

Abstract
Summary: It is common for computational analyses to generate large amounts of complex data that are difficult to process and share with collaborators. Standard methods are needed to transform such data into a more useful and intuitive format. We present ReportingTools, a Bioconductor package, that automatically recognizes and transforms the output of many common Bioconductor packages into rich, interactive, HTML-based reports. Reports are not generic, but have been individually designed to reflect content specific to the result type detected. Tabular output included in reports is sortable, filterable and searchable and contains context-relevant hyperlinks to external databases. Additionally, in-line graphics have been developed for specific analysis types and are embedded by default within table rows, providing a useful visual summary of underlying raw data. ReportingTools is highly flexible and reports can be easily customized for specific applications using the well-defined API.
Availability: The ReportingTools package is implemented in R and available from Bioconductor (version ≥ 2.11) at the URL: http://bioconductor.org/packages/release/bioc/html/ReportingTools.html. Installation instructions and usage documentation can also be found at the above URL.
Contact: hackney.jason@gene.com or kaminker.josh@gene.com
1 INTRODUCTION
Computational analyses often generate large and complex datasets that need to be shared with collaborators. As such, methods are needed to produce standardized reports that summarize analysis results and can be easily and interactively explored. ReportingTools performs this role by providing methods that automatically detect output from many common Bioconductor packages (Gentleman et al., 2004) and transforms the content into intuitively designed rich HTML documents.
Although the use of HTML to present data is not novel (Gehlenborg et al., 2013; Lecoutre, 2003; Pau and Huber, 2009; White, 2010), the development of software to recognize result types from common Bioconductor packages and generate specific summary reports based on the result type is new, and provides a powerful tool for analyzing data. ReportingTools automatically detects the output of many genomic analysis packages and produces an HTML report reflecting specific scientific content from the analysis. Packages for which outputs are automatically detected include limma for analysis of microarray expression data, GSEAlm for gene set analysis, GOstats for ontology analyses and edgeR, DESeq, DESeq2, DEXSeq for RNA-seq-based analyses (Falcon and Gentleman, 2007; Oron et al., 2008; Robinson et al., 2010; Smyth, 2004). Additional analysis packages can be easily accommodated by extending the ReportingTools code base.
ReportingTools provides a common programmatic interface to process and generate reports from many standard computational analyses. A single method call is needed to detect and process results from known analysis types, and two additional lines of code are needed to generate the interactive HTML report that can be shared with collaborators. Finally, for added flexibility in presenting output, ReportingTools has been integrated with knitr and shiny (RStudio, 2013; Xie, 2013). ReportingTools provides important functionality for users generating highly customized analysis reports, or for core facilities that regularly present standardized automated output to collaborators.
2 METHODS
ReportingTools was initially developed as a toolkit to automatically fetch and present gene annotations for gene expression studies. The annotations included hyperlinks to corresponding Entrez gene pages to aid in the navigation and exploration of the analysis results. ReportingTools was subsequently extended to include analysis-specific in-line graphics to provide a visual summary of underlying raw data. Combining the statistical analysis results with the in-line graphics and hyper-linked annotations gave rise to a rich HTML report document. Finally, with the addition of flexible open-source JavaScript libraries and css styling, the report became a highly stylized and interactive document with search, filter and sort functionalities.
ReportingTools can also process generic tabular data and automatically generate user-specified entities for the final report. For more complex analysis types not yet supported by ReportingTools, it is straightforward to extend ReportingTools to specify the content of a new analysis-specific report.
This utility of ReportingTools is best illustrated by describing the default report generated from an RNA expression analysis using the limma package (see Fig. 1 or online examples at http://research-pub.gene.com/ReportingTools). In this example, using only three lines of code (see later in the text), a limma result is recognized, processed and returned as a limma-specific rich HTML report that can be presented on a web server or emailed to collaborators:
report <- HTMLReport (output_file_name, report_title, output_directory)
publish (limma_fit, report, eSet = eSet, factor = eSet$group, coef = 1)
finish(report)
Fig. 1.
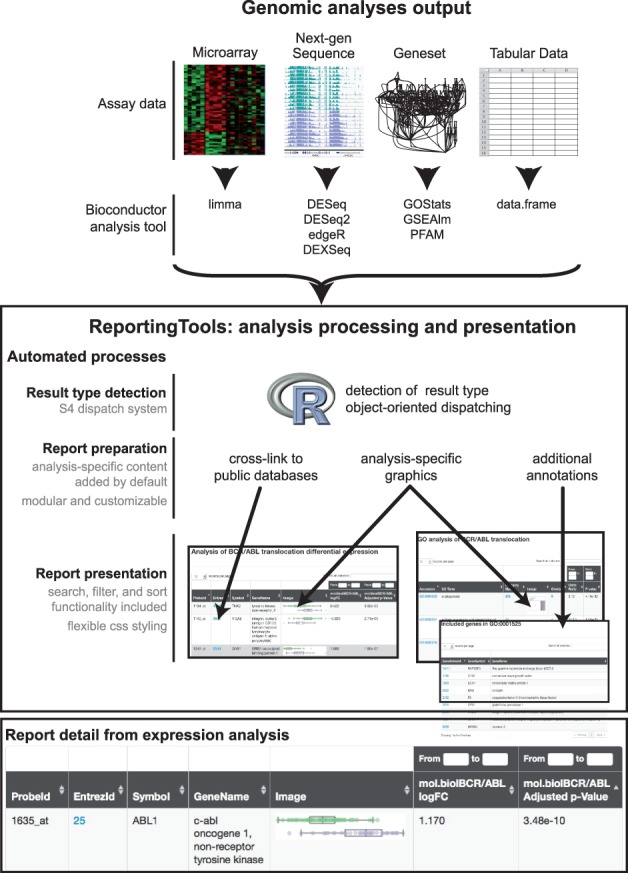
ReportingTools dispatches on results from upstream analyses and automatically prepares analysis-specific graphics, cross-links, annotations, statistics and data for presentation. The final report presents the analysis results and related data in an HTML or knitr document with additional search, filter and sort functionality. For more examples, including fully interactive reports, see http://research-pub.gene.com/ReportingTools
Each row of this report corresponds to an Entrez gene identifier, and descriptive columns are included for Entrez gene identifier, gene name, gene description, fold-change and P-value. In addition, ReportingTools output for limma includes hyperlinks for each gene to the corresponding NCBI Entrez Gene page. Finally, each row includes an in-line box plot with individual datapoints overlaid to allow users to quickly visualize the raw underlying expression values of a gene across all samples in the analysis to help recognize biases that might impact the interpretation of results. Note that as this automatically generated report was specifically designed to present limma data, other automatically generated reports from other Bioconductor packages will not necessarily include the same content.
3 CONCLUSION
ReportingTools offers numerous advantages for analysis workflows. First, ReportingTools automatically detects the R object classes produced by many common Bioconductor packages, reducing the overhead needed to extract, process and present the data in a standardized analysis-specific manner. This encourages and facilitates reproducible research (Gentleman, 2005) by reducing the amount of manual intervention and one-off coding to go from raw data to consumable report. Second, the inclusion of in-line graphics provides a highly effective way to summarize underlying raw data. Third, it is straightforward to extend the code with the flexible low-level interface to support additional result types. Reports can also be highly customized by overriding package defaults and further customized using the lower level interfaces we make available to the users. Fourth, the integration of ReportingTools with knitr and shiny provides many additional and useful options for presenting results. Reports generated by ReportingTools are easily reproduced and provide a consistent point of entry to the results of computational analyses. This facilitates data sharing and enables analysts and collaborators to spend more of their effort inspecting and exploring analysis results.
ACKNOWLEDGEMENTS
The authors thank Anneleen Daemen, Brad Friedman, Sarah Kummerfeld, Matt Brauer, Peter Haverty, Thomas Sandmann, Peng Yue, Gregoire Pau, Gerard Manning and three anonymous reviewers for their helpful comments and feedback during the development of the ReportingTools package and manuscript. They also thank Allison Bruce for her contributions in the graphical design of the manuscript figures.
Conflicts of Interest: none declared.
REFERENCES
- Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- Gehlenborg N, et al. Nozzle: a report generation toolkit for data analysis pipelines. Bioinformatics. 2013;29:1089–1091. doi: 10.1093/bioinformatics/btt085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R. Reproducible research: a bioinformatics case study. Stat. Appl. Genet. Mol. Biol. 2005;4 doi: 10.2202/1544-6115.1034. Article 2. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoutre E. The R2HTML package. R News. 2003;3:33–36. [Google Scholar]
- Oron AP, et al. Gene set enrichment analysis using linear models and diagnostics. Bioinformatics. 2008;24:2586–2591. doi: 10.1093/bioinformatics/btn465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pau G, Huber W. The hwriter package. R. J. 2009;1:22–24. [Google Scholar]
- Robinson MD, et al. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio I. shiny: web application framework for R. R package. 2013 http://www.rstudio.com/shiny/ (16 October 2013, date last accessed) [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- White J. SortableHTMLTables: turns a data frame into an HTML file containing a sortable table. R package. 2010 http://cran.r-project.org/web/packages/SortableHTMLTables/index.html (16 October 2013, date last accessed) [Google Scholar]
- Xie Y. Dynamic Documents with R and knitr. London, UK: Chapman and Hall/CRC; 2013. ISBN 978-1482203530. [Google Scholar]