yield.
3.1.4.12. Methyl(S)-2-(4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propoxy)phenyl) acetate (28b)
Using the synthetic method for 7, compound 4-methoxybenzoic acid and 27b gave 28b as a white solid, 52% yield.
3.1.4.13. (S)-N-(3-(1H-indol-3-yl)-1-(methoxy(methyl)amino)-1-oxopropan-2-yl)-4-methoxybenzamide (31)
Using the synthetic method for 7, compound 14 and N,O-dimethylhydroxylamine gave 31 as a white solid, 78% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 8.52 (d, J = 7.6 Hz, 1H), 7.83 (d, J = 8.8 Hz, 2H), 7.59 (d, J = 7.7 Hz, 1H), 7.32 (d, J = 7.9 Hz, 1H), 7.24 (d, J = 2.1 Hz, 1H), 7.06 (t, J = 7.1 Hz, 1H), 7.02–6.96 (m, 3H), 5.11 (s, 1H), 3.82 (s, 3H), 3.80 (s, 3H),3.16 (s, 3H), 3.14–3.10 (m, 2H).
3.1.4.14. Tert-butyl (S)-(2-(3-(1H-indol-3-yl)-2-(4 methoxybenzamido)propanamido)ethyl) carbamate (39a)
Using the synthetic method for 7, compound 14 and 38a gave 39a as a white solid, 70% yield.
3.1.4.15. Tert-butyl (S)-(3-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)propyl) carbamate (39b)
Using the synthetic method for 7, compound 14 and 38b gave 39b as a white solid, 65% yield.
3.1.5. General procedure for the preparation of 8, 27a, 27b and 37
3.1.5.1. Methyl(S)-4-((2-amino-3-(1H-indol-3-yl)propanamido)methyl)benzoate hydrochloride (8)
Compound 7 (2.0 g, 4.4 mmol) was dissolved in a solution of EtOAc (20 mL) saturated by dry HCl gas. The solution was stirred at room temperature overnight. The filtered precipitate, was washed by diethyl ether to give the compound 8, a white solid powder (1.4 g, 80%). 1H NMR (400 MHz, DMSO-d6) δ 11.09 (d, J = 1.6 Hz, 1H), 9.14 (t, J = 5.8 Hz, 1H), 8.35 (s, 2H), 7.84 (d, J = 8.3 Hz, 2H), 7.67, (d, J = 7.9 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.22–7.19 (m, 3H), 7.13–7.06 (m, 1H), 7.04–6.97 (m, 1H), 4.40 (dd, J = 15.9, 6.0 Hz, 1H), 4.31 (dd, J = 15.9, 5.6 Hz, 1H), 4.06 (s, 1H), 3.85 (s, 3H), 3.31–3.15 (m, 2H). ESI-MS m/z: 352.5 [M+H]+.
3.1.5.2. Methyl (S)-4-(2-amino-3-(1H-indol-3-yl)propoxy)benzoate hydrochloride (27a)
Using the synthetic method for 7, compound 26a gave 27a as a white solid, 78% yield.
3.1.5.3. Methyl (S)-2-(4-(2-amino-3-(1H-indol-3-yl)propoxy)phenyl) acetate (27b)
Using the synthetic method for 7, compound 26b gave 27b as a white solid, 82% yield.
3.1.5.4. (S)-4-(((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propyl) amino)methyl)-N-(2-aminophenyl)benzamide dihydrochloride (37)
Using the synthetic method for 7, compound 36 gave 37 as a white solid, 78% yield. Mp: 180–182 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.92 (s, 1H), 10.59 (s, 1H), 9.43 (s, 2H), 8.55 (d, J = 8.1 Hz, 1H), 8.13 (d, J = 8.1 Hz, 2H), 7.94 (d, J = 8.7 Hz, 2H), 7.69–7.65 (m, 3H), 7.60 (d, J = 7.5 Hz, 1H), 7.49 (d, J = 7.4 Hz, 1H), 7.41–7.29 (m, 3H), 7.19 (d, J = 1.6 Hz, 1H), 7.07 (t, J = 7.4 Hz, 1H), 7.01–6.98 (m, 3H), 4.66 (s, 1H), 4.31–4.28 (m, 1H), 4.21–4.16 (m, 1H), 3.81 (s, 5H), 3.13–3.05 (m, 1H), 3.02–2.97 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.69, 165.60, 162.18, 136.67, 135.94, 134.50, 130.55, 129.96, 128.70, 127.79, 127.75, 127.08, 126.92, 124.07, 123.92, 121.47, 118.98, 118.82, 113.80, 111.90, 110.49, 55.86, 50.22, 49.95, 47.82, 28.84, 21.54. HRMS (AP-ESI) m/z calcd for C33H33N5O3 [M+H]+ 548.2656, found 548.2659.
3.1.6. General procedure for the preparation of 32
3.1.6.1. (S)-N-(1-(1H-indol-3-yl)-3-oxopropan-2-yl)-4-methoxybenzamide (32)
At −40 °C, to a solution of 31 (3.81 g, 10 mmol) in anhydrous THF (150 mL) was added LiAlH4 (0.45 g, 12 mmol) in batch. After 4 h, the reaction was quenched by adding 1 M aqueous citric acid slowly. Then THF was evaporated with the residue being extracted by EtOAc (3 × 50 mL). The EtOAc solution was washed with saturated Na2CO3 (3 × 30 mL) and brine (3 × 30 mL), dried over MgSO4 and evaporated under vacuum to afford 32 (2.58 g, 8 mmol), the crude product was used for the following reaction without any purification.
3.1.7. General procedure for the preparation of 33
3.1.7.1. Methyl(S)-4-(((3-(1H-indol-3-yl)-2-(4-methoxybenzamido) propyl)amino) methyl)benzoate (33)
Solution of compound 6 (1.6 g, 8 mmol) in 50 mL CH3OH, was added with Et3N (0.81 g, 8 mmol) followed by 32 (2.58 g, 8 mmol). After the addition of AcOH (0.48 g, 8 mmol), NaBH3CN (0.75 g, 12 mmol) was added in batch. The reaction was stirred at room temperature overnight. After the reaction finished, the solvent was evaporated under vacuum and the crude material was purified via flash chromatography to afford the compound to yield 33 (1.51 g, 40%). ESI-MS m/z: 472.5 [M+H]+.
3.1.8. General procedure for the preparation of 29
3.1.8.1. Methyl(S)-4-(((3-(1H-indol-3-yl)-2-(4-methoxybenzamido) propyl)(tert-butoxy carbonyl)amino)methyl)benzoate (34)
At room temperature, to a solution of 33 (1.41 g, 3 mmol) in anhydrous CH2Cl2 (20 mL), was added Et3N (0.91 g, 9 mmol) followed by (Boc)2O (0.78 g, 3.6 mmol). After stirring the mixture at room temperature for 8 h, the solution was washed with 1 M aqueous citric acid (3 × 10 mL) and brine (3 × 10 mL), dried over MgSO4, and evaporated under vacuum to yield the crude product compound 34 (1.54 g, 90% yield) as a white solid. This product was used for the following reaction without further purification. ESI-MS m/z: 572.4 [M+H]+.
3.1.9. General procedure for the preparation of 2, 10a-10 g, 14, 18, 21, 29a, 29b, 35
3.1.9.1. (S)-4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)benzoic acid (2)
Compound 1 (0.56 g, 1.2 mmol) was dissolved in 5 mL CH3OH, then 2 mL 3 M KOH aqueous solution was added. The mixture was refluxed at 85 °C for 2 h. After the reaction completed, it was evaporated under vacuum. The residue was acidified with 1 N HCl to pH 3-4, then filtered. The precipitate was 2, a white solid, 85% yield.
3.1.9.2. (S)-4-((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)methyl)benzoic acid (10a)
Using the synthetic method for 2, compound 9a gave 10a as a white solid, 87% yield. 1H NMR (400 MHz, DMSO-d6) δ 12.83 (s, 1H), 10.81 (s, 1H), 8.68 (t, J = 5.9 Hz, 1H), 8.42 (d, J = 7.9 Hz, 1H), 7.90–7.80 (m, 4H), 7.69 (d, J = 7.8 Hz, 1H), 7.33–7.28 (m, 3H), 7.21 (d, J = 2.1 Hz, 1H), 7.05 (t, J = 7.2 Hz, 1H), 6.99–6.96 (m, 3H), 4.79–4.73 (m, 1H), 4.38–4.37 (m, 2H), 3.80 (s, 3H), 3.28–3.12 (m, 2H). ESI-MS m/z: 472.3 [M+H]+.
3.1.9.3. (S)-4-((2-(4-(dimethylamino)benzamido)-3-(1H-indol-3-yl)propanamido)methyl) benzoic acid (10b)
Using the synthetic method for 2, compound 9b gave 10b as a white solid, 87% yield.
3.1.9.4. (S)-4-((3-(1H-indol-3-yl)-2-(4-(trifluoromethyl)benzamido)propanamido)methyl) benzoic acid (10c)
Using the synthetic method for 2, compound 9c gave 10c as a white solid, 80% yield.
3.1.9.5. (S)-4-((3-(1H-indol-3-yl)-2-(picolinamido)propanamido)methyl)benzoic acid (10d)
Using the synthetic method for 2, compound 9d gave 10d as a white solid, 85% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 8.76 (t, J = 5.9 Hz, 1H), 8.67 (d, J = 8.2 Hz, 1H), 8.63 (d, J = 4.6 Hz, 1H), 8.06–7.95 (m, 2H), 7.84 (d, J = 8.2 Hz, 2H), 7.65–7.57 (m, 2H), 7.33 (d, J = 8.1 Hz, 1H), 7.24 (d, J = 8.2 Hz, 2H), 7.13 (d, J = 2.2 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.94 (t, J = 7.4 Hz, 1H), 4.88–4.83 (m, 1H), 4.43–4.27 (m, 2H), 3.27–3.14 (m, 2H). ESI-MS m/z: 443.5 [M+H]+.
3.1.9.6. (S)-4-((3-(1H-indol-3-yl)-2-(pyrazine-2-carboxamido)propanamido)methyl) benzoic acid (10e)
Using the synthetic method for 2, compound 9e gave 10e as a white solid, 83% yield. 1H NMR (400 MHz, DMSO-d6) δ 12.66 (s, 1H), 10.85 (s, 1H), 9.16 (d, J = 1.3 Hz, 1H), 8.87 (d, J = 2.5 Hz, 1H), 8.76 (t, J = 5.9 Hz, 1H), 8.72–8.71 (m, 1H), 8.69 (d, J = 8.2 Hz, 1H), 7.85 (d, J = 8.2 Hz, 2H), 7.60 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 8.1 Hz, 1H), 7.27 (d, J = 8.2 Hz, 2H), 7.14 (d, J = 2.1 Hz, 1H), 7.05 (t, J = 7.2 Hz, 1H), 6.93 (t, J = 7.3 Hz, 1H), 4.89–4.83 (m, 1H), 4.36 (d, J = 5.8 Hz, 2H), 3.31–3.26 (m, 2H). ESI-MS m/z: 444.5 [M+H]+.
3.1.9.7. (S)-4-((3-(1H-indol-3-yl)-2-(thiophene-2-carboxamido)propanamido)methyl)benzoic acid (10f)
Using the synthetic method for 2, compound 9f gave 10f as a white solid, 86% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 8.73 (t, J = 5.9 Hz, 1H), 8.66 (d, J = 8.1 Hz, 1H), 7.89 (d, J = 2.9 Hz, 1H), 7.85 (d, J = 8.2 Hz, 2H), 7.73 (dd, J = 5.0, 0.7 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.26 (d, J = 8.2 Hz, 2H), 7.20 (d, J = 2.1 Hz, 1H), 7.13 (dd, J = 4.9, 3.8 Hz, 1H), 7.06 (t, J = 7.1 Hz, 1H), 6.98 (t, J = 7.1 Hz, 1H), 4.77–4.71 (m, 1H), 4.37 (t, J = 5.8 Hz, 2H), 3.20–3.10 (m, 2H). ESI-MS m/z: 448.4 [M+H]+.
3.1.9.8. (S)-4-((2-(furan-2-carboxamido)-3-(1H-indol-3-yl)propanamido)methyl)benzoic acid (10 g)
Using the synthetic method for 2, compound 9g gave 10g as a white solid, 82% yield. 1H NMR (400 MHz, DMSO-d6) δ 12.84 (s, 1H), 10.81 (s, 1H), 8.71 (t, J = 5.9 Hz, 1H), 8.31 (d, J = 8.1 Hz, 1H), 7.86–7.81 (m, 3H), 7.65 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 8.1 Hz, 1H), 7.28 (d, J = 2H), 8.2 Hz, 7.18–7.15 (m, 2H), 7.05 (t, J = 11.2, 7.2 Hz, 1H), 6.97 (t, J = 7.2 Hz, 1H), 6.60 (dd, J = 3.4, 1.7 Hz, 1H), 4.78–4.72 (m, 1H), 4.36 (d, J = 5.9 Hz, 2H), 3.29–3.24 (m, 1H), 3.19–3.13 (m, 1H). ESI-MS m/z: 432.4 [M+H]+.
3.1.9.9. (4-methoxybenzoyl)-L-tryptophan (14)
Using the synthetic method for 2, compound 13 gave 14 as a white solid, 78% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 8.34 (d, J = 7.7 Hz, 1H), 7.82–7.76 (m, 2H), 7.59 (d, J = 7.8 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 7.18 (d, J = 2.2 Hz, 1H), 7.07–7.01 (m, 1H), 6.97–6.93 (m, 3H), 4.63–4.57 (m, 1H), 3.79 (s, 3H), 3.35–3.30 (m, 1H), 3.26–3.16 (m, 1H).
3.1.9.10. (S)-6-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)hexanoic acid (18)
Using the synthetic method for 2, compound 17 gave 18 as a white solid, 83% yield. 1H NMR (400 MHz, DMSO-d6) δ 12.03 (s, 1H), 10.76 (s, 1H), 8.28 (d, J = 8.1 Hz, 1H), 8.02 (t, J = 5.5 Hz, 1H), 7.81 (d, J = 8.8 Hz, 2H), 7.67 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 2.1 Hz, 1H), 7.04 (dd, J = 11.0, 3.9 Hz, 1H), 6.99–6.95 (m, 3H), 4.69–4.63 (m, 1H), 3.80 (s, 3H), 3.23–2.97 (m, 4H), 2.17 (t, J = 7.4 Hz, 2H), 1.55–1.43 (m, 2H), 1.41–1.34 (m, 2H), 1.26–1.20 (m, 2H). ESI-MS m/z: 474.4 [M+Na]+.
3.1.9.11. (S,E)-3-(4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propoxy)phenyl)acrylic acid (21)
Using the synthetic method for 2, compound 20 gave 21 as a white solid, 80% yield. ESI-MS m/z: 471.5 [M+H]+.
3.1.9.12. (S)-4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propoxy)benzoic acid (29a)
Using the synthetic method for 2, compound 28a gave 29a as a white solid, 83% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 8.37 (d, J = 7.9 Hz, 1H), 7.84 (dd, J = 8.9, 2.5 Hz, 4H), 7.64 (d, J = 7.8 Hz, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.18 (d, 1H), (t, J = 2.2 Hz, 7.06 J = 7.5 Hz, 1H), 7.00–6.94 (m, 5H), 4.59 (dd, J = 12.9, 6.5 Hz, 1H), 4.21 (dd, J = 9.8, 6.5 Hz, 1H), 4.10 (dd, J = 9.9, 5.1 Hz, 1H), 3.80 (s, 3H), 3.10 (d, J = 6.9 Hz, 2H). ESI-MS m/z: 445.6 [M+H]+.
3.1.9.13. (S)-2-(4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propoxy)phenyl)acetic acid (29b)
Using the synthetic method for 2, compound 28b gave 29b as a white solid, 80% yield.
3.1.9.14. (S)-4-(((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propyl)(tertbutoxycarbonyl) amino)methyl)benzoic acid (35)
Using the synthetic method for 2, compound 34 gave 35 as a white solid, 80% yield.
3.1.10. General procedure for the preparation of 39 and 45
3.1.10.1. Tert-butyl (4-bromo-2-nitrophenyl)carbamate (39)
At room temperature, to a solution of 38 (10.85 g, 50 mmol) in anhydrous CH2Cl2 (300 mL), Et3N (15.17 g, 150 mmol) was added followed by (Boc)2O (2.18 g, 100 mmol) and DMAP (4-dimethylaminopyridine) (0.06 g, 0.5 mmol). After stirring the mixture at room temperature for 2 h, the solution was washed with 1 M aqueous citric acid (3 × 100 mL) and brine (3 × 100 mL), dried over MgSO4, and evaporated under vacuum to get the crude product. The crude product was recrystallized by EtOAc and petroleum ether to achieve a white pure solid (8.71 g, 55%). 1H NMR (400 MHz, DMSO-d6) δ 8.34 (d, J = 2.3 Hz, 1H), 8.02 (dd, J = 8.5, 2.3 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 1.33 (s, 9H).
3.1.10.2. Di-tert-butyl (5-fluoro-2-nitrophenyl)carbamate (45)
Using the synthetic method for 39, compound 44 gave 45 as a white solid, 87% yield. ESI-MS m/z: 357.3 [M+H]+.
3.1.11. General procedure for the preparation of 46
3.1.11.1. Tert-butyl (5-fluoro-2-nitrophenyl)carbamate (46)
A solution of 45 (3.57 g, 10 mmol) in CH2Cl2 (100 mL) at room temperature was treated with TFA (1.14 g, 10 mmol) drop wise, and the resulting mixture was stirred at this temperature for 80 min before being treated with a saturated NaHCO3 aqueous solution. The layers were separated, and the organic phase was washed with a saturated NaHCO3 aqueous solution, dried over Na2SO4 concentrated in vacuo to give 46 (0.23 g, 90%) as a yellow solid, which was used in the next step without further purification. ESI-MS m/z: 255.4 [M − H]−.
3.1.12. General procedure for the preparation of 40a and 40b
3.1.12.1. Tert-butyl (2-nitro-4-(thiophen-2-yl)phenyl)carbamate (40a)
To a solution of compound 39 (6.34 g, 20 mmol), thiophene-2-boronic acid (3.07 g, 24 mmol), PPh3 (0.11 g, 0.4 mmol), Pd(OAc)2 (0.04 g, 0.2 mmol) in 20 mL DMSO Na2CO3 (4.24 g, 40 mmol) was added dissolving in 15 mL of water under N2 atmosphere. The N2 protected reaction mixture was heated at 100 °C for 5 h, after the reaction finished, the mixture was poured on 200 mL water and extracted by EtOAc. The organic phase was washed with saturated 1 M HCl (2 × 30 mL), NaHCO3 (2 × 30 mL) and brine (2 × 30 mL), dried over Na2SO4 overnight, and the solvent was evaporated under vacuum. The crude material was purified via flash chromatography to afford the compound 40a (3.71 g, 58%). 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 1H), 8.15 (d, J = 2.2 Hz, 1H), 7.95 (dd, J = 8.6, 2.2 Hz, 1H), 7.70 (d, J = 8.6 Hz, 1H), 7.66–7.59 (m, 2H), 7.17 (dd, J = 5.0, 3.7 Hz, 1H), 1.46 (s, 9H). ESI-MS m/z: 343.3 [M + Na]+.
3.1.12.2. Tert-butyl (3-nitro-[1,1′-biphenyl]-4-yl)carbamate (40b)
Using the synthetic method for 40a, compound 39 and phenylboronic acid gave 40b as a white solid, 48% yield. ESI-MS m/z: 337.6 [M + Na]+.
3.1.13. General procedure for the preparation of 41a, 41b and 47
3.1.13.1. Tert-butyl (2-amino-4-(thiophen-2-yl)phenyl)carbamate (41a)
To a solution of 40a (3.43 g, 10 mmol) in CH3OH (200 mL), was added dry palladium-carbon (0.34 g), the mixture was stirred under a hydrogen atmosphere overnight. The palladium-carbon was filtered off over celite, after evaporating the solvent, the residue was dried under vacuum to give 41a (2.46 g, 85%), a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.35 (s, 1H), 7.45–7.43 (m, 1H), 7.30 (dd, J = 3.6, 1.1 Hz, 1H), 7.27 (d, J = 8.2 Hz, 1H), 7.08 (dd, J = 5.1, 3.6 Hz, 1H), 6.98 (d, J = 2.1 Hz, 1H), 6.85 (dd, J 8.2, 2.1 Hz, 1H), 5.01 (s, 2H), 1.47 (s, 9H). ESI-MS m/z: 291.3 [M+H]+.
3.1.13.2. Tert-butyl (3-amino-[1,1′-biphenyl]-4-yl)carbamate (41b)
Using the synthetic method for 41a, compound 40b gave 41b as a white solid, 86% yield. ESI-MS m/z: 307.4 [M + Na]+.
3.1.13.3. Tert-butyl (2-amino-5-fluorophenyl)carbamate (47)
Using the synthetic method for 41a, compound 46 gave 47 as a white solid, 80% yield. ESI-MS m/z: 227.2 [M+H]+.
3.1.14. General procedure for the preparation of 51a and 51b
3.1.14.1. 4-(2,2,2-trifluoroacetamido)benzoic acid (51a)
50 (2.74 g, 20 mmol) was dissolved in 10 mL TFA slowly at 0 °C, the solution was stirred at room temperature for 2 h. After the reaction finished, the mixture was quenched by adding 100 mL ice water and filtered, the residue was dried under infrared drying oven to afford 51a (4.43 g, 95%).
3.1.14.2. 4-((2,2,2-trifluoroacetamido)methyl)benzoic acid (51b)
Using the synthetic method for 51a, compound 6 gave 51b as a white solid, 98% yield. mp:214-215 °C, 1HNMR (400 MHz DMSO-d6): δ 4.47 (d, J = 6.0 Hz, 2H), 7.40 (d, J = 8.2 Hz, 2H), 7.94 (d, J = 8.2 Hz, 2H), 10.07 (s, 1H), 12.94 (s, 1H). ESI-MS m/z: 248.5 [M+H]+.
3.1.15. General procedure for the preparation of 53a-53c, 57a and 57b
3.1.15.1. Tert-butyl (4-(thiophen-2-yl)-2-(4-(2,2,2-trifluoroacetamido)benzamido)phenyl)carbamate (53a)
Compound 51a (1.16 g, 5 mmol) was dissolved in 15 mL SOCl2, the mixed solution was refluxed at 80 °C for 5 h. The solvent was evaporated under vacuum, the product (52a) was dissolved with anhydrous CH2Cl2 and was dropped in the CH2Cl2 solution of 41a (1.45 g, 5 mmol) at 0 °C followed by Et3N (0.51 g, 5 mmol). The mixture reacted at room temperature overnight, the solution was washed with 1 N HCl (2 × 30 mL), saturated NaHCO3 (2 × 30 mL) and brine (2 × 30 mL), dried over Na2SO4 overnight, and the solvent was evaporated under vacuum. The crude product was recrystallized by EtOAc and petroleum ether to achieve a white pure solid (1.57 g, 62%).1H NMR (400 MHz, DMSO-d6) δ 11.54 (s, 1H), 9.93 (s, 1H), 8.75 (s, 1H), 8.04 (d, J = 8.7 Hz, 2H), 7.86 (d, J = 8.8 Hz, 2H), 7.82 (d, J = 2.2 Hz, 7.63 (d, J = 8.5 Hz, 1H), 7.55–7.49 (m, 2H), 7.46 (dd, J = 3.6, 1.0 Hz, 1H), 7.14 (dd, J = 5.1, 3.6 Hz, 1H), 1.46 (s, 9H). ESI-MS m/z: 506.5 [M+H]+.
3.1.15.2. Tert-butyl(4-(thiophen-2-yl)-2-(4-((2,2,2-trifluoroacetamido)methyl)benzamido)phenyl)carbamate (53b)
Using the synthetic method for 53a, compound 51b and 41a gave 53b as a white solid, 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.11 (t, J = 6.0 Hz, 1H), 9.91 (s, 1H), 8.75 (s, 1H), 7.97 (d, J = 8.3 Hz, 2H), 7.83 (d, J = 2.1 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.55–7.50 (m, 2H), 7.46–7.44 (m, 3H), 7.14 (dd, J = 5.0, 3.6 Hz, 1H), 4.49 (d, J = 6.0 Hz, 2H), 1.46 (s, 9H).
3.1.15.3. Tert-butyl (3-(4-((2,2,2-trifluoroacetamido)methyl)benzamido)-[1,1′-biphenyl]-4-yl)carbamate (53c)
Using the synthetic method for 53a, compound 51b and 41b gave 53c as a white solid, 58% yield. ESI-MS m/z: 514.5 [M+H]+.
3.1.15.4. Tert-butyl (5-fluoro-2-(4-(2,2,2-trifluoroacetamido)benzamido)phenyl)carbamate (57a)
Using the synthetic method for 53a, compound 52a and 47 gave 57a as a white solid, 56% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.52 (s, 1H), 9.79 (s, 1H), 8.79 (s, 1H), 8.01 (t, J = 6.9 Hz, 2H), 7.82 (t, J = 8.2 Hz, 2H), 7.54 (dd, J = 11.2, 2.8 Hz, 1H), 7.45 (dd, J = 8.8, 6.3 Hz, 1H), 6.97 (td, J = 8.4, 2.9 Hz, 1H), 1.45 (s, 9H). ESI-MS m/z: 442.4 [M+H]+.
3.1.15.5. Tert-butyl(5-fluoro-2-(4-((2,2,2-trifluoroacetamido)methyl)benzamido)phenyl)carbamate (57b)
Using the synthetic method for 53a, compound 52b and 47 gave 57b as a white solid, 48% yield. ESI-MS m/z: 456.4 [M+H]+.
3.1.16. General procedure of compound 54a-54c, 58a and 58b
3.1.16.1. Tert-butyl (2-(4-aminobenzamido)-4-(thiophen-2-yl)phenyl)carbamate (54a)
To a solution of 53a (1.51 g, 3 mmol) in the mixture of 10 mL CH3OH and 10 mL H2O, K2CO3 (1.24 g, 9 mmol) was added. The mixture was then stirred at room temperature overnight, and the CH3OH was evaporated under vacuum. The residue was extracted by EtOAc. The organic phase was washed with brine (2 × 30 mL), dried over Na2SO4 overnight, and the solvent was evaporated under vacuum to afford 54a (0.98 g, 80%). 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H), 8.71 (s, 1H), 7.82 (d, J = 2.1 Hz, 1H), 7.71 (d, J = 8.6 Hz, 2H), 7.55 (d, J = 8.5 Hz, 1H), 7.52 (dd, J = 5.1, 0.9 Hz, 1H), 7.47 (dd, J = 8.5, 2.1 Hz, 1H), 7.44 (dd, J = 3.5, 0.9 Hz, 1H), 7.13 (dd, J = 5.0, 3.6 Hz, 1H), 6.62 (d, J = 8.6 Hz, 2H), 5.85 (s, 2H), 1.47 (s, 9H).
3.1.16.2. Tert-butyl (2-(4-(aminomethyl)benzamido)-4-(thiophen-2-yl)phenyl)carbamate (54b)
Using the synthetic method for 54a, compound 53b gave 54b as a white solid, 79% yield.
3.1.16.3. Tert-butyl (3-(4-(aminomethyl)benzamido)-[1,1′-biphenyl]-4-yl)carbamate (54c)
Using the synthetic method for 54a, compound 53c gave 54c as a white solid, 83% yield.
3.1.16.4. Tert-butyl (2-(4-aminobenzamido)-5-fluorophenyl)carbamate (58a)
Using the synthetic method for 54a, compound 57a gave 58a as a white solid, 78% yield. ESI-MS m/z: 346.3 [M+H]+.
3.1.16.5. Tert-butyl (2-(4-(aminomethyl)benzamido)-5-fluorophenyl)carbamate (58b)
Using the synthetic method for 54a, compound 57b gave 58a as a white solid, 84% yield.
3.1.17. General procedure for the preparation of 3, 11a-11 g, 19, 22, 30a, 30b, 36, 49a, 42b, 48, 55a-55c, 59a and 59b
3.1.17.1. (S)-4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)-N-(2-aminophenyl) benzamide (3)
To a solution of 2 (0.46 g, 1 mmol) in anhydrous THF, 2-(1H-Benzotriazole-1-yl) –1,1,3,3-tetramethyluroniumtetrafluoroborate (TBTU, 0.35 g, 1.1 mmol) was added, followed by Et3N (0.15 g, 1.5 mmol). 30 min later, compound benzene-1,2-diamine (0.12 g, 1.1 mmol) was added. After the reaction finished, the solution was evaporated under vacuum, the residue was dissolved with CH2Cl2, and washed with saturated NaHCO3 (2 × 30 mL) and brine (2 × 30 mL), dried over Na2SO4 overnight, and the solvent was evaporated under vacuum. The crude material was purified via flash chromatography to afford the compound 3 (0.16 g, 30%). Mp: 212–214 °C·1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 10.51 (s, 1H), 9.61 (s, 1H), 8.55 (d, J = 7.7 Hz, 1H), 7.98 (d, J = 8.6 Hz, 2H), 7.86 (d, J = 8.8 Hz, 2H), 7.77 (d, J = 8.7 Hz, 3H), 7.32 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 1.9 Hz, 1H), 7.18 (d, J = 7.4 Hz, 1H), 7.07 (t, J = 7.2 Hz, 1H), 7.03–6.94 (m, 4H), 6.84–6.77 (m, 1H), 6.62 (t, J = 7.1 Hz, 1H), 5.03 (s, 2H), 4.94–4.88 (m, 1H), 3.80 (s, 3H), 3.29–3.20 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.97, 166.40, 165.23, 162.19, 143.26, 142.30, 136.54, 129.86, 129.57, 129.12, 127.74, 127.13, 126.86, 126.58, 124.30, 124.17, 121.42, 119.13, 119.06, 118.71, 117.06, 116.82, 113.89, 111.80, 110.67, 55.83, 55.66, 27.97. HRMS (AP-ESI) m/z calcd for C32H29N5O4 [M+H]+ 548.2292, found 548.2293.
3.1.17.2. (S)-4-((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)methyl)-N-(2-aminophenyl)benzamide (11a)
Using the synthetic method for 3, compound 10a gave 11a as a white solid, 32% yield. Mp: 187–188 °C·1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 9.64 (s, 1H), 8.70 (t, J = 5.9 Hz, 1H), 8.41 (d, J = 7.9 Hz, 1H), 7.92 (d, J = 8.1 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 7.70 (d, J = 7.8 Hz, 1H), 7.39–7.30 (m, 3H), 7.22 (d, J = 2.0 Hz, 1H), 7.18 (d, J = 7.6 Hz 1H), 7.06 (t, J = 7.4 Hz, 1H), 7.01–6.96 (m, 4H), 6.83–6.77 (m, 1H), 6.63 (t, J = 7.4 Hz, 1H), 5.09 (s, 2H), 4.80–4.74 (m, 1H), 4.39 (d, J = 5.8 Hz, 2H), 3.28–3.12 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.57, 166.24, 165.65, 162.11, 143.57, 143.10, 136.58, 133.47, 129.81, 128.22, 127.75, 127.23, 127.13, 126.92, 126.81, 124.18, 124.11, 121.36, 118.98, 118.71, 117.11, 116.86, 113.82, 111.80, 111.04, 55.82, 54.93, 42.39, 28.05. HRMS (AP-ESI) m/z calcd for C33H31N5O4 [M+H]+ 562.2449, found 562.2449.
3.1.17.3. (S)-N-(2-aminophenyl)-4-((2-(4-(dimethylamino)benzamido)-3-(1H-indol-3-yl)propanamido)methyl)benzamide (11b)
Using the synthetic method for 3, compound 10b gave 11b as a white solid, 28% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 9.70 (s, 1H), 8.65 (t, J = 5.9 Hz, 1H), 8.15 (d, J = 7.9 Hz, 1H), 7.92 (d, J = 8.1 Hz, 2H), 7.73 (d, J = 8.9 Hz, 2H), 7.70 (d, J = 7.8 Hz, 1H), 7.35–7.31 (m, 3H), 7.21–7.19 (m, 2H), 7.08–7.97 (m, 3H), 6.85 (d, J = 7.3 Hz, 1H), 6.69–6.67 (m, 3H), 5.53 (s, 2H), 4.78–4.73 (m, 1H), 4.39 (d, J = 5.8 Hz, 2H), 3.22–3.12 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.82, 166.64, 165.70, 152.65, 143.66, 136.57, 133.37, 129.32, 128.23, 127.78, 127.24, 127.19, 126.94, 124.63, 124.15, 121.34, 121.20, 118.97, 118.69, 117.86, 117.36, 111.79, 111.12, 54.80, 42.37, 28.08. HRMS (AP-ESI) m/z calcd for C34H34N6O3 [M+H]+ 575.2765, found 575.2767.
3.1.17.4. (S)-4-((3-(1H-indol-3-yl)-2-(4-(trifluoromethyl)benzamido)propanamido)methyl)-N-(2-aminophenyl)benzamide (11c)
Using the synthetic method for 3, compound 10c gave 11c as a white solid, 34% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 9.68 (s, 1H), 8.89 (d, J = 7.9 Hz, 1H), 8.78 (t, J = 5.8 Hz, 1H), 8.06 (d, J = 8.1 Hz, 2H), 7.93 (d, J = 8.0 Hz, 2H), 7.83 (d, J = 8.2 Hz, 2H), 7.71 (d, J = 7.8 Hz, 1H), 7.35–7.32 (m, 3H), 7.25–7.15 (m, 2H), 7.11–7.04 (m, 1H), 7.01–6.98 (m, 2H), 6.83 (d, J = 7.8 Hz, 1H), 6.66 (t, J = 7.4 Hz, 1H), 5.37 (s, 2H), 4.84–4.79 (m, 1H), 4.41 (d, J = 5.6 Hz, 2H), 3.23–3.11 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.15, 165.63, 143.50, 143.08, 138.29, 136.57, 133.46, 128.90, 128.26, 127.67, 127.20, 126.94, 125.66, 124.22, 124.08, 121.40, 118.98, 118.73, 117.11, 116.86, 111.83, 110.86, 55.16, 42.39, 27.99. HRMS (AP-ESI) m/z calcd for C33H28F3N5O3 [M+H]+ 600.2217, found 600.2220.
3.1.17.5. (S)-N-(1-((4-((2-aminophenyl)carbamoyl)benzyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)picolinamide (11d)
Using the synthetic method for 3, compound 10d gave 11d as a white solid, 31% yield. Mp: 187–189 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.99 (s, 1H), 9.75 (s, 1H), 8.94–8.81 (m, 1H), 8.68 (d, J = 8.2 Hz, 1H), 8.63 (d, J = 4.6 Hz, 1H), 8.04–7.94 (m, 4H), 7.63–7.59 (m, 2H), 7.34 (d, J = 8.0 Hz, 1H), 7.30 (t, J = 7.1 Hz, 2H), 7.20 (d, J = 7.5 Hz, 1H), 7.12–7.10 (m, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.98–6.91 (m, 2H), 6.80 (d, J = 7.6 Hz, 1H), 6.61 (t, J = 7.3 Hz, 1H), 5.05 (s, 2H), 4.90–4.85 (m, 1H), 4.45–4.25 (m, 2H), 3.33–3.28 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.58, 165.62, 163.69, 149.83, 148.94, 143.20, 138.36, 136.62, 133.54, 128.30, 128.12, 127.84, 127.19, 126.81, 124.18, 122.27, 121.36, 119.00, 118.71, 116.89, 116.80, 111.84, 109.91, 54.06, 42.46, 28.74. HRMS (AP-ESI) m/z calcd for C31H28N6O3 [M+H]+ 533.2296, found 533.2299.
3.1.17.6. (S)-N-(1-((4-((2-aminophenyl)carbamoyl)benzyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)pyrazine-2-carboxamide (11e)
Using the synthetic method for 3, compound 10e gave 11e as a white solid, 36% yield. Mp: 206–208 °C. 1H NMR(400 MHz, DMSO-d6) δ 10.84 (s, 1H), 9.64 (s, 1H), 9.17 (s, 1H), 8.87 (d, J = 2.1 Hz, 1H), 8.80 (t, J = 5.7 Hz, 1H), 8.72–8.70 (m, 2H), 7.92 (d, J = 7.9 Hz, 2H), 7.62 (d, J = 7.9 Hz, 1H), 7.37–7.28 (m, 3H), 7.18 (d, J = 7.7 Hz, 1H), 7.15 (s, 1H), 7.05 (t, J = 7.5 Hz, 1H), 7.01–6.90 (m, 2H), 6.80 (d, J = 7.9 Hz, 1H), 6.62 (t, J = 7.4 Hz, 1H), 5.07 (s, 2H), 4.87 (dd, J = 13.8, 7.3 Hz, 1H), 4.38 (d, J = 5.6 Hz, 2H), 3.32–3.26 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.38, 165.63, 162.88, 148.19, 144.70, 143.89, 143.81, 143.29, 143.20, 136.62, 133.59, 128.25, 127.81, 127.36, 127.13, 126.93, 124.23, 124.00, 121.43, 118.97, 118.76, 116.97, 116.77, 111.83, 110.03, 54.18, 42.51, 28.44. HRMS (AP-ESI) m/z calcd for C30H27N7O3 [M+H]+ 534.2248, found 534.2248.
3.1.17.7. (S)-N-(1-((4-((2-aminophenyl)carbamoyl)benzyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)thiophene-2-carboxamide (11f)
Using the synthetic method for 3, compound 10f gave 11f as a white solid, 34% yield. Mp: 134–136 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 9.67 (s, 1H), 8.76 (t, J = 5.9 Hz, 1H), 8.65 (d, J = 8.1 Hz, 1H), 8.00–7.86 (m, 3H), 7.78–7.67 (m, 2H), 7.33 (d, J = 8.1 Hz, 3H), 7.21–7.18 (m, 2H), 7.15–7.13 (m, 1H), 7.06 (t, J = 7.1 Hz, 1H), 6.99 (t, J = 7.4 Hz, 2H), 6.82 (d, J = 7.9 Hz, 1H), 6.65 (t, J = 7.1 Hz, 1H), 5.31 (s, 2H), 4.78–4.73 (m, 1H), 4.39 (d, J = 5.8 Hz, 2H), 3.19–3.13 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.24, 165.67, 161.59, 143.50, 142.70, 140.11, 136.57, 133.47, 131.39, 129.09, 128.31. 128.24, 127.70, 127.25, 127.14, 126.94, 124.31, 124.21, 121.39, 118.98, 118.74, 117.42, 117.06, 111.82, 110.84, 54.83, 42.41, 28.09. HRMS (AP-ESI) m/z calcd for C30H27N5O3S [M+H]+ 538.1907, found 538.1903.
3.1.17.8. (S)-N-(1-((4-((2-aminophenyl)carbamoyl)benzyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)furan-2-carboxamide (11g)
Using the synthetic method for 3, compound 10g gave 11g as a white solid, 36% yield. Mp: 202–204 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 9.64 (s, 1H), 8.74 (t, J = 5.9 Hz, 1H), 8.32 (d, J = 8.1 Hz, 1H), 7.92 (d, J = 8.0 Hz, 2H), 7.85–7.78 (m, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.33 (d, J = 8.1 Hz, 3H), 7.22–7.13 (m, 3H), 7.06 (t, J = 7.4 Hz, 1H), 6.98 (t, J = 7.4 Hz, 2H), 6.80 (d, J = 7.8 Hz, 1H), 6.66–6.57 (m, 2H), 5.01 (s, 2H), 4.79–4.73 (m, 1H), 4.39 (d, J = 5.7 Hz, 2H), 3.24–3.15 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.04, 165.65, 158.10, 148.00, 145.54, 143.37, 136.59, 133.54, 128.23, 127.73, 127.29, 127.13, 126.93, 124.19, 123.99, 121.40, 118.96, 118.74, 116.97, 116.75, 114.22, 112.29, 111.82, 110.64, 54.12, 42.44, 28.13. HRMS (AP-ESI) m/z calcd for C30H27N5O4 [M+H]+ 522.2136, found 522.2137.
3.1.17.9. (S)-N-(1-((6-((2-aminophenyl)amino)-6-oxohexyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (19)
Using the synthetic method for 3, compound 18 gave 19 as a white solid, 31% yield. Mp: 133–135 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 9.13 (s, 1H), 8.29 (d, J = 8.1 Hz, 1H), 8.05 (t, J = 5.3 Hz, 1H), 7.81 (d, J = 8.8 Hz, 2H), 7.68 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.22–7.13 (m, 2H), 7.04 (t, J = 7.4 Hz, 1H), 6.99–6.94 (m, 3H), 6.89 (t, J = 7.1 Hz, 1H), 6.73 (d, J = 8.0 Hz, 1H), 6.55 (t, J = 7.1 Hz, 1H), 5.21 (s, 2H), 4.74–4.58 (m, 1H), 3.79 (s, 3H), 3.23–2.96 (m, 4H), 2.30 (t, J = 7.4 Hz, 2H), 1.62–1.55 (m, 2H), 1.45–1.39 (m, 2H), 1.34–1.21 (m, 2H). 13C NMR(100 MHz, DMSO-d6) δ 172.11, 171.63, 166.04, 162.05, 142.01, 136.53, 129.75, 127.80, 126.87, 126.15, 125.78, 124.25, 124.06, 121.30, 119.01, 118.63, 116.91, 116.53, 113.80, 111.74, 111.10, 55.80, 54.75, 39.07, 36.23, 29.35, 28.23, 26.59, 25.54. HRMS (AP-ESI) m/z calcd for C31H35N5O4 [M+H]+ 542.2762, found 542.2764.
3.1.17.10. (S)-4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propoxy)-N-(2-aminophenyl) benzamide (30a)
Using the synthetic method for 3, compound 29a gave 30a as a white solid, 33% yield. Mp: 106–108 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 9.54 (s, 1H), 8.37 (d, J = 7.9 Hz, 1H), 7.94 (d, J = 8.8 Hz, 2H), 7.84 (d, J = 8.9 Hz, 2H), 7.64 (d, J = 7.9 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.18 (d, J = 2.1 Hz, 1H), 7.15 (d, J = 7.8 Hz, 1H), 7.08–7.04 (m, 3H), 7.01–6.93 (m, 4H), 6.81–6.76 (m, 1H), 6.60 (t, J = 7.5 Hz, 1H), 4.96 (s, 2H), 4.63–4.58 (m, 1H), 4.25–4.10 (m, 2H), 3.80 (s, 3H), 3.17–3.07 (m, 2H). HRMS (AP-ESI) m/z calcd for C32H30N4O4 [M+H]+ 535.2340, found 535.2343.
3.1.17.11. (S)-N-(1-(4-(2-((2-aminophenyl)amino)-2-oxoethyl)phenoxy)-3-(1H-indol-3-yl)propan-2-yl)-4-methoxybenzamide (30b)
Using the synthetic method for 3, compound 29b gave 30b as a white solid, 35% yield. Mp: 194–196 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 9.31 (s, 1H), 8.33 (d, J = 7.8 Hz, 1H), 7.84 (d, J = 8.7 Hz, 2H), 7.63 (d, J = 7.8 Hz, 1H), 7.32 (d, J = 8.1 Hz, 1H), 7.24 (d, J = 8.4 Hz, 2H), 7.17–7.13 (m, 2H), 7.05 (t, J = 7.5 Hz, 1H), 7.00–6.86 (m, 6H), 6.72 (d, J = 7.9 Hz, 1H), 6.54 (t, J = 7.5 Hz, 1H), 4.94 (s, 2H), 4.60–4.55 (m, 1H), 4.15–4.00 (m, 2H), 3.80 (s, 3H), 3.56 (s, 2H), 3.15–3.03 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 169.91, 166.24, 162.00, 157.77, 142.29, 136.65, 130.55, 129.63, 128.92, 127.92, 127.26, 126.33, 125.75, 123.92, 123.82, 121.38, 118.83, 118.77, 116.75, 116.40, 114.92, 113.85, 111.84, 111.41, 69.48, 55.81, 50.27, 42.30, 27.26. HRMS (AP-ESI) m/z calcd for C33H32N4O4 [M+H]+ 549.2496, found 549.2500.
3.1.17.12. (S,E)-N-(1-(4-(3-((2-aminophenyl)amino)-3-oxoprop-1-en-1-yl)phenoxy)-3-(1H-indol-3-yl)propan-2-yl)-4-methoxybenzamide (22)
Using the synthetic method for 3, compound 21 gave 22 as a white solid, 30% yield. Mp: 98–100 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 9.33 (s, 1H), 8.36 (d, J = 7.9 Hz, 1H), 7.85 (d, J = 8.8 Hz, 2H), 7.64 (d, J = 7.8 Hz, 1H), 7.56–7.48 (m, 3H), 7.34 (d, J = 8.1 Hz, 2H), 7.18 (d, J = 2.0 Hz, 1H), 7.09–6.89 (m, 7H), 6.77–6.74 (m, 2H), 6.59 (t, J = 8.0 Hz, 1H), 5.07 (s, 2H), 4.62–4.57 (m, 1H), 4.22–4.07 (m, 2H), 3.80 (s, 3H), 3.14–3.05 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 166.28, 164.29, 162.03, 160.27, 141.88, 139.81, 136.67, 129.72, 129.64, 128.04, 127.90, 127.21, 126.15, 125.16, 124.24, 123.86, 121.41, 120.32, 118.78, 116.91, 116.58, 115.55, 113.87, 111.87, 111.32, 69.56, 55.82, 50.22, 27.23. HRMS (AP-ESI) m/z calcd for C34H32N4O4 [M+H]+ 561.2496, found 561.2495.
3.1.17.13. Tert-butyl (S)-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propyl)(4-((2-aminophenyl)carbamoyl)benzyl)carbamate (36)
Using the synthetic method for 3, compound 35 gave 36 as a white solid, 38% yield. ESI-MS m/z: 648.6 [M+H]+.
3.1.17.14. Tert-butyl (S)-(2-(6-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido) hexanamido)-4-(thiophen-2-yl)phenyl)carbamate (42a)
Using the synthetic method for 3, compound 18 and 41a gave 42a as a white solid, 65% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 9.49 (s, 1H), 8.44 (s, 1H), 8.28 (d, J = 8.1 Hz, 1H), 8.05 (t, J = 5.5 Hz, 1H), 7.81 (d, J = 8.8 Hz, 2H), 7.74 (d, J = 2.0 Hz, 1H), 7.67 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.51 (dd, J = 5.1, 1.0 Hz, 1H), 7.44 (dd, J = 8.5, 2.1 Hz, 1H), 7.41 (dd, J = 3.5, 0.8 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 2.2 Hz, 1H), 7.11 (dd, J = 5.1, 3.6 Hz, 1H), 7.07–7.02 (m, 1H), 7.00–6.93 (m, 3H), 4.70–4.64 (m, 1H), 3.79 (s, 3H), 3.19–3.06 (m, 4H), 2.36 (t, J = 7.4 Hz, 2H), 1.69–1.56 (m, 2H), 1.50–1.40 (m, 11H), 1.36–1.25 (m, 2H).
3.1.17.15. Tert-butyl (S)-(3-(6-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)hexanamido)-[1,1’-biphenyl]-4-yl)carbamate (42b)
Using the synthetic method for 3, compound 18 and 41b gave 42b as a white solid, 63% yield.
3.1.17.16. Tert-butyl (S)-(2-(6-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)hexanamido)-5-fluorophenyl)carbamate (48)
Using the synthetic method for 3, compound 18 and 47 gave 48 as a white solid, 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 9.37 (s, 1H), 8.49 (s, 1H), 8.28 (d, J = 8.1 Hz, 1H), 8.05 (t, J = 5.5 Hz, 1H), 7.81 (d, J = 8.8 Hz, 2H), 7.67 (d, J = 7.8 Hz, 1H), 7.49 (dd, J = 11.1, 2.8 Hz, 1H), 7.36 (dd, J = 8.9, 6.2 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.18 (d, J = 2.1 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 6.99–6.94 (m, 3H), 6.86 (td, J = 8.5, 3.0 Hz, 1H), 4.71–4.61 (m, 1H), 3.79 (s, 3H), 3.19–3.05 (m, 4H), 2.32 (t, J = 7.4 Hz, 2H), 1.61 -1.55 (m, 2H), 1.51–1.37 (m, 11H), 1.35–1.21 (m, 2H). ESI-MS m/z: 660.6 [M+H]+.
3.1.17.17. Tert-butyl (S)-(2-(4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)benzamido)-4-(thiophen-2-yl)phenyl)carbamate (55a)
Using the synthetic method for 3, compound 14 and 54a gave 55a as a white solid, 58% yield. ESI-MS m/z: 730.6 [M+H]+.
3.1.17.18. Tert-butyl(S)-(2-(4-((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)methyl)benzamido)-4-(thiophen-2-yl)phenyl)carbamate (55b)
Using the synthetic method for 3, compound 14 and 54b gave 55b as a white solid, 58% yield.
3.1.17.19. Tert-butyl(S)-(3-(4-((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)methyl)benzamido)-[1,1’-biphenyl]-4-yl)carbamate (55c)
Using the synthetic method for 3, compound 14 and 54c gave 55c as a white solid, 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 9.89 (s, 1H), 8.80 (s, 1H), 8.72 (t, J = 6.0 Hz, 1H), 8.42 (d, J = 7.9 Hz, 1H), 7.91 (d, J = 8.3 Hz, 2H), 7.88 (d, J = 2.0 Hz, 1H), 7.85 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 7.8 Hz, 1H), 7.66–7.63 (m, 3H), 7.52 (dd, J = 8.5, 2.1 Hz, 1H), 7.47 (t, J = 7.7 Hz, 2H), 7.41–7.36 (m, 3H), 7.35–7.30 (m, 1H), 7.23 (d, J = 2.2 Hz, 1H), 7.08–7.04 (m, 1H), 7.01–6.97 (m, 3H), 4.80–4.75 (m, 1H), 4.41 (d, J = 5.8 Hz, 2H), 3.80 (s, 3H), 3.30–3.14 (m, 2H), 1.46 (s, 9H). ESI-MS m/z: 738.7 [M+H]+.
3.1.17.20. Tert-butyl(S)-(2-(4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)benzamido)-5-fluorophenyl)carbamate (59a)
Using the synthetic method for 3, compound 14 and 58a gave 59a as a white solid, 56% yield.
3.1.17.21. Tert-butyl(S)-(2-(4-((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)methyl)benzamido)-5-fluorophenyl)carbamate (59b)
Using the synthetic method for 3, compound 14 and 58b gave 59b as a white solid, 50% yield. ESI-MS m/z: 680.5 [M+H]+.
3.1.18. General procedure of compound 43a, 43b, 49, 56a-56c, 60a and 60b
3.1.18.1. (S)-N-(1-((6-((2-amino-5-(thiophen-2-yl)phenyl)amino)-6-oxohexyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (43a)
To a solution of 42a (0.72 g, 1 mmol) in CH2Cl2, TFA (0.23 g, 2 mmol) was added, the mixture was then reacted overnight. After the reaction finished, Et3N (0.21 g, 2 mmol) was added to neutralize the solution. The solvent was washed with brine and dried over Na2SO4 overnight, and the solvent was evaporated under vacuum to get crude product. The crude product was recrystallized by EtOAc and petroleum ether to achieve a white pure solid (0.40 g, 65%). Mp: 118–120 °C. 1H NMR (400 MHz, DMSO-dg) δ 10.81 (s, 1H), 9.75 (s, 1H), 8.42–8.30 (m, 1H), 8.12 (t, J = 6.0 Hz, 1H), 7.84–7.79 (m, 2H), 7.71–7.65 (m, 1H), 7.64–7.61 (m, 1H), 7.46 (d, J = 4.7 Hz, 1H), 7.39 (d, J = 7.9 Hz, 1H), 7.35 (d, J = 3.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.19 (s, 1H), 7.12–7.01 (m, 3H), 7.00–6.91 (m, 3H), 4.72–4.59 (m, 1H), 3.78 (s, 3H), 3.17–3.09 (m, 4H), 2.38 (t, J = 7.4 Hz, 2H), 1.71–1.54 (m, 2H), 1.50–1.39 (m, 2H), 1.33–1.31 (m, 2H). HRMS (AP-ESI) m/z calcd for C35H37N5O4S [M+H]+ 624.2639, found 624.2642.
3.1.18.2. (S)-N-(1-((6-((4-amino-[1,1’-biphenyl]-3-yl)amino)-6-oxohexyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (43b)
Using the synthetic method for 43a, compound 42b gave 43b as a white solid, 63% yield. Mp: 124–126 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 9.86 (s, 1H), 8.34 (d, J = 8.0 Hz, 1H), 8.10 (s, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.71–7.65 (m, 2H), 7.57 (d, J = 7.5 Hz, 2H), 7.42 (dd, J = 13.7, 6.7 Hz, 3H), 7.30 (d, J = 7.6 Hz, 2H), 7.22–7.13 (m, 2H), 7.07–7.00 (m, 1H), 6.99–6.94 (m, 3H), 4.68–4.65 (m, 1H), 3.79 (s, 3H), 3.16–3.00 (m, 4H), 2.44–2.34 (m, 2H), 1.63–1.61 (m, 2H), 1.44–1.42 (m, 2H), 1.33–1.31 (m, 2H). HRMS (AP-ESI) m/z calcd for C37H39N5O4 [M+H]+ 618.3075, found 618.3077.
3.1.18.3. (S)-N-(1-((6-((2-amino-4-fluorophenyl)amino)-6-oxohexyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methoxybenzamide (49)
Using the synthetic method for 43a, compound 48 gave 49 as a white solid, 49% yield. Mp:172–174 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H), 9.08 (s, 1H), 8.31 (d, J = 8.0 Hz, 1H), 8.07 (t, J = 5.1 Hz, 1H), 7.82 (d, J = 8.4 Hz, 2H), 7.68 (d, J = 7.7 Hz, 1H), 7.30 (d, J = 7.9 Hz, 1H), 7.19 (s, 1H), 7.11 (t, J = 7.4 Hz, 1H), 7.04 (t, J = 7.4 Hz, 1H), 6.99–6.96 (m, 3H), 6.49 (dd, J = 11.1, 2.1 Hz, 1H), 6.28 (td, J = 8.4, 2.3 Hz, 1H), 5.19 (s, 2H), 4.70–4.65 (m, 1H), 3.79 (s, 3H), 3.19–3.02 (m, 4H), 2.29 (t, J = 7.3 Hz, 2H), 1.64–1.51 (m, 2H), 1.49–1.37 (m, 2H), 1.30–1.28 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.13, 171.85, 166.03, 162.05, 159.71, 144.65, 144.53, 136.53, 129.75, 127.80, 127.53, 127.43, 126.87, 124.05, 121.28, 120.13, 119.00, 118.62, 113.80, 111.74, 111.09, 102.48, 102.26, 101.98, 101.73, 55.80, 54.79, 39.06, 36.12, 29.34, 28.24, 26.59, 25.48. HRMS (AP-ESI) m/z calcd for C31H34FN5O4 [M+H]+ 560.2668, found 560.2670.
3.1.18.4. (S)-4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)-N-(2-amino-5-(thiophen-2-yl)phenyl)benzamide (56a)
Using the synthetic method for 43a, compound 55a gave 56a as a white solid, 40% yield. Mp: 150–152 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 10.54 (s, 1H), 9.81 (s, 1H), 8.56 (d, J = 7.6 Hz, 1H), 8.01 (d, J = 8.6 Hz, 2H), 7.86 (d, J = 8.8 Hz, 2H), 7.80–7.76 (m, 3H), 7.53 (s, 1H), 7.40 (d, J = 5.0 Hz, 1H), 7.38–7.25 (m, 4H), 7.08–7.04 (m, 2H), 7.02–6.98 (m, 3H), 6.93 (d, J = 8.2 Hz, 1H), 4.93–4.88 (m, 1H), 3.81 (s, 3H), 3.28–3.23 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.01, 166.39, 165.46, 162.19, 144.40, 142.49, 136.53, 129.86, 129.24, 128.76, 127.73, 126.57, 124.30, 124.20, 121.97, 121.41, 119.14, 119.05, 118.70, 113.89, 111.80, 110.66, 55.83, 27.96. HRMS (AP-ESI) m/z calcd for C36H31N5O4S [M+H]+ 630.2170, found 630.2175.
3.1.18.5. (S)-4-((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)methyl)-N-(2-amino-5-(thiophen-2-yl)phenyl)benzamide (56b)
Using the synthetic method for 43a, compound 55b gave 56b as a white solid, 39% yield. Mp: 158–160 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.16 (s, 1H), 8.74 (t, J = 5.9 Hz, 1H), 8.45 (d, J = 7.9 Hz, 1H), 8.01 (d, J = 8.2 Hz, 2H), 7.86 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 7.8 Hz, 1H), 7.68 (d, J = 1.6 Hz, 1H), 7.50–7.45 (m, 2H), 7.40–7.36 (m, 3H), 7.33 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 1.9 Hz, 1H), 7.17 (d, J = 8.3 Hz, 1H), 7.12–7.10 (m, 1H), 7.06 (t, J = 7.1 Hz, 1H), 7.01–6.96 (m, 3H), 4.81–4.75 (m, 1H), 4.41 (d, J = 5.7 Hz, 2H), 3.80 (s, 3H), 3.31–3.17 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.60, 166.24, 166.01, 162.10, 144.04, 143.49, 136.58, 132.84, 129.83, 128.91, 128.45, 127.75, 127.27, 126.80, 125.29, 124.27, 124.18, 124.10, 123.22, 121.35, 118.97, 118.70, 113.82, 111.82, 111.01, 55.82, 55.02, 42.39, 28.05. HRMS (AP-ESI) m/z calcd for C37H33N5O4S [M+H]+ 644.2326, found 644.2326.
3.1.18.6. (S)-4-((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)methyl)-N-(4-amino-[1,1’-biphenyl]-3-yl)benzamide (56c)
Using the synthetic method for 43a, compound 55c gave 56c as a white solid, 39% yield. Mp: 168–170 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 10.35 (s, 1H), 8.76 (t, J = 5.7 Hz, 1H), 8.47 (d, J = 7.9 Hz, 1H), 8.06 (d, J = 7.8 Hz, 2H), 7.87 (d, J = 8.2 Hz, 2H), 7.79 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.65 (d, J = 7.8 Hz, 2H), 7.55 (d, J = 7.7 Hz, 1H), 7.47 (t, J = 7.4 Hz, 2H), 7.38–7.33 (m, 5H), 7.22 (s, 1H), 7.06 (t, J = 7.5 Hz, 1H), 7.01–6.97 (m, 3H), 4.81–4.76 (m, 1H), 4.41 (d, J = 5.5 Hz, 2H), 3.80 (s, 3H), 3.31–3.18 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.60, 166.24, 166.04, 162.10, 144.11, 139.72, 136.59, 132.77, 129.84, 129.46, 128.49, 127.75, 127.28, 126.81, 126.75, 125.53, 125.09, 124.18, 121.34, 118.98, 118.70, 113.82, 111.83, 110.99, 55.82, 55.04, 42.39, 28.06. HRMS (AP-ESI) m/z calcd for C39H35N5O4 [M+H]+ 638.2762, found 638.2763.
3.1.18.7. (S)-4-(3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)-N-(2-amino-4-fluorophenyl)benzamide (60a)
Using the synthetic method for 43a, compound 59a gave 60a as a white solid, 35% yield. Mp: 170–172 °C. 1H NMR(400 MHz, DMSO-d6) δ 10.83 (s, 1H), 10.56 (s, 1H), 9.54 (s, 1H), 8.59 (d, J = 7.7 Hz, 1H), 7.97 (d, J = 8.5 Hz, 2H), 7.87 (d, J = 8.8 Hz, 2H), 7.78 (d, J = 8.7 Hz, 3H), 7.32 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 1.9 Hz, 1H), 7.14–7.10 (m, 1H), 7.10–7.03 (m, 1H), 7.01–6.97 (m, 3H), 6.55 (dd, J = 11.2, 2.8 Hz, 1H), 6.36 (td, J = 8.5, 2.8 Hz, 1H), 5.23 (s, 2H), 4.90 (dd, J = 14.3, 8.0 Hz, 1H), 3.81 (s, 3H), 3.32–3.23 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.97, 166.37, 165.50, 162.19, 160.26, 145.97, 145.86, 142.34, 136.54, 129.87, 129.46, 129.13, 128.92, 127.75, 126.58, 124.29, 121.40, 119.95, 119.14, 119.05, 118.70, 113.88, 111.80, 110.67, 102.65, 102.42, 102.12, 101.87, 55.83, 55.70, 27.98. HRMS (AP-ESI) m/z calcd for C32H28FN5O4 [M+H]+ 566.2198, found 566.2196.
3.1.18.8. (S)-4-((3-(1H-indol-3-yl)-2-(4-methoxybenzamido)propanamido)methyl)-N-(2-amino-4-fluorophenyl)benzamide (60b)
Using the synthetic method for 43a, compound 59b gave 60b as a white solid, 35% yield. Mp: 152–154 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 9.74 (s, 1H), 8.73 (t, J = 5.8 Hz, 1H), 8.45 (d, J = 7.9 Hz, 1H), 7.94 (d, J = 7.8 Hz, 2H), 7.85 (d, J = 8.7 Hz, 2H), 7.70 (d, J = 7.8 Hz, 1H), 7.35–7.32 (m, 3H), 7.22–7.19 (m, 2H), 7.06 (t, J = 7.2 Hz, 1H), 7.00–6.96 (m, 3H), 6.70–6.67 (m, 1H), 6.50 (s, 1H), 4.80–4.75 (m, 1H), 4.39 (d, J = 5.7 Hz, 2H), 3.79 (s, 3H), 3.30–3.17 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.62, 166.22, 165.92, 162.08, 160.08, 143.67, 136.56, 133.22, 129.85, 129.14, 129.04, 128.30, 127.71, 127.18, 126.75, 124.21, 121.35, 120.71, 118.99, 118.69, 113.80, 111.82, 111.01, 103.78, 102.92, 55.81, 55.01, 42.35, 28.04. HRMS (AP-ESI) m/z calcd for C33H30FN5O4 [M+H]+ 580.2355, found 580.2352.
4. HDAC class I cellular activity
The cellular HDAC assay was based on such assay described on patent WO20080952A1 with minor modifications. Briefly, human prostate cancer cell line HL60 were seeded into a 96-well tissue culture plates (Corning, Germany) at a density of 1.2 × 104 cells/well and allowed to grow for 24 h. Then cells were treated with increasing concentrations of test compounds for 3 h. The reaction was started by adding substrate Boc-Ac-Lys-AMC. 3 h later under cell culture conditions, stop solution (50 mM Tris-HCl (pH 8.0), 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1% NP-40, 2.0 mg/mL trypsin, 10 μM TSA) was added and the mixture was incubated for another 3 h. Fluorescence intensity was measured using a microplate reader at excitation and emission wavelengths of 360 and 470 nm, respectively.
5. In vitro HDACs inhibition fluorescence assay
In vitro HDACs inhibition assays were conducted as previously described.17 In brief, 10 μL of enzyme solution was mixed with different concentrations of tested compound (50 μL). The mixture was incubated at 37 °C for 10 min, then 40 μL fluorogenic substrate (Boc-Lys (acetyl)-AMC was used for HDAC1, HDAC2, HDAC3, HDAC8 and HDAC6 while Boc-Lys (triflouroacetyl)-AMC substrate was used for HDAC4) was added. After incubation at 37 °C for 30 min, the mixture was stopped by addition of 100 μL of developer containing trypsin and Trichostatin A (TSA). 20 min later, fluorescence intensity was measured using a microplate reader at excitation and emission wavelengths of 390 and 460 nm, respectively. The inhibition ratios were calculated from the fluorescence intensity readings of tested wells relative to those of control wells, and the IC50 values were calculated using a regression analysis of the concentration/inhibition data.
6. In vitro antiproliferative assay
In vitro antiproliferative assays were determined by the MTT (3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide) method as previously described.17 All cell lines were maintained in RPMI1640 medium containing 10% FBS at 37 °C in a 5% CO2 humidified incubator. Cells were passaged the day before dosing into a 96-well cell plate, allowed to grow for 12 h, and then treated with different concentrations of compound for 48 h. A 0.5% MTT solution was added to each well. After incubation for another 4 h, formazan formed from MTT was extracted by adding 150 μL of DMSO rocking for 15 min. Absorbance was then determined using an ELISA reader at 570 nm.
7. In vivo antitumor assay against U937 and HCT116 xenograft
All experiments involving laboratory animals were conducted with the approved of the local ethics committee. For in vivo anti-tumor efficacy research, 1 × 107 U937 or HCT116 Cells were inoculated subcutaneously in the right flank of male nude mice (5–6 weeks old, SLAC LABORATORY ANIMAL, Shanghai). Ten days after injection, tumors were palpable and mice were randomized into treatment and control groups (6 or 7 mice per group). The treatment groups received compound 9b, MS275 or SAHA by oral administration, and the blank control group received equal volume of PBS solution containing 5% DMSO. During treatment, subcutaneous tumors were measured with vernier caliper every three days, and body weight was monitored regularly. After treatment, mice were sacrificed and dissected to weigh the tumor tissues and to examine the internal organ injury. Tumor growth inhibition (TGI) and relative increment ratio (T/C) were used as the evaluation indicators to reveal the antitumor effects in tumor weight and tumor volume, respectively. Data were analyzed by Student’s two-tailed t-test. A P level <0.05 was considered statistically significant.
TGI = (the mean tumor weight of control group − the mean tumor weight of treated group)/the mean tumor weight of control group.
Tumor volumes (V) were estimated using equation (V = ab2/2, where a and b stand for the longest and shortest diameter, respectively). T/C was calculated according to the following formula:
T/C = the mean RTV of treated group/the mean RTV of control group.
RTV, namely relative tumor volume = Vt/V0 (Vt: the tumor volume measured at the end of treatment; V0: the tumor volume measured at the beginning of treatment).
8. Western blot analysis
HL60 cells were plated at 500,000 cells/mL, 3 mL in flat bottom 6-well plates and allowed to grow for 12 h, and then treated with different concentrations of compounds. 48 h later, cells were harvested, washed by PBS, and lysed with RIPA buffer which was comprised of 50 mM Tris Base, 150 mM NaCl, 5 mM EDTA, 0.1% (v/v) SDS, 0.5% (v/v) Sodium Deoxycholate, and 1% (v/v) Triton-x-100. After lysing, the suspension was ultra-sonicated and centrifuged at 14000 RPM for 15 min at 4 °C. The mixture of 75 μL of supernatant and 25 μL of β-mercaptoethanol:LDS solution (15:85) was heated at 90 °C for 15 min and normalization according BCA test before loading. Lysates were run on Invitrogen NuPAGE 4–12% Bis-Tris 15 well gels at 170 V for approximately 60 min in MES buffer. Gels were transferred to methylcellulose and ran at 30 V for 3 h. Membranes were incubated at 4 °C overnight with 1/1000 primary antibodies, which were diluted in 2.5% (w/v) milk or 5% (w/v) Bovine Serum Albumin. The membrane was washed twice with TBST buffer before incubated with secondary antibodies. Images were acquired using a GE ImageQuant LAS 4000.
Acknowledgments
This work was supported by National Natural Science Foundation of China (Grant no. 81373282, 21302111), National High-tech R&D Program of China, 863 Program (Grant No. 2014AA020523), Major Project of Science and Technology of Shandong Province (2015ZDJS04001), Young Scholars Program of Shandong University (YSPSDU, Grant NO. 2016 WLI H33). This work was also supported by National Institutes of Health/National Cancer Institute (CA163452 to C.J.C.).
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2017.03.069.
References
- 1.Kristensen LS, Nielsen HM, Hansen LL. Epigenetics and cancer treatment. Eur J Pharmacol. 2009;625:131–142. doi: 10.1016/j.ejphar.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 2.Roche J, Bertrand P. Inside HDACs with more selective HDAC inhibitors. Eur J Med Chem. 2016;121:451–483. doi: 10.1016/j.ejmech.2016.05.047. [DOI] [PubMed] [Google Scholar]
- 3.Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications[J] Mol Cancer Ther. 2009;8:1409–1420. doi: 10.1158/1535-7163.MCT-08-0860. [DOI] [PubMed] [Google Scholar]
- 4.Giannini G, Cabri W, Fattorusso C, Rodriquez M. Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med Chem. 2012;4:1439–1460. doi: 10.4155/fmc.12.80. [DOI] [PubMed] [Google Scholar]
- 5.Kumar A, Chauhan S. How much successful are the medicinal chemists in modulation of SIRT1: a critical review. Eur J Med Chem. 2016;119:45–69. doi: 10.1016/j.ejmech.2016.04.063. [DOI] [PubMed] [Google Scholar]
- 6.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 7.Bhadra U, Mondal T, Bag I, Mukhopadhyay D, Das P, Parida BB, Mainkar PS, Reddy CR, Bhadra MP. HDAC inhibitor misprocesses bantam oncomiRNA, but stimulates hid induced apoptotic pathway. Sci Rep. 2015;5:14747. doi: 10.1038/srep14747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zupkovitz G, Tishler J, Posch M, Sadzak I, Ramsauer K, Egger G, Grausenburger R, Schweifer N, Chiocca S, Decker T, Seiser C. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol Cell Biol. 2006;26:7913–7928. doi: 10.1128/MCB.01220-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kidder BL, Palmer S. HDAC1 regulates pluripotency and lineage specific transcriptional networks in embryonic and trophoblast stem cells. Nucleic Acids Res. 2012;40:2925–2939. doi: 10.1093/nar/gkr1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krämer OH. HDAC2: a critical factor in health and disease. Trends Pharmacol Sci. 2009;30:647–655. doi: 10.1016/j.tips.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Jiang Y, Hsieh J. HDAC3 controls gap 2/mitosis progression in adult neural stem/progenitor cells by regulating CDK1 levels. Proc Natl Acad Sci U S A. 2014;111:13541–13546. doi: 10.1073/pnas.1411939111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waltregny D, Glénisson W, Tran SL, North BJ, Verdin E, Colige A, Castronovo V. HDAC8 associates with smooth muscle R-actin and is essential for smooth muscle cell contractility. FASEB J. 2005;19:966–968. doi: 10.1096/fj.04-2303fje. [DOI] [PubMed] [Google Scholar]
- 13.Parra M. Class IIa HDACs - new insights into their functions in physiology and pathology. FEBS J. 2015;282:1736–1744. doi: 10.1111/febs.13061. [DOI] [PubMed] [Google Scholar]
- 14.Regna NL, Vieson MD, Gojmerac AM, Luo XM, Caudell DL, Reilly CM. HDAC expression and activity is upregulated in diseased lupus-prone mice. Int Immunopharmacol. 2015;29:494–503. doi: 10.1016/j.intimp.2015.10.006. pii: S1567-5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Zhang J, Xie Y, Jiang Y, Yingjie Z, Xu W. Progress of HDAC inhibitor panobinostat in the treatment of cancer. Curr Drug Targets. 2014;15:622–634. doi: 10.2174/1389450115666140306152642. [DOI] [PubMed] [Google Scholar]
- 16.Boumber Y, Younes A, Garcia-Manero G. Mocetinostat (MGCD0103): a review of an isotype-specific histone deacetylase inhibitor. Expert Opin Investig Drugs. 2011;20:823–829. doi: 10.1517/13543784.2011.577737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Inks ES, Li X, Hou J, Chou CJ, Zhang J, Jiang Y, Zhang Y, Xu W. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J Med Chem. 2014;57:3324–3341. doi: 10.1021/jm401877m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Wu J, Li X, Mu W, Liu X, Jin Y, Xu W, Zhang Y. Development of N-hydroxybenzamide derivatives with indole-containing cap group as histone deacetylases inhibitors. Bioorg Med Chem. 2015;23:6258–6270. doi: 10.1016/j.bmc.2015.08.040. [DOI] [PubMed] [Google Scholar]
- 19.Demont EH, Bamborough P, Chung CW, Craggs PD, Fallon D, Gordon LJ, Grandi P, Hobbs CI, Hussain J, Jones EJ, Le Gall A, Michon AM, Mitchell DJ, Prinjha RK, Roberts AD, Sheppard RJ, Watson RJ. 1,3-Dimethyl benzimidazolones are potent, selective inhibitors of the BRPF1 br-omodomain. ACS Med Chem Lett. 2014;5:1190–1195. doi: 10.1021/ml5002932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moradei OM, Mallais TC, Frechette S, Paquin I, Tessier PE, Leit SM, Fournel M, Bonfils C, Trachy-Bourget MC, Liu J, Yan TP, Lu AH, Rahil J, Wang J, Lefebvre S, Li Z, Vaisburg AF, Besterman JM. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhance d potency and selectivity. J Med Chem. 2007;50:5543–5546. doi: 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]
- 21.Jia H, Pallos J, Jacques V, Lau A, Tang B, Cooper A, Syed A, Purcell J, Chen Y, Sharma S, Sangrey GR, Darnell SB, Plasterer H, Sadri-Vakili G, Gottesfeld JM, Thompson LM, Rusche JR, Marsh JL, Thomas EA. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol Dis. 2012;46:351–361. doi: 10.1016/j.nbd.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]