

Abstract
Motivation: Various human pathogens secret effector proteins into hosts cells via the type IV secretion system (T4SS). These proteins play important roles in the interaction between bacteria and hosts. Computational methods for T4SS effector prediction have been developed for screening experimental targets in several isolated bacterial species; however, widely applicable prediction approaches are still unavailable
Results: In this work, four types of distinctive features, namely, amino acid composition, dipeptide composition, .position-specific scoring matrix composition and auto covariance transformation of position-specific scoring matrix, were calculated from primary sequences. A classifier, T4EffPred, was developed using the support vector machine with these features and their different combinations for effector prediction. Various theoretical tests were performed in a newly established dataset, and the results were measured with four indexes. We demonstrated that T4EffPred can discriminate IVA and IVB effectors in benchmark datasets with positive rates of 76.7% and 89.7%, respectively. The overall accuracy of 95.9% shows that the present method is accurate for distinguishing the T4SS effector in unidentified sequences. A classifier ensemble was designed to synthesize all single classifiers. Notable performance improvement was observed using this ensemble system in benchmark tests. To demonstrate the model’s application, a genome-scale prediction of effectors was performed in Bartonella henselae, an important zoonotic pathogen. A number of putative candidates were distinguished.
Availability: A web server implementing the prediction method and the source code are both available at http://bioinfo.tmmu.edu.cn/T4EffPred.
Contact: hoofuquan@yahoo.com.cn
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Bacterial pathogens translocate numerous effector proteins into the extracellular environment of host cells via secretion systems. These proteins play critical roles in virulence and survival. In gram-negative bacteria, secretion systems are classified into seven major evolutionarily and functionally related subclasses, termed types I–VII (Tseng et al., 2009). In recent years, type III and type IV secretion systems (T3SS and T4SS), which allow injection of effectors directly into the host cell cytoplasm, have been widely studied (Cambronne and Roy, 2006; Galan and Wolf-Watz, 2006; Llosa et al., 2009). T4SS consists of transmembrane multi-protein complexes and exists in many zoonotic pathogens, such as Brucella sp., Bartonella henselae, Coxiella burnetii, as well as in some other gram-negative pathogens, such as Bordetella pertussis, Helicobacter pylori and L.pneumophila (Chandran et al., 2009; Fronzes et al., 2009). Many T4SS effectors, including two main subtypes called IVA and IVB, have been confirmed to be involved in the pathogenicity of these species (Llosa et al., 2009). To adapt to different hosts and different survival strategies, the arsenal of known effectors varies widely across bacterial species and even shows distinct differences between bacterial strains.
Recently, a number of novel T4SS effectors have been identified by experimental approaches such as fusion protein report assays (Burstein et al., 2009; Chen et al., 2010; Lockwood et al., 2011; Marchesini et al., 2011; Zhu et al., 2011). In many of these studies, previously developed bioinformatics screening strategies were adopted to predict effector candidates in genomic proteins before experimental translocation verification. Sequence homology to known effectors and conserved domain analysis were used to detect putative effectors in the genome of Brucella abortus (Marchesini et al., 2011). An omnibus method, synthesizing hydropathy profile comparison, eukaryotic domain search, signal peptide identification and sequence similarity alignment, was built to scan genomic sequences of Anaplasma marginale for potential T4SS effectors (Lockwood et al., 2011). Two approaches based on feature extraction and machine learning algorithms were developed for large-scale effector identification in L.pneumophila (Burstein et al., 2009) and C.burnetii (Chen et al., 2010), respectively. A hidden semi-Markov model (HSMM) was constructed to describe C-terminal patterns and predict type-IVB secretion signals with high accuracy (Lifshitz et al., 2013). Established methods for detecting the regulatory elements or C-terminal translocation signals of eukaryotic proteins were applied before screening of the experimental targets, resulting in most of the T4SS effectors being identified in L.pneumophila (Segal, 2013; Zhu et al., 2011).
The computational filters in these existing methods successfully selected a small portion of the genomic proteins as candidates for experimental identification. However, these methods are not exhaustive enough and are not generally applicable for secretion signal detection, especially for modelling type-IVA signal. Homology search based on sequence alignment can only find candidates that are similar to known effectors. Scanning of regulatory elements or C-terminal signals relies on the known motifs in the sequences of effectors and their promoters, which are conserved but specific to effector families. Furthermore, all existing machine learning models are based on sequence features from a small supply of known effectors from particular bacterial species. Therefore, they are often not accurate for genomic effector discovery in other bacterial species. There are <300 experimentally validated effectors in the 1884 known effectors stored in the SecReT4 database (Bi et al., 2013). Although several methods have reported excellent performance in identifying T3SS effectors in a wide variety of bacterial genomes (Arnold et al., 2009; Sato et al., 2011; Wang et al., 2011; Yang et al., 2010), they are not recommended for T4SS effector prediction due to low accuracies. Accurate prediction approaches that are widely applicable to mining putative type-IVA or type-IVB effectors in gram-negative pathogens are still absent.
Several computational approaches based on existing machine learning algorithms, e.g. Naive Bayes, hidden Markov models, artificial neural network and support vector machine (SVM), have successfully predicted T3SS secreted signals (Arnold et al., 2009; Lower and Schneider 2009; Samudrala et al., 2009; Wang et al., 2011; Yang et al., 2010). However, the accuracy of machine learning approach depends on the quantities of authentic negative and positive samples. Studies identifying T3SS effectors are abundant, but those identifying T4SS effectors are rare. The two recently published databases, AtlasT4SS (Souza et al., 2012) and SecRet4 (Bi et al., 2013), have announced freely available datasets for T4SS components and secreted proteins. These datasets provide numerous samples to train and optimize machine learning models for T4SS effectors prediction.
In this article, we propose a SVM model using four types of features calculated from residue composition and position-specific scoring matrix (PSSM) profiles. The machine learning-based model is trained on a set of experimentally validated T4SS effectors and 1132 non-effectors and is used to provide accurate predictions of T4SS secreted proteins in evolutionarily distinct bacteria. We demonstrate that our model can discriminate between IVA effectors (or IVB effectors) and non-effectors, with an accuracy of 93.3% (or 95.9%), via leave-one-tests. To the best of our knowledge, this is the first method to predict IVA effectors in gram-negative bacteria. Genomic-scale prediction of effectors in the zoonotic pathogen B.henselae is also performed and discussed. A list of predicted candidates from B.henselae may be of academic interest in studying human pathogenic bacteria.
2 METHODS
2.1 Dataset
We constructed a dataset of known effectors and a dataset of non-effectors. To collect known effectors, all experimentally validated effector sequences were extracted from the effector dataset in the SecRet4 database. In addition, other T4SS effectors confirmed by experiments were collected from other studies. Four hundred and twenty six effectors (51 IVA sequences and 375 IVB sequences) comprised the initial effector dataset. Because a dataset of experimentally validated non-effectors was unavailable, we searched for genes from 10 T4SS pathogens that are also present as homologous genes in the Escherichia coli genome. These pathogens, including Agrobacterium sp., A.marginale, B.henselae, B.pertussis, Brucella melitensis, C.burnetii, Ehrlichia chaffeensis, H.pylori, L.pneumophila and Ochrobactrum anthropic, secrete a majority of the currently known T4SS effectors. The genes that exist in these organisms and in E.coli are most likely not associated with pathogenicity and are thus expected to be non-effectors. To document these genes, the genomic proteins from the 10 pathogens were compared against E.coli proteins using Basic Local Alignment Search Tool (BLAST). Each pathogen protein with an E-value < 1e-20 and sequence similarity with E.coli > 50% was extracted. The 1000 non-effector proteins collected by Lifshitz et al. (Lifshitz et al., 2013) and a number of proteins in UniProt that have been annotated and reviewed as non-secreted proteins were also appended in the non-effector list. Additionally, all experimentally validated T4SS component proteins derived from the SecRet4 database were included in the non-effector dataset. Furthermore, 471 proteins from the 10 pathogens with unique experimentally validated subcellular localization in ePSORTb (intracellular and extracellular with definite functions) (Rey et al., 2005) were also filtered out and added to the complete non-effector list containing 5649 sequences.
To filter out orthologous and paralogous proteins, two steps were performed successively. First, all sequences in each dataset were clustered using BLAST, with the parameters of 20% sequence identity and 80% coverage. Only one seed sequence in each cluster remained for the next step. Second, an all-against-all comparison of full-length sequences using the Smith–Waterman algorithm was performed. The Water program in the European Molecular Biology Open Software Suite (EMBOSS) package was used to implement this pairwise alignment process. For each pair, the ratio between the pairwise score, Spair, and the self-alignment score, Sself, was computed, and sequences were iteratively grouped if they showed a Spair/Sself value ≥0.15. This measure has excellent sensitivity and is similar to the measure used by Arnold et al. (Arnold et al., 2009) for the detection of putative orthologs in T3SS effector sequences. After these steps, 340 effectors (30 IVA proteins and 310 IVB proteins) and 1132 non-effectors were retained in the final datasets, which are collectively referred to as T4_1472 in this study.
2.2 Composition of amino acids and amino acid pairs
For protein sequence A, we used a 20-D vector {f1, f2, … , f20} and a 400-D vector {d1, d2, … , d400} to represent the composition of 20 amino acids and 400 amino acid pairs, respectively. The 20 elements in {f1, f2, … , f20} represent the number of amino acids normalized with the total number of residues of A. The 400 elements in {d1, d2, … , d400} represent the number of 400 amino acid pairs normalized with the total number of residue pairs of A.
2.3 PSSM profiles and auto covariance transformation
The PSSM of a protein contains the protein’s amino acid substitution scores. Hence, we adopted PSSM profiles, which were used for subcellular localization prediction (Xie et al., 2005) and protein structural classification (Chen et al., 2011; Liu et al., 2010) to calculate the evolutionary features of T4SS secreted proteins. In this study, the PSSM profile of each protein was generated by running Position-Specific Iterative Basic Local Alignment Search Tool (PSI-BLAST) against the NCBI’s non-redundant (nr) protein database (version 2012.12.01) with the parameters h = 0.001 and j = 3. The nr database is freely downloadable at ftp://ftp.ncbi.nih.gov/. The (i, j)th entry of the resulting matrix represents integral score of the amino acid in the ith position of the query sequence mutated to amino acid type j during the evolution process. Two features, namely, PSSM composition and auto covariance transformation, were extracted from the PSSM profiles. The PSSM composition was generated by summing the amino acid rows in the PSSM. The details of how to generate 400 composition features from the original PSSM profiles are shown in Supplementary Figure S1. Because each residue has many physicochemical properties, such as polarity, solvent accessibility, hydrophobicity, sequence profiles and so on, a protein sequence can be represented as a numeric matrix. Auto covariance transformation can measure the correlation of two properties (or the same property) along the protein sequence and transform the matrix into a fixed-length vector (Dong et al., 2009). For the auto covariance transformation, let us denote the PSSM of a protein sequence as:
| (1) |
where Si (i = 1, 2, … , 20) is the column vector of amino acid type i in the matrix. We also denote each column vector as:
| (2) |
where L is the length of the protein sequence and si,j denotes the score of number j residue in position i corresponding to the sequence order. The function of auto covariance transformation is defined as the following:
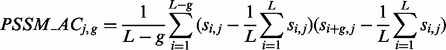 |
(3) |
(j = 1, 2, … , 20, g = 1, 2, … , G). Hence, auto covariance transformation of PSSM (PSSM_AC) has 20*G features, where G is a positive integer that indicates the grouped number of the transformation.
In this study, PSSM_AC transforms the PSSM profile of a sequence into a vector by calculating the correlation of its properties (i.e. evolutionary conservation of 20 residues in each position) between two residues separated by a distance of G along the sequence.
2.4 SVM classifier
SVM is a machine learning algorithm that has been widely used for classification purposes. It is described in many other publications; therefore, it will not be discussed in detail in this article. We used the publicly available software the Library for Support Vector Machines (LIBSVM) for the implementation of our SVM classifier. The LIBSVM toolbox can be freely downloaded at http://www.csie.ntu.edu.tw/∼cjlin/libsvm. We integrated this toolbox in the Matrix Laboratory (MATLAB) workspace to build the prediction system. Here, the radial basis function was chosen as the kernel function and the SVM parameter γ and penalty parameter C optimized using a grid search based on a 10-fold cross-validation.
2.5 Protein sequence representation
We used four types of feature vectors to represent a protein sequence [amino acid composition (AAC), residue pair composition (dipeptide composition, DPC), PSSM profile composition (PSSM) and PSSM_AC]. In addition, the AAC vector and the DPC vector were combined with the PSSM vector and the PSSM_AC vector, respectively, to obtain fixed-length vectors. We examined the SVMs based on different vectors to assess prediction performance in T4_1472. A diagram that describes the computational steps of this study is shown in Supplementary Figure S2.
2.6 Performance assessment
The prediction performance in T4_1472 was assessed by four measures: Acc, Sn, Sp and Matthew’s correlation coefficient (MCC) (Matthews, 1975). These measures are defined as follows:
| (4) |
| (5) |
| (6) |
| (7) |
where TP, FP, TN and FN refer to the number of true positives, false positives, true negatives and false negatives, respectively. A MCC coefficient of one represents a perfect prediction, whereas zero indicates a completely random assignment. Acc is the overall accuracy in discriminating between effectors and non-effector proteins. Sn and Sp are the sensitivity and the specificity values, which measure the ability to correctly predict effectors and correctly reject non-effectors. We also used the receiver-operating characteristic (ROC) curve to measure the present model’s true-positive rate and low false-positive rate during the prediction. Furthermore, the ROC curve can be quantified by the area under the curve (AUC), which is usually more accurate in evaluating learning algorithms.
3 RESULTS
3.1 ACC of effectors and non-effectors
We computed the ACC and the variance in T4_1472 (Fig. 1). We noticed that the residues Ala, Asn, Glu, Gly, Ile, Leu, Lys, Phe, Ser and Val had variances >1. In these residues, Asn, Glu and Lys had higher compositions in IVB effectors. In contrast, Ala, Gly and Val had higher composition in non-effectors. Somewhat differently, Ala, Glu and Ser occurred more frequently in IVA effectors than in non-effectors but Ile, Leu and Phe did not. Some polar amino acids, such as Asp, Cys and His, have small differences between secreted proteins and non-secreted proteins. The detailed data are shown in Supplementary Table S1.
Fig. 1.
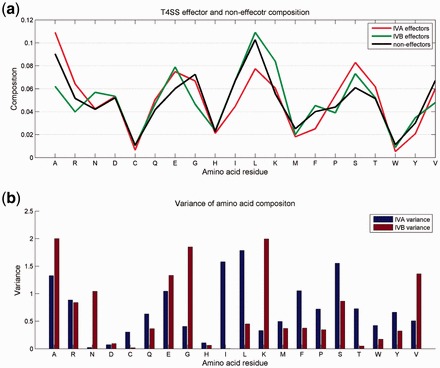
(a) ACC in effectors and non-effectors; (b) variance of 20 amino acid residues between effectors and non-effectors
We also calculated the dipeptide composition and the variance for all 400 possible residue pairs in T4_1472. Those pairs with the topmost variances are listed in Supplementary Tables S2 and S3. The pairs SS, LL and AA showed variances >0.3 between IVA proteins and non-effectors; however, the pairs KE, AA and AG had variances >0.3 between IVB proteins and the non-effectors. Most of the residue pairs with Ala had higher variances than the others. In contrast, some pairs with Cys, His and Tyr, such as YM and IH, had minute composition differences between effectors and non-effectors (data not shown).
3.2 The effect of G for auto covariance transformation
As mentioned in Section 2.3, the theoretical maximum number of G is the length of the shortest sequence in the dataset. G defines the maximum distance between two positions in the target sequence. The optimal value of G varies for different datasets. By setting G from 1 to 20, 20 PSSM_AC vectors with different lengths were calculated for protein representation. We examined the performance of IVB effector discrimination using SVMs with these features in T4_1472 by 5-fold cross-validation tests. The variability curve of the prediction rate is shown in Figure 2. The curve first ascends continuously to a maximum value and then drops slightly with the increase in G. The best accuracy of 93.0% was achieved with G = 10 (see Supplementary Table S4 for details). We also tested the prediction accuracy of PSSM_AC vectors with G > 20 and found that these vectors did not lead to performance improvement. The trend of the performance curve of G for IVA effector prediction was in accordance with the above case (data not shown).The best value for G in T4_1472 was 10, which generated a 200-D vector in the PSSM transformation.
Fig. 2.
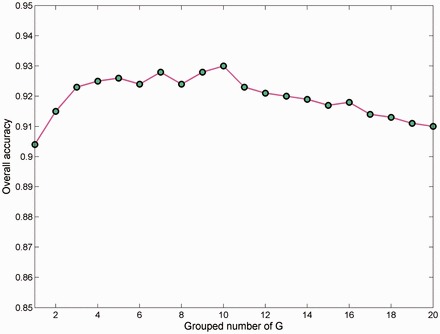
This graph shows how different values of G affect the overall accuracies of the PSSM_AC model for discriminating IVB effectors and non-effectors
3.3 Prediction performance in datasets
Owing to the imbalance between positive samples and negative samples in T4_1472, leave-one-out (LOO) tests were conducted for performance assessment, using radial basis function kernel SVM classifiers with different features as well as their combinations as inputs. We used four single feature vectors and five combination vectors for IVA effector discrimination and IVB effector discrimination. The nine feature vectors and their corresponding results are shown in Table 1. In line with single feature tests, we observed that the performance measures of the four feature categories were different. The classifier using the PSSM composition discriminated IVB effectors and non-effectors with the highest sensitivity (89.4%), which was 6.5% higher than PSSM_AC and 20% higher than the ACC. The PSSM vector is also better than other single vectors for IVA effector prediction. It produced 73.3% sensitivity and 93.3% accuracy with the highest MCC value of 0.782. The capability of two PSSM feature categories, i.e. PSSM and PSSM_AC, is stronger than AAC and DPC. The combined feature tests in Table 1 show that the performance was improved when AAC was combined with PSSM. For IVB effector prediction, the accuracy of AAC improved from 88.7% to 95.9% when it was combined with PSSM. For IVA effector prediction, the highest accuracy of 93.3% and an MCC of 0.784 were achieved by using AAC plus PSSM. We noticed that DPC had the lowest accuracy of all single vectors. DPC predictions did not improve when combined with other features. The sensitivity of these features for IVA effector prediction was generally lower than for IVB effector prediction, but the specificity was higher. This was likely because of a much smaller number of IVA sequences than IVB sequences in T4_1472.
Table 1.
Results of LOO tests using SVM classifiers based on different feature vectors
| Class | Featurea | C|γb | IVA versus non-effectorc |
IVB versus non-effector |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sn | Sp | Acc | MCC | Sn | Sp | Acc | MCC | |||
| Single | AAC(20D) | 10, 0.3 | 0.633 | 0.967 | 0.900 | 0.667 | 0.694 | 0.940 | 0.887 | 0.655 |
| DPC(400D) | 10, 0.0125 | 0.533 | 0.992 | 0.900 | 0.663 | 0.674 | 0.932 | 0.877 | 0.625 | |
| PSSM(400D) | 10, 0.02 | 0.733 | 0.983 | 0.933 | 0.782 | 0.894 | 0.975 | 0.958 | 0.874 | |
| PSSM_AC(200D, G = 10) | 10, 0.1 | 0.667 | 0.967 | 0.907 | 0.691 | 0.829 | 0.957 | 0.929 | 0.790 | |
| Combined | AAC+DPC(420D) | 10, 0.02 | 0.567 | 1.000 | 0.913 | 0.715 | 0.688 | 0.932 | 0.882 | 0.637 |
| PSSM+AAC(420D) | 10, 0.02 | 0.767 | 0.975 | 0.933 | 0.784 | 0.897 | 0.976 | 0.959 | 0.878 | |
| PSSM_AC+AAC(220D) | 10, 0.05 | 0.667 | 0.975 | 0.913 | 0.712 | 0.848 | 0.966 | 0.941 | 0.824 | |
| PSSM+PSSM_AC(600D) | 10, 0.00125 | 0.733 | 0.958 | 0.913 | 0.720 | 0.897 | 0.964 | 0.949 | 0.852 | |
| PSSM+PSSM_AC+AAC(620D) | 10, 0.00125 | 0.700 | 0.983 | 0.927 | 0.759 | 0.897 | 0.964 | 0.950 | 0.853 | |
aAAC: amino acid composition; DPC: dipeptide composition; PSSM: PSSM composition; PSSM_AC: auto covariance transformation of PSSM profiles. The figure in the bracket refers to the dimensions of the features.
bC and γ are the cost and the gamma parameter of the SVM, respectively. They were optimized based on a 10-fold cross-validation grid search.
c120 non-effectors were selected randomly in the non-effector dataset as negative training samples for keeping the ratio of effectors to non-effectors at ∼1:4.
We also executed independent dataset tests to assess the performance of our model for IVA and IVB effector prediction. In these tests, a small portion of positive sequences and negative sequences in T4_1472 was picked up for prediction and others for training. The corresponding results are illustrated in Supplementary Table S5. Supplementary Table S5 shows a similar performance profile to Table 1. For IVB effector prediction, the PSSM vector in combination with AAC outperformed other single vectors and combined vectors. Not surprisingly, the sensitivity of all feature vectors was decreased for IVA effector prediction because of fewer positive training samples and weaker C-terminal signals (Supplementary Fig. S3). PSSM and PSSM_AC were more effective for IVA effector discrimination than other features when they were used alone or in combination with AAC.
The ROC curves in Figure 3 illustrate the performance trends for discriminating IVB effectors and non-effectors using our classification models in 5-fold cross-validation tests. Figure 3a shows that four single feature vectors (i.e. AAC, DPC, PSSM and PSSM_AC) produce different ROCs in 5-fold cross-validation tests. Figure 3b shows ROCs produced by SVMs with four combined vectors. In Figure 3a, the AUC is 0.970, 0.926, 0.904 and 0.892 for PSSM, PSSM_AC, AAC and DPC, respectively. Furthermore, AUCs of 0.970, 0.950 and 0.972 were obtained using four vector combinations [i.e. PSSM plus AAC, PSSM_AC plus AAC, PSSM plus PSSM_AC and AAC plus DPC (see Supplementary Table S6)]. Further analysis indicated that the true-positive rate of PSSM exceeded 85% when the false-positive rate was <3%. Moreover, PSSM plus AAC and PSSM plus PSSM_AC both distinguished IVB effectors with a true-positive rate of over 90% and with a false-positive rate under 4% at the same time.
Fig. 3.
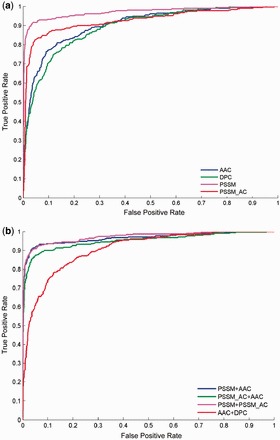
Comparison of ROC curves for IVB effector prediction using different features. The results obtained from amino acid composition (AAC), residue pair composition (DPC), PSSM composition (PSSM) and auto covariance transformation of PSSM profiles (PSSM_AC) as well as the combination of AAC and DPC (AAC+DPC), the combination of AAC and PSSM (PSSM+AAC), the combination of AAC and PSSM_AC (PSSM+ AAC) and the combination of two PSSM feature classes (PSSM+ PSSM_AC) are shown as color curves. (a) Five-fold cross-validation tests in T4_1472 using four single feature classes; (b) 5-fold cross-validation tests in T4_1472 using four combined feature classes
3.4 Prediction performance of the classifier ensemble
We observed that PSSM was the most effective single feature vector for effector prediction. No significant performance improvement was obtained through its combinations with other predictors (shown in Table 1 and Fig. 3). On the other hand, the abilities of the four single vectors to predict T4SS effectors varied. To make full use of these features, we established a classifier ensemble based on four different SVMs, which are shown in Figure 4. We used two voting schemes, 2-in-3 and 3-in-4, to synthesize SVMs outputs and give the final estimate. The 2-in-3 voting system was used for two or more positive predictions in every three classifiers. Likewise the 3-in-4 voting system was used for three or more positive outputs in all four classifiers. We examined the ensemble system via 10-fold cross-validation for IVB effector discrimination in T4_1472. Table 2 shows that the performance of the classifier ensemble is better than independent classifiers. The highest sensitivity and the accuracy of 91.6% and 97.9%, respectively, were obtained using the 2-in-3 voting system based on the three SVM predictors (AAC, PSSM and PSSM_AC) that outperformed the single classifiers in Table 1 and Table 2.
Fig. 4.
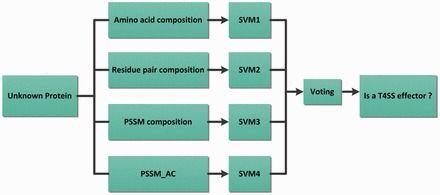
The T4SS effector classifier ensemble based on multi-SVMs with different features
Table 2.
Results for the ensemble prediction of IVB effectors with 10-fold cross-validation tests
| Classifiers | Modela | Votingb | Sn | Sp | Acc | MCC |
|---|---|---|---|---|---|---|
|
1 | — | 0.707 | 0.933 | 0.884 | 0.651 |
| 2 | — | 0.658 | 0.928 | 0.870 | 0.603 | |
| 3 | — | 0.903 | 0.971 | 0.956 | 0.871 | |
| 4 | — | 0.839 | 0.959 | 0.933 | 0.800 | |
| {1,2,3} | 2-in-3 | 0.832 | 0.990 | 0.956 | 0.867 | |
| {1,2,4} | 2-in-3 | 0.816 | 0.984 | 0.948 | 0.842 | |
| {1,3,4} | 2-in-3 | 0.916 | 0.996 | 0.979 | 0.936 | |
| {2,3,4} | 2-in-3 | 0.894 | 0.994 | 0.972 | 0.917 | |
| {1,2,3,4} | 3-in-4 | 0.774 | 0.977 | 0.933 | 0.796 |
aEach term in this column represents the combination form of different classifiers (e.g. {1,2,3} means the ensemble of number 1, 2 and 3 classifiers in the first column).
bThe 2-in-3 voting scheme was adopted for voting on three classifiers and the 3-in-4 voting scheme was adopted for voting on four classifiers. The highest value of each measure is shown in bold.
The characteristics of the classifier ensemble for IVA effector prediction were also detected by LOO tests in T4_1472 (Supplementary Table S7). In the LOO tests, the ensemble of SVM_AAC, SVM_PSSM and SVM_PSSM_AC surpassed all single classifiers. While discriminating negative sequences with 100% accuracy rate, this classifier correctly predicted 76.7% of IVA effectors.
3.5 Comparison with existing methods
Two research groups reported the ability to implement genomic-scale prediction of T4SS effectors in L.pneumophila (Burstein et al., 2009; Lifshitz et al., 2013), predicting experimentally identifying dozens of secreted signals. The method presented by Burstein et al. used multiple sequence features and used a machine learning-based classifier ensemble to distinguish effectors from the genomic proteins. This method was also adopted by Chen et al. to predict effectors in the genome of C.burnetii. In a dataset of 134 known effectors and 670 non-effectors, this method achieved an accuracy of 95.9% with an AUC of 0.980. Another method developed by Lifshitza et al. used the HSMM to construct a C-terminal profile of known effectors and to search those proteins for significant signals. It correctly predicted 92.9% effectors of 283 known effectors against a background of 1000 non-effectors with an AUC of 0.881. Based on these datasets, we performed LOO tests to measure the prediction rate of our model (Supplementary Table S8). Compared with Burstein et al.’s ensemble predictor, our ensemble model had higher accuracy and a higher value for the AUC, although other single models resulted in lower AUCs. In the dataset from Lifshitz et al., HSMM had a higher sensitivity than all other single models; however, our ensemble predictor produced better performance measures in the LOO tests.
3.6 Genome-scale prediction in B.henselae
To assess the ability of our prediction system to discriminate T4SS effectors from genomic proteins, we used several independent classifiers and a classifier ensemble to predict effectors in 1488 genomic proteins from B.henselae strain Houston-1. B.henselae is the major human pathogen in the genus Bartonella, which causes cat-scratch disease. VirB/VirD4 T4SS has been identified in this pathogen and was verified to be essential for establishing intraerythrocytic infection. This T4SS secrets dozens of effectors into host cells, and three of them have been included in T4_1472. The majority of these effectors belong to the Bep family. Sequence alignments showed that all these effectors had high homology with seven proteins from B.henselae (BH13370, BH13390, BH13400, BH13410, BH13420, BH13430 and BH13440). We trained our model using T4_1472 without three known effectors (BH13370, BH13410 and BH13440) and then predicted effector candidates from 1488 genomic proteins (Supplementary Table S9). Using IVA sequences for training, 41–102 proteins were predicted as effectors, and at least three of the seven known effectors were correctly recognized. The AAC predictor and the PSSM predictor had better recognition rates to known effectors than others. Using IVB sequences for training, 117–158 candidates were predicted by these models. We noticed that fewer of the known effectors were discovered by the PSSM predictor and the PSSM_AC predictor, which are more sensitive to training data than other models. Although gaps in ACC and dipeptide composition exist between IVA effectors and IVB effectors, both the AAC model and the DPC model recognized more known effectors when using IVB data rather than IVA data for training. Models trained using all samples in the T4_1472 were then used to predict effectors. All seven known effectors were detected by the AAC model, and six were detected by the PSSM model. The number of inferred effectors decreased when classifier ensembles were used for prediction, but at least six of the known effectors were present in each candidate list. Fifty-seven proteins were detected by the ensemble with the 3-in-4 voting scheme, in which seven were known effectors and 50 were putative secreted proteins (see Supplementary Table S10). Of these putative effectors, three were annotated as tonB (BH04980), filamentous hemagglutinin (BH07950) and kroA (BH15560) in UniProt. The others were uncharacterized proteins. BH04980 was inferred as a cell surface protein for potential transporter activity. BH07950 is a putative surface protein, but its function is not clear. BH15560 has a high homology with the korA protein from Bartonella grahamii, which is characterized as a regulatory gene for trw T4SS (Nystedt et al., 2008). C-terminus sequence profiles (Supplementary Fig. S3) show that the probability of amino acid occurrence in IVA effectors or IVB effectors is different from non-effectors. The profiles of the 50 inferred effectors show weak C-terminal patterns, possibly because many sequences do not carry significant signals in the C-terminus, which is expected for IVA effectors. The homologous targets of the 50 putative effectors were examined in the NCBI protein database using BLAST (E-value < 1e-10) (Supplementary Table S10). The majority of homologous proteins (39 of 50, including 25 conserved homologs and 14 self-homologs) were uncharacterized proteins and four were putative membrane proteins. Three homologs (heme exporter, slyX and tonB) are known to share an evolutionary lineage with T4SS proteins in Bartonella (Engel et al., 2011). In summary, these candidates may provide potential targets for functional identification of T4SS pathogenicity.
4 DISCUSSION
The prediction of effectors in bacterial genomes is an important task for functional analysis of the T4SS of human pathogens. Previous studies demonstrated that T4SS effectors in several bacterial species contained conserved C-terminal signals and eukaryotic domains (McDermott et al., 2011; Segal 2013). Differences in GC content between the effector genes and the non-effector genes were also discovered (Burstein et al., 2009). Xu et al. reported a hidden Markov model-based method to evaluate the distribution pattern of EPIYA motifs in a broad range of biological species to predict potential T3SS effectors, but this motif was not widely distributed from T4SS secreted proteins (Xu et al., 2010). Burstein et al. presented a machine learning method based on known features to predict candidates for genome-wide effector identification in L.pneumophila (Burstein et al., 2009). This approach was effective for screening experimental targets but was specific to certain well-known bacterial species because few general features exist for extensive prediction. The HSMM model presented by Lifshitz et al. captured IVB secreted signals with a high success rate but was not usable for the detection of IVA effectors (Lifshitz et al., 2013). We developed a widely applicable method for accurate prediction of different subtypes of T4SS effectors in gram-negative pathogens. To accomplish this, we calculated ACCs, residue pair compositions, PSSM profiles and their auto covariance transformations. We used these features to train SVM-based predictors for discrimination of effectors from non-effectors.
To assess the performance of our prediction system, two reusable datasets were constructed for experimentally validated effectors and potential non-effectors. ACC analysis showed that hydrophobic residues, especially aliphatic Ala, Gly and Val, occur with higher frequencies in non-effectors than in effectors. On the contrary, hydrophilic and polar residues are more common components of effector sequences. As most of the known secreted proteins exist in aqueous environments, such as the cytoplasm of host cells, residue pairs with hydrophilic residues would be expected to have higher compositions in effectors than in non-effectors. We inferred that some hydrophilic and polar groups may be important for effector protein functions.
Performance tests showed that two classes of PSSM-based features were more helpful for discrimination of effectors than using ACC. The PSSM revealed remote relevance between function-related proteins. This relevance was conserved throughout evolution, although amino acids continuously mutated. For the two PSSM feature classes that we used, PSSM composition indicated the position-specific information of each amino acid type in the query sequence. On the other hand, auto covariance transformation of the PSSM described the sequence-order position-specific information. The former method performed better than the latter method in benchmark tests. Feature combination is a frequently used approach for improving classification performance. We combined four different feature vectors and found that more accurate results were produced using vector combinations. The prediction accuracy for IVB effectors was higher than for IVA effectors. This can be attributed to different numbers of positive samples and more abundant signals within IVB sequences, such as C-terminal motifs, which are more informative than the known motifs in IVA effectors. We noticed that taxonomic and functional biases in the training data impacted the assessment and application of our method. In T4_1472, 30 IVA sequences were derived from the genomic proteins of nine species, i.e. A.marginale (4), Anaplasma phagocytophilum (2), Bartonella sp.(3), H.pylori (1), Agrobacterium tumefacien (3), Agrobacterium rhizogenes (4), Brucella sp.(8), B.pertussis (4) and E.chaffeensis (1); 310 IVB effectors were selected from L.pneumophila (258) and C.burnetii (52). The taxonomic distribution of IVA sequences showed no obvious bias, but bias was present for IVB sequences. Although all the effectors were experimentally validated to be secreted by T4SS, the functions of most of them in host cells have not been identified. Therefore, the functional bias of these effectors is still unknown. To reduce bias for negative sequences, we collected non-effectors through multiple channels. Though biases in datasets are inevitable, the results of theoretical validations in several datasets showed that our predictor was both accurate and robust for distinguishing T4SS effectors. This indicates that our method may be useful for IVA effector prediction in various T4SS pathogens but should be restricted to Legionella sp., Coxiella sp. and their related species for IVB effector prediction. It is noteworthy that characteristic signals exist in both some T4SS effectors and some T3SS effectors (Xu et al., 2010), which may have similar amino acid distributions and evolutionary conservation profiles. Thus, the present method may not completely discriminate between T4SS effectors and those secreted through the T3SS.
We improved the prediction accuracy using multi-classifier ensembles with simple voting strategies. The 2-in-3 voting and 3-in-4 voting systems are linear discrimination systems that use condition relaxation. Therefore, accurate prediction does not indicate identical prediction performance in pathogenic genomes. Accurate discrimination of effectors in bacterial genomes is the ultimate goal of developing these prediction models. The prediction results for the B.henselae genome demonstrate that our method is effective at distinguishing unknown T4SS effectors from genomic sequences. A small list of predicted candidates will help to easily confirm experimental targets. In the B.henselae genome, seven genes encoding known effector proteins were all predicted by our classifier ensemble. Furthermore, several proteins located at BH14310–BH14330 and BH14510–BH14590 were repeatedly predicted as positives by multi-classifiers, indicating strong potential T4SS effectors. In Supplementary Table S10, 47 out of 50 putative T4SS candidates are annotated as ‘hypothetical proteins’ in the B.henselae genome. The left three are inferred to be relevant to T4SS from Bartonella. Most homologs of these proteins in gram-negative species are still uncharacterized and the annotated information in available public databases shows some of the candidates are most likely secreted proteins. A maximum likelihood phylogenic tree was constructed to reveal the evolutionary relationship between the 26 IVA effectors and the 50 inferred effectors using MUSCLE and MEGA5 (Supplementary Fig. S4). Conversely, no phylogenic trees for IVB effectors could be established by any evolutionary algorithms because no common sites in these sequences were found when computing the distances. Supplementary Figure S4 shows that several distinct groups (YP_414449, YP_153762, YP_153570 and YP_505319) were formed by multiple predicted effectors and an embedded IVA protein. The seven known effectors of B.henselae were grouped in a sub-tree, sharing a most recent common ancestor with BH07480 and displaying close relationships to the putative effector BH13840. The arrangement of each branch in the tree indicates that the predicted effectors in each sub-tree were most likely captured by our model based on the evolutionary information of their nearest IVA effectors. We hope that these predicted effectors may serve as a useful resource for the research community.
By taking into account all of our results and analyses, we can conclude that the present method can detect T4SS effectors within unidentified sequences with great power. As the features used in our system are sequence-based and are commonly calculated across both genus and species, we believe that our method can be widely used for T4SS effector screening for 10 confirmed pathogens as well as for other gram-negative bacterial species.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Kang Chen and Supeng Yin for their technical assistance and constructive suggestions. They are particularly grateful to Prof. Hongyu Ou for providing the experimentally validated effector list for the SecRet4 database.
Funding: National Natural Science Foundation of China (grant no. 31301097).
Conflict of Interest: none declared.
REFERENCES
- Arnold R, et al. Sequence-based prediction of type III secreted proteins. PLoS Pathog. 2009;5:e1000376. doi: 10.1371/journal.ppat.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi D, et al. SecReT4: a web-based bacterial type IV secretion system TF4resource. Nucleic Acids Res. 2013;41:D660–D665. doi: 10.1093/nar/gks1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein D, et al. Genome-scale identification of Legionella pneumophila effectors using a machine learning approach. PLoS Pathog. 2009;5:e1000508. doi: 10.1371/journal.ppat.1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambronne ED, Roy CR. Recognition and delivery of effector proteins into eukaryotic cells by bacterial secretion systems. Traffic. 2006;7:929–939. doi: 10.1111/j.1600-0854.2006.00446.x. [DOI] [PubMed] [Google Scholar]
- Chandran V, et al. Structure of the outer membrane complex of a type IV secretion system. Nature. 2009;462:1011–1015. doi: 10.1038/nature08588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, et al. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc. Natl Acad. Sci. USA. 2010;107:21755–21760. doi: 10.1073/pnas.1010485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SA, et al. Prediction of transporter targets using efficient RBF networks with PSSM profiles and biochemical properties. Bioinformatics. 2011;27:2062–2067. doi: 10.1093/bioinformatics/btr340. [DOI] [PubMed] [Google Scholar]
- Dong Q, et al. A new taxonomy-based protein fold recognition approach based on autocross-covariance transformation. Bioinformatics. 2009;25:2655–2662. doi: 10.1093/bioinformatics/btp500. [DOI] [PubMed] [Google Scholar]
- Engel P, et al. Parallel evolution of a type IV secretion system in radiating lineages of the host-restricted bacterial pathogen Bartonella. PLoS Genet. 2011;7:e1001296. doi: 10.1371/journal.pgen.1001296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fronzes R, et al. Structure of a type IV secretion system core complex. Science. 2009;323:266–268. doi: 10.1126/science.1166101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JE, Wolf-Watz H. Protein delivery into eukaryotic cells by type III secretion machines. Nature. 2006;444:567–573. doi: 10.1038/nature05272. [DOI] [PubMed] [Google Scholar]
- Lifshitz Z, et al. Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proc. Natl Acad. Sci. USA. 2013;110:E707–E715. doi: 10.1073/pnas.1215278110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, et al. Prediction of protein structural class for low-similarity sequences using support vector machine and PSI-BLAST profile. Biochimie. 2010;92:1330–1334. doi: 10.1016/j.biochi.2010.06.013. [DOI] [PubMed] [Google Scholar]
- Llosa M, et al. Bacterial type IV secretion systems in human disease. Mol. Microbiol. 2009;73:141–151. doi: 10.1111/j.1365-2958.2009.06751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood S, et al. Identification of Anaplasma marginale type IV secretion system effector proteins. PLoS One. 2011;6:e27724. doi: 10.1371/journal.pone.0027724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lower M, Schneider G. Prediction of type III secretion signals in genomes of gram-negative bacteria. PLoS One. 2009;4:e5917. doi: 10.1371/journal.pone.0005917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesini MI, et al. In search of Brucella abortus type IV secretion substrates: screening and identification of four proteins translocated into host cells through VirB system. Cell. Microbiol. 2011;13:1261–1274. doi: 10.1111/j.1462-5822.2011.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- McDermott JE, et al. Computational prediction of type III and IV secreted effectors in gram-negative bacteria. Infect. Immun. 2011;79:23–32. doi: 10.1128/IAI.00537-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystedt B, et al. Diversifying selection and concerted evolution of a type IV secretion system in Bartonella. Mol. Biol. Evol. 2008;25:287–300. doi: 10.1093/molbev/msm252. [DOI] [PubMed] [Google Scholar]
- Rey S, et al. PSORTdb: a protein subcellular localization database for bacteria. Nucleic Acids Res. 2005;33:D164–D168. doi: 10.1093/nar/gki027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samudrala R, et al. Accurate prediction of secreted substrates and identification of a conserved putative secretion signal for type III secretion systems. PLoS Pathog. 2009;5:e1000375. doi: 10.1371/journal.ppat.1000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, et al. Meta-analytic approach to the accurate prediction of secreted virulence effectors in gram-negative bacteria. BMC Bioinformatics. 2011;12:442. doi: 10.1186/1471-2105-12-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal G. Identification of legionella effectors using bioinformatic approaches. Methods Mol. Biol. 2013;954:595–602. doi: 10.1007/978-1-62703-161-5_37. [DOI] [PubMed] [Google Scholar]
- Souza RC, et al. AtlasT4SS: A curated database for type IV secretion systems. BMC Microbiol. 2012;12:172. doi: 10.1186/1471-2180-12-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng TT, et al. Protein secretion systems in bacterial-host associations, and their description in the Gene Ontology. BMC Microbiol. 2009;9(Suppl. 1):S2. doi: 10.1186/1471-2180-9-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. High-accuracy prediction of bacterial type III secreted effectors based on position-specific amino acid composition profiles. Bioinformatics. 2011;27:777–784. doi: 10.1093/bioinformatics/btr021. [DOI] [PubMed] [Google Scholar]
- Xie D, et al. LOCSVMPSI: a web server for subcellular localization of eukaryotic proteins using SVM and profile of PSI-BLAST. Nucleic Acids Res. 2005;33:W105–W110. doi: 10.1093/nar/gki359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, et al. Effector prediction in host-pathogen interaction based on a Markov model of a ubiquitous EPIYA motif. BMC Genomics. 2010;11(Suppl. 3):S1. doi: 10.1186/1471-2164-11-S3-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. Computational prediction of type III secreted proteins from gram-negative bacteria. BMC Bioinformatics. 2010;11(Suppl. 1):S47. doi: 10.1186/1471-2105-11-S1-S47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, et al. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One. 2011;6:e17638. doi: 10.1371/journal.pone.0017638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.