

Summary
Necroptosis is an important form of lytic cell death triggered by injury and infection, yet whether mixed lineage kinase domain-like (MLKL) is sufficient to execute this pathway is unknown. In a genetic selection for human cell mutants defective for MLKL-dependent necroptosis, we identified mutations in IPMK and ITPK1, which encode inositol phosphate (IP) kinases that regulate the IP code of soluble molecules. We show that IP kinases are essential for necroptosis triggered by death receptor activation, herpesvirus infection, or a pro-necrotic MLKL mutant. In IP kinase mutant cells, MLKL failed to oligomerize and localize to membranes despite proper receptor-interacting protein kinase-3 (RIPK3)-dependent phosphorylation. We demonstrate that necroptosis requires IP-specific kinase activity and that a highly phosphorylated product, but not a lowly phosphorylated precursor, potently displaces the MLKL auto-inhibitory brace region. These observations reveal control of MLKL-mediated necroptosis by a metabolite and identify a key molecular mechanism underlying regulated cell death.
Graphical abstract
Dovey et al. report an unexpected layer of regulation governing a form of cell death termed necroptosis. They find that the inositol phosphate code controls the executioner protein MLKL to induce cell lysis. This study deepens our understanding of cell death mechanisms important for infection, inflammation, and cancer.

Introduction
Cell death is essential for the development of multicellular organisms, the maintenance of homeostasis, and the defense against microbial infection (Fiers et al., 1999). Necroptosis is a genetically programmed, lytic cell death mechanism that can occur as an alternative to apoptosis under conditions of caspase block or compromise (Linkermann and Green, 2014). In contrast to apoptosis, this form of cell death is characterized by the permeabilization of the plasma membrane and release of damage-associated molecular patterns (DAMPs) which carry profound immunological consequences (Pasparakis and Vandenabeele, 2015). Necroptosis is linked to numerous disease states including ischemia–reperfusion injury, viral and bacterial infections, neurodegenerative disorders (e.g., amyotrophic lateral sclerosis and Alzheimer's Disease), and cancer (Caccamo et al., 2017; Gonzalez-Juarbe et al., 2015; Ito et al., 2016; Kitur et al., 2015; Mocarski et al., 2015; Strilic et al., 2016). Further understanding of mechanisms regulating necroptosis are likely to lead to additional markers as well as interventions for inflammatory disease and infection.
Necroptosis can be initiated in response to cytokines, danger signals, and infection with pathogens (Linkermann and Green, 2014; Mocarski et al., 2015; Pasparakis and Vandenabeele, 2015). Upon induction, a complex upstream signaling cascade ultimately converges on the activation of receptor-interacting protein kinase-3 (RIPK3). RIPK3 binds and directly phosphorylates the pseudokinase MLKL, a critical effector of necroptosis that is the most downstream component known to be required for this pathway (He et al., 2009; Sun et al., 2012; Zhao et al., 2012). MLKL is organized into an N-terminal helical bundle domain and a C-terminal pseudokinase domain which contains the RIPK3 phosphorylation site. These domains are bridged together by an auto-inhibitory brace region, which is well positioned to convey structural changes induced by phosphorylation of the C-terminal domain to activate the N-terminal helical bundle (Murphy et al., 2013; Su et al., 2014). This phosphorylation-induced switch mechanism triggers MLKL to oligomerize and translocate to the plasma membrane, ultimately resulting in the lysis of the cell (Cai et al., 2014; Dondelinger et al., 2014; Murphy et al., 2013; Wang et al., 2014).
MLKL phosphorylation by RIPK3 is a hallmark of the necroptotic program. However, key questions remain about the mechanisms by which MLKL induces cell death, including whether, once phosphorylated by RIPK3, MLKL is sufficient to execute cell lysis. When overexpressed, MLKL mutants mimicking the phosphorylated state (i.e. phosphomimetics) can bypass RIPK3 activation and induce necroptosis, showing that MLKL can execute cell death independently of upstream pathway induction (Murphy et al., 2013; Sun et al., 2012). Moreover, purified MLKL can bind to certain lipids and trigger membrane disruption in defined in vitro liposome systems (Dondelinger et al., 2014; Quarato et al., 2016; Su et al., 2014; Wang et al., 2014), suggesting that MLKL is the putative executioner of plasma membrane rupture in necroptosis. Nonetheless, whether MLKL acts alone or in conjunction with additional death effector proteins or pathways remains unknown (Wallach et al., 2016).
Here, using an unbiased, genome-scale screening approach, we identified an essential role for the kinase-dependent inositol phosphate (IP) messenger pathway in execution of necroptosis by MLKL. We show that IP kinases are critical for necroptosis induced by cytokine-dependent death receptor activation and viral infection. Specifically, genetic disruption of IP kinases to abolish production of higher order inositol phosphates blocked necroptosis downstream of MLKL phosphorylation by RIPK3 and prevented the oligomerization and plasma membrane recruitment of MLKL. We found that binding of a higher order IP, but not a lower order IP, promoted an active conformation of MLKL by mediating the structural rearrangement of the N-terminal auto-inhibitory brace. This work deepens our understanding of the molecular mechanisms underlying pro-inflammatory cell death by revealing a critical function for soluble inositol metabolites in necroptosis.
Results
Genome-scale knockout screen for critical necroptotic death effectors identifies inositol phosphate kinases
To identify death effector molecules necessary for MLKL-mediated necroptosis, we pursued an unbiased, forward-genetic screening strategy using the haploid human cell line HAP1 (Carette et al., 2009; Carette et al., 2011b). We developed a necroptosis induction strategy in these cells allowing for execution of cell death independently of the upstream pathway and RIPK3 activation. Doxycycline (DOX)-inducible expression of the human MLKL phosphomimetic mutant T357E/S358D was employed to drive the final steps in necroptosis (Murphy et al., 2013; Sun et al., 2012). These HAP1 Tet-On-MLKLTE/SD cells triggered cell death that was insensitive to the pan-caspase inhibitor zVAD, yet sensitive to the MLKL inhibitor necrosulfonamide (NSA), demonstrating that this cell line specifically undergoes MLKL-induced necroptosis as expected (Figure S1A).
We next performed a selection experiment for necroptosis-resistant mutants among a genome-scale knock out (KO) library in HAP1 Tet-On-MLKLTE/SD cells (100 million clone complexity) (Figure 1A). Mutants surviving DOX-enforced expression of phosphomimetic MLKL were pooled and compared to unselected control cells (not treated with DOX) by deep sequencing of gene trap insertions (Figure 1B and Table S1). The enrichment of multiple subunits of the Mediator (MED) transcription complex was expected because the Tet-On system relies on the recruitment of these proteins by the VP16 transcriptional activation domain. Additionally, we observed significant enrichment of a small subset of other genes that are presumed to be non-specific because they have been previously detected in screens that rely on the Tet-On system (Mandal et al., 2014; Uhlmann et al., 2007). Strikingly, inositol polyphosphate multikinase (IPMK) and inositol-tetrakisphosphate 1-kinase (ITPK1), two critical kinases of the pathway controlling synthesis of cellular inositol phosphates (IPs) (Hatch and York, 2010) were highly enriched and unique to this screen. We chose to pursue these genes for further study due to their related function within in a conserved biochemical pathway (Hatch and York, 2010) and their high degree of statistical enrichment in the screen (IPMK, P=1.56×10-49 and ITPK1, P=1.68×10-24; 58 and 36 independent gene trap insertions within each gene, respectively). We confirmed the importance of these genes for execution of necroptosis in the inducible MLKL system by generating independently-derived ΔIPMK and ΔITPK1 CRISPR mutants in HAP1 Tet-On-MLKLTE/SD cells. These mutants were highly resistant to necroptosis induced by phosphomimetic mutant MLKL (Figure S1), validating the screen results.
Figure 1. Genetic selection for mutants defective for MLKL-mediated necroptosis reveals a critical requirement for inositol phosphate kinases.

(A) Overview of the inducible MLKL cell line and selection strategy. (B) Bubble plot illustrating the enrichment of gene trap insertions following KO library selection on DOX relative to the unselected control (P-value determined by one-sided Fisher exact test). Bubble area represents the number of unique insertions, noted in parentheses. Significantly-enriched genes (P < 10-10) are colored and grouped by predicted function; all other genes are randomly distributed along the X-axis. See Table S1 for the full list of genes. (C-E) Clonal mutants in human HT-29, a model cell line for TNF-induced necroptosis, were independently generated using CRISPR and stably complemented in trans. Cells were co-treated with TNF-α (10 ng/mL), Smac mimetic (100 nM), and zVAD (20 μM) (TSZ) for 24 h to induce necroptosis. Cells stably expressing scrambled (sc) or MLKL shRNA were included as a control. Viability was measured by (C, D) cellular ATP assay at 24 h and (E) kinetic analysis of Sytox Green (SG)-positive cells using IncuCyte live-cell imaging over 24 h (mean ± SD of n = 3 replicates; Data shown are representative of three independent experiments).
We next tested the effects of disrupting IPMK and ITPK1 in human HT-29 cells, a robust model for necroptosis induced by cytokines in the tumor necrosis factor (TNF) family (He et al., 2009; Li et al., 2004). Multiple, independently-derived CRISPR mutant clones for each gene (HT-29 ΔIPMK and ΔITPK1) were extremely resistant to TNF-α-induced necroptosis to an extent that was comparable to knock down of MLKL (Figures 1C-E and Figure S2). Importantly, both mutants were complemented by stably expressing the respective genes in trans. Clonal IPMK mutant human Jurkat cells were also resistant to TNF-α-induced necroptosis, a phenotype that was restored through complementation in trans (Figure S3A). Taken together, these data demonstrate that IP kinases are required for necroptosis in the contexts of both forced induction of the downstream pathway through phosphomimetic MLKL as well as activation of the endogenous pathway by TNF-α.
IP kinases are essential for activated phospho-MLKL to oligomerize and localize to the plasma membrane during necroptosis
To determine the stage or stages at which MLKL necroptotic function depends on the IP pathway, we assessed two hallmarks of MLKL activation in our IP kinase mutants: RIPK3-dependent MLKL phosphorylation and membrane association of phospho-MLKL. Following induction of necroptosis, IPMK and ITPK1 mutant cells exhibited no apparent defect in MLKL phosphorylation at S358, indicating the correct functioning of upstream necroptotic signaling through RIPK3 (Figures 2A and B). MLKL protein levels were also similar in wild-type and mutant cells. Remarkably, however, both ΔIPMK and ΔITPK1 were defective in membrane association of phospho-MLKL (Figures 2C and D) and phospho-MLKL punctae formation at the plasma membrane (Figures 2E-H) during necroptosis. Membrane recruitment of phospho-MLKL was restored in the complemented cell lines, underscoring the specificity of this phenotype for IPMK and ITPK1.
Figure 2. IP kinase mutants fail to localize MLKL to membranes despite proper RIPK3-dependent MLKL phosphorylation during necroptosis.

(A and B) Phospho-MLKL (S358) and total MLKL were detected in HT-29 whole-cell lysates by reducing SDS-PAGE and Western blot following 6 h TSZ stimulation to induce necroptosis. (C and D) HT-29 lysates were fractionated into aqueous (AQ) and detergent-soluble fractions (DET) and blotted for pMLKL, GAPDH (soluble marker) and IMMT (membrane-associated marker). (E and G) Representative images of pMLKL immunofluorescence detection following 6 h TSZ treatment (scale bars: 8 μm). White arrowheads indicate pMLKL punctae. (F and H) TSZ-treated cells containing plasma membrane-associated pMLKL punctae were quantified by blinded assessment of n = 3 fields of 80-140 cells per field (mean ± SD; **P < 0.01 by student's t-test).
Since phospho-MLKL oligomerization is believed to be a requisite for recruitment to the plasma membrane (Cai et al., 2014; Dondelinger et al., 2014; Quarato et al., 2016; Wang et al., 2014), we next sought to test the ability of MLKL to form oligomers in our mutant cell lines. Surprisingly, ΔIPMK and ΔITPK1 were severely defective for oligomeric assembly of MLKL despite its phosphorylation at S358 (Figures 3A and B). The oligomerization phenotype was also fully reversed in the complemented IPMK and ITPK1 cell lines. IPMK mutant Jurkat cells were also defective for formation of MLKL oligomers during necroptosis (despite phosphorylation of MLKL at S358), a phenotype that was reversed through complementation in trans (Figure S3B). Disruption of the IP pathway therefore decouples MLKL phosphorylation from its oligomerization and membrane localization, suggesting an unanticipated layer of regulation at a stage in the necroptotic activation pathway downstream of RIPK3.
Figure 3. IP kinases are required for MLKL oligomerization during necroptosis.
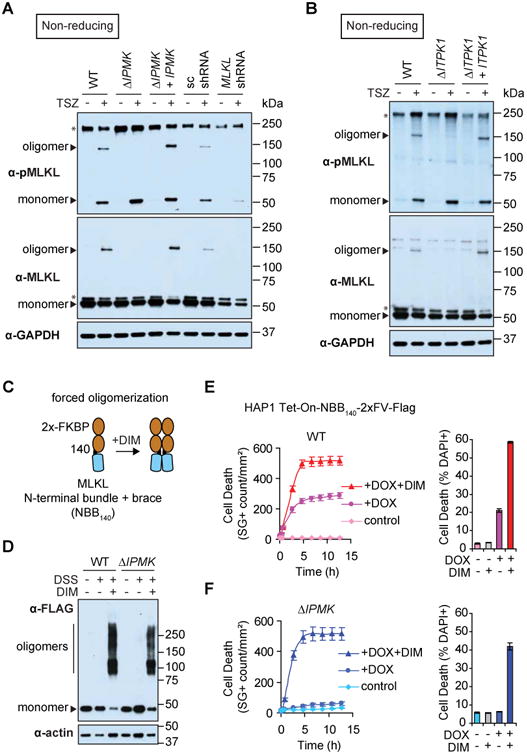
(A and B) MLKL oligomers were resolved from monomers by non-reducing SDS-PAGE and Western blot following TSZ treatment of HT-29 for 6 h. Non-specific bands are denoted with asterisks. (C) Schematic for forced oligomerization of the MLKL N-terminal region (NBB140) fused to 2×-FKBP (2×FV) (Quarato et al., 2016). (D) The NBB140-2×FV-Flag construct was expressed under DOX in WT or ΔIPMK HAP1 Tet-On cells. Following forced oligomerization with DIM for 10 min, oligomers were crosslinked with disuccinimidyl suberate (DSS)-crosslinked and resolved by Western blot. (E and F) Cell death was monitored following inducible MLKL oligomerization with DIM (50 nM) starting at t = 0 h (with or without DOX pre-treatment for 6 h to induce fusion protein expression). Kinetic analysis of cell death was performed by IncuCyte live-cell imaging of Sytox Green (SG)-positive cells or by flow cytometry for DAPI-positive cells at t = 2.5 h (mean ± SD of n = 3 replicates; Data shown are representative of three independent experiments.)
To further clarify our results, we hypothesized that if IP kinases are specifically required at the stage of MLKL oligomerization, selectively bypassing this step should restore necroptosis in IP kinase mutant cells. Thus, we employed a minimal MLKL N-terminal region fused to 2×-FKBP in which oligomerization can be forced by the addition of dimerizer (DIM) (Quarato et al., 2016) (Figures 3C and D). As expected, DIM-enforced MLKL oligomerization accelerated death in wild-type cells expressing the fusion protein (HAP1 Tet-On-NBB140-2×FV-Flag). Remarkably, forced oligomerization of MLKL restored necroptosis in IPMK mutant cells, although to a slightly lesser percentage of the population compared to wild-type cells (Figures 3E and F). These data indicate that the IP kinase pathway is required specifically for MLKL oligomerization during necroptosis.
IP kinases are critical for DAI-dependent necroptosis triggered during infection with HSV-1 (FmutRHIM)
To further explore the functional relevance of the IP kinase pathway during necroptotic death, we tested the effects of disrupting this pathway on cell death during infection with herpes simplex virus-1 (HSV-1). Cells that express sufficient levels of DNA-dependent activator of interferon-regulatory factors (DAI/ZBP1/DLM1) can undergo necroptosis triggered by infection with the ICP6 RHIM mutant virus HSV-1 FmutRHIM, but not the wild-type virus (Guo et al., submitted). As expected, HT-29 cells stably expressing DAI but not empty vector (EV) controls underwent necroptosis in response to infection with HSV-1 FmutRHIM. Importantly, disruption of the IP pathway conferred resistance to infection-induced necroptosis in these cells (Figure 4A), yet sensitivity was fully restored in genetically complemented cells.
Figure 4. IPMK is critical for DAI-dependent necroptosis and MLKL oligomerization triggered by infection with HSV-1 (FmutRHIM).

(A) Wild-type, ΔIPMK, and IPMK complemented HT-29 were stably transduced with a constitutive DAI expression vector or empty vector (EV) control. Infections were performed with the indicated HSV-1 strain at MOI = 5 and cell viability was measured by ATP assay at 24 h post-infection (mean ± SD of n = 3 replicates). The RIPK3 inhibitor GSK840 (3 μM) was included as a control. (B and C) Western blots showing bulk MLKL phosphorylation (B, reducing conditions) and phospho-MLKL oligomerization (C, non-reducing) at 10 h post-infection. HSV-1 infection was confirmed by detection of Infected Cell Polypeptide 0 (ICP0). Non-specific bands are denoted with asterisks.
We hypothesized that, similar to the TNF-induced setting, disruption of the IP pathway should block necroptosis downstream of MLKL phosphorylation by RIPK3 in mutant virus-infected cells. To test our hypothesis, we measured MLKL phosphorylation and oligomerization in IP kinase mutant cells infected with HSV-1 FmutRHIM. DAI-expressing wild-type cells phosphorylated MLKL at S358 in response to infection, further confirming the specific activation of MLKL-dependent necroptosis (Figure 4B). Additionally, disruption of the IP pathway did not affect bulk expression or phosphorylation of MLKL (S358) in infected cells. However, consistent with the TNF-α setting, these cells were clearly defective for oligomerization of phosphorylated MLKL (Figure 4C). Together, our results suggest that the IP pathway is essential for the oligomerization of phospho-MLKL in the contexts of cytokine-dependent death receptor activation and viral infection.
Inositol phosphate kinase activity is essential for necroptosis
IPMK and ITPK1 are conserved enzymes that perform an important role in the phosphorylation of IPs, water-soluble signaling molecules and structural cofactors found in all eukaryotes (Irvine and Schell, 2001; Otto et al., 2007; Shears, 2009). All cellular IPs are derived from soluble inositol trisphosphate (IP3), which is sequentially phosphorylated to generate different species that carry out specific functions in the cell (Hatch and York, 2010). For example, IP3 is a classical second messenger (Streb et al., 1983) and inositol hexakisphosphate (IP6) is an essential structural cofactor for the RNA-editing enzyme ADAR2 (Macbeth et al., 2005). Together, the cellular composition of distinctly phosphorylated IP molecules is termed the IP code; this code critically depends on the activity of IP kinases (Hatch and York, 2010; Otto et al., 2007).
To test whether IPMK and ITPK1 kinase activity is important for necroptosis, we expressed kinase-dead (KD) mutant alleles of the kinases in HT-29 ΔIPMK and ΔITPK1. Cells expressing IPMK(KD) and ITPK1(KD) were unable to execute necroptosis, indicating a crucial requirement for IP kinase activity (Figures 5A and B). In addition to its well-described role as an IP kinase, a physiologic phosphoinositide (PI) 3-kinase activity has also been reported for IPMK (Maag et al., 2011). Thus, to clarify whether the IP kinase function of this enzyme is specifically required for necroptosis, we tested an Arabidopsis thaliana IPMK orthologue IPK2β (At-IPKβ) that has conserved IP kinase activity but lacks PI3-kinase activity (Maag et al., 2011). Importantly, selective complementation of the IP pathway by At-IPK2β fully rescued the IPMK mutant (Figure 5A). These observations strongly suggest that the phosphorylation of soluble inositol metabolites via IP-specific kinase activity is important for MLKL-induced necroptosis.
Figure 5. Inositol phosphate kinase activity is essential for necroptosis.

(A, B) ΔIPMK stably-expressing KD IPMK (D144N/K146A) or A. thaliana IPMK orthologue IPK2β or ΔITPK1 stably-expressing KD ITPK1 (D295A) were treated with TSZ for 24 h and cell viability was measured by cellular ATP assay (mean ± SD of n = 3 replicates; Data shown are representative of three independent experiments). KD, kinase-dead. (C) Inositol phosphate profiles of wild-type and IP kinase mutant HT-29 were determined by anion-exchange HPLC of extracts derived from myo- [3H]-inositol-labeled cells. See also Table S3.
To empirically determine how genetic mutation of IPMK and ITPK1 influences the phosphorylation of IPs in our human cell mutants, we used HPLC to examine extracts from cells cultured with radiolabeled inositol. Compared to wild-type HT-29, both ΔIPMK and ΔAITPK1 had comparable levels of IP3 and IP4 but were noticeably deficient in IP5 and IP6. Complementation with the wild-type genes or the A. thaliana IPMK orthologue, but not KD mutants, restored production of IP5 and IP6 (Figure 5C and Table S3). These observations indicate that IPMK and ITPK1 are both required to produce highly phosphorylated IPs from lowly phosphorylated precursors in these cells. Our metabolic data are consistent with important functions of the human enzymes in the cellular synthesis route(s) from IP3 to IP5. Taken together, these data establish a previously unrecognized link between IP metabolism and necroptosis and suggest, in particular, a role for highly phosphorylated IPs.
MLKL directly binds a highly phosphorylated inositol product
We previously employed certain lowly phosphorylated inositol molecules in a study of MLKL-membrane interactions. We used IP3 as a soluble proxy for the polar head group of the phospholipid PIP2, showing that MLKL can directly bind IP3 (Quarato et al., 2016). However, at that time, the relevance of MLKL interaction with soluble inositol molecules was unclear. In light of our current observations suggesting an important role of highly phosphorylated IPs in necroptosis, we hypothesized that these molecules could directly bind MLKL and modulate its function. To test whether MLKL can bind highly phosphorylated IPs, as demonstrated previously for lowly phosphorylated IPs, we first employed an affinity purification strategy. We expressed and purified Flag-tagged wild-type hMLKL from unstimulated human cells (Figure 6A) and synthesized an inositol hexakisphosphate (IP6) affinity reagent as reported (Figure 6B) (Wu et al., 2016). In pull down experiments, we found that purified MLKL specifically bound the IP6 affinity reagent but not a phosphate control reagent (Figure 6C), suggesting that MLKL directly binds IP6. We next asked whether the binding of MLKL to IP6 is altered upon phosphorylation of MLKL by RIPK3. To address this question, we treated HT-29 cells to stimulate TNF-induced necroptosis and incubated these cell lysates (and untreated controls) with our IP6 affinity probe. Our results show that endogenous MLKL bound to the IP6 affinity reagent specifically compared to the phosphate control, and that binding occurred whether MLKL is phosphorylated at S358 or not (Figure 6D). Together, these data suggested that highly phosphorylated inositol products of the IP kinases could control necroptosis by directly binding MLKL and regulating its function.
Figure 6. Inositol hexakisphosphate (IP6) directly binds MLKL and active phospho-MLKL.

(A) Flag-tagged MLKL was expressed and purified from human HAP1 cell lysates under native conditions and resolved using denaturing SDS-PAGE and Coomassie Brilliant Blue protein stain. (B) Molecular structures of the IP6 reagent and phosphate control reagent (ctrl) synthesized for use in affinity pull down experiments. (C) Affinity reagents were used for pull-downs with purified MLKL (panel A) and analyzed by Western blotting. (D) HT-29 cells were untreated or treated with TSZ for 6 h to induce MLKL phosphorylation and initiate necroptosis. Pull downs using the IP6 affinity reagent or phosphate control reagent (ctrl) were performed from cell lysates and analyzed by Western blotting. GAPDH is a loading marker for the input samples and a control for non-specific protein binding to the affinity reagent.
IP6 dissociates the auto-inhibitory region from the N-terminal bundle of MLKL
The MLKL N-terminal domain consists of a helical bundle and brace (Murphy et al., 2013). It has membrane disrupting activity in vitro and can induce necroptosis in cells when expressed independently of the C-terminal domain (Dondelinger et al., 2014; Hildebrand et al., 2014; Su et al., 2014; Wang et al., 2014), consistent with a critical executioner role for this domain. In our previous study, we showed that IP3 interacts with the N-terminal bundle and brace (NBB) through basic, positively charged residues, using an NMR-based structural approach (Quarato et al., 2016). Here we pursued a similar approach to explore how MLKL binding to IP6 controls its activation. We hypothesized that IP6, similarly to IP3, might engage with electropositive regions within NBB. We assigned the backbone amides of a minimal, well-folded construct expressed in E. coli (NBB149) and confirmed its helical secondary structure from the Cα chemical shift deviations from random coil (Figures 7A and S4A). We then measured changes in the spectra of 15N-NBB149 in the presence of IP6 or IP3 (Figures 7B and S4B, C). IP6 induced multiple significant changes in the fast exchange NMR regime (traceable chemical shift perturbations [CSP]), indicating direct binding of NBB149 to IP6. Notably, IP6 also induced widespread CSPs in the intermediate exchange NMR regime (broadening of resonances), consistent with a robust IP6-NBB interaction (Figure 7C). In contrast, the CSPs induced by IP3 were less widespread, much weaker on average (∼0.02 ppm compared to ∼0.05 ppm), and did not occur within the intermediate exchange NMR regime, suggesting generally less dramatic effects of IP3 binding (Figure S4D). To further characterize the binding landscape of IP6, we mapped IP6-induced CSPs on the backbone amides of residues 1-149 in the three-dimensional structure of human NBB (Figure 7D). IP6-responsive regions clustered into three surface-exposed interfaces, including electropositive residues within helices α1, α2, α3, α5 and the loop between α2 and α3 (interface 1), α2, α6 and the loop between α1 and α2 (interface 2), and α4, α6 and the loop between α4 and α5 (interface 3). These interfaces encompassed and extended well beyond IP3-responsive regions, which were arranged mainly within α2 and α6 (Figure S4E). Overall, its striking engagement across multiple interfaces suggested that IP6 could induce important structural rearrangements within the MLKL N-terminal domain, and that this molecule may mediate different effects than its lowly phosphorylated precursor, IP3.
Figure 7.
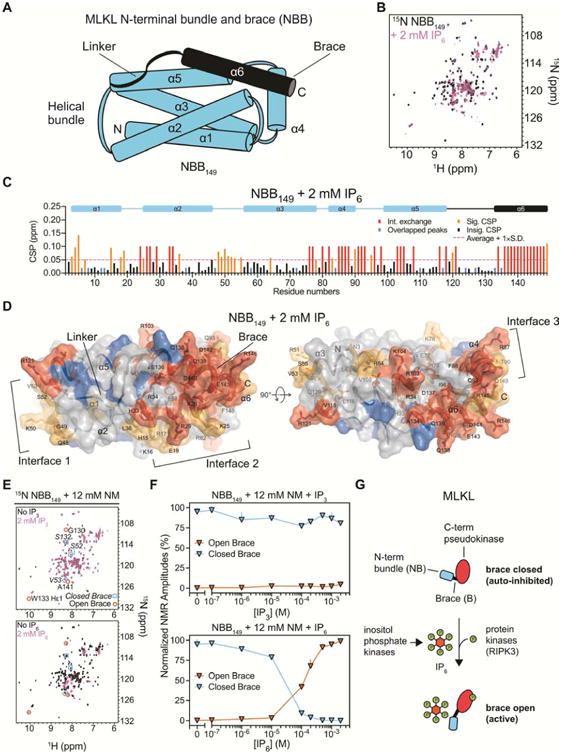
IP6, but not IP3, potently displaces the N-terminal auto-inhibitory brace of MLKL. (A) Diagram of the MLKL N-terminal domain helical structure. (B) Superimposed 1H-15N SOFAST-HMQC nuclear magnetic resonance (NMR) spectra of the MLKL N-terminal bundle and brace (NBB149) in the absence (black) or presence of 2 mM IP6 (pink). (C) Chemical shift perturbation (CSP) induced by IP6 for each backbone amide of NBB149 were calculated with the loss of approximately one third of NBB149 peaks, indicating binding in the intermediate exchange NMR regime. Intermediate CSPs (red) were assigned the value of 0.1 ppm. Overlapping backbone amide resonances (blue) were assigned 0.02 ppm in this and panel D. Significant CSPs (orange) were defined at a threshold of the average CSP plus one standard deviation (dashed line) for the remaining residues. (D) CSPs were mapped onto the putty/surface structure of NBB residues 1-149, which is color-coded as in panel C with thickness of the putty proportional to the CSPs. Surface-exposed interfaces that interact with IP6 are indicated with brackets. See also Figure S4 for the equivalent analysis with IP3. (E) Superimposed NMR spectra of NBB149 with (pink) or without (black) IP3 and IP6 in the presence of nonyl maltoside (NM) detergent. Three NMR resonances associated with the closed or open conformations of the inhibitory region are indicated in squares and circles, respectively. (F) Normalized amplitudes of the resonances highlighted in panel E are plotted over a titration of IP3 (top) and IP6 (bottom) (mean ± SD). See Figure S5 for complete corresponding spectra. (G) Model for combined activation of MLKL by RIPK3-mediated phosphorylation of the C-terminal pseudokinase domain and IP6-mediated displacement of the N-terminal auto-inhibitory brace.
Studies by our group and others have implicated the release of an auto-inhibitory region, containing the linker and brace helix α6 of the N-terminal domain, in the activation of MLKL (Davies et al., 2018; Murphy et al., 2013; Quarato et al., 2016; Su et al., 2014). We therefore sought to determine how binding of IP6 and IP3 may structurally affect this region, using a previously established assay which monitors brace displacement and unfolding from a closed, soluble state to an open, disordered state by NMR (Quarato et al., 2016). Remarkably, we found that IP6, but not IP3, potently displaced the MLKL auto-inhibitory region in the presence of an enabling detergent (Figure 7E, F, and Figure S5). Titration analysis revealed that the transition between apo and brace-displaced forms of NBB149 occurred at concentrations between 10–200 μM IP6. In contrast, even at 2 mM concentration, IP3 was unable to displace the brace in this assay. We therefore propose that highly phosphorylated IPs such as IP6 act as selective molecular switches able to destabilize the inhibitory region of MLKL. In particular, the electronegative charges of highly phosphorylated inositol species could have the unique capacity to disrupt electrostatic interactions that stabilize NBB149 in a dormant, inactive state. Our results suggest that by binding MLKL and displacing the auto-inhibitory region, IP6, but not IP3, can promote an active conformation of MLKL that allows it to unleash necroptosis. These data are consistent with our observations that production of highly phosphorylated IPs through IP kinase activity is required for MLKL necroptotic function in the cell.
Discussion
We conclude that inositol phosphate metabolites perform a critical role in necroptosis. Our results suggest that phosphorylation of MLKL by RIPK3, a hallmark of commitment to the necroptotic program (Cai et al., 2014; Dondelinger et al., 2014; Hildebrand et al., 2014; Murphy et al., 2013; Quarato et al., 2016; Su et al., 2014; Sun et al., 2012; Wang et al., 2014; Zhao et al., 2012), is insufficient for execution of necroptosis in the human cell. Indeed, our data reveal that highly phosphorylated IPs are an additional, crucial requirement for MLKL death effector function acting directly on MLKL (Figure 7G). These observations challenge the model that MLKL oligomerization occurs autonomously following a RIPK3-induced phosphorylation switch mechanism. Our findings may also have practical consequences for the study of necroptosis in disease. For example, IP levels could be an important consideration, even in tissues expressing established necroptotic markers (e.g. phospho-MLKL).
The soluble IP messengers perform key functions in a number of fundamental cellular processes including translation, transcription, and phosphate metabolism (Hatch and York, 2010; Irvine and Schell, 2001; Otto et al., 2007; Shears, 2009). Our work identifies an unexpected, essential role for the IP code in the necroptotic response to cytokines or infection with pathogens. An interesting possibility arising from this work is that the IP code, analogously to essential components acting upstream (e.g. RIP kinases), may be dysregulated during infection or inflammatory diseases. Indeed, some reports have indicated that pathogens can manipulate the IP code (Minisini et al., 2003; Ruschkowski et al., 1992; Zhou et al., 2001), although the consequences of such dysregulation should be further explored.
Informed by reconstituted in vitro systems and structural analysis, a model has emerged in which the brace region of MLKL performs an inhibitory function to keep the N-terminal domain in check (Davies et al., 2018; Murphy et al., 2013; Quarato et al., 2016; Su et al., 2014). Disruption of the interactions between the inhibitory region and the N-terminal helix bundle results in activation of MLKL to execute plasma membrane rupture (Quarato et al., 2016; Su et al., 2014). The brace has also been implicated in influencing the oligomerization status of MLKL, either by directly contributing oligomeric contacts (Quarato et al., 2016) or by allowing oligomeric assembly of the N-terminal bundle after its release during membrane insertion (Su et al., 2014). Our results provide important structural insights into the activation mechanisms of MLKL by showing that highly phosphorylated, soluble IPs can directly destabilize the inhibitory region, consistent with the requirement of these molecules for the downstream activation of MLKL oligomerization and membrane recruitment in cells. Together, our results support a model in which MLKL activation requires not only RIPK3-dependent phosphorylation of the C-terminal domain but also binding of highly phosphorylated IPs to the N-terminal domain (Figure 7G).
In addition to the well-studied role of IP second messengers (Hatch and York, 2010; Irvine and Schell, 2001), IPs can act as structural cofactors that stabilize proteins and protein complexes. For example, inositol tetrakisphosphate (IP4) acts as “intermolecular glue” for binding of the histone deacetylase HDAC3 to its corepressor (Watson et al., 2012) and IP6 binds the tetrameric TRPA1 receptor at a region important for stability of its oligomeric contacts (Paulsen et al., 2015). One possibility is that MLKL may similarly depend on binding of IPs to maintain its oligomeric status during necroptosis. Additionally, while our results indicate an important role for highly phosphorylated IPs as activators of MLKL function, they do not exclude a potential role for other IPs. Indeed, this study confirms that MLKL can bind to IP3, as reported (Quarato et al., 2016). In principle, different IP species could exert combinatorial control of MLKL activity by competing for binding sites or influencing other contacts.
It is interesting to consider our findings in the context of the contribution of phospholipids in necroptosis. These membrane-associated phosphatidylinositol phosphate (PIP) lipids mediate MLKL membrane localization and permeabilization of reconstituted membranes. MLKL binds to certain PIPs at least in part via recognition of their IP polar head group, and membrane PIP constituency may be an important determinant of selective targeting of MLKL to cellular membranes (Dondelinger et al., 2014; Quarato et al., 2016; Wang et al., 2014). We note, however, that PIP phosphorylation is regulated by PIP kinases (e.g. PI3K) that are distinct from the IP kinases identified in our genetic screen. Importantly, our results demonstrate that necroptosis specifically requires the IP kinase activity of IPMK and ITPK1. Our data indicate a critical role of soluble IPs in MLKL oligomerization preceding membrane recruitment mediated by PIP lipids. Moreover, we show that IP6 is a bonafide soluble activator that does not require phospholipids to mediate structural rearrangements in MLKL. Nevertheless, it is intriguing that IP6 can bind to regions known to mediate interactions with PIPs (Dondelinger et al., 2014; Quarato et al., 2016), suggesting that these regions could serve as shared binding sites. Engagement of IPs and PIPs may occur at distinct stages of MLKL activation, promoting MLKL oligomerization, membrane recruitment, and downstream effector function during necroptosis. Future studies are needed to dissect the precise dynamics of these regulatory mechanisms.
An unbiased proteomic analysis of the IP6 interactome in yeast revealed a strong enrichment for phosphoproteins, suggesting important regulatory roles for highly phosphorylated IPs in signaling pathways that rely on protein kinases (Wu et al., 2016). Our study highlights necroptosis as one such pathway in human cells that is regulated by both direct protein phosphorylation (dependent on RIPK3) and interaction with highly phosphorylated IPs (produced by IP kinases). It will be fascinating to investigate how MLKL regulation by IPs has evolved, especially considering species-specific differences in MLKL (Tanzer et al., 2016). Notably, there are significant differences between the human and mouse MLKL in the regulatory brace region (Davies et al., 2018; Murphy et al., 2013), which may impact these mechanisms. Classically, cellular IP metabolism has been studied using IP kinase knockout model organisms (e.g., yeast, flies, and mice) (Hatch and York, 2010; Irvine and Schell, 2001) but not with human cell knockouts. For example, a study using stem cells from IPMK knockout mouse embryos showed lowered but detectable production of IP6 (Frederick et al., 2005), indicating limited IPMK-independent synthesis in mouse. Additional investigations of IP kinases in different vertebrates are needed to explore the evolutionary relationship of IPs and MLKL. Nonetheless, our study provides an extensive analysis of IP-mediated control of necroptosis in human cells, beginning with an unbiased genetic screen in human cell knockouts. This work reveals a critical layer of regulation of necroptosis that may have implications in inflammatory disease and infection and could be exploited for therapeutic intervention.
Star Methods
Contact For Reagent And Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jan Carette (carette@stanford.edu).
Experimental Model and Subject Details
Cell culture
HAP1 (sex: male; source: Thijn Brummelkamp laboratory), HT-29 (sex: female; source: William Kaiser laboratory), and FADD-deficient Jurkat clone I 2.1 (sex: male; source: ATCC CRL-2572) were cultured at 37 °C in 5% CO 2 in IMDM, DMEM, and RPMI-1640 (Hyclone), respectively. Media was supplemented with 10% fetal bovine serum and L-glutamine/penicillin/streptomycin (Sigma Aldrich). Cell lines were routinely inspected to visually confirm morphological and growth characteristics indicative of that cell line. HAP1 were routinely tested for ploidy by DNA content analysis of propidium iodide-stained nuclei. All cell lines were confirmed to be mycoplasma negative by PCR testing. No additional cell line authentication was performed.
XL10-Gold E. coli
Ligation-independent cloning (LIC) was carried out using XL10-Gold competent cells (Agilent Technologies) transformed according to the manufacturer's or published protocols, respectively, and grown on LB medium and LB-agar plates at 37 °C overnight (Savitsky et al., 2010).
BL21 Star (DE3) E. coli
For protein expression, BL21 Star (DE3) E. coli (Thermo Fisher Scientific) containing NBB149 in the pNIC28-Bsa4 LIC vector was grown in LB medium (Thermo Fisher Scientific) supplemented with 50 mg/L kanamycin (Gold Biotechnology) to an OD600 of 0.8-1.2 before induction with 0.42 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) (Gold Biotechnology) at 18-22 °C overnight. For iso tope labeling, WT NBB149-expressing E. coli was grown in MOPS-based minimal media supplemented with 15NH4Cl and either 13C-glucose or unlabeled glucose (Neidhardt et al., 1974) under similar growth and induction conditions.
Method Details
Reagents and antibodies
Recombinant human TNF-α (Invivogen, rhtnf-a), Smac mimetic RMT5265 (also referred to as SMAC007) (Li et al., 2004), and zVAD (Z-VAD-FMK, Santa Cruz, sc-311561) (TSZ) were used at 10 ng/mL, 10 nM, and 20 μM, respectively. HSV-1 infections were performed at MOI = 5. Doxycycline (DOX) was used between 100 ng/mL and 500 ng/mL, as specified for each experiment. The RIPK3 inhibitor GSK840 (GlaxoSmithKline) was used at 3 μM and the MLKL inhibitor NSA (Calbiochem, #480073) was used at 0.2 μM. The following antibodies were used: Anti-FLAG M2-HRP (Sigma, A8592), Anti-MLKL (Abcam, ab184718), Anti-MLKL phospho S358 (Abcam, ab187091), Anti-Myc (Sigma, M4439), Anti-ITPK1 (GeneTex, GTX107974), Anti-HSV-1 ICP0 (Santa Cruz, 11060), Anti-GAPDH (Santa Cruz, sc-32233), Anti-IMMT (Protein Tech, 10179-1-AP), and Anti-Actin (Sigma, A2066).
Cell viability measurements
HAP1 and HT-29 cells were seeded at 20,000 or 5,000 cells per well, respectively, in 96-well tissue culture plates (Greiner Bio-One) the day before the experiment. Jurkat cells were seeded at 200,000 cells/mL at the start of each experiment. Following necroptosis induction, cell viability was assayed using the CellTiter-Glo assay (Promega) according to the manufacturer's recommendations. Viability data are presented as a percentage compared to untreated control cells. Live cell imaging experiments were performed on black clear-bottom 96-well plates (Corning, 3904) in the presence of 20 nM Sytox Green (ThermoFisher, S7020) by IncuCyte ZOOM imaging (Essen Bioscience). Imaging analysis was performed using a custom analysis routine that applied the following settings to detect green fluorescent objects: minimum size: 20 μm2, maximum size: 750 μm2, maximum eccentricity: 0.9, exposure time: 400 ms. Flow cytometry was conducted on DAPI-stained cells using the BD LSRFortessa flow cytometer with BD FACSDiva software. All experiments were performed three times with n = 3 biological replicates each, with a representative experiment shown.
HSV-1 infections
Wild-type, ΔIPMK, and ΔIPMK complemented HT-29 were stably transduced with a constitutive DAI expression vector (pQCXIH-hDAI) or empty vector (EV) control (pQCXIH). Infections were performed with either HSV-1 F strain (Li et al., 2011) or HSV-1 FmutRHIM (Huang et al., 2015) strain at MOI = 5. After one hour of infection, virus-containing medium was replaced with fresh medium, and cell viability was measured at 24 h post-infection using the CellTiter-Glo assay (Promega) according to the manufacturer's recommendations.
Haploid genetic screen
Inducible MLKL cell line
The haploid cell line HAP1 Tet-On-MLKLTE/SD was generated as follows: N-terminallyFLAG-tagged human MLKL phosphomimetic mutant T357E/S358D was Gibson cloned (NEB) into the pLenti CMVTRE3G Puro DEST (w811-1) inducible expression system (gift of Eric Campeau, Addgene #27565) using the EcoRV site. The forward and reverse primers CAGTGTGGTGGAATTCTGCAGATACCATGGATTACAAGGATGACGAC and GCGGCCGCCACTGTGCTGGATCTACTTAGAAAAGGTGGAGAGTTTC were used for overlap extension PCR in combination with the mutagenic primers GAGTTGAGGAAAACACAGGAGGACATGAGTTTGGGAACTACGAG and CTCGTAGTTCCCAAACTCATGTCCTCCTGTGTTTTCCTCAACTC to introduce the T357E/S358D mutation. The cloned construct (pCD603) was introduced into HAP1 stably expressing the reverse tetracycline repressor-VP16 transactivator fusion protein (pLenti CMV rtTA3G Blast (R980-M38-658), gift of Dominic Esposito, Addgene #31797) using lentiviral delivery. Stable transductants were selected with puromycin and single-cell sorted to isolate the HAP1 Tet-On-MLKLTE/SD clonal cell line.
Gene trap mutagenesis
One hundred million HAP1 Tet-On-MLKLTE/SD cells were mutagenized with the GFP gene trap retrovirus, as described (Carette et al., 2011a). Five T-175 flasks of 70-80% confluent 293FT cells were co-transfected with pgt-GFP-0, pgt-GFP-1, and pgt-GFP-2 (Carette et al., 2009) (2.2 μg of each vector per flask) and standard retroviral packaging plasmids. The viral supernatant was harvested in the morning and evening two days post-transfection, with care taken not to disturb the monolayer. After filtration through a 0.45 μm filter, the viral supernatant was concentrated by centrifugation in an Optima L-90K Ultracentrifuge (Beckman Coulter) at 23,000 RPM for 1.5 h at 4 °C. The day before the transduction, 60 million HAP1 Tet-On-MLKLTE/SD were seeded (30 million cells per flask in two T175 flasks). The concentrated supernatant was diluted in 50 mL complete HAP1 growth medium containing 8 μg/mL protamine sulphate. The day of the transduction, half of the viral medium was added to the flasks containing approximately 120 million cells in the morning and replaced with the remaining viral medium in the evening. Two days following transduction, the occurrence of GFP-positive gene trap mutants was confirmed by flow cytometry. Cells were pooled and expanded over the five days following transduction without discarding any cells, to maintain diversity. (Note: At this point, the cells can be frozen in cryovials for later use, although the experiments described below were conducted with the freshly prepared library.)
Genetic screen and sequencing
Seven days after transduction, the HAP1 Tet-On-MLKLTE/SD library was seeded at 180 million total cells (30 million cells per flask in six flasks). The next day, the selection was begun by enforcing MLKLTE/SD expression under 200 ng/mL DOX. Selection was continued over five days and, subsequently, DOX was withdrawn and surviving colonies were allowed to grow out for another four days. Colonies were then pooled together and genomic DNA was extracted from the pool (Qiagen, Cat# 51306). Gene trap insertion sites were amplified using a linear amplification (LAM) PCR protocol as described (Carette et al., 2011a). Briefly, genomic DNA was digested separately with either MseI or SpeI and then combined in equal parts. The LAM-PCR reaction was conducted on the digested template DNA with a 5′-biotinylated packaging-signal specific primer (3LTRflanking-biot1: GGTCTCCAAATCTCGGTGGAAC) to elongate through the adjacent proviral LTR to the flanking genomic sequence. Seven replicates were conducted in addition to a negative control sample containing no gene trap insertions. LAM PCR products were purified with streptavidin-coated beads (Dynal kilobaseBINDER Kit, Thermo Fisher). Next, a 5′ phosphorylated and 3′ Dideoxy-C DNA linker (TCGTATGCCGTCTTCTGCTTGACTCAGTAGTTGTGCGATGGATTGATG) was ligated to the 3′ ends of the purified LAM PCR products using CircLigase II (EPICENTRE). The ligated fragments were then PCR amplified using the primers LTRSolexaI (AATGATACGGCGACCACCGAGATCTGATGGTTCTCTAGCTTGCC) and Solexa adaptor II (CAAGCAGAAGACGGCATACGA) and AccuPrime Taq DNA Polymerase, High Fidelity (Invitrogen). The PCR products were then pooled, sequenced using the Genome Analyzer II (Illumina) with the primer SolexaSeqFlank (CTAGCTTGCCAAACCTACAGGTGGGGTCTTTCA), and aligned to the human genome. Finally, inactivating insertion events in the selected sample dataset were compared with the unselected control dataset to calculate an enrichment score for each gene. A P-value for enrichment (corrected for false-discovery rate) was calculated using the one-sided Fisher exact test, as described (Carette et al., 2011a). See Table S1 for a complete list of genes identified in the screen.
Generation of mutants
CRISPR mutant cell lines were generated using pSpCas9(BB)-2A-GFP (PX458), a gift from Feng Zhang (Addgene #48138). Specific sgRNA sequences for IPMK (GCCCGGCCACCTGATGCGAG) and ITPK1 (GAACCTTAGCCGGCCGATCG) were designed using the tool at http://crispr.mit.edu/ and cloned into PX458 (IPMK CRISPR construct: pCD623; ITPK1 CRISPR construct: pCD624). Following transfection with X-tremeGENE HP reagent (Roche) for HAP1 or Lipofectamine LTX reagent (ThermoFisher) for HT-29, positively transfected cells were single-cell sorted based on the transiently-expressed GFP marker using a BD Influx cell sorter. The presence of single colonies was visually confirmed several weeks after sorting. Jurkat cells were transfected using the Amaxa Cell Line Nucleofector Kit V, following the manufacturer's instructions (including the recommended centrifugation speed of 90 × g) before single- cell sorting as above. Both HT-29 and Jurkat were cultured in sterile-filtered, conditioned media for optimal single-cell outgrowth.
Mutant clones were confirmed by PCR sequencing from genomic DNA prior to phenotypic analysis, as described (Marceau et al., 2016). See Table S2 for a complete list of sequences. Briefly, an approximately 500 bp genomic region encompassing the CRISPR sgRNA target site was PCR amplified and sequenced by Sanger sequencing. We note that in HT-29, we routinely sequenced multiple non-identical mutations at the target site in a single clone. This finding was consistent with CRISPR-based gene editing of other aneuploid cell lines (Marceau et al., 2016). We also sequenced homozygous identical mutations in a single clone, as has also been reported with this approach (Canver et al., 2014; Horii et al., 2014). Clones were selected in which only frame-shift or large indels occurred at the sgRNA target site. Analysis of indel sequences was aided by CRISP-ID software (Dehairs et al., 2016), following the designer's recommendations.
For MLKL knockdown experiments, pLKO.1-hMLKL (Zhao et al., 2012) or the pLKO.1 control scramble shRNA expressing lentiviral vector were transfected along with psPAX2 and VSV-G-expressing plasmids into 293T cells. The virus-containing medium was harvested 48 hours later for transduction. HT-29 cells were transduced with the lentivirus and selected with 2 μg/ml puromycin (Invitrogen).
Genetic complementation of mutants
The cDNAs for N-terminally Myc-tagged human IPMK (GenBank: BC065709) and human ITPK1 (GenBank: BC018192) were cloned into retroviral expression plasmid pMXs-IRES-blasticidin (Cell Biolabs), and the stably-transduced population was selected with Blasticidin. Myc-tagged hIPMK was originally cloned into pENTR/D-TOPO (ThermoFisher) using primers CACCATGGAGCAGAAGCTGATCTCCGAGGAGGACCTGAAGGATCCGAGCTCGAAT TCCACCATGGCAACAGAGCCACCATC and GACTGCAACAGAGGATTCAATTGTC, and then cloned into pMXs-IRES-BLAST-DEST containing a Gateway destination cassette (Marceau et al., 2016) using the Gateway LR reaction (ThermoFisher) (complementation construct: pCD653). hITPK1 was cloned with a similar strategy using the primers CACCATGCAGACCTTTCTGAAAGGGAAG and CTACTGGGAGGAGGCCTTGGTG (complementation construct: pJD017). Kinase-dead(KD) mutations in IPMK (D144N/K146A) (Dubois et al., 2000; Odom et al., 2000;Wickramasinghe et al., 2013) and ITPK1 (D295A) (Miller et al., 2005; Qian et al., 2005)were cloned by overlap extension PCR and stably expressed using the same strategy. For IPMK (KD) (pJD012) mutagenic primers were GCCCTGTATAATGAATGTAGCCATAGGGCAAAAAAGCTATGATCC and GGATCATAGCTTTTTTGCCCTATGGCTACATTCATTATACAGGGC; For ITPK1 (KD) (pCD674), mutagenic primers were GGGCAGCACGCCGTCATTGCCATCAATGCCTTCCCAGGCTACGAG and CTCGTAGCCTGGGAAGGCATTGATGGCAATGACGGCGTGCTGCCC. The N-terminally Myc-tagged A. thaliana IPK2β construct (pMXs-IRES-blasticidin) was a gift from Solomon Snyder.
Inositol phosphate labeling
Prior to labeling, HT-29 cells were seeded at 100,000 cells per well in 6-well tissue culture dishes and starved in inositol-free medium for 24 hours. Cells were labeled with 37.5 μCi 3H inositol until nearly confluent, approximately five days of growth. The cells were then washed in PBS and IPs were extracted with 100μl of 0.5N HCl and stored at -80°C. The cell extracts were subsequently filtered through a .45 μm nylon filter to remove debris, loaded onto a Partisphere SAX 5 4.6mm × 125mm column and eluted with 1.7M Ammonium Phosphate pH 3.5 with the following gradient profile: 10mM for 5 min, a linear gradient for 65 min from 10mM to 1.7M, 1.7M for 30min. 1 mL fractions were collected and mixed with 6 mL scintillation fluid, and DPM was measured with a Liquid Scintillation Analyzer. Two independent experiments were performed and one representative experiment is shown.
Pull downs with the IP6 affinity reagent
To purify hMLKL from human cells, hMLKL was first overexpressed in HAP1 cells (stably tranduced with Tet-On-MLKL, pCD601) in the presence of DOX (100 ng/mL) over night and cell pellets were lysed in NP-40 lysis buffer (20 mM Tris pH 7.5, 250 mM NaCl, 0.5% NP-40, 1 mM EDTA, 1 mM DTT, 1× Halt protease inhibitor cocktail). hMLKL was purified from cell lysates using Flag (M2) magnetic beads (Sigma) according to the manufacturer's recommendations. Flag-tagged MLKL was eluted from the beads under native conditions in TBS pH 7.4 with 200 μg/mL recombinant 3×-Flag peptide (Sigma).
Synthesis of the IP6 affinity reagent was performed essentially as described (Wu et al., 2016). Briefly, all washes were performed with end-over-end rotation for 10 minutes at 4°C, followed by centrifugation at 5000 rpm for 5 minutes and aspiration of the supernatant by pipette. All steps were carried out at 4°C. A 0.5 mL suspension of Affi-Gel 15 resin was washed 3× 1 mL 0.01% Triton X-100 in ultrapure water. To the washed bead resin was added a 0.5 mL solution containing 1.5 mM desired amine-containing affinity reagent (1.5 μmol total phosphate control or IP6, synthesized according to Wu et al., 2016), and 100 mM HEPES pH 7.4, 0.01% (v/v) Triton X-100. Affinity solution was incubated overnight at 4°C with end-over end rotation. After incubation, bead solution was centrifuged, and the supernatant collected. Resin was washed 3× 1 mL in 0.01% Triton X-100, with each wash supernatant saved. Washes and reaction supernatant were combined and lyophilized for 31P NMR analysis. Unreacted amines on the resin were blocked with 1 mL 50 mM ethanolamine in ultrapure water, and incubated by end-over-end rotation at 4°C for 2 hours. Beads were washed 3× 1 mL in ultrapure water, and the supernatants discarded. Blocked resin was then resuspended in 2% sodium azide in ultrapure water to a final volume of 1 mL, for a final composition of 50% v/v slurry. Resin was stored at 4°C until use. (Note that it is important not to freeze the resin.)
Pull-down experiments were also performed essentially as described (Wu et al., 2016). For pull-down experiments starting with purified MLKL, 32 ng hMLKL was incubated with pre-washed 50 nmol IP6 affinity reagent (or phosphate control reagent) in 40 μL binding/wash buffer (20 mM HEPES pH 7.4, 125 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.05% Triton X100) while rotating at 4°C for two hours. The volume of resin was adjusted according to immobilization yield to ensure equimolar IP6 and phosphate control between experiments. The affinity resin was washed three times for five minutes while rotating at room temperature in binding/wash buffer, then eluted by boiling in SDS-PAGE sample buffer. For pull-down experiments from whole cell lysates, HT-29 cells were seeded the day before the experiment at 1 million cells per well in a 6-well dish. The day of the experiment, the cells were treated (or untreated) with TSZ for 6 h. The cells were then washed in PBS, scraped from the dish in ice-cold binding/wash buffer (supplemented with 1× Halt protease inhibitor cocktail and 20 mM β-glycerophosphate), lysed on ice by passing 10 times through a BD 26g1/2 needle, and centrifuged at 20,000 × g at 4°C for 10 min to remove cell debris. From 180 μL total clarified whole cell lysate, 80 μL of lysate was incubated with IP6 affinity reagent (50 nmol), 80 μL with phosphate control reagent (50 nmol), and 20 μL was saved for the input control. The binding, wash, and elution steps were performed as described above.
SDS-PAGE and cell fractionation
For routine Western blot analysis, HT-29 and HAP-1 cells were seeded the day before the experiment at 800,000 and 1.2-1.5 million cells per well, respectively, in 6-well dishes. Jurkat cells were resuspended at 500,000 cells/mL at the start of each experiment. Samples were prepared for denaturing, reducing SDS-PAGE by heating for 3 min at 98°C in Laemmli buffer (Bio-Rad, 1610737) in the presence of freshly prepared 10 mM dithiothreitol (DTT). For non-reducing SDS-PAGE experiments, samples were prepared in the same way in the absence of DTT.
Cell fractionation experiments were performed essentially as described (Wang et al., 2014). Briefly, HT-29 cells were seeded at 800,000 cells per well in a 6-well dish. Twelve hours after seeding, cells were treated with TSZ for 6 hours. Cell pellets were washed in PBS and lysed in 240 μL Triton X-114 lysis buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 2% Triton X-114 (Sigma) with Halt protease inhibitor cocktail (ThermoFisher, 78430) and 20 mM β-glycerophosphate) on ice for 30 min. Lysates were then phase separated into aqueous (AQ) and detergent-soluble fractions (DET) by heating to 30°C, centrifugation at 1,500 × g, and washing, as described (Wang et al., 2014).
Immunofluorescence microscopy
HT-29 cells were seeded onto micro cover glass (VWR) in 24-well plates (Greiner Bio-One) at 180,000 cells per well. Twelve hours after seeding, TSZ was added to appropriate wells, and samples were fixed 6 hours post-treatment with 4% paraformaldehyde (Sigma) for 30 min at RT. Anti-MLKL phospho S358 antibody (1:200) was added to each sample in immunofluorescence blocking buffer (PBS with 3% BSA, 1% saponin, and 1% Triton X-100) and incubated at 4°C overnight. After washing 5× with 1× phosphate buffered saline (PBS), Alexa488 anti-rabbit antibody (1:500) (Life Technologies), DAPI stain (1:300), and Alexa660 phalloidin (1:50) (Life Technologies) in immunofluorescence blocking buffer were added to each sample and incubated for 1 hour at RT. Samples were washed 5× with 1× PBS. Micro cover glasses were then mounted onto microscope slides (Fisher Scientific) with Vectashield (Vector Laboratories Inc). Images were taken with Zeiss LSM 700 confocal microscope and processed with Velocity software.
Forced MLKL oligomerization
The NBB140-2×FV-Flag construct was a gift from Douglas Green (Quarato et al., 2016).The construct was Gibson cloned into pLenti CMVTRE3G Puro DEST (w811-1) using the EcoRV site and primers AGTGTGGTGGAATTCTGCAGATACCATGGAAAATTTGAAGCATATTATCACCC and GCGGCCGCCACTGTGCTGGATTTATTTATCATCATCATCTTTATAATCCTCTCCGC. The construct was introduced into wild-type or ΔIPMK transactivator-positive HAP1 using lentiviral delivery, and a stably-transduced population was selected with puromycin (WT or ΔAIPMK Tet-On-NBB140-2×FV-Flag). Following pre-treatment with DOX (100 ng/mL) for 6 h to induce NBB140-2×FV-Flag fusion protein expression, oligomerization was forced using 50 nM B/B homodimerizer (DIM) (Clontech, 635059) in the absence of DOX. Cell death was monitored by live-cell imaging (IncuCyte ZOOM) and flow cytometry as described in the above section.
For crosslinking experiments, cells were seeded at 1.2 million cells per well in 6-well dishes (Greiner Bio-One). The following day, cells were treated with 100 ng/mL DOX for 2 hours to induce NBB140-2×FV-Flag expression, after which the cells were incubated for 6.5 hours in the absence of DOX. Cells were then treated with 50 nM DIM for 10 min, trypsinized, and washed in PBS. Crosslinking was performed with DSS crosslinking reagent (Thermo, 21658) according to the manufacturer's instructions. Briefly, samples were split into two tubes and either treated with 1.25 mM DSS or untreated for 15 min at 4°C. After quenching the re action in 20 mM Tris pH 7.4 for 15 min, samples were prepared in reducing Laemmli buffer for SDS-PAGE.
MLKL purification for NMR
Human MLKL residues 1-149 (NBB149) was expressed using the pNIC28-Bsa4 vector in BL21 Star (DE3) E. coli. In-house TEV protease was used to cleave the N-terminal His6-tag and protein was purified and stored as previously described (Quarato et al., 2016). Briefly, harvested cells were resuspended at 3 g/mL in 50 mM Tris pH 7.6, 250 mM NaCl, and 5-25 mM imidazole. Cells were passed through an Emulsiflex C3 three times at 17K psi for lysis (Avestin, Inc.) and clarified for 30 min at 33,000×g. Clarified lysates were incubated with nickel agarose resin (Gold Biotechnology) for 1 h with stirring on ice for batch binding. Gravity immobilized metal affinity chromatography used binding, wash, and elution buffers containing 25, 40, and 250 mM imidazole, respectively. Samples were concentrated in 3K molecular-weight cutoff centrifugal filters following digestion with TEV protease at <10:1 molar ratio for 24-36 hours (MilliporeSigma). Size-exclusion chromatography was performed using a HiPrep 16/60 Sephacryl S-100 HR column in 20 mM 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.0 or 20 mM sodium acetate pH 5.5 and 150 mM NaCl. MLKL-containing fractions were concentrated and resolved with 150 mM to 1 M NaCl gradient cation-exchange chromatography on a MonoS 5/50 GL column using an AKTA Pure (GE Healthcare). Samples were buffer exchanged by greater than 15-fold dilution three times in centrifugal filters to 20 mM sodium phosphate pH 6.8 before flash-freezing in liquid nitrogen and storage at -80 °C. Protein concentrations were calculated from extinction coefficients predicted from primary sequences and absorbance measured at 280 nm.
NMR titrations
Titrations of 50 μM human 15N-labeled NBB149 were done with inositol-1,4,5-trisphosphate (IP3) or inositol hexakisphosphate (IP6) in the presence of absence of n-nonyl-β-D-maltopyranoside (NM) using 150 mM HEPES buffer pH 6.8 supplemented with 2 mM deuterated dithiothreitol (dDTT), 2 mM ethylenediaminetetraacetic acid (EDTA), and 10% D2O at 298 K as previously described (Quarato et al., 2016). Chemical shift perturbations were measured in ppm as the root mean square of the 1H and 15N chemical shifts weighted as 1.0 and 0.2, respectively. The backbone of 13C/15N-labeled NBB149 in the absence or presence of 12 mM NM and 2 mM IP6 was assigned using the 15N-1H TROSY spectrum and data collected from the triple-resonance experiments HNCACB and CBCA(CO)NH at 303 K. The detergent-free 13C/15N-labeled NBB149 was measured at 350 μM in 20 mM sodium phosphate pH 6.8 supplemented with 10% D2O, 2 mM deuterated DTT, and 0.1% (w/v) sodium azide. The double-labeled sample supplemented with 12 mM NM and 2 mM IP6 was measured at 275 μM in 150 mM HEPES buffer pH 6.8 with added 2 mM dDTT, 2 mM EDTA, 10% D2O, and 0.1% (w/v) sodium azide. NMR data were collected on a Bruker Avance 600-MHz spectrometer equipped with 5-mm triple-resonance cyroprobe and single-axis pulsed-field gradient, processed in Topspin 3.5, and analyzed in CARA (Keller, 2004).
Comparative NMR titrations measuring soluble, closed brace and micellar, open brace NBB149 resonance amplitudes were performed as previously described with 1H-15N SOFAST-HMQC experiments (Quarato et al., 2016; Schanda et al., 2005). Three well-dispersed apo-NBB149 resonances normalized titrations across conditions. Three resonances assigned for the disordered linker-brace segment (122-149) were averaged as the normalized fraction of 100% micellar NBB149 in the presence of inositol polyphosphates and detergents.
Quantification and Statistical Analysis
Statistical information, including the number of replicates and independent experiments, is addressed specifically within the corresponding figure legend for each experiment when relevant. For cell viability and cell death experiments, all replicates were biological replicates conducted in separate wells. For experiments involving a student t-test, unpaired, two-tailed tests were performed. No statistical methods were used to predetermine sample size or determine whether the data met the assumptions of the statistical approach. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment, with the exception of the quantification of phospho-MLKL punctae (Figures 2F and H), for which the investigators were blinded to sample identity. In Figure 7, summary NMR data are plotted as means with error bars of standard deviation for three normalized NMR amplitudes as indicated.
Data and Software Availability
Raw deep sequencing data for the haploid genetic screen: https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6777/
The raw deep sequencing data for the haploid genetic screen have been deposited in the EMBL-EBI ArrayExpress database under ID code E-MTAB-6777.
Supplementary Material
Document S1: Figures S1–S5 and Tables S2 and S4
Table S1. Ranked List of Genes Identified in a Haploid Genetic Screen for Components of MLKL-Mediated Cell Death, Related to Figure 1. For each gene, insertion counts are listed for the DOX-treated, selected cells and the untreated, unselected control cells. The significance of enrichment (P-value) was determined using the one-sided Fisher exact test corrected for multiple hypothesis testing.
Table S3. Inositol Phosphate Labeling, Related to Figure 5. Inositol phosphate labeling data for HT-29 containing a summary of normalized peak integration for all inositol species detected, raw [3H]-detection values (DPM), and graphs for replicate experiments for each sample. In Figure 5C, the representative dataset “Experiment 2” was presented for each sample.
Highlights.
IP kinases are critical for necroptosis induced by TNF and viral infection
MLKL fails to oligomerize in IP kinase mutants despite phosphorylation by RIPK3
Necroptosis requires IP kinase activity to produce the IP code
IP6—but not IP3—displaces the MLKL auto-inhibitory brace region
Acknowledgments
We thank Jody Puglisi, Karim Majzoub, and Chris Baker for critical reading of the manuscript, Sirika Pillay, Alex Johnson, and Yaw Shin Ooi for discussions, Marcela Alcantara Hernandez for help with flow cytometry, Solomon Snyder for the gift of the A thaliana IPK2β expression vector, Douglas Green for sharing the NBB140-2×FV-Flag construct, and Meredith Weglarz of the Stanford FACS facility for help. This work was supported in part by grants from the NIH: T32AI007328 (C.M.D.), DP2AI104557 (J.E.C.), 5T32GM007347 (A.T.H.), R01AI020211 (to E.S.M.), 1R01GM122923 (to S.J.D.), and R01GM124404 (to J.D.Y.). Further support was provided by The David and Lucile Packard Foundation (J.E.C.), by the St. Jude Academic Programs Office Special Postdoctoral Fellowship (D.E.M.) and American Lebanese Syrian Associated Charities (T.M.).
Footnotes
Author Contributions: C.M.D. and J.E.C. were responsible for overall design of the study. C.M.D. conducted the screen, generated the mutant cell lines, performed necroptosis experiments, conducted cellular fractionation, MLKL phosphorylation, and oligomerization studies, and performed protein purification and pull down experiments. C.M.D and J.D. generated complemented cell lines and performed necroptosis experiments in HAP1. J.D. performed immunofluorescence microscopy experiments. B.P.C. and A.T.H. performed inositol phosphate labeling and analysis. D.E.M., T.M., and C.R.G. performed NMR experiments and analysis. H.G. performed infections with HSV-1 and contributed the DAI expression and MLKL knock down cell lines. N.B. and D.F. synthesized the IP6 affinity reagent. J.Y.C. and S.J.D. contributed expertise in kinetic analysis of cell death. P.J.G. and J.B. contributed the Smac mimetic compound. E.S.M. and W.J.K. contributed expertise in design of the screen and necroptosis experiments. T.M. contributed expertise in design of NMR experiments. J.D.Y. contributed expertise in design of inositol phosphate biochemical experiments. C.M.D. and J.E.C. wrote the manuscript with input from all authors.
Declaration of Interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT, Spangenberg EE, Green KN, et al. Necroptosis activation in Alzheimer's disease. Nat Neurosci. 2017;20:1236–1246. doi: 10.1038/nn.4608. [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver MC, Bauer DE, Dass A, Yien YY, Chung J, Masuda T, Maeda T, Paw BH, Orkin SH. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 2014;289:21312–21324. doi: 10.1074/jbc.M114.564625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- Carette JE, Guimaraes CP, Wuethrich I, Blomen VA, Varadarajan M, Sun C, Bell G, Yuan B, Muellner MK, Nijman SM, et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat Biotechnol. 2011a;29:542–546. doi: 10.1038/nbt.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011b;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KA, Tanzer MC, Griffin MDW, Mok YF, Young SN, Qin R, Petrie EJ, Czabotar PE, Silke J, Murphy JM. The brace helices of MLKL mediate interdomain communication and oligomerisation to regulate cell death by necroptosis. Cell Death Differ. 2018 doi: 10.1038/s41418-018-0061-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehairs J, Talebi A, Cherifi Y, Swinnen JV. CRISP-ID: decoding CRISPR mediated indels by Sanger sequencing. Sci Rep. 2016;6:28973. doi: 10.1038/srep28973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–981. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- Dubois E, Dewaste V, Erneux C, Messenguy F. Inositol polyphosphate kinase activity of Arg82/ArgRIII is not required for the regulation of the arginine metabolism in yeast. FEBS Lett. 2000;486:300–304. doi: 10.1016/s0014-5793(00)02318-8. [DOI] [PubMed] [Google Scholar]
- Fiers W, Beyaert R, Declercq W, Vandenabeele P. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene. 1999;18:7719–7730. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- Frederick JP, Mattiske D, Wofford JA, Megosh LC, Drake LY, Chiou ST, Hogan BL, York JD. An essential role for an inositol polyphosphate multikinase, Ipk2, in mouse embryogenesis and second messenger production. Proc Natl Acad Sci U S A. 2005;102:8454–8459. doi: 10.1073/pnas.0503706102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, Dube PH, Bergman MA, Orihuela CJ. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015;11:e1005337. doi: 10.1371/journal.ppat.1005337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch AJ, York JD. SnapShot: Inositol phosphates. Cell. 2010;143:1030–1030 e1031. doi: 10.1016/j.cell.2010.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RC, Webb AI, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;111:15072–15077. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horii T, Arai Y, Yamazaki M, Morita S, Kimura M, Itoh M, Abe Y, Hatada I. Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci Rep. 2014;4:4513. doi: 10.1038/srep04513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, Wu J, Zhuang Q, Chen C, Li J, et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe. 2015;17:229–242. doi: 10.1016/j.chom.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Irvine RF, Schell MJ. Back in the water: the return of the inositol phosphates. Nat Rev Mol Cell Biol. 2001;2:327–338. doi: 10.1038/35073015. [DOI] [PubMed] [Google Scholar]
- Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603–608. doi: 10.1126/science.aaf6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R. The computer aided resonance assignment tutorial. Goldau, Switzerland: CANTINA Verlag; 2004. [Google Scholar]
- Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, Wachtel S, Bueno S, Prince A. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015;11:e1004820. doi: 10.1371/journal.ppat.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305:1471–1474. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang S, Zhu H, Zheng C. Cloning of the herpes simplex virus type 1 genome as a novel luciferase-tagged infectious bacterial artificial chromosome. Arch Virol. 2011;156:2267–2272. doi: 10.1007/s00705-011-1094-9. [DOI] [PubMed] [Google Scholar]
- Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maag D, Maxwell MJ, Hardesty DA, Boucher KL, Choudhari N, Hanno AG, Ma JF, Snowman AS, Pietropaoli JW, Xu R, et al. Inositol polyphosphate multikinase is a physiologic PI3-kinase that activates Akt/PKB. Proc Natl Acad Sci U S A. 2011;108:1391–1396. doi: 10.1073/pnas.1017831108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature. 2016;535:159–163. doi: 10.1038/nature18631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GJ, Wilson MP, Majerus PW, Hurley JH. Specificity determinants in inositol polyphosphate synthesis: crystal structure of inositol 1,3,4-trisphosphate 5/6-kinase. Mol Cell. 2005;18:201–212. doi: 10.1016/j.molcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Minisini R, Tulone C, Luske A, Michel D, Mertens T, Gierschik P, Moepps B. Constitutive inositol phosphate formation in cytomegalovirus-infected human fibroblasts is due to expression of the chemokine receptor homologue pUS28. J Virol. 2003;77:4489–4501. doi: 10.1128/JVI.77.8.4489-4501.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarski ES, Guo H, Kaiser WJ. Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology. 2015;479-480:160–166. doi: 10.1016/j.virol.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom AR, Stahlberg A, Wente SR, York JD. A role for nuclear inositol 1,4,5-trisphosphate kinase in transcriptional control. Science. 2000;287:2026–2029. doi: 10.1126/science.287.5460.2026. [DOI] [PubMed] [Google Scholar]
- Otto JC, Kelly P, Chiou ST, York JD. Alterations in an inositol phosphate code through synergistic activation of a G protein and inositol phosphate kinases. Proc Natl Acad Sci U S A. 2007;104:15653–15658. doi: 10.1073/pnas.0705729104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- Paulsen CE, Armache JP, Gao Y, Cheng Y, Julius D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature. 2015;525:552. doi: 10.1038/nature14871. [DOI] [PubMed] [Google Scholar]
- Qian X, Mitchell J, Wei SJ, Williams J, Petrovich RM, Shears SB. The Ins (1,3,4)P3 5/6-kinase/Ins (3,4,5,6)P4 1-kinase is not a protein kinase. Biochem J. 2005;389:389–395. doi: 10.1042/BJ20050297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T, Green DR. Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol Cell. 2016;61:589–601. doi: 10.1016/j.molcel.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruschkowski S, Rosenshine I, Finlay BB. Salmonella typhimurium induces an inositol phosphate flux in infected epithelial cells. FEMS Microbiol Lett. 1992;74:121–126. doi: 10.1016/0378-1097(92)90416-l. [DOI] [PubMed] [Google Scholar]
- Savitsky P, Bray J, Cooper CD, Marsden BD, Mahajan P, Burgess-Brown NA, Gileadi O. High-throughput production of human proteins for crystallization: the SGC experience. J Struct Biol. 2010;172:3–13. doi: 10.1016/j.jsb.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanda P, Kupce E, Brutscher B. SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J Biomol NMR. 2005;33:199–211. doi: 10.1007/s10858-005-4425-x. [DOI] [PubMed] [Google Scholar]
- Shears SB. Molecular basis for the integration of inositol phosphate signaling pathways via human ITPK1. Adv Enzyme Regul. 2009;49:87–96. doi: 10.1016/j.advenzreg.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streb H, Irvine RF, Berridge MJ, Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature. 1983;306:67–69. doi: 10.1038/306067a0. [DOI] [PubMed] [Google Scholar]
- Strilic B, Yang L, Albarran-Juarez J, Wachsmuth L, Han K, Muller UC, Pasparakis M, Offermanns S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536:215–218. doi: 10.1038/nature19076. [DOI] [PubMed] [Google Scholar]
- Su L, Quade B, Wang H, Sun L, Wang X, Rizo J. A Plug Release Mechanism for Membrane Permeation by MLKL. Structure. 2014;22:1489–1500. doi: 10.1016/j.str.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Tanzer MC, Matti I, Hildebrand JM, Young SN, Wardak A, Tripaydonis A, Petrie EJ, Mildenhall AL, Vaux DL, Vince JE, et al. Evolutionary divergence of the necroptosis effector MLKL. Cell Death Differ. 2016;23:1185–1197. doi: 10.1038/cdd.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann T, Boeing S, Lehmbacher M, Meisterernst M. The VP16 activation domain establishes an active mediator lacking CDK8 in vivo. J Biol Chem. 2007;282:2163–2173. doi: 10.1074/jbc.M608451200. [DOI] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation. Toward identification of the effector molecules Science. 2016;352:aaf2154. doi: 10.1126/science.aaf2154. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Watson PJ, Fairall L, Santos GM, Schwabe JW. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe VO, Savill JM, Chavali S, Jonsdottir AB, Rajendra E, Gruner T, Laskey RA, Babu MM, Venkitaraman AR. Human inositol polyphosphate multikinase regulates transcript-selective nuclear mRNA export to preserve genome integrity. Mol Cell. 2013;51:737–750. doi: 10.1016/j.molcel.2013.08.031. [DOI] [PubMed] [Google Scholar]
- Wu M, Chong LS, Perlman DH, Resnick AC, Fiedler D. Inositol polyphosphates intersect with signaling and metabolic networks via two distinct mechanisms. Proc Natl Acad Sci U S A. 2016;113:E6757–E6765. doi: 10.1073/pnas.1606853113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109:5322–5327. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Chen LM, Hernandez L, Shears SB, Galan JE. A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol Microbiol. 2001;39:248–259. doi: 10.1046/j.1365-2958.2001.02230.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1: Figures S1–S5 and Tables S2 and S4
Table S1. Ranked List of Genes Identified in a Haploid Genetic Screen for Components of MLKL-Mediated Cell Death, Related to Figure 1. For each gene, insertion counts are listed for the DOX-treated, selected cells and the untreated, unselected control cells. The significance of enrichment (P-value) was determined using the one-sided Fisher exact test corrected for multiple hypothesis testing.
Table S3. Inositol Phosphate Labeling, Related to Figure 5. Inositol phosphate labeling data for HT-29 containing a summary of normalized peak integration for all inositol species detected, raw [3H]-detection values (DPM), and graphs for replicate experiments for each sample. In Figure 5C, the representative dataset “Experiment 2” was presented for each sample.