

SUMMARY
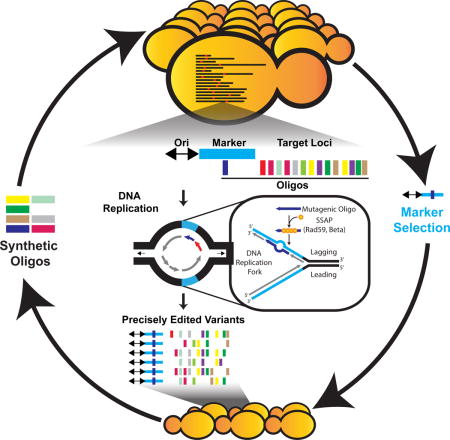
We describe a multiplex genome engineering technology in Saccharomyces cerevisiae based on annealing of synthetic oligonucleotides at the lagging strand of DNA replication. The mechanism is independent of Rad51-directed homologous recombination and avoids the creation of double-strand DNA breaks, enabling precise chromosome modifications at single base-pair resolution with efficiencies >40% without unintended mutagenic changes at the targeted genetic loci. We observed the simultaneous incorporation of up to 12 oligonucleotides with as many as 60 targeted mutations in one transformation. Iterative transformations of a complex pool of oligonucleotides rapidly produced large combinatorial genomic diversity >105. This method was used to diversify a heterologous β-carotene biosynthetic pathway that produced genetic variants with precise mutations in promoters, genes, and terminators, leading to altered carotenoid levels. Our approach of engineering the conserved processes of DNA replication, repair, and recombination could be automated and establishes a general strategy for multiplex combinatorial genome engineering in eukaryotes.
In Brief
Replication forks can be co-opted to introduce multi-site mutations in eukaryotic genomes without the need for double strand breaks.
INTRODUCTION
Most eukaryotic genome editing technologies — zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR-associated endonuclease Cas9 (CRISPR-Cas9) – generate DNA double-strand breaks (DSBs) at targeted loci to introduce genomic modifications (Chandrasegaran and Carroll, 2016; Doudna and Charpentier, 2014). Although ZFNs and TALENs recognize specific DNA sequences through protein-DNA interactions and use the FokI nuclease domain to introduce DSBs at genomic loci, construction of functional ZFNs and TALENs with desired DNA specificity remains laborious, costly, and primarily limited to modifications at a single locus. CRISPR-Cas9 has been broadly adopted for multiplexed targeting of genomic modifications because the nuclease Cas9 uses a short guide RNA to recognize the target DNA via Watson-Crick base-pairing (Jinek et al., 2012) and has been shown to function in many organisms (Cong et al., 2013; Mali et al., 2013). CRISPR-Cas9 is suited for gene disruption applications by non-homologous end joining (NHEJ) (Yang et al., 2015) and gene editing via homology directed repair (HDR) (Doudna and Charpentier, 2014).
For applications that require multisite editing or precise base-pair (bp) level genome modifications by HDR, the DSB mechanism is limiting for three key reasons. First, cleaving the genome is cytotoxic, and cell lethality is magnified when DSBs are introduced across multiple target sites (Jakociunas et al., 2015). Second, in eukaryotes most single bp HDR changes introduced by DSB repair are subject to additional unwanted insertions or deletions (indels) resulting from NHEJ (Inui et al., 2014). These additional mutations result from high tolerance of the targeted nuclease (i.e., Cas9:gRNA) to mismatches in the DSB target which can lead to additional cleavage even after HDR editing has occurred (Fu et al., 2013). For single bp HDR, the inclusion of blocking mutations in the donor DNA is typically required to mask the genomic target site from further cutting by the Cas9:gRNA (Horwitz et al., 2015; Paquet et al., 2016). For many types of genetic elements (e.g., promoters, ncRNAs), the exact DNA sequence dictates function such that additional blocking mutations can be prohibitive. Despite the improved stringencies of engineered Cas9 variants, mismatches are still tolerated for many nonstandard target sites with repetitive regions (Tsai and Joung, 2016). Third, the inefficiency of generating targeted single bp edits with DSBs limits the ability to simultaneously modify many loci in a single cell or across a population to produce combinatorial genetic diversity for exploration of vast genomic landscapes. Efficient DNA base editing without a DSB has been reported using Cas9-guided deamination, but this technique is limited to specific C → T or G → A mutations in an imprecise window of several bps (Komor et al., 2016). Thus, creating precise edits at single bp resolution remains a defining challenge for eukaryotic genome engineering technologies.
Prior work in Escherichia coli demonstrated that targeted chromosomal modifications could be introduced without DSBs using synthetic ssDNA oligodeoxynucleotides (ssODNs) complementary to the lagging strand of the replicating chromosome. High efficiencies (>10%) were achieved by using the phage λ Red Beta ssDNA annealing protein (SSAP) to facilitate ssODN annealing at the lagging strand during DNA replication (Costantino and Court, 2003). With the advent of multiplex automated genome engineering (MAGE), this approach was enhanced to generate multi-site genome modifications with bp precision at increased efficiencies (>30%) and used for pathway diversification (Wang et al., 2009), whole genomic recoding (Lajoie et al., 2013), and molecular evolution of proteins (Amiram et al., 2015). In S. cerevisiae homologous recombination (HR) of ssODNs results in low gene targeting efficiencies (~10−4–10−3 %), which limits the scope of applications to single locus modifications (Kow et al., 2007; Storici and Resnick, 2003). The mechanism of ssODN incorporation in eukaryotic cells is not precisely defined and may involve several factors, which include direction of DNA replication, cell-cycle phase, transcription, DNA mismatch repair (MMR) and HR (Rivera-Torres and Kmiec, 2016). Efforts to develop an analogous MAGE technology in S. cerevisiae have focused on overexpression of HR factors Rad51 and Rad54 in MMR deficient strains and resulted in moderately enhanced allelic replacement frequencies (ARF) (~0.1–2%) (DiCarlo et al., 2013). Thus, no method has been established in eukaryotes for precise, multisite genome modification with ssODNs at efficiencies attained in E. coli.
Here we describe eukaryotic MAGE (eMAGE) for the generation of precise combinatorial genome modifications of the model eukaryote and industrial chassis organism S. cerevisiae. This approach is rooted in biasing the annealing of synthetic ssODNs during DNA replication rather than a Rad51-directed strand-invasion mechanism. We observe single bp precision gene editing with efficiencies >40%. Since eMAGE does not rely on DSBs, the process is highly scalable across a genomic region of interest and can simultaneously generate many precise and diverse modifications of a chromosome. We demonstrate that combinatorial genomic diversity can be generated across a population of cells in a single transformation, genomic landscapes can be traversed through successive iterations of this process, and multiple distal genetic changes can be parallelized and combined through strain mating to sample new phenotypes.
RESULTS
Engineering ssODN annealing at DNA replication forks
Recombinase proteins (e.g., Rad51) catalyze the pairing and exchange of homologous DNA sequences. In S. cerevisiae, additional factors (e.g., Rad52, Rad54, Rad55, Rad57, Rad59) participate in HR by promoting the formation of Rad51-ssDNA filaments or through annealing of ssDNA (San Filippo et al., 2008). Prior work proposed that ssODNs are incorporated in the yeast genome through Rad51-mediated HR (Figure 1A) (DiCarlo et al., 2013; Liu et al., 2004). Along these lines, we tried to enhance ssODN-mediated recombination by increasing expression of HR genes and impairing MMR. We measured ARF for a ssODN containing a single bp mutation in the RPL28 gene, which confers cycloheximide resistance, for a panel of HR genes and MMR knockout (KO) strains (Figures 1B and S1A, B). Overexpression of the three HR factors Rad51, Rad52 and Rad59 increased the ARF at least 10-fold above the negative control, with Rad51 and Rad52 producing the highest ARF (.02%), followed by Rad59 (.01%). Ablation of MMR led to ~100-fold enhancement (0.1%) in the strain msh2Δ. We next combined HR overexpression with MMR KO strains (Figures 1B and S1C–F). Overexpression of Rad51 in msh2Δ, msh6Δ, and mlh1Δ strains increased ARF four-fold above the level of the msh2Δ strain alone, yielding a maximum ARF of 0.4% that is consistent with prior work (DiCarlo et al., 2013). Together, these data suggest that ssODN incorporation by promoting Rad51-dependent HR and disabling MMR may be limited to the ARF previously reported.
Figure 1. Engineering ssODN incorporation at DNA replication forks.
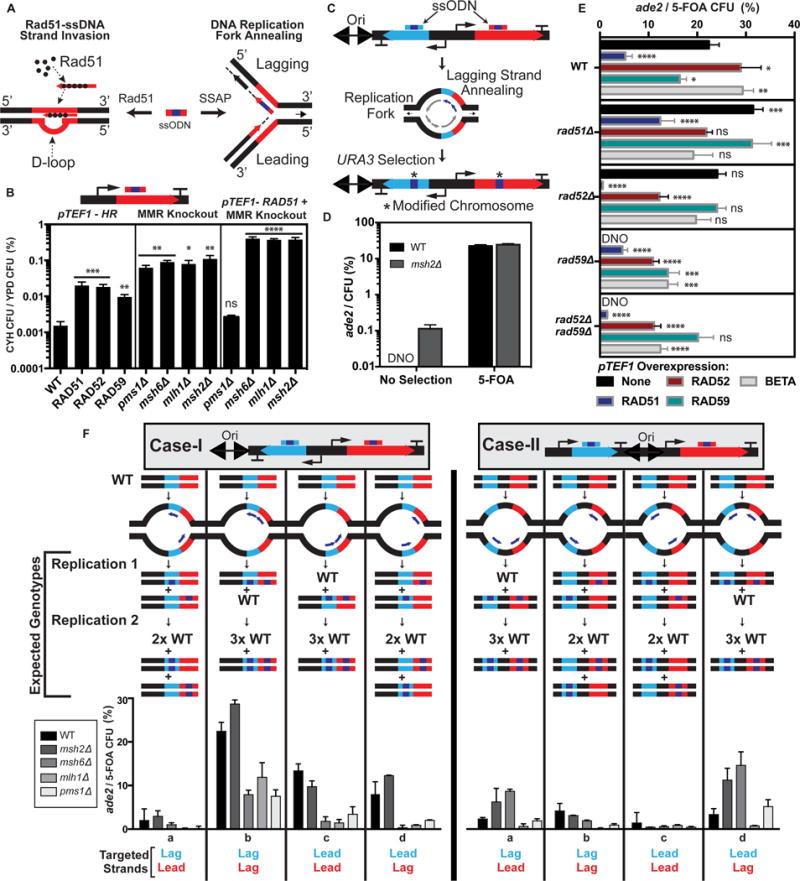
(A) Two pathways for ssODN incorporation in the genome. Rad51-dependent ssDNA strand invasion and SSAP-dependent annealing of ssODNs at the replication fork. (B) ARF for ssODNs with pTEF1 overexpression of HR genes (capitalized), MMR KO strains, and combinations of pTEF1-RAD51 and MMR KO. (C) Schematic for selection of ssODN annealing at the replication fork. (D) Measurement of ADE2 ARF after transformation with ura3 and ade2 ssODNs in WT and msh2Δ on non-selection and 5-FOA selection plates. (E) WT and HR KO strains + overexpression of the indicated gene. (DNO: did not observe). (F) Two cases for URA3 and ADE2 orientation at the Ori. Diagrams for each subcase (a–d) indicate the predicted segregation of the edited ura3 and ade2 alleles after two replications. The number of expected WT genotypes is depicted as # × WT. Plots show ARF for target strand combinations for Case I and Case II. All values represent mean +/− SD for three replicates. p-values from ordinary one-way ANOVA Dunnet’s multiple comparisons test (alpha = 0.05). Comparisons are to WT (B), and WT-None (E). (*p<.05; **p<.005; ***p≤.001;****p≤.0001). See also Figure S1.
We hypothesized that Rad51-dependent ssODN recombination and incorporation of ssODNs at the DNA replication fork are two distinct pathways in eukaryotes (Figure 1A). Sherman and colleagues observed high frequencies of co-transformation for two ssODNs targeted within a cyc1 mutant gene when one of the ssODNs included a selectable mutation (Yamamoto et al., 1992), which suggests that the two ssODNs were incorporated at the same DNA replication fork, and a finding also observed in E. coli (Carr et al., 2012). To test ssODN gene editing at the replication fork, we constructed an experimental locus on chromosome XV with a defined DNA replication direction by placing URA3 proximal to the origin of replication ARS1516 (Ori) directly adjacent to ADE2, which confers a colorimetric phenotype (WT = white; Mutant = red) (Figure 1C). We transformed cells with ssODNs targeting the predicted lagging strand of both URA3 and ADE2 and assayed the frequency of ade2 CFUs. We did not detect ade2 CFUs for the WT strain (limit of detection (LOD) = 10−4 CFU), and we observed ~0.1% ade2 CFUs for msh2Δ (Figure 1D). After 5-FOA selection to enrich for a competent subpopulation in ura3 clones, we observed 22% and 27% of CFUs with ssODN-derived ade2 mutations for WT and msh2Δ, respectively. To determine if the ARF enhancement is due to a coupled replication fork annealing mechanism we tested a marker-target pair (RPL28-ADE2) separated on different chromosomes (Figures S1G, H). After selection for rpl28 mutants, we recovered ade2 mutants in only 0.4% of CFUs, and overexpression of Rad51 led to a ~100% increase (0.8%). Thus, ssODN incorporation is enhanced in ura3-edited competent cells where downstream modifications are kinetically favored along a DNA replication fork in a localized chromosomal region.
To assess the impact of HR factors on the proposed replication fork annealing mechanism, we created a set of KO and overexpression strains for HR genes and assayed ssODN incorporation at the Ori-URA3-ADE2 locus. Unlike the RPL28-ADE2 interchromosomal pair, overexpression of Rad51 decreased ARF from ~22% to ~5%, whereas deletion of Rad51 increased ARF to ~30% (Figure 1E). Application of the Rad51 inhibitor RI-1 also enhanced ARF (Figure S1I). Overexpression of the SSAP Rad52 increased ARF to ~29%, whereas deletion of Rad52 showed a neutral effect. Overexpression of the SSAP Rad59 decreased ARF to ~16%, whereas we did not observe ade2 mutants in rad59Δ (LOD = 10−3 CFU). The effect of rad59Δ was epistatic to rad52Δ (rad52Δrad59Δ). We observed varying degrees of suppression of rad59Δ and rad52Δrad59Δ with overexpression of Rad51, Rad52, Rad59, or λ Red Beta. Since loss of Rad52 impairs Rad51-dependent HR (Sung, 1997), we observed a higher ARF with Rad51 overexpression in rad59Δ (4.6%) than rad52Δ (0.6%) or rad52Δrad59Δ (1.5%). Overexpression of Rad52 showed an equivalent ARF in all three strains (~10%). The effect of Rad59 overexpression was enhanced in rad51Δ, rad52Δ and rad52Δrad59Δ, potentially due to reduced competition from Rad51-dependent HR in these strains. Notably, λ Red Beta enhanced ARF to ~29% in WT, and recovered rad59Δ and rad52Δrad59Δ to ~13%. In summary, our data show that ssODN annealing at the replication fork is distinct from Rad51-mediated ssODN recombination, enhanced in the rad51Δ background, and requires an SSAP (e.g., Rad59, Beta).
To further study ssODN annealing at the replication fork, we tested the effect of leading (lead) versus lagging (lag) strand targeting. Since yeast chromosomes contain multiple origins of replication that fire at varying efficiencies (Friedman et al., 1997), we placed URA3 in two orientations with respect to ADE2 (Figure 1F). In Case-I, URA3 and ADE2 reside on the same side of the Ori, such that they are replicated in the same replication fork. In Case-II, URA3 and ADE2 are positioned on different sides of the Ori, such that they are replicated in opposing directions when the Ori fires. We tested all lead/lag strand combinations in a set of WT (BY4741) and MMR mutant strains (msh2Δ, msh6Δ, mlh1Δ, and pms1Δ). The WT and msh2Δ strains had the highest ARF; however, the number of 5-FOA resistant CFUs was ~100-fold higher for msh2Δ compared to WT (see Figure 1B). In addition to selection for competent cells (Figure 1D), we observed two key factors that enhance ARF: (1) ssODNs that target the lag strand and (2) ssODN pairs that target the same chromosomal strand. The lag strand is favorable due to the prevalence of available ssDNA compared to the lead strand (Rivera-Torres and Kmiec, 2016), and pairs of ssODNs that target the same chromosomal strand result in cosegregation of the ssODN-derived alleles. Consistent with this model, the lag-lag (ura3-ade2) strand combination for Case-I (Case-Ib) satisfies both criteria and showed the highest ARF in all strains except msh6Δ, which was enhanced in Case-II. Such differences in ARF could be due to MMR activity at the replisome or altered replisome dynamics and warrants investigation in future work. Cases Ic (lead-lead), IIa (lag-lead), and IId (lead-lag) satisfy condition 2, and showed elevated ARF. Since the Ori does not always fire, the enhanced ARF for Case-IIa and IId could be explained by replication from adjacent origins in some cells rendering the ssODNs in the same replication fork. Four scenarios (Cases Ia, Id, IIb, and IIc) should not result in cosegregation of the ura3 and ade2 alleles, yet we observed clones with both ssODN-derived alleles. This suggests that either the ssODNs can persist for more than a single cell cycle, or rapid processing of the heteroduplex by MMR or a MMR-independent pathway followed by HR between sister chromatids might explain the cosegregation (San Filippo et al., 2008). For these scenarios, higher ARF occur when the ade2 ssODN is targeted to the lag strand. To validate our findings, we observed equivalent strand bias trends for msh2Δ in both Case-I and Case-II for a distal locus (RPL28) on chromosome VII (Figure S1J). Thus, efficient ssODN incorporation (>10%) can be achieved for marker-target pairs adjacent to or on opposite sides of an Ori through precise targeting at the replication fork.
Investigating replication and repair factors for ssODN incorporation
We conducted three key experiments to investigate the impact of replication dynamics and factors that monitor replication and repair. First, we tested if slowing DNA replication with hydroxyurea (HU) enhances ssODN incorporation. A 30-minute treatment with 500mM HU increased the ARF by ~two-fold with no significant increase in spontaneous ura3 mutations (Figure S2A, B). HU also enhanced ARF by an average of 56% for seven ssODNs containing a single bp mismatch (ssODNs #1,2,5–7), insertion (ssODN #4), or deletion (ssODN #3) in ADE2 (Figure 2A). To validate these data and disconnect the selection step from dNTP metabolism, we observed similar findings with HIS3 selection (Figure S2C). Next, to determine if annealing of the ura3 ssODN induces a DNA damage response that enhances downstream ssODN annealing, we exposed cells to a range of UV irradiation, a general DNA damage event, and did not observe an ARF increase (Figure S2D). Third, replication slowing has been implicated to occur during gene editing (Rivera-Torres and Kmiec, 2016), and we postulated that ssODN annealing might stall the replication fork to enhance ARF at downstream loci. Since Mec1-dependent signaling is associated with stalled replication forks (Branzei and Foiani, 2010), we tested the effect of deleting Mec1 (Figure 2B). Given that null mutants of Mec1 are viable in sml1Δ strains, we measured ARF in sml1Δ and sml1Δmec1Δ. ARF was reduced by ~25% in sml1Δ and by ~60% in sml1Δmec1Δ for both −/+ HU conditions. In summary, slowing replication fork speed with HU increases ARF, ARF enhancement is independent of UV-induced DNA damage, and the Mec1 deletion attenuates ARF.
Figure 2. Investigating replication dynamics for incorporation of diverse mutations with ssODNs.
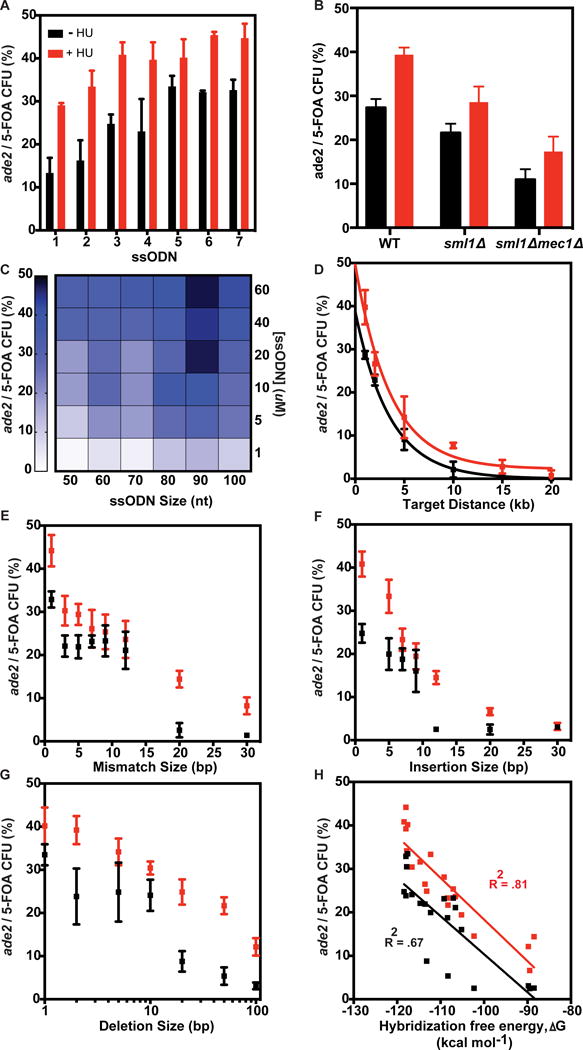
(A) The effect of hydroxyurea (HU) treatment (red) for seven ssODNs each containing a single bp change. (B) Effect of Mec1 deletion −/+ HU. (C) Optimization of ssODN concentration and size for ura3 + ade2 ssODNs. Heatmap shows mean ARF for each ssODN. See Figure S2G for these data as mean +/− SD. (D) ARF for increasing target site distances from the URA3 marker. Non-linear curve fit one-phase decay equation: Y=(Y0 − Z)*exp(−K*X) + Z ; (+HU: Y0 = 49.6, Z=2.25, K= 0.279), (−HU: Y0 = 38.7, Z=2.22*e−16, K=0.2826). (E–G) ARF of various genome modifications at 1.5kb distance in ADE2: (E) Mismatches, (F) Insertions, and (G) Deletions. (H) The Hybridization free energy plotted against the mean ARF for all ssODNs tested in E–G with Pearson correlation coefficients. Values in A,B,D–G represent mean +/− SD for three replicates. See also Figure S2.
Optimizing parameters to enhance genome editing
To maximize ARF, we performed a set of experiments to identify the optimal size and concentration of ssODNs in msh2Δ using 500mM HU. Given that cell survival is crucial for generating large mutant libraries, we tested the effect of increasing ssODN concentration on cell survival one-hour post electroporation, allowing for cell recovery below the doubling-time (>100-minutes for msh2Δ). Robust survival (85%) was obtained for ssODN concentrations from 0 to 20 μM, which decreased to 45% at 80 μM ssODN (Figure S2E). We also tested the effect of ssODN size (50–100nt) and concentration (0.1–60 μM) on the ARF for single (ura3) and double (ura3-ade2) ssODN incorporation. The efficiency of ura3 mutants increased with increasing ssODN size and concentration with the highest ARF observed (0.08%) for 100nt ssODN at 60uM (Figure S1F). For coupled ura3-ade2 targeting, we observed an optimum number of ade2 mutants per 5-FOA CFU (ARF = 47.5%) using a 90nt ssODN at 20uM (Figure 2C, S1G), and used these parameters for all subsequent experiments.
Since yeast origins are ~30kb apart, we tested the effect of target distance for loci at increasing distances from the Ori-URA3 locus (Figure 2D). ARF decreased with target distance from the Ori-URA3 locus, but remained >1% at a distance of 20kb. This decrease in ARF could be due to interference of replication from distal origins and warrants future investigation. To investigate the introduction of diverse mutations, we tested a set of ssODNs containing a contiguous block of mismatches, insertions, or deletions within ADE2 (~1.5kb from the Ori-URA3) for the msh2Δ strain. The ARF was inversely correlated to the number of bp targeted for modification, in which a single bp mismatch, insertion, or deletion was incorporated with an ARF >40% (Figures 2E–G). Mismatches up to 30 bp and deletions up to 100 bp were incorporated at ~10% efficiency (Figures 2E, G). Insertions are the least efficient modification with ARF ≤10% for insertions >12 bps (Figure 2F). The two-state hybridization free energy between the ssODN and genomic target sequence was a better predictor of ARF for ssODNs tested in the +HU condition than −HU (Figure 2H) (Markham and Zuker, 2008). These data demonstrate that ssODN annealing at the replication fork is enhanced in close proximity to the Ori-URA3 locus and can generate diverse mutations required for precision genome editing.
Generation of precise combinatorial genome modifications via multiplexing and cycling
Since the average size of Okazaki fragments in S. cerevisiae (~165nt) (Smith and Whitehouse, 2012) is significantly shorter than that of E. coli (~1–2kb) (Wu et al., 1992), we hypothesized that the introduction of multiple ssODNs at high density at the replication fork could enable a high degree of multiplex gene editing. To test this hypothesis, we targeted 10 loci across the ADE2 gene in WT and msh2Δ strains (Figure 3A). The mean number of ssODNs incorporated per clone was higher in msh2Δ (2.2 per clone) than WT (1.2 per clone). We hypothesized that HU would enhance ssODN multiplexing and tested ssODN pools targeting 2-, 4-, 6-, and 10-sites across the ADE2 gene (Figure 3A). For 10-target multiplexing, HU increased the mean number of ssODNs incorporated in msh2Δ to 3.4 per clone, whereas WT exhibited no multiplex enhancement with HU (Figure S3A). The mean number of ssODNs incorporated plateaued at ~1 mutation per clone for WT and increased as a function of the number of target loci for msh2Δ (Figure 3A). We observed clones with diverse combinations of targeted changes for all multiplex pools (Figure S3A).
Figure 3. Multiplex ssODN incorporation and cycling.
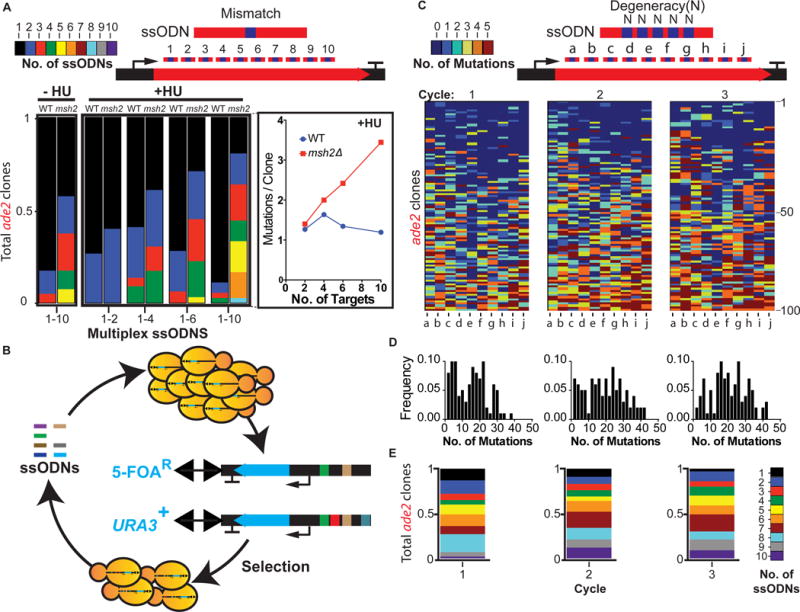
(A) Multiplexed ssODNs harboring single point mutations across ADE2. Pools of 2-, 4-, 6-, and 10-plex ssODNs with HU; 10 ssODNs untreated (−HU). Panel shows msh2Δ compared to WT mean mutations per clone for +HU. Number of clones sequenced (n), n= 20 (2-plex), n= 22 (4-plex), n=32 (6-plex), n=36 (10-plex), n=40 (10-plex −HU condition). (B) Iterative cycling of ssODNs to a population of cells. URA3 is targeted by an ‘OFF’ ssODN in odd cycles and an ‘ON’ ssODN in even cycles. Positive-negative selections enable recovery of diversified chromosomes. (C) Cyclical introduction of 10-plex ssODNs containing 5 degenerate ‘N’ positions per ssODN. Heat maps show sequence data for n = 100 ade2 mutant clones per cycle. ssODN position (columns a–j) and clonal sequence data (rows 1–100). (D) Frequency distributions of ade2 bp mutations per clone for each cycle. (E) Number of ssODNs incorporated per ade2 clone for each cycle. See also Figure S3 and Table S3.
To increase genetic diversity, we developed a strategy for continuous diversification of a yeast cell population by cyclical introduction of a complex pool of ssODNs coupled to +/− URA3 selections (Figure 3B, see methods for details). We designed a set of 10 ssODNs containing 5 degenerate (‘N’) insertion positions spaced 10bp apart in each ssODN. We analyzed 100 ade2 clones by Sanger sequencing after each cycle, and observed a broad distribution of ade2 genotypes (Figure 3C–E; Table S3), ranging from one to five insertions per ssODN. After three cycles of eMAGE with the same pool of ade2 ssODNs, the maximum number of mutations increased from 37 to 42 per clone and the average number of mutations increased from 14.4 to 20.4 per clone (Figure 3D). The average number of ssODNs incorporated increased from 5.1 to 6.0 ssODNs per clone (Figure 3E). All of the sequenced clones contained unique genotypes in cycles 1 and 2, and 76% were unique in cycle 3. We performed whole genome sequencing (WGS) for 12 clones from each cycle in order to understand the effect of the eMAGE protocol on the background mutation rate. Consistent with the reported rate for msh2Δ (7.1×10−8 mutations per bp per generation) (Lang et al., 2013), we observed a mean mutation rate of 8.1×10−8 mutations per bp per generation (Figures S4A–C; Tables S3–S5). These data demonstrate the ability to rapidly create combinatorial genomic diversity by iterative incorporation of a complex pool of ssODNs at the replication fork.
High Throughput Sequencing (HTS) of a Diversified Population
To measure the genetic diversity created by eMAGE and to further study multiplex ssODN incorporation, we performed HTS of a diversified population at a defined region of ADE2 (Figure 4). We transformed a pool of three ssODNs each encoding five degenerate insertions at ADE2 to an initial population of ~108 cells, expecting ~105 edited cells to survive 5-FOA liquid selection (Figure 4A; See Figure S2F for URA3 selection efficiencies). The 15 insertion positions span a 307 bp region of ADE2 such that the sequence diversity could be analyzed with 2×250bp paired-end reads. Using a computational pipeline, we observed ~1.59×105 and ~6.70×105 unique variants for read quality scores of Q30 and Q20, respectively (Figure 4B, S4D–F). The mutants contained one, two, or three ssODNs incorporated, and the two most abundant contained either the most proximal ssODN (ssODN #1) or all three ssODNs (Figure 4C). For the mutants with two ssODNs incorporated, we did not observe a strong preference for adjacent ssODN pairs. We performed a rarefaction analysis and the sequence accumulation plots (Figures 4D, S4D) did not plateau before the number of HTS reads reached its maximum (Amiram et al., 2015; Szpiech et al., 2008). Given this result, we hypothesize that our diversity estimates likely represent lower bounds and expect that the actual complexity can be quantified as HTS technologies improve.
Figure 4. Deep sequencing analysis of a population diversified with eMAGE.
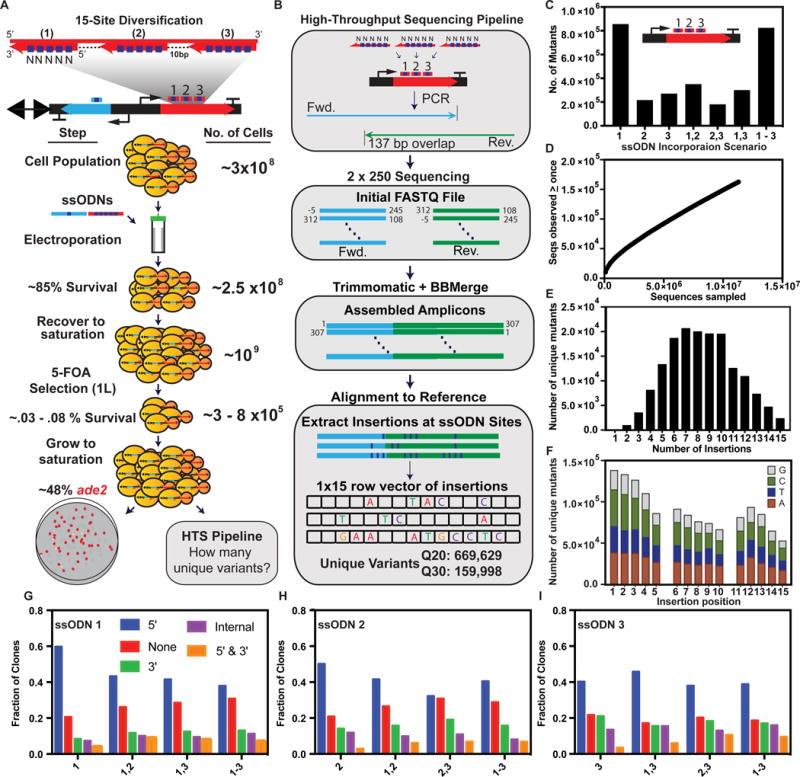
(A) 15-site diversification of the ADE2 gene with three ssODNs containing five degenerate (N) positions each. A population of cells is diversified via electroporation of the ssODN pool and ura3 selection ssODN. After recovery to saturation the population is subjected to liquid selection in 5-FOA media and grown to saturation, a small aliquot is plated to YPD, and a genomic prep is processed by the HTS pipeline. (B) Pipeline for determining the number of unique variants generated by eMAGE. A PCR amplicon containing the diversified locus is deep sequenced using 2×250 sequencing. The overlapping paired end reads are trimmed and processed for quality score and aligned to the reference genome. The ssODN insertions are extracted and analyzed to quantify the total number of unique mutants. (C–I) Plots derived from Q30 sequence data. (C) Abundance of mutants detected with each possible ssODN incorporation scenario. (D) Rarefaction curve illustrating the accumulation of sequences seen at least once as a function of the total number of sequences observed. (E) Number of targeted insertions present in unique mutant sequences. (F) Positional distribution of the targeted insertions with the relative abundance of each base at each ssODN. (G–I) Plots show the frequency and type of processing events for each ssODN in all incorporation scenarios. Colored bars represent the removal of an insertion mutation at the 5′ portion of the ssODN (blue), 3′ (green), both 5′ and 3′ (orange), at an internal position in the ssODN sequence with retained 5′ and 3′ insertions (purple), and incorporation of all five insertions (red). See also Figure S4 and Tables S4, S5.
Consistent with our Sanger sequencing data (Figures S3B,C), we observed a distribution of insertions per ssODN with a 3′ position bias (Figures 4E,F). Prior work (Rodriguez et al., 2012) implicated the Fen1-endonuclease in flap degradation at the 5′ end of ssODNs. We posited that this effect could be partially explained by truncated ssODNs arising from errors in DNA synthesis since ssODNs are synthesized 3′ to 5′. Although we observed a reduction in 3′ bias with a PAGE purified ssODN (Figure S3D), this effect was not completely eliminated with PAGE purification, suggesting that the effect could be a combination of truncated ssODNs from synthesis and native processing. The greater read-depth with HTS allowed us to uncover additional processing events at the 3′, potentially due to proofreading activity of DNA-polymerase δ (Anand et al., 2017), and in some cases we observed clones lacking an internal mutation in the ssODN but retaining the 5′ and 3′ mutations (Figure 4G–I).
Targeted Diversification of a Heterologous Biosynthetic Pathway
To further study our ability to generate multi-site combinatorial genomic variation at bp-level precision, we targeted a heterologous β-carotene pathway for the creation of diverse variants. The pathway consists of four constitutively expressed genes (crtE, crtI, crtYB, and tHMG1), which convert farnesyl diphosphate (FPP) to β-carotene through a series of enzymatic steps in S. cerevisiae (Figure 5A) (Mitchell et al., 2015). We designed a pool of ssODNs to precisely target distinct genetic elements in promoters, open reading frames (ORFs), and terminators (Figure 5B,C, Table S4; See methods for details of ssODN designs). Overall, the ssODN pool consisted of 74 ssODNs encoding targeted mutations at 482 nucleotide positions.
Figure 5. Targeted diversification of a β-carotene pathway.
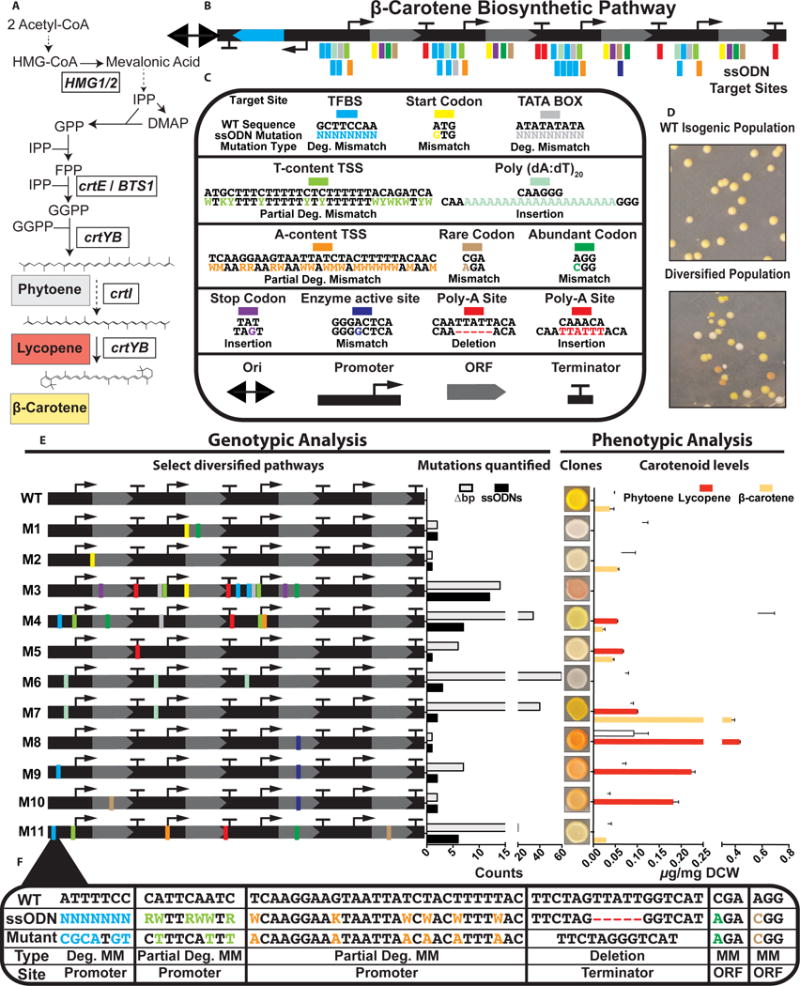
(A) β-carotene biosynthetic pathway constitutively expressed in yeast (B) The genomically integrated β-carotene pathway adjacent to Ori-URA3. ssODN target sites in promoters, ORFs, and terminators. (C) Examples of specific mutations for each sequence element targeted. Degenerate mutations abbreviated as ‘Deg.’, Mismatch as ‘MM’. ‘N’ = mixed bases A,T,G,C; ‘W’ = mixed bases A,T; ‘Y’ = mixed bases C,T; ‘R’ = mixed bases A,G; ‘K’ = mixed bases G,T; ‘M’ = mixed bases A,C. (D) Images showing representative colonies expressing the β-carotene biosynthetic pathway (WT) and diversified phenotypes after eMAGE. (E) Genotypic and phenotypic analysis of select clones containing diversified genotypes and phenotypes uncovered with Sanger sequencing and HPLC analysis. Total number of ssODNs incorporated (black bar) and number of targeted bp changes (light-grey bar). HPLC data for clonal production of β-carotene (orange) lycopene (red), and phytoene (white) (ug/mg dry cell weight). Values represent mean +/− SD for three replicates. (F) Expanded view of clone M11 containing targeted edits in promoters, ORFs, and a terminator. See also Figures S5–S7 and Table S6.
After a single eMAGE cycle with the entire 74 ssODN pool, we observed clones with diverse colorimetric phenotypes that differed from both the ancestral strain and the full set of 15 possible combinatorial pathway gene KOs (Figures 5D,S5A). We selected diverse variants for Sanger sequencing and HPLC analysis to reveal causal genotype-phenotype relationships. The analyzed clones contained a range of 1–60 bp changes and 1–12 ssODNs incorporated (Figures 5E, S6, S7). Consistent with our findings for the ADE2 locus, we observed enhanced ARF for targets more proximal to URA3 and targets with the fewest bp changes (Figure S7). We observed many examples of precise genetic modifications that resulted in distinct phenotypic variation. For example, three clones with varied carotenoid levels contained mutations in the crtE gene element distinct from the crtE KO (KO1): an alternative start codon (M2), polyadenylation signal site insertion (M5), and a rare codon (M35). KO of crtI (KO2) resulted in buildup of phytoene corresponding to a white phenotype, which was indistinguishable from a clone containing an alternate start and an abundant arginine codon in crtI (M1). In contrast, a deletion of 6 bp in the crtI terminator (M39) resulted in β-carotene buildup and no detectable phytoene. Incorporation of nucleosome disfavoring poly(dT)20 sequences in promoters for crtE and crtI resulted in ~seven-fold increase in β-carotene production (M7), whereas an additional poly(dT)20 in the promoter of crtYB led to detection of phytoene only (M6). We also recovered high lycopene variants containing crtYB-D52G and additional gene modifications that altered lycopene levels (M8-10, M12-14). Notably, clone M11 contained 22 bp of targeted mutations derived from 6 distinct ssODNs in all three classes of genetic elements across all four genes (Figure 5E,F). The background mutation rate (6.6×10−8 mutations per bp per generation) for 55 diversified clones measured by WGS was consistent with prior findings (Figure S4 G–I). Since the β-carotene pathway contains four promoters and three terminators found at native loci in the genome, we also checked for ssODN incorporation at these off-target sites and did not observe any ssODN-derived mutations (Table S4, S5). These results demonstrate the ability to sample phenotypic variation through precise bp editing at target sites, which could be applied to any set of genetic elements to elucidate causal links between genotype and phenotype.
We then tested whether targeted edits in genes located on different chromosomes could be generated across haploids in parallel and combined via mating. We constructed a MATα haploid containing the crtE gene adjacent to a URA3 cassette at Ori ARS510 on chromosome V, and a MATa haploid containing the crtI, crtYB, and tHMG1 genes at Ori ARS1516 on chromosome XV (Figure 6A–C). Upon mating, the resultant diploids showed the yellow phenotype indicative of the presence of all four genes of the wildtype β-carotene pathway (Figure 6D). Next, we generated parallel diversity of the haploids with ssODN pools targeting the genes present in each strain, and mated the populations to generate diploid strains with diversified phenotypes resulting from the independent chromosomes targeted (Figure 6E). We repeated the process for crtE at Ori ARS446 on chromosome IV and Ori ARS702 on chromosome VII and observed equivalent results (Figure S5C). These experiments demonstrate the generalizability of replication fork targeting of distinct loci on multiple chromosomes (IV,V,VII, and XV) and subsequent mating of the diversified haploid strains to amplify combinatorial genetic variation in diploids.
Figure 6. Genomic diversification across chromosomes and combined through mating.
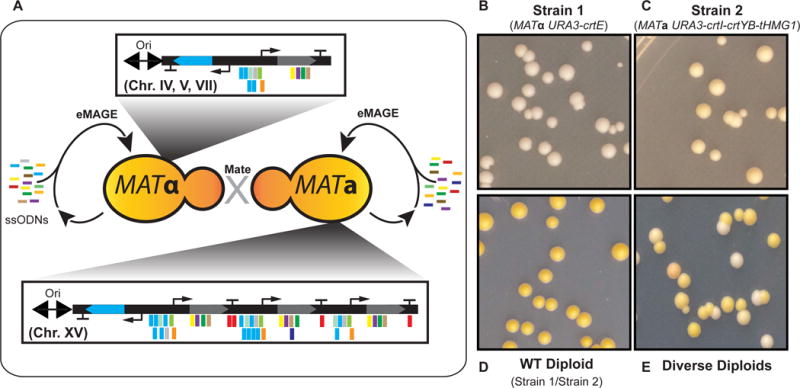
(A) Parallel diversification of the β-carotene pathway split into two haploid strains. URA3-crtE is integrated in three MATα strains at Chr. IV, V, or VII to demonstrate eMAGE targeting on additional chromosomes. URA3-crtI-crtYB-tHMG1is integrated at Chr. XV in MATa. After performing eMAGE on the strains in parallel, the populations are combined with mating to yield diversified diploids. (B) Strain1 genotype is MATα ARS510-URA3-crtE at Chr. V. (C) Strain 2 genotype is MATa ARS1516 URA3-crtI-crtYB-tHMG1 at Chr. XV. (D) Control mating of Strain 1 and 2 shows ancestral phenotype of full pathway. (E) Mating of diversified populations shows altered phenotypes in diploids. See also Figure S5.
Altering Transcriptional Logic with ssODNs
Finally, we tested whether ssODNs can be used to precisely replace transcription factor binding sites (TFBS) to alter regulatory logic. We designed a set of ssODNs to replace native TFBS in the β-carotene pathway with the 18 bp galactose-inducible Gal4 binding sequence (Figure 7A). We transformed cells with the Gal4 ssODNs and a ssODN containing the crtYB-D52G mutation to enhance the detection of new phenotypes. We isolated clones with altered color phenotypes on glucose versus galactose plates and sequenced the targeted loci. Consistent with previous data (Figure 2D,E) at ~1.5kb we observed the insertion of Gal4 TFBS with ARF between 8%–22% and ARF <5% at distances >3kb (Figure 7B). We studied five clones that exhibited color changes when spotted to galactose to confirm the introduction of a Gal4 TFBS (G1–G5) (Figure 7C). RT-qPCR of these clones confirmed that genes with a Gal4 TFBS were induced on galactose and are responsible for the color changes (Figure 7D, Table S7). For example, G4 showed the strongest galactose gene induction of crtI (1130%). We observed galactose induction of some genes that did not contain a Gal4 TFBS (G4, G5), which could be due to altered transcription levels of genes in the pathway. Similar to the native GAL1 gene, induction of crtI expression in G4 was dose-responsive to a range (.01–5%) of galactose (Figure 7E). These data show that eMAGE can introduce sequence elements that can impart galactose-based induction of gene expression capable of altering transcriptional levels. This strategy can be applied to many other transcriptional logic elements (e.g., TetR-tetO) and promoters to create diverse sets of genetic pathways with programmable regulatory properties.
Figure 7. Introduction of Gal4 transcriptional logic sequences with ssODNs.
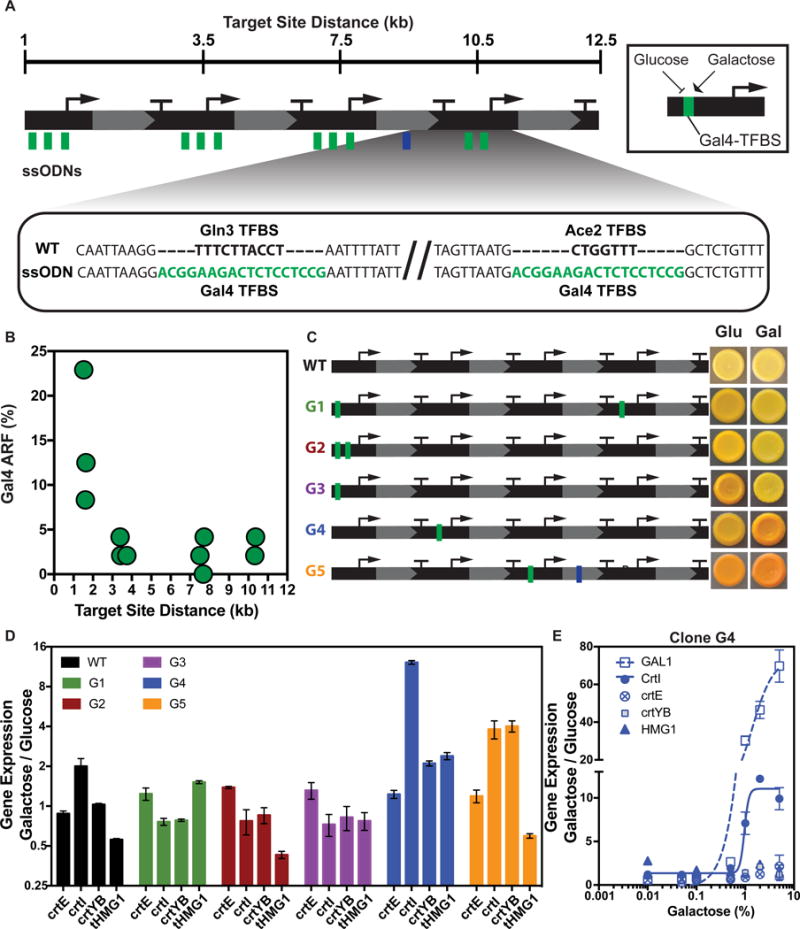
(A) ssODNs containing the 18-nt Gal4 (green) binding site targeted to replace native TFBS (bold) in promoters and a ssODN containing crtYB-D52G mutation (blue) at distances between 1 and 11kb across the pathway. (B) Plot shows observed ARF for each Gal4 ssODN at the indicated distance. n=48 sequenced clones. (C) Mutant pathways containing Gal4 TFBS inserted in promoters and clonal spots show phenotypes of the clones on glucose (Glu) and galactose (Gal). (D) Fold-change in gene expression in galactose vs. glucose. (E) Fold-change in gene expression of clone G4 across a range of galactose at the native GAL1 gene, and the engineered pathway. Non-linear curve fit for GAL1 and crtI. R2 = .97(GAL1) and .96(crtI). Values in D,E represent mean +/− SD for three replicates. See also Table S7.
DISCUSSION
In this study, we developed a eukaryotic genome engineering technology by elucidating a new mechanism that avoids the creation of DSBs by precise annealing of ssODNs at the DNA replication fork to enact bp precision and combinatorial genome editing across many genetic loci. Incorporation of ssODNs at the replication fork is independent of Rad51, and overexpression of Rad51 reduces ARF potentially by competing for ssODN binding (Song and Sung, 2000). Rad51-independent HDR has also been reported for CRISPR-Cas9 (Collonnier et al., 2017). Rad52 and Rad59 are involved in Rad51-independent processing of Okazaki fragments (Lee et al., 2014), and the loss of detectable ARF in rad59Δ and rad52Δrad59Δ compared to the neutral effect of its paralog (rad52Δ) suggests a unique role for Rad59 to promote annealing of ssODNs at the replication fork. Although Rad52 may contribute to ssODN annealing at the replication fork, it also mediates loading of Rad51 to RPA-bound ssDNA (Song and Sung, 2000) whereas Rad59 does not contain a known Rad51 binding domain (Erler et al., 2009). The rescue of rad59Δ and rad52Δrad59Δ by Rad52, Rad59, and λ Red Beta suggests that a ssDNA annealing function is required for the high ARF we observed in this study and supports our model of ssODN annealing at the replication fork. We did not observe further enhancement of rad51Δ with overexpression of SSAPs, which suggests that the replication fork annealing pathway is nearly saturated in rad51Δ. The partial rescue of rad59Δ by Rad51 matched the ARF of Rad51 overexpression in WT, suggesting that these are Rad51-mediated HR events. Surprisingly, Rad51 also partially rescued ARF in rad52Δrad59Δ despite the absence of its mediator Rad52. Thus, Rad51 might exhibit low-level HR or replication fork annealing activity in rad52Δrad59Δ.
Our results reveal several factors that govern ARFs. First, selection for ssODN incorporation at the replication fork enriches for a competent subpopulation. Second, combining the selectable ssODN with a pool of ssODNs targeting proximal loci permits kinetically driven incorporation of ssODNs within the same replication fork in contrast to loci separated on different chromosomes. Third, when the selection marker and target loci reside on the same side of an active Ori, targeting ssODNs to the lagging strand is optimal; if they reside on opposite sides of the Ori then targeting cosegregating strands is favorable. Fourth, slowing replication fork speed with HU increases multiplex genome editing at downstream loci. Fifth, the ARF decrease in sml1Δmec1Δ suggests that Mec1-dependent signaling partially stabilizes the replication fork (Branzei and Foiani, 2010) during ssODN incorporation. Lastly, unlike HU, UV exposure does not amplify ARF, suggesting that ssODN incorporation at the replication fork is not mediated by a UV-induced DNA damage response. Further insights into the ssODN annealing mechanism through studies of Rad59, additional HR factors (e.g., Rad54, Rad55, Rad57, Dmc1), SSAPs, DNA replication or repair gene combinations, and modulation of replication fork kinetics could enhance eMAGE.
For single site editing, we observed similar ARF in WT and msh2Δ strains, however, our data showed that MMR inhibits multiplex gene editing and therefore decreases the generation of genome complexity across the population. Although MMR mutants are not desirable when trying to maintain genome stability, we envision directed evolution and pathway engineering applications, as shown here, that could benefit from an elevated mutation rate. Alternatively, transient disabling of MMR through small molecule inhibitors, dominant-negative mutants, RNAi, or CRISPRi approaches could be used to introduce genetic modifications during a transient relaxed genomic state (disabled MMR) followed by rapid return to a stabilized genomic state (intact MMR).
Although the goal of our study does not aim to maximize titers of small molecule production, our approach to diversify the β-carotene pathway can serve as a blueprint for natural product discovery to investigate and activate cryptic biosynthetic gene clusters or metabolic engineering applications to produce high-value molecules from heterologous genetic sources (Krivoruchko and Nielsen, 2015). Current methods to tune the expression of biosynthetic pathways in yeast rely on the use of promoter swapping strategies (Mitchell et al., 2015; Wingler and Cornish, 2011). Although these approaches are effective, the possible modes of gene expression are limited to quantal units of transcription dictated by the promoter library. Diversification of biosynthetic pathways with eMAGE enables targeting across all associated genetic elements and allows for exploration of greater diversity. In contrast to ~102 variants constructed using current DNA synthesis technologies (Smanski et al., 2014), generating targeted and complex (>105) diversity across multiple genetic loci is uniquely enabled by eMAGE. An equivalent diversification of the β-carotene pathway would not be possible using DSB methods, in which ~41 DSBs are required to target 482 nucleotides and many of the single bp mutations would not sufficiently prevent re-cutting by the DSB machinery. Given prior reports (Jakociunas et al., 2015), we expect that >4 DSBs would be lethal, and thus intractable with DSB-mediated genome editing technologies. Since CRISPR-Cas9 can efficiently introduce large genetic fragments into the genome and eMAGE can enact many precise combinatorial edits, we envision that the two approaches could be used in concert to recombine and diversify pathways.
Eukaryotic MAGE uniquely enables the construction of targeted sets of genetic variants that can be functionally studied to elucidate causal links between genotype and phenotype. The eMAGE process could be automated, employed to uncover new allelic interactions to complement synthetic genetic arrays (Costanzo et al., 2016), and used to hierarchically construct highly modified chimeric genomes from multiple strains. We anticipate that eMAGE capabilities could be included in future synthetic eukaryotic chromosome projects (Annaluru et al., 2014; Boeke et al., 2016) to enable efficient single bp precision editing of designer genomes. Finally, the approach used for S. cerevisiae targets conserved mechanisms and establishes a framework for developing efficient multiplex ssODN annealing methods in multicellular eukaryotes, including plants and animals.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Farren Isaacs (farren.isaacs@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
A complete list of Saccharomyces cerevisiae strains used in this study can be found in Table S1. Strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) was chosen due to its common use as a laboratory strain and for its use in the Saccharomyces Genome Deletion Project (Brachmann et al., 1998; Winzeler et al., 1999). MMR KO strains from the Saccharomyces Genome Deletion Project were purchased (Open Biosystems, Thermo Scientific).
METHOD DETAILS
Strain Construction
Strains harboring the URA3 marker for coupled ssODN selection (eMAGE) were constructed via standard homologous recombination with a dsDNA URA3 PCR product containing 60nt of overlap to the genomic locus followed by selection on CSM-Uracil plates. For Case-I the following primers were used to amplify and integrate URA3. Forward primer with ADE2 locus overlap: 5′-ATAATATTGTCCATTTAGTTCTTAATAAAAGGTCAGCAAGAGTCAATCACTTA GTATTACGATGTAGAAAAGGATTAAAGATGCTAAGAGATAGTGA-3′ Reverse primer with ARS1516 locus overlap: 5′-TTAATTATGATACATTTCTTACGTCATGATTGATTATTACAGCTATGCTGACA AATGACTCAATGCGTCCATCTTTACAGTCCTG-3′.
For Case-II, Forward primer: 5′-TCCATCTGACATTACTATTTTGCATTTTAATTTAATTAGAACTTGACTAGCGCA CTACCAGATGTAGAAAAGGATTAAAGATGCTAAGAGATAGTGA-3′. Reverse primer: 5′-TGAAGTTTCTTTTATAATAACCTGGTCAAAAGCTTTCAATATATAATACATTT GGTATTTCAATGCGTCCATCTTTACAGTCCTG-3′. The β-carotene pathway is derived from Xanthophyllomyces dendrorhous and was amplified by PCR from plasmid pJC178 (Mitchell, 2015), then genomically incorporated at ARS1516 or at the indicated ARS location for mating experiments (Figure 5,6, S5–7, Table S1). The β-carotene pathway PCR product was genomically integrated using a CRISPR-Cas9 genome integration. First, the WT strain BY4741 was transformed with a constitutive expression Cas9-NAT plasmid, a gift from Yong-Su Jin (Addgene #64329) (Zhang et al., 2014) by standard PEG/Lithium acetate transformation (Gietz, 2014) and positive clones were selected via Nourseothricin (clonNat) selection. Second, the gRNA plasmid, pRPR1_gRNA_handle_RPR1t, a gift from Timothy Lu, (Addgene #49014) (Farzadfard et al., 2013) containing gRNA target site sequence ‘CTTGTTGCATGGCTACGAAC’ located at chrXV:566360 was transformed along with the β-carotene pathway PCR product. Positive clones were selected using leucine auxotrophic selection, inspection for yellow colored phenotype, and sequencing confirmation. Third, the URA3 selection marker was introduced between the ARS1516 and β-carotene pathway in the Case-I orientation as described above with sequence overlap to the β-carotene pathway. Fourth, MSH2 was deleted using a dsDNA recombination of an hphMX cassette conferring hygromycin B resistance. All modified loci were sequence verified in the final strain EMB294 used in this study.
Media
For general strain manipulation cells were grown in YPADU liquid medium, which consists of YPD (10g/L Yeast Extract, 20g/L Peptone, 20g/L Dextrose), supplemented with 40mg/L adenine hemisulfate, and 40mg/L uracil. For URA3-coupled ssODN experiments (eMAGE), strains were grown in CSM-Ura medium during odd numbered cycles and CSM-Ura+5-FOA (1g/L) + uracil (50mg/L) medium during even numbered cycles. For HR gene overexpression experiments, CEN/ARS plasmids were maintained with the hygromycin B resistance marker. After electroporation, cells were allowed to recover in YPADU/0.5M Sorbitol (Recovery Medium).
Plasmid Assembly for overexpression of HR Genes and SSAPs
To clone pTEF1 expression plasmids for HR genes, all HR gene ORFs were PCR amplified from BY4741 genomic DNA prepared via a standard glass bead yeast genomic preparation protocol. The λ Red Beta gene was purchased as a dsDNA fragment (IDT) containing the SV40 nuclear localization sequence (NLS) at the N-terminus. The Forward primers for each ORF contained 40 bases of 5′ overhang with sequence identity to 3′ end of the TEF1 promoter. Reverse primers for each ORF contained 5′ overhang with 40 bases of cyc1T terminator identity. Gibson assembly cloning (Gibson, 2011) was used to assemble a hygromycin B resistant (hphMX) CEN/ARS plasmid backbone (pRCVS6H) for each ORF to generate (pRCVS6H-pTEF1-ORF-CycT-hphMX plasmids). All plasmids were sequence verified.
Yeast ssODN Electroporation with Rad51-Dependent HR
A 2mL culture was inoculated with a single colony and grown to saturation overnight in YPADU or YPADU + Hygromycin B (200ug/mL) for plasmid maintenance during HR overexpression experiments. The next day a 10mL culture was inoculated at OD600 ~0.1 and grown for 6 hours in a roller drum at 30 °C until OD600 ~0.7–1.0 (~3×107 cells/mL). Cells were pelleted at 2,900× g for 3 minutes and washed twice with 40mL of room temperature dH2O. Cells were pre-treated with 1mL of TE pH 8 containing 500mM Lithium Acetate/25mM DTT (Pretreatment Buffer) for 30 minutes in the roller drum at 30 °C. Cells were washed 1× with 1mL ice cold dH2O and 1× with 1mL ice cold 1M sorbitol. Cells were gently suspended in 200μL of 1M sorbitol + 2μM of total ssODN for each transformation, and added to a pre-chilled electroporation cuvette (0.2cm) on ice. ~2μM of ssODN was previously determined to be optimal for Rad51-dependent HR (DiCarlo et al., 2013). Electroporation was performed with the following parameters: 1500V, 25uF, 200Ω. Immediately after pulsing, the cells were recovered in 6mL of Recovery Medium for 12 hours in the roller drum at 30°C.
ssODN transformations for eMAGE
A list of ssODNs used for figures 1–4 in this study can be found in Table S2 and for figures 5–7 the ssODNs can be found in Table S6. All ssODNs were designed as 90nt in length, with slight (1–3nt) length deviations for some ssODNs in an attempt to minimize secondary structure. For eMAGE experiments, a single colony was inoculated in 2mL CSM-Ura medium and grown overnight to saturation. The next day the culture was diluted 1:50 in 10mL of CSM-Ura and grown for 6 hours prior to electroporation. The ssODN concentration and size were determined from optimization experiments performed in Figure S2. A total of 20μM of 90nt ssODNs consisting of 50% selection ssODN 50% target ssODN(s) was used for eMAGE transformations. The optimal HU concentration was determined from figure S2A and HU was used from figures 2–7. The eMAGE pretreatment mixture contained 500mM HU in TE pH 8 / 500mM Lithium Acetate / 25mM DTT. Electroporation parameters for eMAGE were as described above. After 12 hours of recovery, ~105 cells were plated on 5-FOA selection plates for MMR mutant strains and ~107 cells were plated for strains with WT MMR. The resultant 5-FOA plates contained ~100–200 colonies which were subject to screening for target ssODN incorporation. The indicated ARF values represent mean +/− SD for three replicates unless otherwise indicated. For the Rad51 chemical inhibitor experiment (Figure S1I) the recovery medium was supplemented with inhibitor RI-1 (Abcam ab144558) + 1.5% DMSO for solubility and the untreated sample also contained 1.5% DMSO to control for any DMSO effects.
UV Irradiation
UV irradiation was performed using a Stratalinker® UV crosslinker model 1800. After DTT treatment prior to the electroporation step, cells were washed and suspended in dH2O, irradiated with varying doses (0 – 106 uJ) of UV, and immediately electroporated with ssODNs as described above.
Target Distance Efficiency Determination
Target mutation distance is reported as the distance between the URA3 marker mutation incorporated by the selection-ssODN and the mutation incorporated by the target-ssODN. For target distances of 1.5 and 2kb, ssODNs ade290RC and ade2_Mult10 were used within the ADE2 gene and the ARF for these sites were determined by red/white phenotype screening. For targets at 5, 10, 15, and 20kb distances from the Ori-URA3, target sites were chosen that differ by only a single bp from an EcoRI restriction site. The target ssODN was designed to incorporate a bp change to create the EcoRI restriction site. For each target assayed, 96 clones were analyzed by colony PCR (ssODNS and Primers for the amplicons analyzed are listed in Table S2) coupled to EcoRI (NEB) digestion of the amplicon for 1hr at 37 °C. The percentage of amplicons cut vs. uncut is represented as the ARF for each distance site. Each distance experiment was performed in triplicate +/− HU treatment. ARF values represent mean +/− SD.
Multiplex Incorporation of ssODNs and Cycling
For multiplex eMAGE experiments targeting ADE2, red clones that grew on 5-FOA plates were assayed via yeast colony PCR and Sanger sequencing (Genewiz). Insertions were chosen for easy sequence detection since degenerate mismatches would contain WT sequence positions. For cycling experiments, cells were recovered in recovery media after electroporation as described above. After recovery in nonselective media, the population was subjected to liquid selection. For odd numbered cycles, the selection-ssODN creates a non-sense mutation in ura3 for negative selection with 5-FOA, and the even cycle selection-ssODN restores the functional URA3 gene for positive selection in uracil-dropout media. Selections were performed for 500uL of recovered culture seeded into 50mL of selection medium and grown to saturation at 30 °C (~2 days). After the first 1:100 selection the population was diluted 1:50 in selection media and grown for 6 hours for the next electroporation step. The process was repeated for 3 cycles. A total of 100 red clones were sequenced after each cycle.
Whole genome sequencing sample prep
For each clone sequenced, 10mL of cell culture (OD600 = 1) grown in YPADU was pelleted and treated with 100U of Zymolyase in buffer (1M Sorbitol in Tris pH = 7.4, 100mM EDTA, 14mM β-mercaptoethanol) for 90 minutes in a roller drum at 30 °C. The resultant spheroplasts were used as the input for the DNeasy Blood & Tissue Kit (Qiagen) to isolate genomic DNA for whole genome sequencing library preparation. 96 genomic libraries were prepped using the TruSeq DNA PCR-Free HT Kit (Illumina). The libraries were pooled and sequenced by the Yale Center for Genome Analysis using the Illumina Hiseq4000 with 2×100bp paired-end reads.
Generation of mutant population for HTS diversity analysis
Approximately 3×108 cells were electroporated in a 20μM solution containing ssODNs designed to introduce at total of 15 insertions at ADE2 targeted sites and the ura3190 ssODN for selection. The three ade2 ssODNs (Nade2MULTB, Nade2MULTC, and Nade2MULTD) each encode five insertions. After electroporation and recovery the entire population was then seeded in 1L of liquid 5-FOA selection media and grown to saturation over 2 days. 10mL of this selected population was used for the genome prep and 10μg or approximately 5×109 genomic copies were seeded into the PCR reaction.
HTS diversity sample preparation and sequencing
A 307–322 bp region (depending on the number of insertions introduced) including the bases targeted for mutagenesis was PCR amplified with primers that added five degenerate bps on each end. Forward primer: 5′-(N)(N)(N)(N)(N)TCCAATCCTCTTGATATCGAAAAACTAGCTGA-3′; Reverse primer: 5′-(N)(N)(N)(N)(N)CATCGTATGCCAAAGTCCTCGACTTC-3′. The addition of degenerate bases aided in initial base calling during sequencing and reduced the need to add an increased fraction of phiX. The number of PCR cycles was limited to 12 in order to reduce the introduction of errors and bias. The PCR product was then gel purified and sent to the Yale Center for Genome Analysis for adaptor ligation and 2×250 paired-end sequencing on an Illumina HiSeq4000. This allowed for coverage of the entire amplicon and sufficient overlap between the paired-end reads to assemble phased sequences for each observed variant.
Design of ssODNs for β-carotene Pathway Diversification
See Table S6 for a complete list of ssODNs used to diversify the β-carotene pathway along with details regarding the specific mutation design and outcomes for each ssODN. We designed ssODNs to target known regulatory elements in promoters, ORFs, and terminators (Lubliner et al., 2013). For promoters, we targeted annotated transcription factor binding sites (TFBS), TATA boxes, insertion of nucleosome-disfavoring (dT)20 sequences, and altered the A and T sequence content near the transcription start signal (TSS). Mutations to TFBS and TATA boxes were designed as ‘N’ degenerate and LOGO-inspired sequences to create the potential for both highly divergent sequences and single-bp changes. For each ORF, the ssODNs encoded an alternate start codon (GTG), a common codon, a rare codon, and a frame-shift knockout (KO) mutation. In addition to mutations that alter gene expression, we also included a protein sequence change in the crtYB lycopene-cyclase domain known to increase lycopene production (Xie et al., 2015). Lastly, we targeted terminators at putative poly-A signal sites.
HPLC Characterization of Carotenoids
Each of the analyzed clones was grown for 3 days at 30°C in 5mL YPADU media. Carotenoids were harvested from 1mL of cell culture. 1mL of cells was pelleted via centrifugation and washed twice with water. The resulting pellet was extracted with 200uL of hexane using glass bead disruption with the Beadbeater cell homogenizer (3× 45s at 7,000rpm). After centrifugation, 120uL of the hexane carotenoid mixture was transferred to a glass vial and dried with a speedvac machine for 1 hour. The sample was then resuspended in 50/50 Hexane/Ethyl Acetate and filtered before HPLC analysis. 20uL of sample was injected for HPLC using an Agilent Poroshell 120 EC-C18 2.7 um 3.0 × 50mm column. Peaks were detected using an isocratic elution with 50/50 Methanol/Acetonitrile (containing 0.1% Formic Acid). Analytical standards were used for quantification of β-carotene (detected at 475nm), phytoene (286nm), and lycopene (475nm). Carotenoid quantifications were calculated in relation to dry cell weight (DCW) for 100uL of cell culture dried for 2 days and weighed. Additional carotenoid peaks were observed beyond the three carotenoid peaks analyzed, which likely contribute to clone color in some cases. HPLC experiments were carried out as three replicates for all clones.
Isolation of RNA and RT-qPCR Conditions
For RT-qPCR in Figure 7, three replicates of each clone were grown at 30°C in 5mL YP containing either 2% Galactose, 2% Dextrose, or a range of galactose (.01–5%) for 16 hours. For galactose concentrations less than 2% (Figure 7D), the remainder of the carbon source was supplemented by addition of raffinose to a total of 2%. Total RNA was harvested from each sample using the RNeasy RNA isolation kit (Qiagen) with an input of approximately 3×107 cells (1mL of culture at OD600 = 1). Total RNA was quantified using a Qubit fluorometer. Typical yield was approximately 200 ng/uL. For all samples a total of 10ng of RNA was used in each 20μL reaction. For RT-qPCR we used the Luna one-step universal RT-qPCR kit (NEB) run on a CFX Connect Real-Time System (Bio-Rad). The RT-qPCR reaction cycle consisted of the following steps: (1) 55°C for 10 min (2) 95°C for 1 min (3) 95°C for 10 seconds (4) 60°C for 30 seconds (5) Measure SYBR (6) Go to Step 3, 45X (7) Melt curve analysis 60°C to 95°C.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Notes
For all statistical analysis and curve fitting of experimental samples we used Graphpad Prism 7 software. All data sets for Figures 1,2, 5E(HPLC), and 7D, 7E(RT-qPCR) were carried out as three replicates. For Figures 1–2 each condition or strain was tested three independent times. For Figure 5E each clone was grown as three separate replicates, harvested as described, and the relevant pigments were measured by HPLC for each replicate. For Figures 7D,E each clone was grown as three independent replicates under each condition (glucose or galactose), RNA was harvested, and RT-qPCR was executed on each of the replicates. Where error bars are shown the data is reported as the Mean +/−SD. Statistical significance cutoff for all relevant experiments is * p<.05. For all additional figures the number of samples ‘n’ is indicated in the figure caption.
Calculation of ARF
The allelic replacement frequency (ARF) for homologous recombination of ssODNs at the RPL28 gene (Figure 1B, S1A–F) was calculated by measuring the number of CFUs present on YPD-Cycloheximide agar plates divided by the number of CFUs present on YPD agar plates. When assaying for red ade2 mutants without selection (Figure 1D) we plated ~103 colonies on 10 YPD plates such that ~104 colonies could be counted for each data point in the triplicate set. The ARF for single-plex ura3 targeting (Figure S2F) was calculated by measuring the number of CFUs on CSM-uracil dropout plates divided by the number of CFUs on YPD plates. For all experiments where the ARF for ade2 is reported, the ARF represents the number of ade2 mutant (red) CFUs divided by the total number of 5-FOA resistant CFUs (or divided by YPD-Cycloheximide CFUs in Figure S1G, S1H; or divided by HIS3+ CFUs in Figure S2C). For Figure S1J the ARF for ura3 after rpl28 mutant selection was determined by first plating to YPD-Cycloheximide and then replica plating the surviving CFUs to 5-FOA plates. The ARF is the number of 5-FOA CFUs divided by the number of YPD-Cycloheximide CFUs. For all experiments the ARF are reported as a percent with the mean +/− SD shown.
Calculation of ssODN hybridization free energies
Hybridization free energies were calculated for all ssODNs used in Figure 2E–G using UNAfold two-state melting software available at http://unafold.rna.albany.edu/?q=DINAMelt/Two-state-melting. The following parameters were used: 30°C, [Na+]= 1M, [Mg2+] = 0M, and strand concentration = 0.00001M. Each ssODN was hybridized the ADE2 WT sequence. The Hybridization free energies were plotted against the mean ARF for each ssODN and analyzed with a linear regression using Graphpad Prism 7 and Pearson correlation coefficients are displayed in Figure 2H.
Unique Mutants from Multiplex Incorporation of ssODNs and Cycling
For figure 3C, in cycles 1 and 2 we observed 200 unique genotypes out of 200 sequenced, but for cycle 3 we observed 76 unique genotypes out of the 100 clones sequenced. Given that the degeneracy of the ssODN pool largely out-scales the number of clones assayed we would not expect any redundant genotypes to arise for independent clones in any cycle assayed. The 24 redundant clones observed in cycle 3 were comprised of 5 genotypes. These clones observed in cycle 3 are due to an enrichment of those genotypes that occurred from selection after cycle 2. For future experiments improved selection capabilities are necessary in order to ensure maintenance of high population diversity between multiple cycles. For the purposes of this small-scale demonstration the liquid selections we performed were seeded with 500uL of culture (~103–104 edited genotypes) between each cycle. For applications requiring large library sizes, larger scale selections (Liters) could be employed to maintain the population complexity generated after electroporation.
Whole genome sequencing read filtering, mapping, and variant calling
The sequencing reads generated for each sample were first filtered using Trimmomatic with the parameters “LEADING:3 TRAILING:3 SLIDINGWINDOW:2:30” in order to remove low quality bases at the ends of each read and to truncate reads containing consecutive bases with an average quality score below 30 (Bolger et al., 2014). The reads for each strain were then independently mapped to the current version of the SC288 reference genome available at the Saccharomyces Genome Database using BWA-mem (Li, 2013). Reads were also mapped to a modified genome that incorporated the addition of the β-carotene pathway and KO of MSH2. Duplicates were marked in the resulting BAM file using Picard’s MarkDuplicates tool (https://github.com/broadinstitute/picard). The reads were then realigned using the RealignerTargetCreator and IndelRealigner tools from GATK (Van der Auwera et al., 2013). Variants were called using the GATK’s HaplotypeCaller with ploidy=1 and when relevant with -comp option in order to identify strains-specific variants by filtering out variants shared with the ancestor. The GATK tools SelectVariants and VariantFiltration were then used to filter SNP and Indel call sets and yield a final variant set.
HTS diversity filtering and processing of paired end reads into merged sequences
The sequencing process generated 49,714,782 paired end reads. Trimmomatic was used to remove the first five degenerate bases added by the primers and trim reads with low quality bases. A sliding window requiring an average quality score of either Q20, Q25, or Q30 over two bases was used to trim the ends of lower quality reads. This resulted in 49,384,456, 48,711,289, and 47,505,196 paired end reads r3espectively passing quality control. BBMerge (sourceforge.net/projects/bbmap/) was then used to assemble the overlapping paired end reads into full-length amplicons. The “strict” stringency setting was used during this step. This resulted in 37,227,893, 24,116,204, and 12,065,464 fully assembled amplicons of which 16,704,731, 12,952,187, and 9,048,602 were wild type length of 307 bp. BBMerge seeks to reduce the error rate in the assembled amplicons by considering the quality score of the bases in the overlapping sequences during merging and when there is a mismatch between the two reads selecting the base with the higher quality score. Similar numbers of reads pass the quality trimming step at Q20 and Q30, but the number of assembled amplicons is dramatically different between the two cutoffs. This suggests that while a large number of reads are passing quality control at Q30 they are being trimmed to a greater extent and this is resulting in read pairs that no longer overlap and are unable to be assembled.
HTS diversity computational analysis
The merged reads were then arranged in the same orientation and aligned to a WT copy of the edited genomic locus using the BWA-mem algorithm. Picard tools were used to calculate the experimental substitution error rate (https://github.com/broadinstitute/picard). Custom scripts then utilized the CIGAR and MD strings in the resulting SAM file to extract the position and base introduced when insertions occurred at the targeted sites. Calculations of the distribution of the number of insertions introduced and the positional insertion distribution were then performed on these vectors containing the base introduced by each targeted insertion in an amplicon.
Determination of Primer Efficiency for RT-qPCR Analysis
Detailed information regarding the RT-qPCR parameters including primers and calculation of primer efficiencies are found in Table S7. For determination of primer efficiencies we used purified total RNA from the EMB294 strain grown in glucose and galactose conditions. ACT1 was used as a normalizing control gene, and primer pairs were designed for crtE, crtI, crtYB, and tHMG1 (Table S7). Each primer pair was tested against a 10-fold dilution series of the RNA template from 100pg–100ng of total RNA in the RT-qPCR reaction and analyzed with a linear regression from a plot of log[RNA] vs. Cq. The linear equations and R2 values for each primer pair in each condition are listed in Table S7.
Measurement of Relative Gene Expression with RT-qPCR
For calculation of gene expression fold-change between glucose and galactose growth conditions we used the ΔΔCq method (Schmittgen and Livak, 2008) with ACT1 as the normalizing gene. Reactions were performed using 10ng of total RNA, which is in the linear regime for the optimized conditions. GraphPad Prism 7 software was used for nonlinear curve fitting for GAL1 and crtI of clone G4 using a four-parameter variable-slope dose-response model.
DATA AND SOFTWARE AVAILABILITY
Datasets containing the raw sequence reads from WGS (Figure S4, Tables S3–S5) and deep sequencing (Figures 4, S4) experiments are available at the NIH Sequence Read Archive (SRA) BioProject ID PRJNA413161.
Supplementary Material
Figure S1. Enhancement of ssODN incorporation efficiencies (ARF) through constitutive expression of HR genes and knockout of MMR, related to Figure 1. (A) Measurement of RPL28 ARF with overexpression of HR genes from pTEF1 promoter with a CEN/ARS plasmid in BY4741 (WT) strain, (B) mismatch repair (MMR) mutant strains, and (C–F) combinations of pTEF1-HR gene overexpression with four MMR mutant strain backgrounds. (G) Schematic illustrating co-transformation of two ssODNs targeted to loci on separate chromosomes to determine if the high ARFs (>10%) observed for Ori-URA3-ADE2 require targeting within a contiguous chromosome. The ARF for ADE2 is measured before and after selection with cycloheximide. (H) Measurement of ARF at RPL28 (+,−), ADE2 (−,+), and the ARF for ADE2 after prior selection for rpl28 mutants (+,+). Strains are msh2 and msh2 pTEF1-RAD51. (I) Dose-dependent fold-increase in ADE2 mutants recovered with Rad51 inhibitor (RI-1) in YPADU 0.5M sorbitol + 1.5% DMSO, compared to vehicle control recovered in media containing only YPADU 0.5M sorbitol + 1.5% DMSO. Strain is EMB259. (J) Additional mechanistic evidence for ssODN incorporation at replication forks. Strand targeting for marker-target orientation cases (I and II) at the RPL28 locus. The RPL28 gene contains an embedded origin of replication located at Chr. VII ~311600 (Xu et al., 2006), which enabled construction of Case-I and Case-II by placing URA3 upstream or downstream of RPL28. The strand targeting efficiency (ARF %) trends are equivalent to those observed at the ARS1516 locus. Origin of replication indicated by double arrow. Strain is EMB259. (n=3 for all data points).
Figure S2. Characterization of HU treatment conditions and ssODN transformation parameters in the msh2Δ strain, related to Figure 2. (A) Concentration optimization for 30-minute HU treatment prior to electroporation with ssODNs targeting URA3 and ADE2. (B) Comparison of spontaneous 5-FOA resistant mutants +/− 500mM HU treatment. (C) The HU enhancement effect for recovering ADE2 targeted mutants is observed when using a HIS3 marker containing premature stop codon. Transformants are selected via the ssODN reversion of the his3* stop codon and subsequent selection on histidine auxotrophic media. (D) Measurement of ADE2 ARF after treatment with the indicated dose of UV irradiation. (E) Characterization of cell survival post electroporation for a range of ssODN concentrations. (F) The effect of ssODN size and concentration for single-plex targeting of the URA3 marker. The number of 5-FOA resistant CFUs is normalized to CFUs on YPD (nonselective media). Cells are treated with 500mM HU. (G) The effect of ssODN size and concentration for coupled targeting of ADE2 and URA3. Shown is the number of ade2 mutant (red) CFUs per 5-FOA resistant CFU. Cells are treated with 500mM HU. This is the same data that is represented in the heat map showing the mean ARF in Figure 2C. N=3 for all data.
Figure S3. Characterization of multiplex mutagenesis with ssODNs, related to Figure 3. (A) Color-maps indicating multiplex ssODN incorporation within the ADE2 gene in WT and msh2 strains. Data illustrates genotypes of ade2 mutant clones resulting from multiplexed ssODN sets targeting 4,6, or 10 target sites (4-plex, 6-plex, 10-plex) with HU treatment, and 10 ssODNs untreated (10-plex −HU). Clones are represented in rows, and the columns (indicated numerically) represent each ssODN target position across the ADE2 gene. A red bar at a given position indicates incorporation of a targeted point mutation, and black indicates WT sequence at the locus Notably, C-C mismatch mutations at position 6 were enriched by an average of 7.3-fold in WT cells, which is consistent with prior work showing that C-C mismatches evade MMR (Detloff et al., 1991). Number of clones sequenced (n), n= 22 (4 ssODNs), n=32 (6 ssODNs), n=36 (10 ssODNs), n=40 (10 ssODNs –HU condition). 10-pex +/− HU data is summarized on the adjacent plot as the average mutations per clone +/− HU. (B) Analysis of degenerate-insertion positional incorporation frequencies. Single cycle positional insertion frequencies for 10-plex ssODNs. The ssODNs are shown 3′ to 5′ to indicate directionality of incorporation at the lagging strand. Each ssODN is targeted to a distinct site within the ADE2 gene indicated by a representative letter (a–j). Each insertion in the ssODN is indicated by “N” with subscript indicating the position of the insertion. (C) Total insertion incorporation efficiency for all 10 ssODNS at each position within each ssODN from 100 clones sequenced after cycle 1. Statistical significance of 3′ bias with ordinary one-way ANOVA comparison of individual positions N5-N2 with the 5′ position being N1. (D) The 3′ bias trend was not statistically significant for ssODNs (b–d), however PAGE purification qualitatively reduced the 3′ bias effect.
Figure S4. WGS analysis of background mutation rates and HTS analysis of diversity generated by eMAGE, related to Figures 3–5. (A) Mutation rates for 12 ade2 mutant clones per eMAGE cycle. (B) SNP rates for 12 ade2 mutant clones per eMAGE cycle. (C) Indel rates for 12 ade2 mutant clones per eMAGE cycle. (D) Rarefaction curve for Q20 quality score HTS reads. (E) Q20 distribution of insertion mutations observed.
(F) Q20 positional frequencies of insertion mutations within each ssODN. (G) Mutation rates for 55 diversified β-carotene pathway clones. (H) SNP rates for 55 diversified β-carotene pathway clones. (I) Indel rates for 55 diversified β-carotene pathway clones.
Figure S5. Clonal phenotypes for combinatorial gene knockouts set and inter-chromosomal targeting with mating to combine the diversity between haploid strains, related to Figures 5 and 6. (A) Corresponding clone phenotype generated via a single ssODN transformation experiment. (B) HPLC data for clonal production of β-carotene (orange) lycopene (red), and phytoene (white) (ug/mg dry cell weight). Values represent mean +/− SD for three replicates. (C) Two MATα haploids with URA3-crtE at ARS446 on chromosome IV and ARS702 on chromosome VII (Left most panel). Control cross with MATa haploid containing the crtI, crtyb, and tHMG1 at ARS1516 (Middle panel). Cross of diversified MATa and MATα haploids (Right most panel)
Figure S6. Set of clones analyzed after diversification, related to Figure 5. Genotypic and phenotypic analysis of variant clones after Sanger sequencing and HPLC analysis. Total number of ssODNs incorporated (black bar) and number of targeted base-pair changes (light-grey bar). HPLC data for clonal production of β-carotene (orange) lycopene (red), and phytoene (white) (ug/mg dry cell weight).
Figure S7. Quantitative data for ssODNs used in diversification, related to Figure 5. Quantitative data for ARFs associated with each ssODN. Heat map represents the ARF determined by prevalence of ssODN-derived mutation at each target site for the clones analyzed.
Table S1. Complete list of strains and genotypes used in this study, related to Figures 1–7.
Table S2. List of ssODNs for URA3, RPL28, ADE2, HIS3, and primers for target distance experiments, related to Figures 1–7.
Table S3. Statistics associated with ADE2 Sanger sequencing and whole genome sequencing of background mutations for eMAGE cycling, related to Figure 3.
Table S4. List of background indels and annotations from whole genome sequencing, related to Figures 3–5.
Table S5. List of background SNPs and annotations from whole genome sequencing, related to Figures 3–5.
Table S6. List of ssODNs designed for the β–Carotene pathway diversification, related to Figures 5–7. Each targeted sequence is indicated with the Watson Strand sequence for the WT (Column 4) and the ssODN mutation design (Column 5). The sequence type (Column 6), and notes on the mutation outcome (Column 7) are listed.
Abbreviations: Transcription factor binding site (TFBS), Transcription start site (TSS). Mutation annotations for ssODNs: ‘N’ = mixed bases A,T,G,C; ‘W’ = mixed bases A,T; ‘Y’ = mixed bases C,T; ‘R’ = mixed bases A,G; ‘K’ = mixed bases G,T; ‘M’ = mixed bases A,C. All sequences are listed in the 5′ to 3′ direction.
Table S7. Parameters for RT-qPCR analysis, related to Figure 7. This table includes the RT-qPCR parameters for the gene expression analysis in Figure 7D,E. (A) Primers for each target gene. (B) Determination of primer efficiencies. Mean and standard deviation of Cq values for each primer set across a diluted RNA template series for strain EMB294 grown in glucose and galactose conditions. (C) Determination of primer efficiencies using linear regression analysis for each primer set in each condition.
Highlights.
Yeast multiplex genome engineering technology that avoids DNA double strand breaks
Rad51-independent mechanism of gene editing by ssODN annealing at DNA replication
Silencing DNA repair and slowing replication enhances multiplex editing by ssODNs
Generation of combinatorial genetic variants at base pair precision in eukaryotes
Acknowledgments
We thank P. Sung, S. Dellaporta, M. Hochstrasser, T. Xu, J. Rinehart, the anonymous reviewers, and members of the Isaacs Lab for valuable feedback. E.M.B. is funded by NSF-GRFP (1122492) and NIH Predoctoral Training Grant in Genetics. F.J.I. gratefully acknowledges support from DARPA (HR0011-15-C-0091, N66001-12-C-4020), DOE (DE-FG02-02ER63445), NIH (1R01GM117230-01, 1U54CA209992-01), and the Arnold and Mabel Beckman Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: F.J.I. and E.M.B. conceived the study, designed the experiments and wrote the manuscript. E.M.B. performed the experiments with assistance from B.O.A., P.M. and C.M.Y. P.M. performed the HTS analysis.
References
- Amiram M, Haimovich AD, Fan C, Wang YS, Aerni HR, Ntai I, Moonan DW, Ma NJ, Rovner AJ, Hong SH, et al. Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nature biotechnology. 2015;33:1272–1279. doi: 10.1038/nbt.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Beach A, Li K, Haber J. Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature. 2017;544:377–380. doi: 10.1038/nature22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annaluru N, Muller H, Mitchell LA, Ramalingam S, Stracquadanio G, Richardson SM, Dymond JS, Kuang Z, Scheifele LZ, Cooper EM, et al. Total synthesis of a functional designer eukaryotic chromosome. Science. 2014;344:55–58. doi: 10.1126/science.1249252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, Church G, Hessel A, Kelley NJ, Arkin A, Cai Y, Carlson R, Chakravarti A, Cornish VW, Holt L, et al. GENOME ENGINEERING. The Genome Project-Write. Science. 2016;353:126–127. doi: 10.1126/science.aaf6850. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- Carr PA, Wang HH, Sterling B, Isaacs FJ, Lajoie MJ, Xu G, Church GM, Jacobson JM. Enhanced multiplex genome engineering through co-operative oligonucleotide co-selection. Nucleic acids research. 2012;40:e132. doi: 10.1093/nar/gks455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasegaran S, Carroll D. Origins of Programmable Nucleases for Genome Engineering. J Mol Biol. 2016;428:963–989. doi: 10.1016/j.jmb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collonnier C, Epert A, Mara K, Maclot F, Guyon-Debast A, Charlot F, White C, Schaefer DG, Nogue F. CRISPR-Cas9-mediated efficient directed mutagenesis and RAD51-dependent and RAD51-independent gene targeting in the moss Physcomitrella patens. Plant Biotechnol J. 2017;15:122–131. doi: 10.1111/pbi.12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino N, Court DL. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15748–15753. doi: 10.1073/pnas.2434959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353 doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detloff P, Sieber J, Petes TD. Repair of specific base pair mismatches formed during meiotic recombination in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:737–745. doi: 10.1128/mcb.11.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo JE, Conley AJ, Penttila M, Jantti J, Wang HH, Church GM. Yeast oligo-mediated genome engineering (YOGE) ACS Synth Biol. 2013;2:741–749. doi: 10.1021/sb400117c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Erler A, Wegmann S, Elie-Caille C, Bradshaw CR, Maresca M, Seidel R, Habermann B, Muller DJ, Stewart AF. Conformational adaptability of Redbeta during DNA annealing and implications for its structural relationship with Rad52. J Mol Biol. 2009;391:586–598. doi: 10.1016/j.jmb.2009.06.030. [DOI] [PubMed] [Google Scholar]
- Farzadfard F, Perli SD, Lu TK. Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth Biol. 2013;2:604–613. doi: 10.1021/sb400081r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman KL, Brewer BJ, Fangman WL. Replication profile of Saccharomyces cerevisiae chromosome VI. Genes Cells. 1997;2:667–678. doi: 10.1046/j.1365-2443.1997.1520350.x. [DOI] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature biotechnology. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG. Enzymatic assembly of overlapping DNA fragments. Methods in enzymology. 2011;498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD. Yeast transformation by the LiAc/SS carrier DNA/PEG method. Methods Mol Biol. 2014;1205:1–12. doi: 10.1007/978-1-4939-1363-3_1. [DOI] [PubMed] [Google Scholar]
- Horwitz AA, Walter JM, Schubert MG, Kung SH, Hawkins K, Platt DM, Hernday AD, Mahatdejkul-Meadows T, Szeto W, Chandran SS, et al. Efficient Multiplexed Integration of Synergistic Alleles and Metabolic Pathways in Yeasts via CRISPR-Cas. Cell Syst. 2015;1:88–96. doi: 10.1016/j.cels.2015.02.001. [DOI] [PubMed] [Google Scholar]
- Inui M, Miyado M, Igarashi M, Tamano M, Kubo A, Yamashita S, Asahara H, Fukami M, Takada S. Rapid generation of mouse models with defined point mutations by the CRISPR/Cas9 system. Sci Rep. 2014;4:5396. doi: 10.1038/srep05396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakociunas T, Bonde I, Herrgard M, Harrison SJ, Kristensen M, Pedersen LE, Jensen MK, Keasling JD. Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae. Metab Eng. 2015;28:213–222. doi: 10.1016/j.ymben.2015.01.008. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016 doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kow YW, Bao G, Reeves JW, Jinks-Robertson S, Crouse GF. Oligonucleotide transformation of yeast reveals mismatch repair complexes to be differentially active on DNA replication strands. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11352–11357. doi: 10.1073/pnas.0704695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivoruchko A, Nielsen J. Production of natural products through metabolic engineering of Saccharomyces cerevisiae. Curr Opin Biotechnol. 2015;35:7–15. doi: 10.1016/j.copbio.2014.12.004. [DOI] [PubMed] [Google Scholar]
- Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, et al. Genomically recoded organisms expand biological functions. Science. 2013;342:357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Parsons L, Gammie AE. Mutation rates, spectra, and genome-wide distribution of spontaneous mutations in mismatch repair deficient yeast. G3 (Bethesda) 2013;3:1453–1465. doi: 10.1534/g3.113.006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Lee CH, Demin AA, Munashingha PR, Amangyeld T, Kwon B, Formosa T, Seo YS. Rad52/Rad59-dependent recombination as a means to rectify faulty Okazaki fragment processing. J Biol Chem. 2014;289:15064–15079. doi: 10.1074/jbc.M114.548388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 13033997v1 [q-bioGN] [Google Scholar]
- Liu L, Maguire KK, Kmiec EB. Genetic re-engineering of Saccharomyces cerevisiae RAD51 leads to a significant increase in the frequency of gene repair in vivo. Nucleic acids research. 2004;32:2093–2101. doi: 10.1093/nar/gkh506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubliner S, Keren L, Segal E. Sequence features of yeast and human core promoters that are predictive of maximal promoter activity. Nucleic acids research. 2013;41:5569–5581. doi: 10.1093/nar/gkt256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol. 2008;453:3–31. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- Mitchell LA, Chuang J, Agmon N, Khunsriraksakul C, Phillips NA, Cai Y, Truong DM, Veerakumar A, Wang Y, Mayorga M, et al. Versatile genetic assembly system (VEGAS) to assemble pathways for expression in S. cerevisiae. Nucleic acids research. 2015;43:6620–6630. doi: 10.1093/nar/gkv466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- Rivera-Torres N, Kmiec EB. Genetic spell-checking: gene editing using single-stranded DNA oligonucleotides. Plant Biotechnol J. 2016;14:463–470. doi: 10.1111/pbi.12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GP, Song JB, Crouse GF. Transformation with oligonucleotides creating clustered changes in the yeast genome. PloS one. 2012;7:e42905. doi: 10.1371/journal.pone.0042905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annual review of biochemistry. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Smanski MJ, Bhatia S, Zhao D, Park Y, L BAW, Giannoukos G, Ciulla D, Busby M, Calderon J, Nicol R, et al. Functional optimization of gene clusters by combinatorial design and assembly. Nature biotechnology. 2014;32:1241–1249. doi: 10.1038/nbt.3063. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Whitehouse I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 2012;483:434–438. doi: 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Sung P. Functional interactions among yeast Rad51 recombinase, Rad52 mediator, and replication protein A in DNA strand exchange. J Biol Chem. 2000;275:15895–15904. doi: 10.1074/jbc.M910244199. [DOI] [PubMed] [Google Scholar]
- Storici F, Resnick MA. Delitto perfetto targeted mutagenesis in yeast with oligonucleotides. Genet Eng (N Y) 2003;25:189–207. [PubMed] [Google Scholar]
- Sung P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem. 1997;272:28194–28197. doi: 10.1074/jbc.272.45.28194. [DOI] [PubMed] [Google Scholar]
- Szpiech ZA, Jakobsson M, Rosenberg NA. ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics. 2008;24:2498–2504. doi: 10.1093/bioinformatics/btn478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Joung JK. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat Rev Genet. 2016;17:300–312. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 10 11–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler LM, Cornish VW. Reiterative Recombination for the in vivo assembly of libraries of multigene pathways. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15135–15140. doi: 10.1073/pnas.1100507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Wu CA, Zechner EL, Marians KJ. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. I. Multiple effectors act to modulate Okazaki fragment size. J Biol Chem. 1992;267:4030–4044. [PubMed] [Google Scholar]
- Xie W, Lv X, Ye L, Zhou P, Yu H. Construction of lycopene-overproducing Saccharomyces cerevisiae by combining directed evolution and metabolic engineering. Metab Eng. 2015;30:69–78. doi: 10.1016/j.ymben.2015.04.009. [DOI] [PubMed] [Google Scholar]
- Xu W, Aparicio JG, Aparicio OM, Tavare S. Genome-wide mapping of ORC and Mcm2p binding sites on tiling arrays and identification of essential ARS consensus sequences in S. cerevisiae. BMC Genomics. 2006;7:276. doi: 10.1186/1471-2164-7-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Moerschell RP, Wakem LP, Ferguson D, Sherman F. Parameters affecting the frequencies of transformation and co-transformation with synthetic oligonucleotides in yeast. Yeast. 1992;8:935–948. doi: 10.1002/yea.320081104. [DOI] [PubMed] [Google Scholar]
- Yang L, Guell M, Niu D, George H, Lesha E, Grishin D, Aach J, Shrock E, Xu W, Poci J, et al. Genome-wide inactivation of porcine endogenous retroviruses (PERVs) Science. 2015;350:1101–1104. doi: 10.1126/science.aad1191. [DOI] [PubMed] [Google Scholar]
- Zhang GC, Kong II, Kim H, Liu JJ, Cate JH, Jin YS. Construction of a quadruple auxotrophic mutant of an industrial polyploid saccharomyces cerevisiae strain by using RNA-guided Cas9 nuclease. Appl Environ Microbiol. 2014;80:7694–7701. doi: 10.1128/AEM.02310-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Enhancement of ssODN incorporation efficiencies (ARF) through constitutive expression of HR genes and knockout of MMR, related to Figure 1. (A) Measurement of RPL28 ARF with overexpression of HR genes from pTEF1 promoter with a CEN/ARS plasmid in BY4741 (WT) strain, (B) mismatch repair (MMR) mutant strains, and (C–F) combinations of pTEF1-HR gene overexpression with four MMR mutant strain backgrounds. (G) Schematic illustrating co-transformation of two ssODNs targeted to loci on separate chromosomes to determine if the high ARFs (>10%) observed for Ori-URA3-ADE2 require targeting within a contiguous chromosome. The ARF for ADE2 is measured before and after selection with cycloheximide. (H) Measurement of ARF at RPL28 (+,−), ADE2 (−,+), and the ARF for ADE2 after prior selection for rpl28 mutants (+,+). Strains are msh2 and msh2 pTEF1-RAD51. (I) Dose-dependent fold-increase in ADE2 mutants recovered with Rad51 inhibitor (RI-1) in YPADU 0.5M sorbitol + 1.5% DMSO, compared to vehicle control recovered in media containing only YPADU 0.5M sorbitol + 1.5% DMSO. Strain is EMB259. (J) Additional mechanistic evidence for ssODN incorporation at replication forks. Strand targeting for marker-target orientation cases (I and II) at the RPL28 locus. The RPL28 gene contains an embedded origin of replication located at Chr. VII ~311600 (Xu et al., 2006), which enabled construction of Case-I and Case-II by placing URA3 upstream or downstream of RPL28. The strand targeting efficiency (ARF %) trends are equivalent to those observed at the ARS1516 locus. Origin of replication indicated by double arrow. Strain is EMB259. (n=3 for all data points).
Figure S2. Characterization of HU treatment conditions and ssODN transformation parameters in the msh2Δ strain, related to Figure 2. (A) Concentration optimization for 30-minute HU treatment prior to electroporation with ssODNs targeting URA3 and ADE2. (B) Comparison of spontaneous 5-FOA resistant mutants +/− 500mM HU treatment. (C) The HU enhancement effect for recovering ADE2 targeted mutants is observed when using a HIS3 marker containing premature stop codon. Transformants are selected via the ssODN reversion of the his3* stop codon and subsequent selection on histidine auxotrophic media. (D) Measurement of ADE2 ARF after treatment with the indicated dose of UV irradiation. (E) Characterization of cell survival post electroporation for a range of ssODN concentrations. (F) The effect of ssODN size and concentration for single-plex targeting of the URA3 marker. The number of 5-FOA resistant CFUs is normalized to CFUs on YPD (nonselective media). Cells are treated with 500mM HU. (G) The effect of ssODN size and concentration for coupled targeting of ADE2 and URA3. Shown is the number of ade2 mutant (red) CFUs per 5-FOA resistant CFU. Cells are treated with 500mM HU. This is the same data that is represented in the heat map showing the mean ARF in Figure 2C. N=3 for all data.
Figure S3. Characterization of multiplex mutagenesis with ssODNs, related to Figure 3. (A) Color-maps indicating multiplex ssODN incorporation within the ADE2 gene in WT and msh2 strains. Data illustrates genotypes of ade2 mutant clones resulting from multiplexed ssODN sets targeting 4,6, or 10 target sites (4-plex, 6-plex, 10-plex) with HU treatment, and 10 ssODNs untreated (10-plex −HU). Clones are represented in rows, and the columns (indicated numerically) represent each ssODN target position across the ADE2 gene. A red bar at a given position indicates incorporation of a targeted point mutation, and black indicates WT sequence at the locus Notably, C-C mismatch mutations at position 6 were enriched by an average of 7.3-fold in WT cells, which is consistent with prior work showing that C-C mismatches evade MMR (Detloff et al., 1991). Number of clones sequenced (n), n= 22 (4 ssODNs), n=32 (6 ssODNs), n=36 (10 ssODNs), n=40 (10 ssODNs –HU condition). 10-pex +/− HU data is summarized on the adjacent plot as the average mutations per clone +/− HU. (B) Analysis of degenerate-insertion positional incorporation frequencies. Single cycle positional insertion frequencies for 10-plex ssODNs. The ssODNs are shown 3′ to 5′ to indicate directionality of incorporation at the lagging strand. Each ssODN is targeted to a distinct site within the ADE2 gene indicated by a representative letter (a–j). Each insertion in the ssODN is indicated by “N” with subscript indicating the position of the insertion. (C) Total insertion incorporation efficiency for all 10 ssODNS at each position within each ssODN from 100 clones sequenced after cycle 1. Statistical significance of 3′ bias with ordinary one-way ANOVA comparison of individual positions N5-N2 with the 5′ position being N1. (D) The 3′ bias trend was not statistically significant for ssODNs (b–d), however PAGE purification qualitatively reduced the 3′ bias effect.
Figure S4. WGS analysis of background mutation rates and HTS analysis of diversity generated by eMAGE, related to Figures 3–5. (A) Mutation rates for 12 ade2 mutant clones per eMAGE cycle. (B) SNP rates for 12 ade2 mutant clones per eMAGE cycle. (C) Indel rates for 12 ade2 mutant clones per eMAGE cycle. (D) Rarefaction curve for Q20 quality score HTS reads. (E) Q20 distribution of insertion mutations observed.
(F) Q20 positional frequencies of insertion mutations within each ssODN. (G) Mutation rates for 55 diversified β-carotene pathway clones. (H) SNP rates for 55 diversified β-carotene pathway clones. (I) Indel rates for 55 diversified β-carotene pathway clones.
Figure S5. Clonal phenotypes for combinatorial gene knockouts set and inter-chromosomal targeting with mating to combine the diversity between haploid strains, related to Figures 5 and 6. (A) Corresponding clone phenotype generated via a single ssODN transformation experiment. (B) HPLC data for clonal production of β-carotene (orange) lycopene (red), and phytoene (white) (ug/mg dry cell weight). Values represent mean +/− SD for three replicates. (C) Two MATα haploids with URA3-crtE at ARS446 on chromosome IV and ARS702 on chromosome VII (Left most panel). Control cross with MATa haploid containing the crtI, crtyb, and tHMG1 at ARS1516 (Middle panel). Cross of diversified MATa and MATα haploids (Right most panel)
Figure S6. Set of clones analyzed after diversification, related to Figure 5. Genotypic and phenotypic analysis of variant clones after Sanger sequencing and HPLC analysis. Total number of ssODNs incorporated (black bar) and number of targeted base-pair changes (light-grey bar). HPLC data for clonal production of β-carotene (orange) lycopene (red), and phytoene (white) (ug/mg dry cell weight).
Figure S7. Quantitative data for ssODNs used in diversification, related to Figure 5. Quantitative data for ARFs associated with each ssODN. Heat map represents the ARF determined by prevalence of ssODN-derived mutation at each target site for the clones analyzed.
Table S1. Complete list of strains and genotypes used in this study, related to Figures 1–7.
Table S2. List of ssODNs for URA3, RPL28, ADE2, HIS3, and primers for target distance experiments, related to Figures 1–7.
Table S3. Statistics associated with ADE2 Sanger sequencing and whole genome sequencing of background mutations for eMAGE cycling, related to Figure 3.
Table S4. List of background indels and annotations from whole genome sequencing, related to Figures 3–5.
Table S5. List of background SNPs and annotations from whole genome sequencing, related to Figures 3–5.
Table S6. List of ssODNs designed for the β–Carotene pathway diversification, related to Figures 5–7. Each targeted sequence is indicated with the Watson Strand sequence for the WT (Column 4) and the ssODN mutation design (Column 5). The sequence type (Column 6), and notes on the mutation outcome (Column 7) are listed.
Abbreviations: Transcription factor binding site (TFBS), Transcription start site (TSS). Mutation annotations for ssODNs: ‘N’ = mixed bases A,T,G,C; ‘W’ = mixed bases A,T; ‘Y’ = mixed bases C,T; ‘R’ = mixed bases A,G; ‘K’ = mixed bases G,T; ‘M’ = mixed bases A,C. All sequences are listed in the 5′ to 3′ direction.
Table S7. Parameters for RT-qPCR analysis, related to Figure 7. This table includes the RT-qPCR parameters for the gene expression analysis in Figure 7D,E. (A) Primers for each target gene. (B) Determination of primer efficiencies. Mean and standard deviation of Cq values for each primer set across a diluted RNA template series for strain EMB294 grown in glucose and galactose conditions. (C) Determination of primer efficiencies using linear regression analysis for each primer set in each condition.