

Abstract
Motivation: Prediction of protein residue contacts, even at the coarse-grain level, can help in finding solutions to the protein structure prediction problem. Unlike α-helices that are locally stabilized, β-sheets result from pairwise hydrogen bonding of two or more disjoint regions of the protein backbone. The problem of predicting contacts among β-strands in proteins has been addressed by several supervised computational approaches. Recently, prediction of residue contacts based on correlated mutations has been greatly improved and finally allows the prediction of 3D structures of the proteins.
Results: In this article, we describe BCov, which is the first unsupervised method to predict the β-sheet topology starting from the protein sequence and its secondary structure. BCov takes advantage of the sparse inverse covariance estimation to define β-strand partner scores. Then an optimization based on integer programming is carried out to predict the β-sheet connectivity. When tested on the prediction of β-strand pairing, BCov scores with average values of Matthews Correlation Coefficient (MCC) and F1 equal to 0.56 and 0.61, respectively, on a non-redundant dataset of 916 protein chains known with atomic resolution. Our approach well compares with the state-of-the-art methods trained so far for this specific task.
Availability and implementation: The method is freely available under General Public License at http://biocomp.unibo.it/savojard/bcov/bcov-1.0.tar.gz. The new dataset BetaSheet1452 can be downloaded at http://biocomp.unibo.it/savojard/bcov/BetaSheet1452.dat.
Contact: piero.fariselli@unibo.it
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
β-Sheets are widespread motifs of local structure found in over 80% of the protein structures presently available in the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). β-Sheets are generated by the pairing of two or more β-strands held together by characteristic patterns of hydrogen bonds running in a parallel or antiparallel fashion (Zhang and Kim, 2000). Prediction of the β-sheet topology from the covalent structure of the protein is useful for predicting its tertiary structure, for designing new proteins and for understanding folding pathways. The first method to predict β-strand pairing was based on a statistical potential approach (Hubbard, 1994). Prediction of β-residue contacts was addressed by Baldi et al. (2000) using an elaborate method based on neural networks. Steward and Thornton (2002) adopted an information theoretic approach to predict β-residue pairwise interaction. Cheng and Baldi (2005) pioneered the idea of predicting β-sheet topologies when the protein secondary structure is known and set the standard for this type of task. Their method BetaPro is based on a 2D-recursive neural network (Baldi and Pollastri, 2003) trained to predict pairing probabilities of interstrand β-residue pairs. Then an algorithm finds alignments between all pairs of β-strands, and a weighted-graph matching algorithm predicts the β-sheet topologies. Lippi and Frasconi (2009) introduced an alternative approach based on Markov logic networks (MLNs). This approach exploits β-sheet structural constraints defined as logical formulas whose weights can be learned from examples (Lippi and Frasconi, 2009). The prediction of β-residue contacts was also applied to the problem of predicting the 3D structure of proteins using integer linear optimization (Rajgaria et al., 2010). Aydin et al. (2011) combined BetaPro outputs with a Bayesian approach and tested it on subsets of the Cheng and Baldi benchmark set (Aydin et al., 2011). Recently, a new method to predict protein β-sheet contacts using a maximum entropy-based correlated mutation measure (CMM) has been introduced and compared with the state-of-the-art methods (Burkoff et al., 2013). CMM achieves performances similar to BetaPro and MLNs (Burkoff et al., 2013).
In recent years, the main breakthroughs in residue contact prediction have concerned improvements in the exploitation of information from multiple sequence alignments (MSA). The degree of coevolution of pair of sites in the MSA can be used to infer the closeness of the corresponding residues in the 3D structure. Different approaches have been described to elucidate the coevolution of columns in an MSA, including standard correlated mutations analysis (Olmea and Valencia, 1997) and information theoretic measures (Dunn et al., 2008). Recently some powerful methods based on the extraction of direct coupling information from MSAs have been introduced to predict protein contacts both in globular (Cocco et al. 2013; Ekeberg et al. 2013; Jones et al., 2012; Marks et al., 2011; Morcos et al., 2001; Weigt et al. 2009) and membrane proteins (Hopf et al., 2012; Nugent and Jones 2012). These new contact prediction methods are not only improved with respect the previous approaches but also finally allow to predict 3D structures of the proteins (for review see de Juan et al., 2013; Marks et al., 2012; Taylor et al., 2013).
In this article, we describe BCov, a new approach for β-sheet topology prediction based on sparse inverse covariance estimation and integer programming. BCov is the first unsupervised method that addresses this task. We use the residue-contact propensities provided by the Protein Sparse Inverse COVariance (PSICOV) method (Jones et al., 2012) to compute the scores of the β-strand pairings. We selected PSICOV because it is freely available and does not require proprietary software to run. Then we apply an integer programming optimization to assign the β-sheet topology. BCov well compares with the performances of the state-of-the-art algorithms (BetaPro, CMMs and MLNs) that integrate supervised techniques to address the same task.
2 METHODS
2.1 The BetaSheet916 dataset
The BetaSheet916 dataset was first introduced by Cheng and Baldi (2005) to evaluate their BetaPro method for β-sheet prediction and then adopted by other authors (Burkoff et al., 2013; Lippi and Frasconi, 2009). The set is routinely adopted as benchmark set for β-sheet prediction methods and for sake of comparison we also use it in this article. The atomic coordinates of 916 protein chains (with X-ray diffraction at a resolution ≤2.5 Å) were extracted from the Protein Data Bank (PDB) as to May, 2004 (Cheng and Baldi, 2005). Secondary structure assignments were computed by means of the Define Secondary Structure of Proteins (DSSP) program (Kabsch and Sander, 1983). Both extended β-strands (marked by E in the DSSP output) and isolated β-bridges (B in the DSSP output) were considered β-residues. The β-contact maps were defined using information about β-partnership available from the DSSP output. Statistics of the dataset are shown in Table 1.
Table 1.
Statistics of the datasets
| Feature | BetaSheet916a | BetaSheet1452b |
|---|---|---|
| Number of chains | 916 | 1452 |
| Total number of residues | 187 516 | 361 668 |
| Total number of β-residues | 48 996 | 88 352 |
| Number of β-residue contacts | 31 638 | 56 552 |
| Number of β-strands | 10 745 | 19 186 |
| Number of β-strand pairs | 8172 | 14 241 |
| Number of anti-parallel β-strand pairs | 4519 | 3937 |
| Number of parallel β-strand pairs | 2214 | 7892 |
| Number of isolated β-bridges | 1439 | 2412 |
| Number of β-sheets | 2533 | 4894 |
aDerived from Cheng and Baldi (2005).
bA non-redundant complement of the BetaSheet916 set comprising 1452 protein chains deposited in the PDB after May, 2004 (see Section 2.2 for a complete description).
2.2 The BetaSheet1452 dataset
To complement the BetaSheet916 dataset, we constructed a new dataset of more recently deposited high-resolution proteins. We extracted from the PDB a set of protein chains whose structures were obtained by X-ray diffraction with a resolution ≤ 2.5 Å. We restricted our search to PDB entries deposited after May, 2004 to exclude chains already present in the BetaSheet916 dataset. We filtered out protein sequences at 20% of sequence identity level to obtain a non-redundant dataset. More importantly, we also removed sequences with identity >20% with any of the proteins contained in the BetaSheet916 set. We used DSSP to assign secondary structures, and we filtered out sequences having <3 distinct extended β-strands to exclude trivial cases. We also discharged protein chains shorter than 50 residues or having backbone interruptions or non-standard amino acids. The final dataset of proteins, referred to as BetaSheet1452, contained 1452 protein chains. Statistics are shown in Table 1.
2.3 CASP 2010 dataset
For sake of comparison, we also considered protein chains from the Critical Assessment of protein Structure Prediction (CASP) 2010 experiment. The original set comprised 116 targets. We used the same procedure described in Burkoff et al. (2013) to filter out sequences with a number of β-residues ≤ 10. The final set consisted of 92 protein chains. Secondary structures have been assigned using DSSP.
2.4 MSA construction
For each sequence in the datasets described earlier in the text, we obtained an MSA using the jackhmmer program that is part of HMMER 3.0 package (http://hmmer.org). Given a target protein sequence, homologous sequences were found by running three iterations of jackhmmer against the UNIREF90 database (Magrane and the Uniprot Consortium, 2011) setting the E-value threshold to 1e-3. The corresponding MSA has been obtained from jackhmmer output.
2.5 BCov general description
BCov consists of three main steps: (i) compute the residue contact propensity with PSICOV; (ii) compute the score of each possible β-strand pairing; (iii) compute the β-sheet topology using an integer programming optimization to find the best solution according to the β-pairing scores and constraints. Below we report the details of these three major steps.
2.6 Computing the residue contact propensity with PSICOV
For sake of clarity, in this section we provide a brief description of the PSICOV method. We refer to Jones et al. (2012) for further details about the method.
Starting from an MSA with m columns, PSICOV first computes a sample 21 m-by-21 m (also gaps are considered) covariance matrix C using observed single and pair amino acid occurrence frequencies:
| (1) |
where fi,j(a,b), fi(a) and fj(b) are the sample relative frequency of amino acid pair ab at sites ij, the relative frequency of amino acid a at column i and the frequency of amino acid b at column j, respectively.
The inverse of the sample covariance matrix is computed with the graphical lasso method (Banerjee et al., 2008; Friedman et al., 2008). This algorithm allows estimating a sparse inverse covariance matrix Θ by minimizing the following objective function:
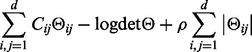 |
(2) |
where C is a d-by-d covariance matrix, Θ is the inverse covariance matrix and the last term is a regularization term (the ℓ1-norm of the inverse matrix) that governs the sparsity of the solutions. ρ is the sparsity hyper-parameter: the greater is ρ the sparser is the solution.
The sparse inverse covariance matrix Θ is used, in turn, to derive a contact score between residues at positions i and j by computing the ℓ1-norm of the 20-by-20 sub-matrix of Θ corresponding to all possible pairs of amino acid at position i and j:
| (3) |
The raw contact score computed as in Equation 3 is finally corrected as follows:
| (4) |
where 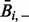 is the mean raw contact score between the i-th position and all other positions (analogously
is the mean raw contact score between the i-th position and all other positions (analogously 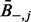 for the j-th position) and
for the j-th position) and 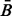 is the overall mean contact score. This correction, referred to as average product correction, allows reducing both entropic and phylogenetic biases that are major source of noise in MSAs (Dunn et al., 2008).
is the overall mean contact score. This correction, referred to as average product correction, allows reducing both entropic and phylogenetic biases that are major source of noise in MSAs (Dunn et al., 2008).
For each possible pairs of residues belonging to two different β-sheets, we then adopt the score computed in Equation 4 as a measure of the β-residue contact strength.
2.7 β-contact and β-sheet topology prediction
BCov consists of an integrated approach to predict both β-residue contacts and β-sheet topology i.e. the specification of the β-strand pairings, including the directions of interactions (parallel, antiparallel or isolated β-bridge). The method takes advantage of β-contact scores as computed by PSICOV and integer programming for β-sheet topology prediction.
Consider a protein sequence with n distinct β-strands and m β-residues. As mentioned earlier in the text, PSICOV is used to compute contact scores between all residue pairs in the protein sequence (even for non-β residues). Then we extract the submatrix obtained by considering only β-residue pairs. After this step, we end up with an m-by-m symmetric matrix B whose Bi,j components can be interpreted as a propensity value for β-residues i and j to form a β-contact.
Our algorithm proceeds by computing an n-by-n matrix S whose entries represent interaction scores between pairs of strands. The matrix S is defined as follows:
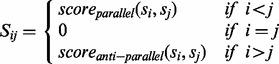 |
(5) |
where, for strands si and sj with i < j, Sij contains the best alignment score between the two strands in the parallel direction, whereas Sji stores the best alignment score in the antiparallel direction. Here, without loss of generality, we use the upper diagonal part of S for parallel scores and the lower diagonal part for antiparallel scores. To compute alignments, we use β-contact propensities Bi,j obtained from PSICOV as local residue match scores. We restrict the search space for optimal alignments by considering only solutions that can be obtained by sliding one strand along the other without gaps and with at most one unmatched residue at the shortest segment ends. With these constraints, for two strands of length r and t, respectively, assuming r > t, there are 2 × (r − t + 3) possible alignments (see Fig. 1 for an example).
Fig. 1.
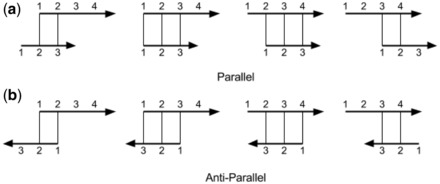
All possible alignments of two β-strands of length 4 and 3, respectively
Strand interaction scores stored in the matrix S are then used to assign the β-sheet topology. Because a naïf approach that enumerates all possible β -sheet pairings is infeasible because of the combinatorial nature of the problem (Zhang and Kim, 2000), several alternative solutions were adopted such as graph matching or maximum spanning-tree algorithms (Cheng and Baldi, 2005).
In this article, we tackle this problem using integer programming. In particular, the optimal β-strand pairing pattern can be obtained by solving the following integer program:
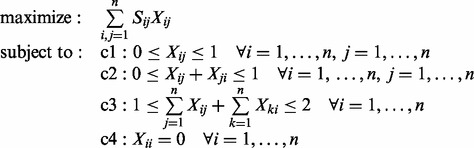 |
The solution X is an n-by-n integer matrix that maximizes the overall β-sheet sum of scores. The matrix X is binary (c1 constraints) and its non-zero entries identify interacting β-strands. c2 constraints ensure the consistency of the solution by enforcing the assignment of either parallel or antiparallel pairing direction for each strand pair (i, j). Furthermore, a given β-strand can pair with at most two distinct β-strands (c3 and c4 constraints). Figure 2 shows an example of the overall procedure for a protein with five β-strands. We also evaluate a version of BCov that takes into consideration the geometric constraint of generating parallel β-sheets at short sequence separation. In protein structures, the formation of parallel β-sheets with few interleaving residues between two strands is difficult and it requires a strong unfavorable free energy contribution. For instance, in the Cheng and Baldi (2005) dataset almost all pairing β-strands separated by <6 residues are antiparallel. For this reason, we introduce a modified version of our algorithm, referred to as BCov6, which always assigns antiparallel directions to pairs of β-strands whose sequence separation is <6 residues.
Fig. 2.
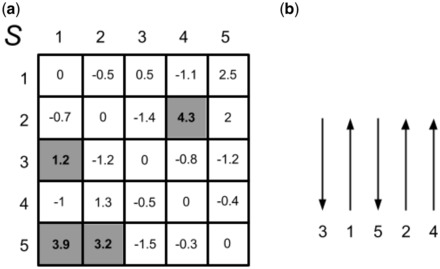
Example of an assignment of the β-sheet topology for a protein with five β-strands. (a) The weight matrix S of the integer programming optimization. Each element of S corresponds to the strand interaction score. Strands interaction scores may be derived from parallel (upper diagonal part of the matrix) or antiparallel (lower diagonal part of the matrix) strand alignments. (b) The assigned β-sheet topology {(1,3,A), (1,5,A), (2,4,P), (2,5,A)} (A = antiparallel, P = parallel) obtained from the integer program solution and corresponding strand pairing directions
2.8 Method implementation
BCov has been developed in the C programming language (available at http://biocomp.unibo.it/savojard/bcov/bcov-1.0.tar.gz). The source code is released under the terms of the GNU General Public License (GPL) version 3. BCov is linked with the PSICOV source code (available at http://bioinfadmin.cs.ucl.ac.uk/downloads/PSICOV/ and also released under GPL terms) and the graphical lasso FORTRAN package for sparse inverse covariance estimation (Friedman et al., 2008). For integer linear optimization program, we used the C application program interface of the GNU Linear Programming Kit (http://www.gnu.org/software/glpk/).
2.9 Measures of performance
We evaluated the performance of the method at both β-residue contact level (i.e. contact maps) and β-strand pairing level (coarse contact maps). In either case, we computed the canonical scoring measures, which include:
| (6) |
| (7) |
| (8) |
| (9) |
where TP, TN, FP and FN are, respectively, true positives, true negatives, false positives and false negatives. We also evaluate the methods using qualitative measures introduced by Lippi end Frasconi (2009). In particular,
Precision:
Recall:
F1-score:
Matthews Correlation Coefficient:
F1≥ 70, which measures the percentage of chains for which β-strands are predicted with F1 ≥ 70% on coarse map,
F1 on parallel strands,
F1 on antiparallel strands,
Correct directions : percentage of correct pairing directions over correctly predicted strand pairs,
CβCC: percentage of correct β-sheet connected components.
3 RESULTS AND DISCUSSION
3.1 PSICOV performance on β-residue contacts
PSICOV has been described as one of the most promising methods to address the prediction of residue contact in proteins (Jones et al., 2012). The accuracy of PSICOV depends on both the number and the quality of the aligned sequences (Jones et al., 2012). In the case of BetaSheet916 dataset, the number of sequences in each MSA ranged between 2 and 190576 (see Fig. 3). Here, we address a slightly different goal, namely we want to evaluate the predictions on the subsets of the contact map identified by the β-strand regions. In Table 2, we report the PSICOV accuracy on the whole BetaSheet916 dataset and also on two disjoint subsets: a subset of proteins whose number of aligned sequences are ≥1000 and the subset of proteins whose number of aligned sequences are <1000. Results listed in Table 2 indicate that a low number of aligned sequences can affect the accuracy. The performances of PSICOV on this task are lower than those obtained by the state-of-the-art methods (see Table 3, rows 1–4). However, it must be noticed that PSICOV is an unsupervised method (differently from the others) and that here we evaluate it only on the portion of β-residue contacts.
Fig. 3.
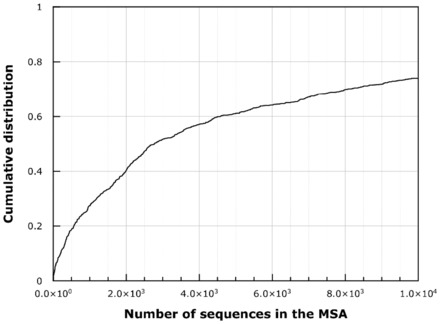
Cumulative frequency distribution of the number of sequences in MSAs. In all, 251 of 916 chains (∼27%) in the BetaSheet916 dataset have <1000 aligned sequences in the corresponding MSA
Table 2.
PSICOV performance on the β-residue contacts
aThe whole BetaSheet916 dataset is used.
bThe score is evaluated only on the subsets of the proteins whose number of aligned sequences (NAS) is ≥1000.
cThe score is evaluated only on the subsets of the proteins whose NAS is <1000.
Note: P(L*x) = precision evaluated selecting the L*x highest scoring β-residue pairs.
Table 3.
BCov and Bcov6 performances at residue level on the BetaSheet916 dataset
| Method | R | P | F1 | MCC |
|---|---|---|---|---|
| CMM | 44.0 | 44.0 | 44.0 | 0.43 |
| MLN-2S | 42.7 | 47.3 | 44.9 | 0.44 |
| MLN | 39.3 | 46.1 | 42.4 | 0.42 |
| BetaPro | 44.1 | 38.0 | 40.8 | 0.40 |
| BCov | 42.4 | 40.9 | 41.6 | 0.40 |
| Bcov ≥ 1000 | 47.3 | 45.8 | 46.5 | 0.45 |
| Bcov < 1000 | 27.7 | 26.5 | 27.1 | 0.25 |
| Bcov6 | 43.9 | 42.4 | 43.1 | 0.42 |
| Bcov6 ≥ 1000 | 48.7 | 47.2 | 47.9 | 0.47 |
| Bcov6 < 1000 | 29.4 | 28.2 | 28.8 | 0.27 |
Note: The values of CMM, BetaPro and MLN are taken from the corresponding articles. CMM = Burkoff et al., 2013. BetaPro = Cheng and Baldi, 2005. MLN = Lippi and Frasconi, 2009, MLN-2S = BetaPro + MLN Lippi and Frasconi, 2009. BCov and BCov6 are also evaluated on the subsets of the proteins whose number of aligned sequences (NAS) are ≥1000 or <1000, respectively. For the indices see Section 3.2.
3.2 BCov performance on β-residue contacts
As described in Section 2, BCov optimizes the PSICOV outputs using integer programming approaches. BCov assigns the β-sheet topology and also all segment pairings including β-sheet directions. For each pair of β-strand partners the best score obtained by the segment pairing (see Section 2.4) can be used to assign β-residue contacts. With this procedure, the performances are significantly higher than those obtained using PSICOV alone (compare Table 2 with Table 3). When the predicted β-sheet contacts are used to reconstruct the corresponding 3D structures with FT-COMAR (Vassura et al., 2008), the improvement of BCov over PSICOV leads to a better reconstruction of the protein fold (see Supplementary Fig. S1 of the Supplementary Materials).
In Table 3, we report the performances of BCov and BCov6 with respect to the state-of-the-art methods on the BetaSheet916 dataset. It is evident that when we do not allow parallel pairing of β-strand segments whose sequence separation is <6 residues (a simple geometric constraint) the accuracy increases, as highlighted by the two percentage points of the correlation coefficient (MCC). BCov and BCov6 performances compare well with the single supervised methods (CMM, BetaPro, MLN) and are slightly worse than the combination of two methods (MLN-2S).
3.3 BCov performance at strand level
The evaluation of the methods at strand level (detecting the correct β-sheet topology and β-strand pairings) is the most relevant benchmark, as this is the main goal of all the approaches developed so far (BetaPro, NLN, CMM). In Table 4, we report the comparison of BCov with the state-of-the-art methods on the BetaSheet916 dataset. It is worth noticing that in this task BCov (which is not trained on the dataset) compares well with the other methods achieving a correlation coefficient value of 0.56 (MCC). This indicates that the information extracted by PSICOV is relevant also for the problem at hand, and if coupled with an optimization procedure the overall accuracy can improve significantly. Furthermore, it is evident that when the number of aligned sequences is sufficiently high (Bcov ≥ 1000) the performances increases by 5 percentage points and with an MCC of 7 percentage points higher than the best state-of-the-art method. As a consequence, we may expect that BCov performance will improve as new sequences will be made available by mass sequencing efforts.
Table 4.
BCov performance at strand level on the BetaSheet916 dataset
| Method | R | P | F1 | MCC |
|---|---|---|---|---|
| CMM | 55.0 | 61.0 | 58.0 | 0.53 |
| MLN-2S | 59.8 | 58.4 | 59.1 | 0.54 |
| MLN | 55.5 | 59.8 | 57.6 | 0.53 |
| BetaPro | 59.7 | 53.1 | 56.2 | 0.51 |
| BCov | 62.0 | 59.5 | 60.7 | 0.56 |
| Bcov ≥ 1000 | 66.0 | 64.1 | 65.1 | 0.61 |
| Bcov < 1000 | 48.9 | 45.2 | 47.0 | 0.39 |
Note: For the legend see Table 3.
In Table 5, we show scoring of the different methods obtained with the qualitative measures introduced by Lippi and Frasconi (2009) (see Section 2.6). It is interesting to see that BCov outperforms all other methods with respect to all the reported indices with the exception of the prediction of the correct directions of the correctly predicted β-sheets. This indicates that with respect to the other methods, BCov is better at guessing the segment pairings and β-sheet topology but less effective at detecting the contacts at residue level.
Table 5.
BCov and BCov6 qualitative comparison at strand level on the BetaSheet916 dataset
| Method | F1 ≥ 70% | F1 on parallel strands | F1 on antiparallel strands | Correct directions | CβCC |
|---|---|---|---|---|---|
| CMM | 35.0 | — | — | — | — |
| MLN-2S | 36.2 | 52.8 | 60.9 | 95.6 | 24.8 |
| MLN | 33.7 | 49.5 | 59.9 | 95.6 | 24.3 |
| BetaPro | 31.7 | 50.2 | 57.0 | 93.0 | 17.1 |
| BCov0 | 44.2 | 68.3 | 65.7 | 77.6 | 24.9 |
| Bcov ≥ 1000 | 51.3 | 71.9 | 71.0 | 79.3 | 25.8 |
| Bcov < 1000 | 25.5 | 54.1 | 50.8 | 70.1 | 21.9 |
| BCov6 | 44.2 | 66.1 | 67.2 | 84.2 | 25.0 |
| Bcov6 ≥ 1000 | 51.3 | 70.5 | 72.0 | 84.7 | 25.9 |
| Bcov6 < 1000 | 25.5 | 46.0 | 54.1 | 82.1 | 21.9 |
Note: For legend see Table 3.
3.4 Performance on the CASP 2010 dataset
To evaluate the BCov performances at the strand level on another benchmark, we used the CASP 2010 dataset. With this choice we can also compare the BCov performance at the strand level with other two state-of-the-art predictors (BetaPro and CMM). We downloaded the protein structures provided during the CASP 2010 experiment, and we followed the same procedure described in Burkoff et al. (2013) to filter out sequences with a number of β-residues ≤ 10. The final set consisted of 92 protein chains. The results reported in Table 6 are really encouraging, as it appears that BCov6 (an unsupervised method) compares well with the state-of-the-art methods BetaPro and CMM [as reported from the data taken from the supplementary material of Burkoff et al. (2013)]. It is also worth noticing that BCov6 MCC and F1 performances are almost unaffected by the dataset change (compare Table 6 with indexes reported in Tables 4 and 5).
Table 6.
BCov6 performance on the CASP 2010 dataset
| Index | BCov6 | CMM | BetaPro |
|---|---|---|---|
| R | 61.4 | 54.2 | 57.1 |
| P | 58.0 | 53.1 | 44.1 |
| F1 | 59.6 | 53.7 | 50.0 |
| MCC | 0.56 | 0.50 | 0.45 |
| Chains with F1 ≥ 70% | 46.1 | 32.6 | 32.6 |
Note: Results for CMM and BetaPro taken from Burkoff et al. (2013). For the indices see Section 3.2.
3.5 Performance on the new BetaSheet1452 dataset
To evaluate the BCov performance on a larger and newer dataset, in Table 7 we report the performances both at residue and at strand level. It is encouraging that the performances are consistent and comparable with those obtained using the classical BetaSheet916 dataset (compare with Tables 3–5) and also the CASP 2010 dataset (compare with Table 6). Because BetaSheet1452 is larger than the previous dataset (far larger than any CASP datasets, at least so far), the fact that the performances do not degrade indicates that BCov is a robust predictor of β-sheet topologies. In Table 7 we also report the performance of the most recently introduced CMM method (Burkoff et al., 2013) that was trained on the BetaSheet916 dataset. It is worth noticing that BCov6 performs similarly at residue level, but generally better at strand level indicating that BCov more efficient than CMM at locating the β-sheet segment pairing (see MCC and F1 measure in Table 7).
Table 7.
BCov6 performance on the BetaSheet1452 dataset
| Index | BCov6 | CMM |
|---|---|---|
| R at residue level | 45 | 35 |
| P at residue level | 42 | 56 |
| F1 at residue level | 43 | 43 |
| MCC at residue level | 0.43 | 0.43 |
| R at strand level | 63 | 50 |
| P at strand level | 59 | 61 |
| F1 at strand level | 61 | 55 |
| F1 ≥ 70% | 44 | 36 |
| MCC at strand level | 0.57 | 0.51 |
Note: For the indices see Section 3.2.
4 CONCLUSIONS
In this article, we presented BCov, a new unsupervised method which integrates correlated mutation analysis and integer programming for β-sheet topology prediction. BCov well compares with the performances of the state-of-the-art methods, such as BetaPro, CMMs and MLNs. The main advantage of BCov is in the identification of the β-strand pairing and the prediction of the β-sheet topology. This is also confirmed when BCov is compared with the most recently introduced method to address this task (MCC by Burkoff et al., 2013) on the new BetaSheet1452 dataset (Table 7). It is also worth noticing that BCov might be coupled or incorporated into more complex machine-learning frameworks such as recurrent neural networks or MLNs, as all the information used by BCov (or its components) is obtained at the sequence level without training. Finally, all the experiments carried out in this article showed once more the strength of the correlated mutation analysis for residue contact prediction, especially when this is performed using a sparse inverse covariance approach as the one implemented by PSICOV (Jones et al. 2012) or related approaches (Cocco et al., 2013; Ekeberg et al. 2013; Marks et al., 2011, 2012; Morcos et al., 2001; Weigt et al. 2009).
Supplementary Material
ACKNOWLEDGEMENT
The authors thank Dr. Moreno Marzolla for having introduced them to GNU Linear Programming Kit.
Funding: PRIN 2009 project 009WXT45Y (to R.C.) (Italian Ministry for University and Research: MIUR); PRIN 2010-2011 project 20108XYHJS (to P.L.M.) (Italian Ministry for University and Research: MIUR); COST BMBS Action TD1101 (European Union RTD Framework Program); PON project PON01_02249 (Italian Ministry for University and Research).
Conflicts of Interest: none declared.
REFERENCES
- Aydin Z, et al. Bayesian models and algorithms for protein beta-sheet prediction. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2011;8:395–409. doi: 10.1109/TCBB.2008.140. [DOI] [PubMed] [Google Scholar]
- Baldi P, et al. Matching protein beta-sheet partners by feed-forward and recurrent neural networks. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2000;8:25–36. [PubMed] [Google Scholar]
- Baldi P, Pollastri G. The principled design of large-scale recursive neural network architectures DAG-RNNs and the protein structure prediction problem. J. Mach. Learn. Res. 2003;4:575–602. [Google Scholar]
- Banerjee O, et al. Model selection through sparse maximum likelihood estimation. J. Mach. Learn. Res. 2008;9:485–516. [Google Scholar]
- Burkoff NS, et al. Predicting protein β-sheet contacts using a maximum entropy-based correlated mutation measure. Bioinformatics. 2013;5:580–587. doi: 10.1093/bioinformatics/btt005. [DOI] [PubMed] [Google Scholar]
- Cheng J, Baldi P. Three-stage prediction of protein beta-sheets by neuralnetworks, alignments and graph algorithms. Bioinformatics. 2005;21:i75–i84. doi: 10.1093/bioinformatics/bti1004. [DOI] [PubMed] [Google Scholar]
- Cocco S, et al. From principal component to direct coupling analysis of coevolution in proteins: low-eigenvalue modes are needed for structure prediction. PLoS Comput. Biol. 2013;9:e1003176. doi: 10.1371/journal.pcbi.1003176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Juan D, et al. Emerging methods in protein co-evolution. Nat. Rev. Genet. 2013;14:249–261. doi: 10.1038/nrg3414. [DOI] [PubMed] [Google Scholar]
- Dunn SD, et al. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics. 2008;24:333–340. doi: 10.1093/bioinformatics/btm604. [DOI] [PubMed] [Google Scholar]
- Ekeberg M, et al. Improved contact prediction in proteins: using pseudolikelihoods to infer Potts models. Phys. Rev. E. 2013;87:012707. doi: 10.1103/PhysRevE.87.012707. [DOI] [PubMed] [Google Scholar]
- Friedman J, et al. Sparse inverse covariance estimation with the graphical Lasso. Biostatistics. 2008;9:432–441. doi: 10.1093/biostatistics/kxm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf TA, et al. Three-dimensional structures of membrane proteins from genomic sequencing. Cell. 2012;149:1607–1621. doi: 10.1016/j.cell.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard TJ, Park J. Proceedings of the 27th Hawaii Int’l Conf. System Sciences. Maui, HI, USA. 1994. Use of beta-strand interaction pseudo potentials in protein structure and modelling; pp. 336–344. [Google Scholar]
- Jones DT, et al. PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012;28:184–190. doi: 10.1093/bioinformatics/btr638. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Lippi M, Frasconi P. Prediction of protein-residue contacts by Markov logic networks with grounding-specific weights. Bioinformatics. 2009;25:2326–2333. doi: 10.1093/bioinformatics/btp421. [DOI] [PubMed] [Google Scholar]
- Magrane M The UniProt Consortium. UniProt knowledge base: a hub of integrated protein data. Database. 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks DS, et al. Protein 3D structure computed from evolutionary sequence variation. PLoS One. 2011;6:e28766. doi: 10.1371/journal.pone.0028766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks DS, et al. Protein structure prediction from sequence variation. Nat. Biotechnol. 2012;30:1072–1080. doi: 10.1038/nbt.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcos F, et al. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl Acad. Sci. USA. 2011;108:E1293–E1301. doi: 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent T, Jones DT. Accurate de novo structure prediction of large transmembrane protein domains using fragment-assembly and correlated mutation analysis. Proc. Natl Acad. Sci. USA. 2012;109:E1540–E1547. doi: 10.1073/pnas.1120036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmea O, Valencia A. Improving contact predictions by the combination of correlated mutations and other sources of sequence information. Fold Des. 1997;2:S25–S32. doi: 10.1016/s1359-0278(97)00060-6. [DOI] [PubMed] [Google Scholar]
- Rajgaria R, et al. Contact prediction for beta and alpha-beta proteins using integer linear optimization and its impact on the first principles 3D structure prediction method ASTRO-FOLD. Proteins. 2010;78:1825–1846. doi: 10.1002/prot.22696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward RE, Thornton JM. Prediction of strand pairing in antiparallel and parallel beta-sheets using information theory. Proteins. 2002;48:178–191. doi: 10.1002/prot.10152. [DOI] [PubMed] [Google Scholar]
- Taylor WR, et al. Prediction of contacts from correlated sequence substitutions. Curr. Opin. Struct. Biol. 2013;23:473–479. doi: 10.1016/j.sbi.2013.04.001. [DOI] [PubMed] [Google Scholar]
- Vassura M, et al. FT-COMAR: fault tolerant three-dimensional structure reconstruction from protein contact maps. Bioinformatics. 2008;24:1313–1315. doi: 10.1093/bioinformatics/btn115. [DOI] [PubMed] [Google Scholar]
- Weigt M, et al. Identification of direct residue contacts in protein-protein interaction by message passing. Proc. Natl Acad. Sci. USA. 2009;106:67–72. doi: 10.1073/pnas.0805923106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Kim S. The anatomy of protein beta-sheet topology. J. Mol. Biol. 2000;2:1075–1089. doi: 10.1006/jmbi.2000.3678. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.