

SUMMARY
Animal cells have a remarkable capacity to adopt durable and heritable gene expression programs or epigenetic states that define the physical properties and diversity of somatic cell types. The maintenance of epigenetic programs depends on poorly understood pathways that prevent gain or loss of inherited signals. In the germline, epigenetic factors are enriched in liquid-like perinuclear condensates called nuage. Here we identify the deeply conserved helicase-domain protein, ZNFX-1, as an epigenetic regulator and component of nuage that interacts with Argonaute systems to balance epigenetic inheritance. Our findings suggest that ZNFX-1 promotes the 3′ recruitment of machinery that propagates the small RNA epigenetic signal and thus counteracts a tendency for Argonaute-targeting to shift 5′ along the mRNA. These functional insights support the idea that recently identified subdomains of nuage, including ZNFX-1 granules or “Z-granules,” may define spatial and temporal zones of molecular activity during epigenetic regulation.
eTOC Blurb
Ishidate et al. identify ZNFX-1, a highly conserved helicase protein, as a factor required for epigenetic inheritance in C. elegans. Their findings indicate that ZNFX-1 localizes within nuage where it interacts with Argonaute systems and RNA-dependent RNA polymerase (RdRP) to ensure balanced amplification of small RNA signals along germline mRNAs.
INTRODUCTION
Numerous and well-documented examples of epigenetic inheritance in both plants and animals have begun to reveal the underlying mechanisms of epigenetic regulation (Castel and Martienssen, 2013). Rather than relying on sequence-specific binding sites, epigenetic mechanisms often achieve regulation by modifying features common to all genes, including modifications to chromatin, direct DNA methylation, and in some instances, Argonuate/small RNA-mediated targeting of mRNA or nascent transcript. Epigenetic effects are often linked to mechanisms that drive amplification and the spreading of regulation in cis or trans, e.g., from a nearby transposon to an adjacent gene. Indeed transposons, which make up more than half the genome in many organisms are well-established targets and mediators of epigenetic variation (McClintock, 1984).
In many organisms, Argonaute/small RNA mechanisms can promote both histone and DNA modifications to regulate gene expression (Brennecke et al., 2008; Dalmay et al., 2000; Gu et al., 2012a; Guang et al., 2010; Hamilton and Baulcombe, 1999; Jones et al., 1999; Mette et al., 1999; Mochizuki et al., 2002; Volpe et al., 2002). In the C. elegans germline, Argonaute pathways can initiate and maintain remarkably stable epigenetic silencing of transgenes (Ashe et al., 2012; Bagijn et al., 2012; Buckley et al., 2012; Lee et al., 2012; Shirayama et al., 2012). So-called RNA-induced epigenetic silencing (RNAe) is initiated by the Piwi Argonaute, PRG-1, and maintained by cytoplasmic and nuclear worm-specific Argonautes (WAGOs) that direct post-transcriptional and transcriptional silencing mechanisms. Transcriptional silencing involves conserved components of the Polycomb repressive complex and histone H3K9 methyltransferase (Burkhart et al., 2011; Guang et al., 2010; Guang et al., 2008).
Recent studies reveal that the entire germline transcriptome is under surveillance by the PRG-1/piRNA pathway (Shen et al., 2018). PRG-1 engages genomically-encoded piRNAs transcribed by RNA polymerase II from over ten thousand tiny genes (Batista et al., 2008; Das et al., 2008), and the resulting piRNA silencing complex (piRISC) targets mRNAs using miRNA-like pairing rules (Shen et al., 2018). piRISC binding can recruit RNA-dependent RNA polymerase (RdRP) which synthesizes secondary antisense siRNAs (termed 22G-RNAs) that engage worm-specific Argonaute (WAGO) family members. Cytoplasmic and nuclear WAGOs in turn mediate post-transcriptional and transcriptional silencing of the target gene. WAGOs in turn are thought to recruit RdRP to maintain and amplify the silencing signal. WAGOs and their 22G-cofactors are transmitted via egg and sperm to propagate RNAe from one generation to the next.
Given the potent and ubiquitous nature of piRNA scanning in the worm germline, mechanisms must exist to prevent piRNA-induced silencing of essential germline genes. One such mechanism involves the Argonaute CSR-1, which engages RdRP-derived 22G-RNAs templated from essentially all germline expressed mRNAs. CSR-1 targeting correlates with suppression of both piRISC and WAGO targeting (Shen et al., 2018). The precise mechanism remains unknown, but the CSR-1 pathway can counteract and sometimes reverse silencing induced by the PRG-1/WAGO pathway (Conine et al., 2013; Seth et al., 2013; Wedeles et al., 2013). For example, foreign DNA, such as the jellyfish green fluorescent protein (GFP) coding sequence, is prone to piRISC silencing presumably because it is not recognized by CSR-1. However, foreign sequences in a transgene can sometimes gain CSR-1 recognition (Seth et al., 2013), and the acquisition of CSR-1 targeting correlates with resistance to RNAe. CSR-1 and its associated 22G-RNAs are abundantly localized in mature gametes to transmit, from parent to offspring, a memory of self-gene expression (and resistance to piRNA silencing).
All three Argonaute systems involved in transcriptome surveillance localize to cytoplasmic foci called nuage or P-granules (Kawasaki et al., 1998; Phillips et al., 2012). In transcriptionally active germ cells, P-granules are perinuclear, juxtaposed to nuclear pores (Pitt et al., 2000; Sheth et al., 2010). The perinuclear association of the P-granules depends in part on CSR-1 activity and may reflect the coalescence of Argonaute pathways that survey nascent transcripts as they emerge from nuclear pores.
To understand mechanisms of epigenetic inheritance, we have performed a search for mutants that restore the expression of an epigenetically silenced transgene in C. elegans. Here we report the identification of a previously uncharacterized gene, znfx-1, that encodes a conserved protein with a “superfamily 1” (SF1) helicase domain and six repeats of a putative cysteine-rich zinc-finger domain. Mutations in znfx-1 not only cause activation of silent transgenes, but also cause silencing or variegation of active transgenes. We show that ZNFX-1 localizes prominently to P-granules and interacts with CSR-1, PRG-1, WAGO-1, and RdRP. Analysis of Argonaute small RNA signals in znfx-1 mutant reveals a shift of targeting from 3′ toward the 5′ ends of mRNAs, suggesting that ZNFX-1 translocates toward the 3′ to position RdRP for maintenance of epigenetic silencing.
RESULTS
Identification of mutants defective in the maintenance of transgene silencing
To identify genes required for maintenance of RNAe, we devised a screen wherein activation of a silent cdk-1∷gfp transgene rescues a conditional lethal allele of cdk-1(ne2257ts). We mutagenized this strain and searched for worms that were viable at the restrictive temperature (24°C) and that also expressed nuclear GFP in all g erm cells. To enhance the detection of de-silencing mutants, the strain also contained a silent gfp∷csr-1 transgene that when activated produces a bright perinuclear GFP signal throughout the germline.
Worms mutagenized with N-ethyl-N-nitrosourea were propagated at the permissive temperature of 15°C for 1, 2, 3, or 4 generations. These cultivation intervals at permissive temperature were chosen to allow for the segregation of mutations in the silencing machinery and for the conversion of epigenetic silencing to active expression (Figure 1A). At the end of each interval, we shifted populations to the non-permissive temperature of 24°C to select for viable worms. Many genes required for maintenance of RNAe are also required for germline RNAi. Indeed, ~200 mutants viable at 24°C were resistant to RNAi targeting the essential maternal gene pos-1. Most of these mutants likely define known RNAi-deficient mutants, because the pos-1 RNAi-deficient screens are near saturation (Chen et al., 2005), and were discarded. Among the RNAi sensitive mutants, we identified a total of 57 RNAe defective mutants (Figure S1A) that expressed nuclear CDK-1∷GFP and perinuclear GFP∷CSR-1 (Figure 1B). Interestingly, among them, we identified three mutants that were sensitive to RNAi at room temperature, but exhibited a mild RNAi deficiency at 15°C due to delayed onset of RNAi response (Table 1 and Figure 1D). Cold-sensitive RNAi (csRNAi)-deficient mutants were not identified by any of our previous screens.
Figure 1. Identification of mutants defective in the maintenance of transgene silencing.
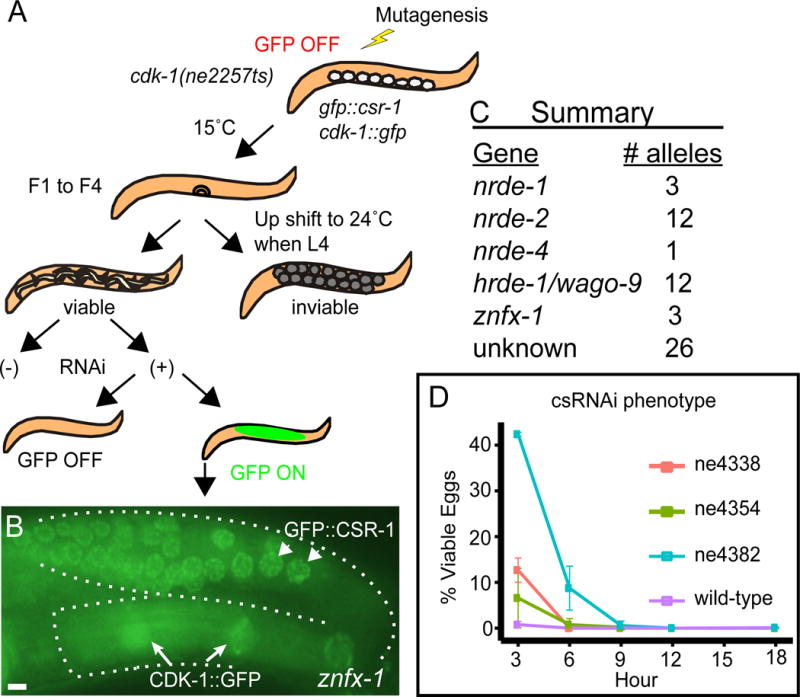
(A) Schematic diagram depicting the screening strategy.
(B) Fluorescence micrographs of representative germlines (outlined with broken lines) in a znfx-1(ne4338) mutant. Arrows indicate nuclear CDK-1∷GFP and arrowheads indicate perinuclear speckles of GFP∷CSR-1. Scale bar represents 10 μm.
(C) Summary of identified genes and the number of independently isolated alleles.
(D) Graphic representation of hatching rate in % when animals carrying various znfx-1 alleles indicated are exposed to pos-1 RNAi at 15°C, in 3 hour windows.
See also Figure S1.
Table 1.
Cold-sensitive RNAi defect of znfx-1(ne4338) to germ line RNAi
| Genotype | unc - 22 food % twitching | pos - 1 food % dead eggs | ||
|---|---|---|---|---|
| 15°C | 25°C | 15°C | 25°C | |
| wild - type | 100 (29) | 100 (39) | 100 (723) | 100 (837) |
| ne4338 | 100 (35) | 100 (35) | 89.6 (1889) | 98.1 (1698) |
| ne4354 | 100 (30) | 100 (25) | 99.4 (1698) | 100 (1389) |
% of animals (unc-22 food) or eggs (pos-1 food) exhibiting the indicated phenotypes at two different temperatures are shown. Numbers in parentheses indicate the numbers of animals (unc-22 food) or eggs (pos-1 food) scored. See also Table S4.
Previous studies have identified 12 genes that are required for the maintenance of RNAe (Figure S1B). To rapidly identify alleles of these genes among the 57 mutants sensitive to RNAi at room temperature, we amplified the genomic region of each gene from each mutant and used high-throughput sequencing to simultaneously sequence each locus. Sequencing barcodes were used to identify each mutant strain. This analysis identified candidate lesions in nrde-1, nrde-2, nrde-4, and hrde-1/wago-9 in 28 of the mutants (Figure 1C and Figure S1C). The remaining 29 mutants, including the cold-sensitive RNAi-deficient mutants, appear to define new complementation groups.
One of these complementation groups comprised three alleles, including a csRNAi-deficient ne4338 allele (Table 1 and Figure 1D). After outcrossing the ne4338 allele, we performed whole-genome sequencing (see experimental procedures) and identified lesions in several candidate genes. Sanger sequencing of these candidate genes revealed that all three mutants contained lesions in zk1067.2, a conserved gene henceforth referred to as znfx-1.
ZNFX-1 helicase and cysteine-rich domains are required for epigenetic inheritance
Worm ZNFX-1 contains a central UPF1-like “superfamily 1” helicase domain (Jankowsky, 2011), and six copies of a cysteine-rich motif also found in the conserved transcriptional repressor NF-X1 (nuclear transcription factor, X-box binding 1; Figure 2A). znfx-1 is the only worm gene that encodes both motifs, and ZNFX-1 orthologs can be found in many animals, including humans.
Figure 2. ZNFX-1 helicase and cysteine-rich domains are required for epigenetic inheritance.
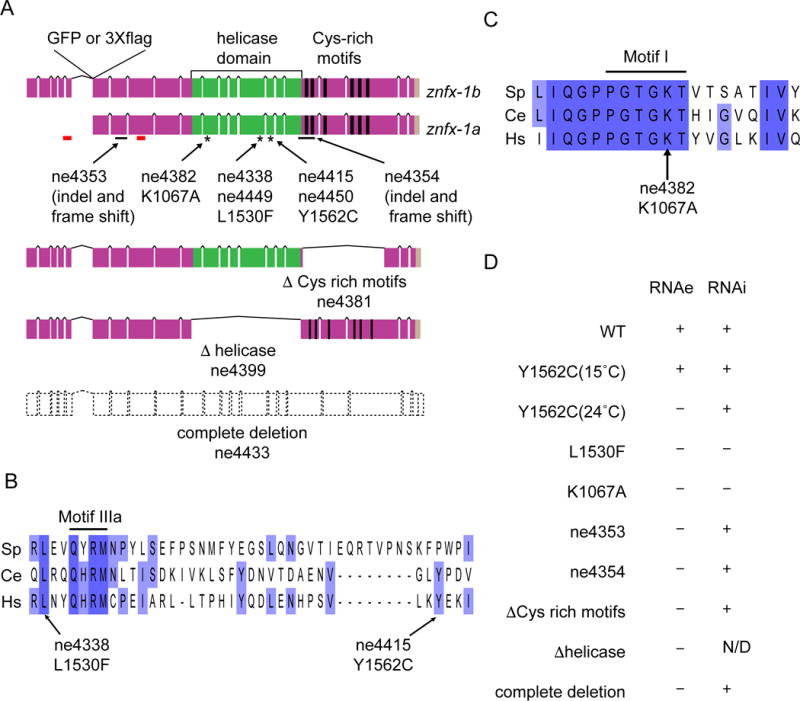
(A) Schematic diagram depicting the structure of znfx-1 locus and locations of predicted domains. Positions of GFP or 3×flag tag insertion, indel (solid black lines) or point (asterisks) mutations used in the study, regions targeted in RNAi in Figure S3B (red lines), as well as the structures of in-frame mutations deleting Cys-rich motifs or helicase domain and of complete deletion mutation, are shown.
(B) Alignment of a region encompassing conserved motif IIIa of helicase domains (solid black line) from Schizosaccharomyces pombe (Sp), Caenorhabditis elegans (Ce) and Homo sapiens (Hs), and the positions of point mutations identified in the screen are shown. Purple, lighter purple, and no background indicate 100%, 75%, 0% conservation.
(C) Alignment of a region encompassing conserved motif I (solid black line) of helicase domains represented in (B). The position of K1067A mutation used in the study is shown.
(D) Table summarizing the phenotypes of alleles used in the study. Defects in RNAe or cold sensitive RNAi are indicated by + (wild-type) or - (defective). N/D represents (not determined). See also Figures S2 and S3.
Though highly conserved, all three znfx-1 mutants recovered in our screen were viable and fertile as homozygotes. Two of the alleles are missense mutations (ne4338, L1530F and ne4415, Y1562C) that change conserved amino acids in the helicase domain (Figure 2A, and B). The third allele (ne4354) is an insertion/deletion/frameshift predicted to truncate the protein after the helicase domain (Figure 2A). Because these alleles might not be null mutations, and because they differed with respect to the csRNAi defect, we used genome editing to create seven additional alleles (Figure 2A and C): a clean deletion (ne4433) that removes the entire znfx-1 open reading frame; in-frame deletions of the helicase (Δhelicase; ne4399) or cysteine-rich (ΔZF; ne4381) domains; an independent L1530F mutation (ne4449), an independent Y1562C mutation (ne4450); an ATP-binding site mutation in the helicase domain (ne4382, K1067A); and a deletion/frameshift allele (ne4353) predicted to truncate ZNFX-1 near the N-terminus but upstream of the helicase domain.
All ten znfx-1 alleles exhibited a common set of phenotypes that we interpret as complete or partial loss-of-function (Figure 2D). Complete deletion of znfx-1 (i.e., ne4433) activated the silent RNAe transgene one generation earlier than the other alleles (Figure S2A, B and data not shown). Only the L1530F mutants and the K1067A ATP-binding site mutant exhibited the cold-sensitive RNAi defect. Thus, the cold-sensitive deficiency in RNAi might reflect a neomorphic or antimorphic activity of defective ZNFX-1 proteins. Interestingly, the csRNAi defect was suppressed when prg-1 mutation was introduced in the strain (Table S4), suggesting that defective ZNFX-1 protein competes with the prg-1 pathway for shared components of RNAi machinery (see Discussion). Finally, both of the Y1562C point mutants caused a temperature-sensitive RNAe defect; transgenes were de-silenced only when Y1562C worms were cultured at the elevated temperature of 24°C and reverted back to a silenced state in three generations when the culture temperature was lowered to 15°C (Figure 2D and S2C). Taken together, these findings indicate that ZNFX-1, including its helicase and cysteine-rich domains, is required for maintenance of RNAe, and suggest that helicase domain mutations predicted to compromise ATPase activity can interfere in subtle ways with other Argonaute-dependent silencing pathways.
We next explored the dynamics and temperature dependence of de-silencing in the znfx-1 mutants. The RNAe transgene was activated at least one generation sooner in the clean deletion mutant than in the other znfx-1 mutants, but in all znfx-1 mutants, expression was unstable for the first few generations. When cultured at 25°C, stably expressing strains were identified in the F5 generation for the complete deletion, K1067A, and L1530F mutants (Figure S2A and B and data not shown), and in the F6 generation for the Y1562C mutant (Figure S2C). In contrast, when cultured at 15°C, GFP expression never stabilized during the course of our studies (more than 10 generations), and only became stable after shifting the mutants to 25°C for several generations. These findings suggest that ZNFX-1 promotes the robustness of germline epigenetic programs to temperature.
ZNFX-1 localizes to perinuclear granules in germ cells
To examine the expression and localization of ZNFX-1, we inserted sequences encoding green fluorescent protein (GFP) or three copies of the FLAG peptide at the beginning of exon 5, which is shared by the two predicted splice variants of znfx-1 (Figure 2A). The resulting strains produced fusion proteins that coincide with the expected sizes for both ZFNX-1 isoforms: 250-kD and 280-kD GFP-tagged ZNFX-1 isoforms, and 220-kD and 250-kD FLAG-tagged isoforms (Figure S3A and B). To examine the effect of our znfx-1 mutations on ZNFX-1 protein levels, we introduced the K1067A, L1530F, and Y1562C point mutations, and the ΔZF and Δhelicase mutations into otherwise wild-type GFP-tagged ZNFX-1. Western blot analyses (using anti-GFP antibody) revealed that the K1067A, L1530F, Y1562C and ΔZF proteins were expressed at reduced levels (10% to 25%) compared to the wild-type (Figure S3C). The Δhelicase protein was undetectable.
Live imaging revealed that GFP∷ZNFX-1 protein localizes to perinuclear and cytoplasmic foci in germ cells at all stages of development (Figure S3D to H). The mutant GFP∷ZNFX-1 proteins showed a similar localization pattern, though the perinuclear fluorescence signals were dimmer in K1067A, L1530F, and ΔZF mutants compared to wild type (Figure S3I to L). In Y1562C mutants, which exhibit a temperature-sensitive RNAe defect, perinuclear GFP∷ZNFX-1 signal was lower in worms reared at 24°C compared to 15°C (Figure S3M and N). Western blot analysis revealed that the Y1562C protein is significantly reduced at 24°C (Figure S3O), suggesting that the mutant protein is less stable at the restrictive temperature.
The perinuclear and cytoplasmic localization of ZNFX-1 resembled that of P-granules— the germline nuage of C. elegans (Kawasaki et al., 1998). P-granules are liquid-like condensates composed of RNAs and RNA-binding proteins, including GLH-1, PGL-1 and the Argonaute proteins CSR-1, WAGO-1, and PRG-1. Confocal microscopy of GFP∷ZNFX-1 in strains that also express mCherry-tagged PGL-1 or CSR-1 confirmed the P-granule localization of ZNFX-1 (Figure 3A and 3B). Moreover, we found that localization of GFP∷ZNFX-1 was disrupted in csr-1 and glh-1 mutant strains, where P-granule formation and localization does not occur properly (Figures 3C and 3D) (Claycomb et al., 2009; Kawasaki et al., 1998). On the other hand, PGL-1 and CSR-1 proteins localized normally in znfx-1 mutants. Thus ZNFX-1 localizes to P-granules, but is not required for P-granule formation and localization.
Figure 3. ZNFX-1 localizes to perinuclear granules in germ cells.
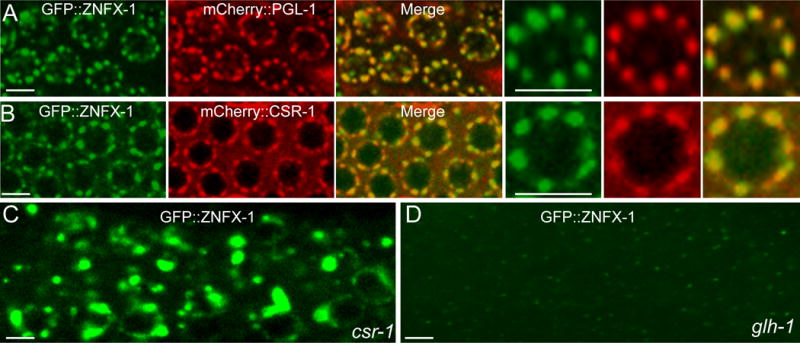
(A) Confocal images of adult germline in a strain expressing GFP∷ZNFX-1 and mCherry∷PGL-1. Magnified images of one nucleus are shown in the right three panels. Scale bars represent 5 μm.
(B) Confocal images of adult germline in a strain expressing GFP∷ZNFX-1 and mCherry∷CSR-1. Magnified images of one nucleus are shown in the right three panels. Scale bars represent 5 μm.
(C) Confocal images of adult germline in csr-1(tm892) expressing GFP∷ZNFX-1. Scale bars represent 5 μm.
(D) Confocal images of adult germline in glh-1(ok439) expressing GFP∷ZNFX-1. Scale bars represent 5 μm.
znfx-1 mutations cause instability of otherwise stably expressed genes
Transgenes that share sequence identity can act in trans to negatively or positively influence each other’s expression (Seth et al., 2013; Shirayama et al., 2012). In some strains, however, the silent and active states of each transgene can be maintained within the same cell (Seth et al., 2018). This balanced situation is remarkably stable in wild-type animals and has proven useful for measuring genetic perturbations that weaken or strengthen silencing. For example removing prg-1(+) activity shifts the balance toward bright and stable expression of the silent transgene, while conversely artificially increasing the piRNA targeting of the expressed transgene shifts the balance toward stable silencing (Seth et al., 2018). We therefore explored how znfx-1 mutants affect this balance. For example, if ZNFX-1 is primarily required for silencing, we would expect znfx-1 mutations to tip the balance toward activation of the silent transgene. We crossed the znfx-1(ne4338) L1530F mutant bearing an expressed cdk-1∷gfp transgene into a sensor strain with balanced expression states: oma-1∷gfp[ON]; cdk-1∷gfp[ON]; gfp∷csr-1[OFF] (Figure 4A). In F1 heterozygotes and in F2 to F4 homozygotes, we found that the transgenes maintained their initial expression status: oma-1∷gfp and cdk-1∷gfp remained ON, and gfp∷csr-1 OFF (Figure 4A). At the F5 generation, however, the expression status of each transgene became unstable (Figure 4A–E). While 8 out 39 worms maintained the initial expression status (Figure 4A), OMA-1∷GFP and CDK-1∷GFP was silenced in 16 of 39 worms (Figure 4B), OMA-1∷GFP alone was expressed in 5 out 39 worms (Figure 4C), CDK-1∷GFP alone was expressed in 9 out 39 worms (Figure 4E) and GFP∷CSR-1 was activated in 1 of 39 worms (Figure 4D). Remarkably, the expression states could switch back and forth between silent and expressed from one generation to the next. An originally active transgene that went silent in one generation could reactivate in the next generation, and an originally silent transgene that was expressed in one generation could go silent again in the next generation (Figure S4).
Figure 4. znfx-1 mutation cause variegation of an otherwise stably expressed gene.
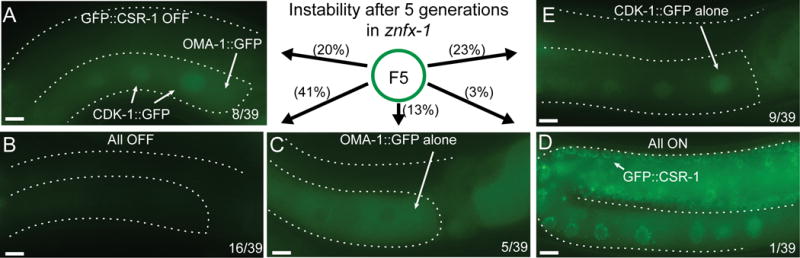
(A-E) Fluorescence micrographs of variegating GFP signals in the adult germline of F5 animals after the introduction of znfx-1 mutation. Numbers and % of animals having the phenotype shown in the pictures are indicated. Scale bars represent 10 μm. See also Figure S4.
These findings suggest that ZNFX-1 is not simply required for epigenetic silencing, but rather for balanced inheritance of both expressed and silent epigenetic states. We have never seen this type of instability before, but it will be important in the future to carefully examine additional alleles of znfx-1 and of other genes involved in epigenetic control.
De-silencing in znfx-1 mutants does not require prg-1
One possible explanation for the complex phenotype of znfx-1 mutants is that ZNFX-1 functions primarily in the CSR-1 pathway to help protect expressed mRNAs from PRG-1 targeting (Seth et al., 2013; Wedeles et al., 2013). In the absence of ZNFX-1 activity, mis-targeting by PRG-1 could dilute the available WAGO pathway silencing machinery and disrupt silencing activity. If so, the RNAe defect of znfx-1 mutants should require prg-1(+) activity. We could directly test this model because PRG-1 is required to initiate, but not to maintain RNAe on most transgenes (Shirayama et al., 2012). We generated wild-type and prg-1 mutant strains bearing a silent gfp∷cdk-1(RNAe) transgene, and then we deleted znfx-1 in both strains and followed the expression of gfp∷cdk-1 over several generations. Deletion of znfx-1 activated the gfp∷cdk-1 transgene by the third generation in the presence or absence of PRG-1 in 5 out of 5 generated lines, suggesting that the RNAe defect of znfx-1 mutants is not caused by PRG-1 mis-targeting. Rather, these findings are consistent with the idea that ZNFX-1 promotes the maintenance of epigenetic memory in both the WAGO and CSR-1 pathways.
ZNFX-1 binds RdRP and the Argonautes CSR-1, PRG-1, and WAGO-1
The helicase domain of ZNFX-1 is similar to the S. pombe helicase, Hrr1, which was originally isolated as a component of the RDRC (RNA-directed RNA polymerase complex) (Motamedi et al., 2004). We therefore tested whether the ZNFX-1 interacts with the RdRP EGO-1, which functions in both the CSR-1 and WAGO pathways (Claycomb et al., 2009; Gu et al., 2009). Indeed, co-immunoprecipitation assays revealed a strong and specific physical interaction between GFP∷EGO-1 and FLAG∷ZNFX-1 fusion proteins (Figure 5A). This interaction resisted treatment with RNase I, suggesting that ZNFX-1 exists in a complex with EGO-1 that is not bridged by single-stranded RNA.
Figure 5. ZNFX-1 forms stable complex with an RdRP, EGO-1, and argonautes in both gene-silencing and gene-activating pathways.
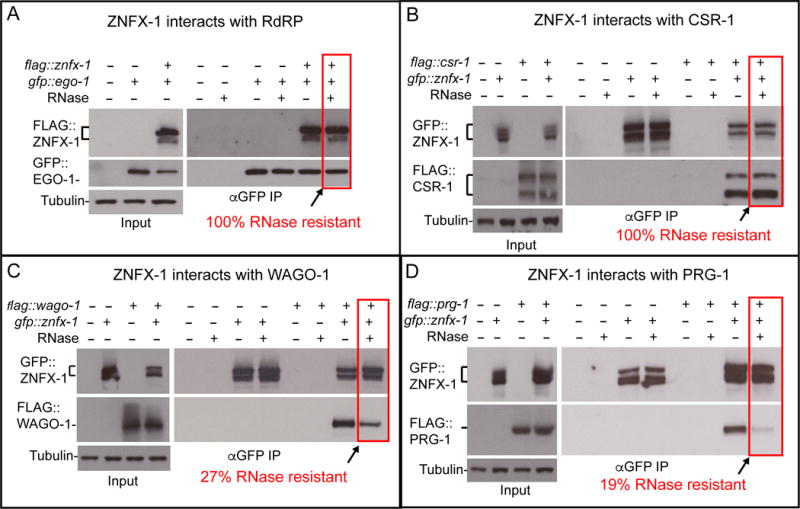
(A-D) Co-immunoprecipitation experiments showing physical interaction between ZNFX-1 and EGO-1 (A), CSR-1 (B), WAGO-1(C) or PRG-1(D). Presence (+) or absence (-) of the tagged proteins or RNase I treatment are indicated. Immunoprecipitation was performed using α-GFP antibody, and the blots were probed with α -GFP or α -flag antibodies, and with α -tubulin antibody for loading control. Brackets indicates the positions of two major isoforms of znfx-1 and csr-1. % of RNase resistant complex as determined by the relative amounts of proteins in ZNFX-1 immunoprecipiates with or without the RNase treatment, are indicated. Level of co-precipitating protein was normalized to the amount of ZNFX-1 in the immunoprecipitate.
In yeast, RDRC also associates physically with the RNAi effector complex called RITS, which contains the Argonaute protein Ago1 (Verdel et al., 2004). We therefore tested pair-wise interactions between GFP∷ZNFX-1 and four different FLAG-tagged germline Argonautes: CSR-1, PRG-1, WAGO-1, and WAGO-9. Whereas CSR-1, PRG-1, and WAGO-1 localize to P-granules, WAGO-9 is nuclear. Consistent with its P-granule localization, ZNFX-1 co-immunoprecipitated with CSR-1, WAGO-1, and PRG-1 (Figure 5B–D), but not with WAGO-9 (data not shown). The interaction between ZNFX-1 and CSR-1 was resistant to RNase I treatment, and the ZNFX-1 interactions with WAGO-1 and PRG-1 were partially sensitive to RNase I. These findings further support the idea that ZNFX-1 functions in multiple, even opposing, epigenetic programs.
znfx-1 mutants exhibit altered patterns of RdRP-dependent small RNAs
To gain insight into the molecular mechanisms underlying the znfx-1 phenotype, we used deep-sequencing to analyze the total small RNA profile and small RNAs bound to CSR-1 or WAGO-9 in wild-type and znfx-1(ne4354) mutant worms. Although its phenotype is slightly less severe than the complete deletion, we chose to analyze the ne4354 allele because it was molecularly the strongest allele (a stop codon after the helicase domain) that was backcrossed and available at the time we initiated the analysis. We analyzed small RNAs recovered from three independent populations of znfx-1 mutant and wild-type animals that express FLAG∷CSR-1 or FLAG∷WAGO-9 from the endogenous csr-1 and wago-9 loci. Consistent with the relatively subtle phenotype of znfx-1 mutants, the overall number of small RNA reads mapping to various small RNA categories, was similar in znfx-1 and wild-type worms (Figure S5A). However, we noticed a small, but reproducible increase of approximately 12% in total 22G-RNA levels in znfx-1, when normalized to total read counts. This increase was most significant among WAGO targets (Table S2). Interestingly, whereas wild-type replicates (and znfx-1 replicates) exhibited a fairly uniform distribution of WAGO 22G-RNAs, when compared to each other, we found that the mutant and wild-type populations exhibited a much wider spread in 22G-RNA levels targeting annotated WAGO targets (Figure 6A to C, and Figure S5B to G). Strikingly, very reproducible subsets of WAGO targets exhibited either increased or decreased cumulative 22G-RNA levels in znfx-1 mutants. This pattern was also observed in the population of 22G-RNAs recovered in the WAGO-9 IP from znfx-1 mutants (Figure S6A to C). Thus, subsets of WAGO target genes exhibit consistent increases or decreases in 22G-RNA levels in znfx-1 mutants. By contrast, the overall levels of 22G-RNAs on annotated CSR-1 targets (and in CSR-1 IPs) was similar between wild-type and znfx-1 (Figure S6D to F and Figure S6G to I).
Figure 6. znfx-1 mutants exhibit altered patterns of RdRP-dependent small RNAs.
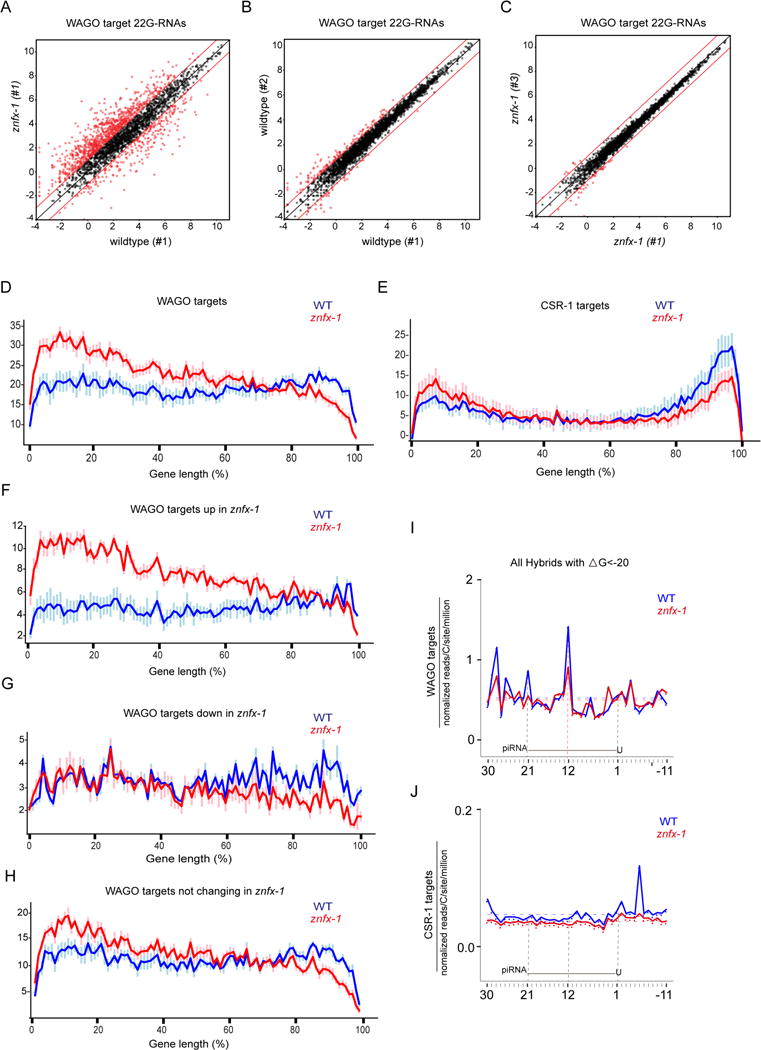
(A) Scatter plot comparing the numbers of small RNA reads (log2) for each WAGO target cloned from wildtype or znfx-1 worms. Each dot represents a WAGO target gene. Red dots represent genes whose 22G-RNAs increase or decrease at least 2-fold (indicated by lines above and below x=y).
(B) Scatter plot as in (A) but comparing two independent wild-type populations.
(C) Scatter plot as in (A) but comparing two independent znfx-1 populations.
(D and E) Distribution of small RNAs along the length of genes in 100 segments (x-axis) on WAGO targets (D) and CSR-1 targets (E). Y-axis represents number of 22Gs in each segment in arbitrary units after normalization to overall 22G levels. Data represented are average values of 6 replicates with standard deviations.
(F-H) Distribution of small RNAs along the length of genes in 100 segments (x-axis) on WAGO targets whose 22Gs goes up (F), goes down (G) or remain unchanged (H). Y-axis represents number of 22Gs in each segment in arbitrary units after normalization to overall 22G levels. Data represented are average values of 6 replicates with standard deviations.
(I and J) 22G-RNA 5′ prime ends mapped at single-nucleotide resolution within a 40-nt window around piRNA target sites determined by prg-1 CLASH in wild-type (blue) or znfx-1 mutant (red) worms, for hybrids with ΔG < -20 kcal/mol. The plots are centered on 21-nt piRNA ± 10 nt shown schematically in each graph. WAGO targets (I) and CSR-1 targets (J) were analyzed separately.
See also Figures S6 and S7.
Because ZNFX-1 interacts with RdRP, we asked whether znfx-1 mutations affect the distribution 22G-RNAs—i.e., where they are synthesized—along the length of WAGO and CSR-1 target mRNAs. For ease of comparison, we divided each cohort of mRNAs into 100 segments and normalized the 22G-RNA levels to the total read count for that gene to prevent heavily targeted mRNAs from skewing the analysis. This analysis revealed a striking shift in the distribution of 22G-RNAs toward the 5′ ends of both WAGO and CSR-1 targets (Figure 6D and E).
The above analysis averages 22G levels over thousands of genes, and thus could not tell us whether most or all genes were affected similarly, or alternatively, whether a subset of genes was responsible for the apparent shift. To determine what fraction of WAGO and CSR-1 targets contribute to the observed shift, we plotted normalized read density in the 5′ and 3′ 10% of each gene in WT and zfnx-1 mutants. This analysis revealed that in znfx-1 mutants 46.39% of WAGO targets and 28.55% of CSR-1 targets had significantly increased 22G-RNA levels at the 5′ end, while similar proportions, 44.87% and 31.55% for WAGO and CSR-1 targets respectively, had decreased levels at their 3′ end (Figure S7A–D and Table S3). Interestingly, even the subsets of WAGO targets that were up and down in overall levels could also be explained by a shift in targeting—the up genes had a pronounced 5′ increase (Figure 6F) and the down genes had a 3′ decrease in levels (Figure 6G). Most of the genes with normal levels of 22G-RNAs in znfx-1 mutants showed compensatory changes at opposite ends (Figure 6H). Why the former classes of targets do not exhibit this compensatory change in 5′ and 3′ levels of 22G-RNAs remains unclear (see Discussion). These findings suggest that the wild-type activity of ZNFX-1 prevents the 5′ drift of 22G-RNA targeting along mRNAs.
Finally, we wished to examine the effects of znfx-1 mutants on the patterns of 22G-RNA accumulation near piRNA target sites. To do this we aligned both the input and IP data from wild-type and znfx-1 mutant animals to piRNA binding sites in WAGO and CSR-1 target mRNAs (Shen et al., 2018). This analysis revealed that the peaks of 22G-RNA-association visible in WT, were reduced, or completely absent in znfx-1 (Figure 6I and J). Thus znfx-1 appears to be important to maintain the focused targeting of 22G-RNA biogenesis near piRNA target sites.
DISCUSSION
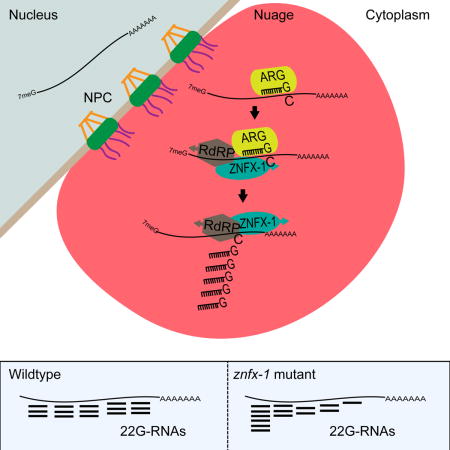
Here we used a genetic screen to identify ZNFX-1 as a factor required to maintain a piRNA-induced silent epigenetic state (RNAe) in C. elegans. We have shown that ZNFX-1 localizes prominently to germline nuage where it associates with at least three different Argonautes and with an RdRP involved in propagating RNAe. Strikingly however, our findings support a model in which ZNFX-1 is not merely required for silencing, but instead functions to maintain balanced epigenetic regulation by preventing the spread of epigenetic signals toward the 5′ ends of target mRNAs (Figure 7).
Figure 7.
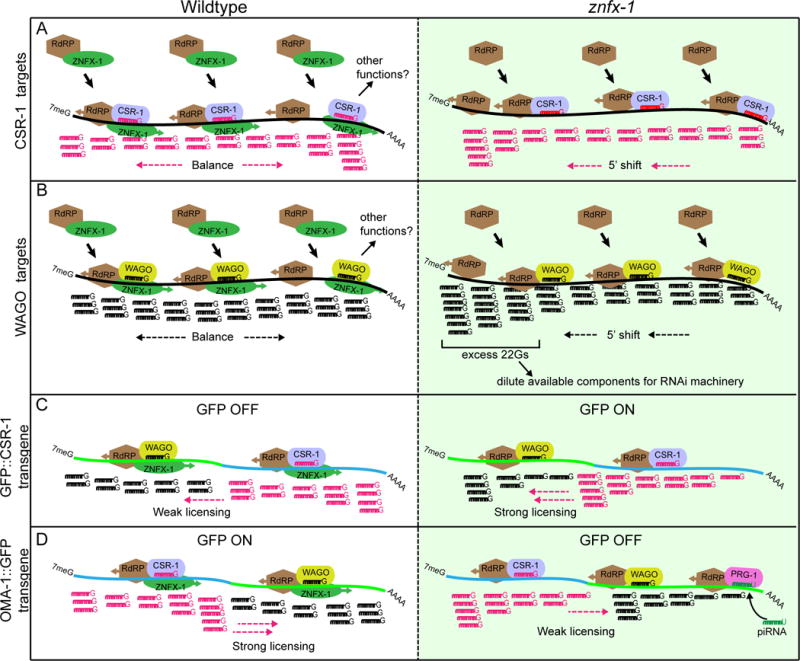
Model
Our genetic analysis of ZNFX-1 suggests that its helicase and cysteine-rich (NFX1-type zinc finger) domains are both required for function. In the fission yeast S. pombe, the Hrr1 protein, which contains a closely related helicase domain but lacks the cysteine-rich domain, co-purifies with both RdRP and Ago1 (Motamedi et al., 2004) suggesting a conserved role for the ZNFX-1 helicase domain in promoting RdRP-dependent maintenance of epigenetic silencing (Figure 7). The marked tendency of 22G-RNAs to accumulate toward the 5′ ends of WAGO and CSR-1 target mRNAs in znfx-1 mutants suggests a possible role for the ZNFX-1 helicase domain in re-positioning RdRP toward the 3′ end after each round or Argonaute targeting. Binding of ZNFX-1 just to the 5′ side of an Argonaute binding site on the target mRNA might allow ATP-dependent translocation of a ZNFX-1-RdRP complex toward the 3′ end of the mRNA, unwinding the small RNA-mRNA duplex, displacing the Argonaute, and positioning RdRP to template new 22G-RNA transcription. If precisely tuned, this mechanism would ensure that distributions of 22G-RNAs remain balanced and focused on the original target region over time (Figure 7A and B, left panels).
Unlike 22G-RNAs, piRNAs are not templated from their targets but rather are transcribed as independent genes by pol II. Thus piRNA targeting is determined solely by complementarity between the piRNA and the target mRNA and is not subject to spreading along a transcript. Our findings suggest that ZNFX-1 plays a role in maintaining the focused production of 22G-RNAs near piRNA target sites. In the absence of ZNFX-1, although the overall levels of 22G-RNAs remain high, instead of well-defined local peaks, piRNA targeting drives a shift in the production or accumulation of 22G-RNAs toward the 5′ end of the mRNA (Figure 7A and B, right panels).
We do not know why subsets of WAGO targets lack the bimodal change in 22G-RNAs (5′ increase and 3′ decrease) observed on many WAGO and most CSR-1 targets. This could be due in part to mis-annotation. For example, genes that appear to lack 3′ (or 5′) excursions in 22G-RNA levels, might actually have non-annotated extensions where these excursions occur. Since we map our data to annotations we are blind to this possibility. Indeed WAGO targets are unusual genes, including many apparent pseudo genes, and are often expressed at very low levels, so experimental evidence for validating annotations may be lacking. However future studies may also identify any number of other more interesting explanations for these subsets of unusual genes.
Unstable epigenetic states in znfx-1 mutants
The role of znfx-1 in preventing the 5′ drift of 22G-RNA accumulation suggests some possible explanations for a number of curious previous observations, and could also explain how znfx-1 mutations can render the expression of both silenced and expressed transgenes unstable (Figure 7C and D). For example, it has long been known that gfp insertions at the 5′ ends of fusion genes are more prone to silencing (in wild-type animals) than are insertions at the 3′ end (Shirayama et al., 2012). If the tendency of ZNFX-1 to translocate RdRP toward the 3′ end exceeds the default tendency of RdRP to drift toward the 5′ end, then in wild-type animals transgenes bearing a CSR-1 targeted region fused to a 3′ gfp will tend to acquire CSR-1 protection while transgenes with 5′ fusions will be less likely to acquire this protection. In znfx-1 mutants this polarity could shift, such that a normally silencing resistant transgene such as oma-1∷gfp now becomes sensitive to silencing.
Another mechanism that could contribute to the znfx-1 de-silencing phenotype is dilution of the WAGO machinery. Previous studies support the idea that WAGOs are limiting for the silencing pathways that employ these Argonautes. These pathways include the canonical RNAi-dsRNA pathway, the PRG-1 pathway, and the Eri-WAGO pathway. The WAGO Argonautes themselves have been amplified during evolution and comprise 12 genes that appear to have partially overlapping functions (Shirayama et al., 2012; Yigit et al., 2006). Indeed, artificially over-expressing a WAGO Argonaute (Yigit et al., 2006) or removing upstream components of a competing endogenous pathway (the Eri pathway) enhances the sensitivity of worms to RNAi (Duchaine et al., 2006; Kennedy et al., 2004; Yigit et al., 2006). The mild cold-sensitive RNAi defect of certain non-null znfx-1 alleles is consistent with this competition model. Overproduction of 22G-RNAs on PRG-1 targets (perhaps increased by a neomorphic or antimorphic ZNFX-1 protein) could saturate the WAGO machinery exhausting the silencing capacity available for RNAi. Consistent with this idea, prg-1 mutants suppress the cold-sensitive RNAi defect of znfx-1 mutants. RNA sequencing studies on these special alleles of znfx-1 may shed light on this question.
Phase-separated domains of mRNA regulation
A fascinating aspect of ZNFX-1 biology is its localization within nuage (P-granules) in C. elegans germ cells. P-granules are molecularly complex organelles with liquid-like properties (Brangwynne et al., 2009). Throughout the worm life cycle, P-granules contain all three major Argonaute pathways discussed in this study, as well as the literally millions of distinct small RNA species that guide Argonaute surveillance. P-granules are thought to transiently contain mRNAs as they export to the cytoplasm (Updike and Strome, 2010). The perinuclear association of P-granules is driven in part by CSR-1, an Argonaute that targets nearly all germline-expressed mRNAs (Claycomb et al., 2009). Thus, Argonaute surveillance of mRNAs likely represents a key function of P-granules (Seth et al., 2018).
A parallel study that independently identified ZNFX-1 as a factor required for epigenetic silencing reports a temporal subdomain of nuage, where ZNFX-1 levels are enriched (Wan et al., In Press). This domain—dubbed the “Z-granule”—resides between a nuclear-proximal zone and a more distal zone containing RdRP. The latter zone is known as the mutator focus (Phillips et al., 2012). This pattern of localization is strikingly consistent with our genetic and molecular findings which suggest (i) that Argonautes recruit ZNFX-1 to essentially all mRNAs; (ii) that ZNFX-1 then translocates from 5′ to 3′, (iii) that ZNFX-1 positions RdRP on the mRNA to initiate new small RNA synthesis. Thus, our findings suggest that the nuage subdomains observed by Wan et al. may define regions of concerted molecular activity. When transcription initiates in embryonic germ cells where Z granules first appear, the synchronous initiation of the molecular events discussed here, Argonaute scanning, ZNFX-1 translocation, and RdRP recruitment may proceed on such a massive scale that they are visible temporarily as phase-separated domains within cytoplasmic nuage.
Other functions for ZNFX-1 family members
Finally, although mutations within the cysteine-rich domain of ZNFX-1 disrupt its function, we do not know how this domain contributes to ZNFX-1 activity. This repeated motif was first identified in the conserved transcription factor NF-X1 (Song et al., 1994), which like ZNFX-1 contains six NFX1-type zinc fingers. All six of the fingers in human NF-X1 are required for sequence-specific binding to the conserved X-box motif of class II major histocompatibility complex (MHC) genes. Thus perhaps direct DNA-binding by ZNFX-1 helps couple post-transcriptional and transcriptional modes of epigenetic silencing. Our genetic, biochemical, and small RNA sequencing studies support the idea that ZNFX-1 prevents the drift of epigenetic regulation along target mRNAs, by using its helicase activity to position a key enzyme, RdRP, that amplifies and helps maintain epigenetic states in C. elegans. Nevertheless, ZNFX-1 orthologs also exist in animals—including humans—that lack RdRP. In the mouse testes, ZNFX-1 is expressed in pachytene spermatocytes (Wu and Zamore, unpublished result), where mouse Piwi homologs are also abundantly expressed. Thus it will be interesting to learn if the deep conservation of ZNFX-1 reflects additional RdRP-independent functions for ZNFX-1 in gene regulation, epigenetics or other germline mechanisms.
Considering the conservation of ZNFX-1 protein, it is surprising that znfx-1 mutants are so healthy and fertile. Although the de-silencing phenotype of znfx-1 is stronger at elevated temperatures, the animals remain fertile over at least several generations. Perhaps the deep conservation of ZNFX-1 reflects a role for this factor in maintaining latent epigenetic programs that are not important during laboratory growth, but rather allow animals in the wild to adapt rapidly to changing environmental conditions such as exposure to varying food sources and pathogens.
STAR METHODS TEXT
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Craig Mello (Craig.Mello @umassmed.edu).
Experimental Model and Subject Details
C.elegans strains and genetics
Strains and alleles used in this study are listed in Supplementary Table S1. C.elegans culture and genetics were performed essentially as described (Brenner, 1974).
Method Details
Mutagenesis and screening
A synchronized population (~500,000 worms) of the starting strain (WM462) was exposed to N-ethyl-N-nitrosourea (0.5mM) at late L4 to young adult stage in 6 ml of M9 buffer for 4 hours on a rocker at 20°C, washed 4 times with M9, re-plated onto NGM plates, and propagated at 15°C for successive generations, with bleaching at each generation to synchronize the population. About 1 million animals at each generation, up to F4 generation, were shifted to 24°C when L4 on 35 mm or 150 mm plates (500 or 100,000 animals/plate) and incubated continuously for about 2 weeks at 24°C to perform F1, F2, F3 and F4 screen, respectively. Plates having growing animals were then identified and one line was established from each plate that had viable animals. The mutants were triaged by first exposing the animals to pos-1 RNAi by bacterial feeding to identify RNAi sensitive mutants and then scoring each RNAi sensitive mutants under a high-power fluorescent microscope to identify those with germline GFP signals.
Mapping of mutants
Mutants with de-silenced cdk-1∷gfp and gfp∷csr-1 transgenes were outcrossed to dpy-10 marked MosSCI lines harboring silenced gfp∷cdk-1 (RNAe) transgene on LGII or gfp∷csr-1 (RNAe) transgene on LGIV. After the cross, all the transgenes in F1 cross progeny were silenced due to the trans-silencing activity of the RNAe alleles. In F2, homozygous animals for each of the marker genes including lin-11 on LGI, dpy-10 on LGII, cdk-1ts(ne2257) on LGIII and gfp∷csr-1 transgene on LGIV were individually picked and allowed to self for the next several generations. At each generation, transgene reactivation was scored and the linkage between the respective marker genes and the mutation that de-silenced the transgene was analyzed. In many cases, transgenes became reactivated at F3 generation, and the linkages to LGII or LGIII were observed. Sanger sequencing of nrde-2 (LGII) and wago-9 (LGIII) were then performed and molecular lesion in those genes, if any, were identified. In other instances, the reactivation did not occur by F3, in which case further mapping was not attempted and instead the mutants were subjected to bulk PCR amplicon deep-sequencing strategy described below.
Preparation of genomic DNA libraries from PCR amplicons of candidate genes and bulk PCR amplicon deep-sequencing strategy to identify mutations
Genomic DNA fragments encompassing nrde-1, nrde-2, nrde-4, wago-1, wago-9/hrde-1, mes-2, mes-3, mes-4, mes-6, hpl-2, set-25, set-32 were PCR amplified, spin-column purified using E.Z.N.A. Gel-Extraction Kit (Omega bio-tek), and suspended in 150μl of TE at 10ng/μl. The DNA was then sheared using Bioruptor (Diagenode) to approximately 300 bp long fragments (5 cycles of 30 sec. ON and 30 sec. OFF cycle, with intensity of M), and precipitated with ethanol precipitation. The ends of the DNA were fixed with End Repair Kit (Epicenter) following the instructions of the manufacturer, size selected using Agencourt AMPure XP size selection and purification kit (Beckman) following the kit instructions, and then A-tailed using Klenow (exo-) fragment (NEB). After AMPure purification, the fragments were ligated to pre-annealed Y-shaped adaptor (5′-P-GATCGGAAGAGCGGTTCAGCAGGAATGCCGAG-3′, and 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′ from IDT) using Fastlink Ligation Kit (Epicenter), purified again using the AMPure purification kit, and then subjected to PCR amplification using barcoded primers. PCR amplicons were purified by spin column (Omega biotek), mixed at equimolar concentration, and subjected to deep sequencing using Illumina MiSeq system (UMASS Medical School Deep Sequencing Core Facility).
Whole Genome Sequencing and Analysis
Genomic DNA used for whole genome sequencing was prepared as follows. First, mutant was outcrossed to the starting strain two times. In the second outcross, 40 F2 re-segregant lines homozygous for the mutation were phenotypically identified. After harvesting and mixing equal number of worms from the 40 lines in M9 buffer, the worm pellet was washed three times with M9 buffer and once with TEN (20mM Tris-HCl pH7.5, 50mM EDTA, 100mM NaCl), and the pellet was resuspended in the total volume of 0.5ml of TEN and frozen at −20°C. To the thawed pellet, 25 μl of 10% SDS, 2.5 μl of 20mg/ml Proteinase K and 1 μl of β-mercaptoethanol was added, and the mixture was incubated in 50°C water bath. After 1 hour, another 2.5 μl of proteinase K was added and incubation was continued for another 2 hours. Then the mixture was extracted two times with 0.5 ml of Phenol/Chloroform/Isoamylalcohol, and the DNA was ethanol precipitated with 1.2 ml of ethanol at room temperature. After washing the pellet with 70% ethanol, the pellet was resuspended in 0.5 ml of TEN. 3 μl of 10mg/ml RNAse A was added to the DNA solution and incubated at 37°C for 2 hours. Extraction with Phenol/Chloroform/Isoamylalcohol followed by ethanol precipitation was repeated and the pellet was resuspended in 100 μl of TE. Genomic DNA library was then prepared essentially the same way as the PCR amplicon library described above, and subjected to deep sequencing using Illumina HiSeq system (UMASS Medical School Deep Sequencing Core Facility), together with a control library prepared by the same way from non-mutagenized starting strain. The deep sequencing data from mutant and control were each analyzed using Cloudmap Unmapped Mutant Workflow on Galaxy (https://usegalaxy.org), and homozygous variants from the control was subtracted from homozygous variants from the mutant(Minevich et al., 2012). The resulting file was then subjected to Cloudmap EMS Variant Density Mapping workflow(Minevich et al., 2012) to determine a linked region on chromosome II and candidate changes that affect protein coding potential were identified.
Gene editing by CRISPR
GFP tagged znfx-1 was generated by the co-CRISPR strategy using unc-22 sgRNA as a co-injection marker to enrich CRISPR/CAS9-mediated genome editing events (Kim et al., 2014). The vector expressing rol-6 (su1006), a dominant allele conferring a roller phenotype, was used as a co-injection marker. GFP positive animals were identified among the progeny of F1 rollers that segregated F2 twitchers. All the other znfx-1 mutants including clean deletion were generated using an approach injecting pre-assembled Cas9 ribonucleoprotein complex using oligos as template essentially as described (Paix et al., 2015) with modifications including employment of rol-6 (su1006) as the injection marker and genome editing events were identified among the F1 rollers.
RNAi
RNAi was performed by feeding worms E.coli strain HT115(DE3) transformed with the control vector L4440 or L4440 subcloned with gene fragments of pos-1 or znfx-1. Bacteria were grown overnight in LB with antibiotic (ampicillin) overnight at 37°C and seeded onto NGM plates supplemented with 1mM isopropyl β-d-thiogalactoside and ampicillin (100 μg/ml). The plates were dried overnight at room temperature to which L1 larvae were seeded. For pos-1 RNAi, pos-1 genomic fragment amplified by primers (5′-ACGCTAATACTTCATCCAAA-3′ and 5′-GCTGATTACGAGAAATTTCA-3′) were cloned in L4440. For RNAi targeting specifically the long form of znfx-1, the genomic fragment specific to the long form (znfx-1b) was amplified by the primers (5′-GTCCAGAGATAAACCACCTC-3′ and 5′-TTGTGCGATCTGCTTGTGAG-3′) and cloned into L4440. For RNAi targeting both forms of znfx-1, the zk1067.2 clone from the Ahringer RNAi library (Kamath et al., 2003) was used.
Microscopy and Imaging
For wide-field live images, transgenic worms were mounted in H2O on RITE-ON glass slides (Beckton Dickinson) in the presence of 1mM levamisole. Epi-fluorescence and differential interference contrast (DIC) microscopy were performed using an Axio Imager M2 Microscope (Zeiss). Images were captured with an ORCA-Flash 4.0 digital camera (Hamamatsu) and processed using Zen software (Zeiss). Confocal images were taken using Zeiss Axiovert 200M microscope equipped with a Yokogawa CSU21 spinning disk confocal scan head and custom laser launch, and relay optics (Solamere Technology Group). Images were obtained and processed using Micro-Manager and Image-J programs.
Immunoprecipitation and western blotting
Gravid adult worms growing on 15 cm NGM plate (100,000 animals) were collected and washed three times in M9 buffer and then washed once and suspended in 750 μl of lysis buffer (10mM Tris-Cl pH7.4, 150mM NaCl, 1% NP-40, 1mM DTT supplemented with Mini Protease Inhibitor Cocktail Tablet (Roche)). Worms were homogenized using FastPrep-24 benchtop homogenizer (MP Biomedicals). Lysate were centrifuged twice at 20,000g for 15 minutes at 4°C. Immunoprecipitation was performed as follows. Lysates (~10mg total protein) were precleared with 50 μl of protein-A Dynabeads (Invitrogen) for 1 hour at 4°C and then incubated with 1 μg of anti-GFP monoclonal antibody (Wako Chemicals USA) or anti-flag M2 monoclonal antibody (Sigma) on ice for 1 hour. Immune-complex were incubated with 50 μl of protein-A Dynabeads for 1 hour at 4°C and precipitated on a magnet. The immune-complex were then split in two and incubated at 37°C for 15 minutes with or without RNase If (NEB) in NEB3.1 buffer, washed with ice cold lysis buffer three times, once with ice cold H2O, and eluted with NuPAGE LDS sample buffer (Thermo Fisher Scientific) by heating at 70°C for 10 minutes. Samples were loaded onto the precast NuPAGE Novex Tris-Acetate polyacrylamide gel (Thermo Fisher Scientific), subjected to electrophoresis using XCell SureLock Mini-Cell (Thermo Fisher Scientific), and transferred onto the PVDF membrane (Bio-Rad) using Mini Trans-Blot Cell (Bio-rad) at 15V overnight. The blot was incubated with polyclonal anti-gfp (GenScript, A01704) antibody and then with goat-anti-Rabbit HRP (Abcam) secondary antibody for GFP western blot, with HRP conjugated anti-flag antibody (Sigma) for flag western blot, and HRP conjugated anti-tubulin antibody (Thermo Fisher Scientific) for tubulin western blot.
Total Small RNA cloning
Total RNA was extracted from adult worms using Trizol (Molecular Research Center). Small RNAs were further enriched using MirVana Kit (Thermo Fisher Scientific). Samples from wild-type and mutant were pretreated with a homemade recombinant 5′ Polyphosphatase PIR-1 to remove the γ and β phosphates from triphosphorylated 5′ ends of 22G-RNAs (Gu et al., 2009). The resulting 5′-monophosphorylated small RNAs were ligated to a 3′ adaptor linker 1 (5′ rAppAGATCGGAAGAGCACACGTCTGAACTCCAGTCA/3ddC/3′, IDT) using T4 RNA ligase 2 (NEB). Subsequently the 5′ adaptor (rArCrArCrUrCrUrUrUrCrCrCrUrArCrArCrGrArCrGrCrUrCrUrUrCrCrGrArUrCrU) was ligated using T4 RNA ligase 1 (NEB). Ligation products were converted to cDNA using SuperScript III (Thermo Fisher Scientific). Libraries were amplified by PCR and sequenced using the HiSeq (Illumina) at the UMass Medical School Deep Sequencing facility.
IP small RNA cloning
Gravid adult worms growing on 15 cm NGM plate (100,000 animals) were collected and washed three times in M9 buffer without centrifugation and then washed once and suspended in 750 μl of lysis buffer (110mM potassium acetate, 20mM HEPES pH7.3, 0.5% Trition X-100, 0.1% Tween 20, 1mM EDTA for CSR-1 IP or 110mM potassium acetate, 20mM HEPES pH7.3, 0.5% Trition X-100, 0.1% Tween 20, 250mM trisodium citrate, 1mM EDTA for WAGO-9 IP) supplemented with Mini Protease Inhibitor Cocktail Tablet (Roche). Worms were homogenized using FastPrep-24 benchtop homogenizer (MP Biomedicals). Lysate were centrifuged twice at 20,000g for 15 minutes at 4°C. Immunoprecipitation was performed as follows. Lysates (~10 mg total protein) were precleared with 50 μl of protein-A Dynabeads (Invitrogen) for 1 hour at 4°C and then incubated with 3.9 μg of anti-flag M2 monoclonal antibody (Sigma) on ice for 1 hour. The Immune-complex were incubated with 50 μl of protein-A Dynabeads for 1 hour at 4°C and precipitated on a magnet. The immune-complex was then washed three times with ice cold lysis buffer by rotating in 4°C for 30 minutes, washed once with Wash buffer (50mM Tris pH 7.5, 150mM NaCl, 0.2% Tween 20, 2mM MgCl2), and resuspended in 100 μl of TE. The suspension was extracted twice with 100μl of Phenol/Chloroform/Isoamylalcohol pH6.7, and RNA was precipitated from the aqueous phase by adding 10μl of 3M Sodium Acetate, 2 μl of GlycoBlue (Thermo Fisher Scientific) and 100 μl of isopropanol, incubating at -20°C overnight, and centrifuging at 20,000g for 30 minutes. The pellet was washed once with 70% ethanol and dissolved in 3.5 μl of H2O. Small RNA cloning was performed starting from the PIR-1 treatment step as described above in the total small RNA cloning method, using 50% (1.75 μl) of the sample.
Small RNA Data Analysis
Small RNA sequencing results were analyzed using an established pipeline (Gu et al., 2012b). Briefly, sequencing reads were sorted according to barcode sequences, and both 5′ and 3′ adaptor sequences were removed using a custom Perl script. Reads starting with G and 21~23 nt in length were mapped to WormBase WS215 allowing at most two mismatches and normalized to non-structural RNA reads. To account for differences in sequencing depth between samples, each read were normalized to total number of reads. Normalized counts were visualized in the UCSC genome browser. All scripts are available upon request.
Generation of scatter plots
IP-enriched 22Gs targeting protein coding genes were binned into their target genes and their read counts normalized to input. Enrichment criteria is having at least 2 ppm per gene in IP data, and minimum 2-fold change comparing IP to input data. The normalized read counts in log2 from wild-type or znfx-1 mutant in triplicates were then plotted against each other.
Analysis of distribution of 22Gs along the gene length
We created five categories: all CSR-1 targeted genes, all WAGO targeted genes, genes whose 22Gs go up in znfx-1 compared to wildtype, genes whose 22Gs go down in znfx-1 compared to wildtype, and genes whose 22Gs do not change between znfx-1 and wildtype. For each category, we filtered for genes having at least 5ppm overall 22G targeting, and normalized each 22G to total 22G abundance within that gene. We binned normalized 22G values over 100 equal-length hypothetical segments. Then we plotted the cumulative 22G values for each percentile. The plots show average values from six independently grown worm populations with error bars representing standard deviations.
Bioinformatic Analysis of P-CLASH Data
Total small RNA sequencing data were processed as follows. Adapter sequences and low-quality reads were removed using Trimmomatic (version 0.33). The short reads were then mapped to the genome using STAR (version 2.4.2a) and normalized to sequencing depth. Short reads 21-to-23-bp long with 5′G were defined as 22G-RNAs. Their 5′-end signals were aggregated for each position around the reported piRNA target sites (Shen et al., 2018) and normalized to the number of transcriptomic C at these positions. The analyses were repeated on all piRNA-target hybrids, as well as on piRNA-target hybrids with the changes in Gibbs free energy (ΔG) < -20 calculated using RNAfold in Vienna RNA Package (version 2.3.5).
Quantification and Statistical Analysis
T-test is used in Figure S6 in order to calculate the significance of comparison. P-values and other details can be found in figure legends.
Data and Software Availability
Small RNA sequencing data can be accessed through GEO (GEO: GSE112708). Bowtie-2 is used for alignments. Custom in-house scripts are utilized to do further analysis and generate plots. These scripts are available upon request.
Supplementary Material
Highlights.
-
–
Genetic identification of ZNFX-1, a conserved epigenetic inheritance factor.
-
–
ZNFX-1 balances the transgenerational inheritance of epigenetic information.
-
–
ZNFX-1 interacts with RdRP and three distinct Argonaute systems in germline nuage.
-
–
ZNFX-1 promotes balanced amplification of small-RNA signals along germline mRNAs.
Acknowledgments
We thank members of Mello, Ambros, Zamore labs for discussions and suggestions; G. Wan and S. Kennedy for sharing unpublished results; P. Wu and P. Zamore for sharing unpublished results; D. Conte, Jr for critical review of the manuscript; W. Gu for providing the reagents for library preparation and technical support for the targeted PCR amplicon bulk sequencing; E. Kittler and the UMass Deep Sequencing Core for Illumina sequencing; K. McJunkin for Galaxy analysis; E. Hudson and V. Balog for initial mapping of mutants, Y. Yang for image processing and G. Konar Jr. and I. Quiles for technical assistance. Some of the strains were provided by the Caenorhabditis Genetics Center supported by NIH (P40 OD010440). The work was supported by Hughes Medical Institute and NIH grants (GM058800 and HD078253) to C.C.M. C.C.M. is a Howard Hughes Medical Institute Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Mendeley Data: https://data.mendeley.com/datasets/fwfj6fc23k/draft?a=9b6b91ed-1693-4f74-9764-39fe1391feaf
ARTHUR CONTRIBUTIONS
Conceptualization, T.I. and C.C.M.; Investigation, T.I., D.J.D., and R.S.; Visualization, T.I. D.J.D., A.R.O. E.Z.S., H.C. and C.C.M.; Software, A.R.O. E.Z.S. and H.C.; Formal Analysis, A.R.O., E.Z.S., H.C. and C.C.M.; Methodology and Resources, T.I., M.S., M.S., H.C. and E.Z.S.; Writing-Original draft, T.I. and C.C.M.; Writing-Review & Editing, T.I. and C.C.M.; Supervision, C.C.M.
DECLARATION OF INTEREST
The authors declare no competing interests.
References
- Ashe A, Sapetschnig A, Weick EM, Mitchell J, Bagijn MP, Cording AC, Doebley AL, Goldstein LD, Lehrbach NJ, Le Pen J, et al. piRNAs can trigger a multigenerational epigenetic memory in the germline of C. elegans. Cell. 2012;150:88–99. doi: 10.1016/j.cell.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagijn MP, Goldstein LD, Sapetschnig A, Weick EM, Bouasker S, Lehrbach NJ, Simard MJ, Miska EA. Function, targets, and evolution of Caenorhabditis elegans piRNAs. Science. 2012;337:574–578. doi: 10.1126/science.1220952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Ruby JG, Claycomb JM, Chiang R, Fahlgren N, Kasschau KD, Chaves DA, Gu W, Vasale JJ, Duan S, et al. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol Cell. 2008;31:67–78. doi: 10.1016/j.molcel.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Julicher F, Hyman AA. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Malone CD, Aravin AA, Sachidanandam R, Stark A, Hannon GJ. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science. 2008;322:1387–1392. doi: 10.1126/science.1165171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley BA, Burkhart KB, Gu SG, Spracklin G, Kershner A, Fritz H, Kimble J, Fire A, Kennedy S. A nuclear Argonaute promotes multigenerational epigenetic inheritance and germline immortality. Nature. 2012;489:447–451. doi: 10.1038/nature11352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart KB, Guang S, Buckley BA, Wong L, Bochner AF, Kennedy S. A Pre-mRNA–Associating Factor Links Endogenous siRNAs to Chromatin Regulation. PLOS Genetics. 2011;7:e1002249. doi: 10.1371/journal.pgen.1002249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel SE, Martienssen RA. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet. 2013;14:100–112. doi: 10.1038/nrg3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Simard MJ, Tabara H, Brownell DR, McCollough JA, Mello CC. A member of the polymerase beta nucleotidyltransferase superfamily is required for RNA interference in C. elegans. Curr Biol. 2005;15:378–383. doi: 10.1016/j.cub.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Claycomb JM, Batista PJ, Pang KM, Gu W, Vasale JJ, van Wolfswinkel JC, Chaves DA, Shirayama M, Mitani S, Ketting RF, et al. The Argonaute CSR-1 and its 22G-RNA cofactors are required for holocentric chromosome segregation. Cell. 2009;139:123–134. doi: 10.1016/j.cell.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conine CC, Moresco JJ, Gu W, Shirayama M, Conte D, Jr, Yates JR, 3rd, Mello CC. Argonautes promote male fertility and provide a paternal memory of germline gene expression in C. elegans. Cell. 2013;155:1532–1544. doi: 10.1016/j.cell.2013.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmay T, Hamilton A, Mueller E, Baulcombe DC. Potato virus X amplicons in arabidopsis mediate genetic and epigenetic gene silencing. Plant Cell. 2000;12:369–379. doi: 10.1105/tpc.12.3.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das PP, Bagijn MP, Goldstein LD, Woolford JR, Lehrbach NJ, Sapetschnig A, Buhecha HR, Gilchrist MJ, Howe KL, Stark R, et al. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol Cell. 2008;31:79–90. doi: 10.1016/j.molcel.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchaine TF, Wohlschlegel JA, Kennedy S, Bei Y, Conte D, Jr, Pang K, Brownell DR, Harding S, Mitani S, Ruvkun G, et al. Functional proteomics reveals the biochemical niche of C. elegans DCR-1 in multiple small-RNA-mediated pathways. Cell. 2006;124:343–354. doi: 10.1016/j.cell.2005.11.036. [DOI] [PubMed] [Google Scholar]
- Gu SG, Pak J, Guang S, Maniar JM, Kennedy S, Fire A. Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint. Nat Genet. 2012a;44:157–164. doi: 10.1038/ng.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Lee HC, Chaves D, Youngman EM, Pazour GJ, Conte D, Jr, Mello CC. CapSeq and CIP-TAP identify Pol II start sites and reveal capped small RNAs as C. elegans piRNA precursors. Cell. 2012b;151:1488–1500. doi: 10.1016/j.cell.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Shirayama M, Conte D, Jr, Vasale J, Batista PJ, Claycomb JM, Moresco JJ, Youngman EM, Keys J, Stoltz MJ, et al. Distinct argonaute-mediated 22G-RNA pathways direct genome surveillance in the C. elegans germline. Mol Cell. 2009;36:231–244. doi: 10.1016/j.molcel.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang S, Bochner AF, Burkhart KB, Burton N, Pavelec DM, Kennedy S. Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature. 2010;465:1097–1101. doi: 10.1038/nature09095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang S, Bochner AF, Pavelec DM, Burkhart KB, Harding S, Lachowiec J, Kennedy S. An Argonaute transports siRNAs from the cytoplasm to the nucleus. Science. 2008;321:537–541. doi: 10.1126/science.1157647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999;286:950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem Sci. 2011;36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L, Hamilton AJ, Voinnet O, Thomas CL, Maule AJ, Baulcombe DC. RNA-DNA interactions and DNA methylation in post-transcriptional gene silencing. Plant Cell. 1999;11:2291–2301. doi: 10.1105/tpc.11.12.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kawasaki I, Shim YH, Kirchner J, Kaminker J, Wood WB, Strome S. PGL-1, a predicted RNA-binding component of germ granules, is essential for fertility in C. elegans. Cell. 1998;94:635–645. doi: 10.1016/s0092-8674(00)81605-0. [DOI] [PubMed] [Google Scholar]
- Kennedy S, Wang D, Ruvkun G. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature. 2004;427:645. doi: 10.1038/nature02302. [DOI] [PubMed] [Google Scholar]
- Kim H, Ishidate T, Ghanta KS, Seth M, Conte D, Jr, Shirayama M, Mello CC. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 2014;197:1069–1080. doi: 10.1534/genetics.114.166389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Gu W, Shirayama M, Youngman E, Conte D, Jr, Mello CC. C. elegans piRNAs mediate the genome-wide surveillance of germline transcripts. Cell. 2012;150:78–87. doi: 10.1016/j.cell.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. The significance of responses of the genome to challenge. Science. 1984;226:792–801. doi: 10.1126/science.15739260. [DOI] [PubMed] [Google Scholar]
- Mette MF, van der Winden J, Matzke MA, Matzke AJ. Production of aberrant promoter transcripts contributes to methylation and silencing of unlinked homologous promotersin trans. EMBO J. 1999;18:241–248. doi: 10.1093/emboj/18.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minevich G, Park DS, Blankenberg D, Poole RJ, Hobert O. CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics. 2012;192:1249–1269. doi: 10.1534/genetics.112.144204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Fine NA, Fujisawa T, Gorovsky MA. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in tetrahymena. Cell. 2002;110:689–699. doi: 10.1016/s0092-8674(02)00909-1. [DOI] [PubMed] [Google Scholar]
- Motamedi MR, Verdel A, Colmenares SU, Gerber SA, Gygi SP, Moazed D. Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding centromeric RNAs. Cell. 2004;119:789–802. doi: 10.1016/j.cell.2004.11.034. [DOI] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Rasoloson D, Seydoux G. High Efficiency, Homology-Directed Genome Editing in Caenorhabditis elegans Using CRISPR-Cas9 Ribonucleoprotein Complexes. Genetics. 2015;201:47–54. doi: 10.1534/genetics.115.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CM, Montgomery TA, Breen PC, Ruvkun G. MUT-16 promotes formation of perinuclear mutator foci required for RNA silencing in the C. elegans germline. Genes Dev. 2012;26:1433–1444. doi: 10.1101/gad.193904.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt JN, Schisa JA, Priess JR. P granules in the germ cells of Caenorhabditis elegans adults are associated with clusters of nuclear pores and contain RNA. Dev Biol. 2000;219:315–333. doi: 10.1006/dbio.2000.9607. [DOI] [PubMed] [Google Scholar]
- Seth M, Shirayama M, Gu W, Ishidate T, Conte D, Jr, Mello CC. The C. elegans CSR-1 argonaute pathway counteracts epigenetic silencing to promote germline gene expression. Dev Cell. 2013;27:656–663. doi: 10.1016/j.devcel.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth M, Shirayama M, Tang W, Shen EZ, Tu S, Lee HC, Weng Z, Mello CC. The Coding Regions of Germline mRNAs Confer Sensitivity to Argonaute Regulation in C.elegans. Cell Reports. 2018;22:2254–2264. doi: 10.1016/j.celrep.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen EZ, Chen H, Ozturk AR, Tu S, Shirayama M, Tang W, Ding YH, Dai SY, Weng Z, Mello CC. Identification of piRNA Binding Sites Reveals the Argonaute Regulatory Landscape of the C. elegans Germline. Cell. 2018;172:937–951.e918. doi: 10.1016/j.cell.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Pitt J, Dennis S, Priess JR. Perinuclear P granules are the principal sites of mRNA export in adult C. elegans germ cells. Development. 2010;137:1305–1314. doi: 10.1242/dev.044255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama M, Seth M, Lee HC, Gu W, Ishidate T, Conte D, Jr, Mello CC. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell. 2012;150:65–77. doi: 10.1016/j.cell.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Krishna S, Thanos D, Strominger JL, Ono SJ. A novel cysteine-rich sequence-specific DNA-binding protein interacts with the conserved X-box motif of the human major histocompatibility complex class II genes via a repeated Cys-His domain and functions as a transcriptional repressor. J Exp Med. 1994;180:1763–1774. doi: 10.1084/jem.180.5.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Updike D, Strome S. P granule assembly and function in Caenorhabditis elegans germ cells. J Androl. 2010;31:53–60. doi: 10.2164/jandrol.109.008292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SIS, Moazed D. RNAi-Mediated Targeting of Heterochromatin by the RITS Complex. Science. 2004;303:672–676. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- Wedeles CJ, Wu MZ, Claycomb JM. Protection of germline gene expression by the C. elegans Argonaute CSR-1. Dev Cell. 2013;27:664–671. doi: 10.1016/j.devcel.2013.11.016. [DOI] [PubMed] [Google Scholar]
- Yigit E, Batista PJ, Bei Y, Pang KM, Chen CC, Tolia NH, Joshua-Tor L, Mitani S, Simard MJ, Mello CC. Analysis of the C. elegans Argonaute family reveals that distinct Argonautes act sequentially during RNAi. Cell. 2006;127:747–757. doi: 10.1016/j.cell.2006.09.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.