

Abstract
The direct catalytic C–H amination of arenes is a powerful synthetic strategy with useful applications in pharmaceuticals, agrochemicals, and materials chemistry. Despite the advances in catalytic C–H functionalization, the use of aliphatic amine coupling partners is limited. Described herein is the construction of C–N bonds, using primary amines, by direct C–H functionalization with an acridinium photoredox catalyst under an aerobic atmosphere. A wide variety of primary amines, including amino acids and more complex amines are competent coupling partners. Various electron-rich aromatics and heteroaromatics are useful scaffolds in this reaction, as are complex, biologically active arenes. We also describe the ability to functionalize arenes that are not oxidized by an acridinium catalyst, such as benzene and toluene, thus supporting a reactive amine cation radical intermediate.
Keywords: amines, arenes, organocatalysis, photochemistry, synthetic methods
The construction of C–N bonds is of particular importance because of their prevalence in natural products, pharmaceuticals, agrochemicals, and materials.[1,2] Strategies for the direct functionalization of carbon–hydrogen (C–H) bonds have garnered much attention because of the ability to streamline complex molecule synthesis in an atom-economical manner.[3–5] Specifically, C–H functionalization bypasses the need for employing a preoxidized arene coupling partner required for conventional cross-couplings.
While several methodologies have been developed for C– H amination,[6–8] nitrogen coupling partners are generally limited to electron-poor species, such as amides and imides.[9–12] Significant challenges exist for C–N couplings using aliphatic amines, since many strategies for functionalization of these substrates involve C–C bond construction adjacent to the nitrogen atom.[13–16] Accordingly, C–H amination of arenes with aliphatic amines has been demonstrated only in select examples (Scheme 1).[17] To achieve C–N bond formation, the use of strongly acidic media and prefunctionalized chloroamines, which suffer from limited stability, are often required (Scheme 1a).[18,19] To address these shortcomings, we hoped to develop a direct C–H amination with aliphatic amines using a mild catalytic system.
Scheme 1.
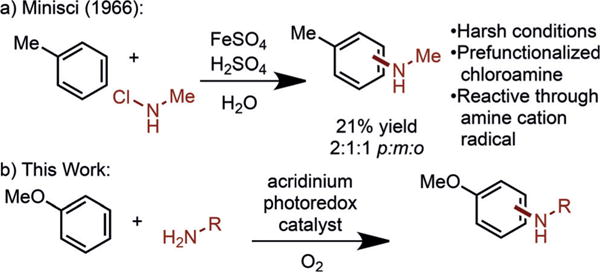
Arene C–H amination with aliphatic nitrogen coupling partners.
Previously, our laboratory reported a method for direct C–H amination of arenes and heteroarenes with nitrogen-containing heterocycles and ammonia surrogates, circumventing the need for pre-oxidized coupling partners.[20] This reaction features a high degree of site selectivity, with monosubstituted arenes favoring functionalization in the para position. This approach exploits an acridinium photoredox catalyst capable of oxidizing arenes through single-electron transfer (SET) to generate the corresponding cation radicals, which then act as electrophilic intermediates for nitrogenous nucleophiles.
In designing a strategy to construct C–N bonds by photoredox catalysis using aliphatic amines (Scheme 1b), it is noteworthy that both aliphatic amines and electron-rich arenes can be oxidized by the acridinium photocatalyst to the corresponding cation radicals (see Table S1 in the Supporting Information).[21] To examine whether aryl amination could occur using aliphatic amines, irradiation (455 nm LEDs) of an arene and an amino ester hydrochloride salt in the presence of the acridinium photocatalyst Me2-Mes-Acr + in a mixture of 1,2-dichloroethane (DCE) and pH 8 phosphate buffer afforded the aminated arene products (Figure 1). A wide variety of amino ester hydrochloride salts participated in the reaction, providing access to N-arylated amino acids under mild reaction conditions. When anisole was used as the arene coupling partner, the ortho isomer was favored in all cases, in moderate to excellent yields (1a–14a), complimentary to the aryl amination previously reported by our laboratory. The regioselectivity could be reversed by employing tert-butyldimethylsilyl (TBS) phenyl ether as the arene, favoring the formation of the para isomer (1b–14b). In addition to amino acids bearing hydrocarbon side chains, the reaction tolerated additional functionality, with protected serine and threonine, N-Boc lysine, and glutamic and aspartic acid esters all reacting smoothly. Furthermore, amination products derived from isoleucine and threonine (5a/b and 11a/b) formed as single diastereomers, as observed by 1H NMR analysis, indicating that amino acids do not epimerize during the reaction. Fourteen amino acids were shown as coupling partners, thus highlighting the generality of this approach.
Figure 1.
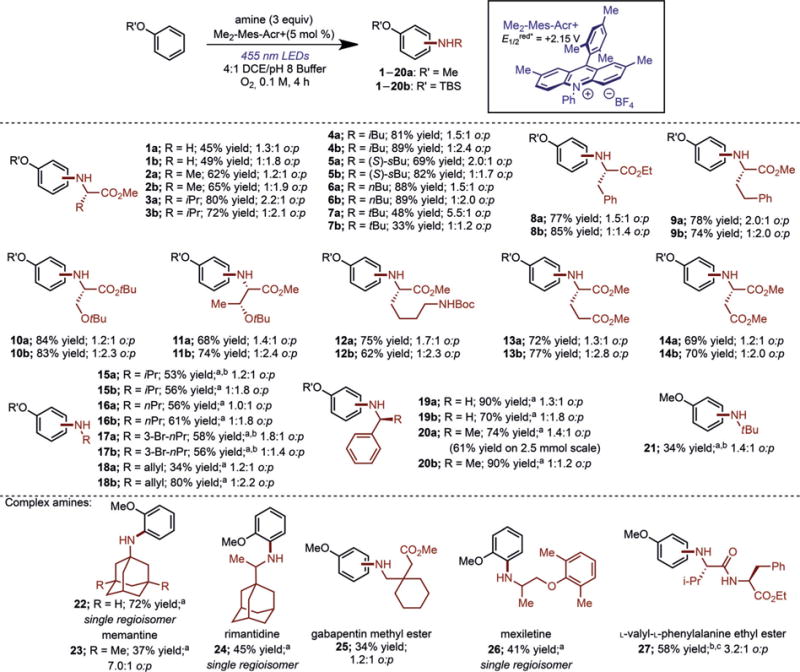
Products of C–H amination illustrating amine scope. Ratios refer to ortho/para (o:p) selectivity for the given adduct. Reactions were run in 4:1 DCE/pH 8 phosphate buffer at 0.1M concentration with respect to the arene limiting reagent. [a] Reaction run in DCE at 0.1M concentration with respect to the arene limiting reagent. [b] Yield of adduct determined by 1H NMR spectroscopy. [c] Reaction run in DCE at 0.1M concentration, with respect to the arene limiting reagent, for 14 h. DCE= 1,2-dichloroethane, TBS =tert-butyldimethylsilyl.
By applying these reaction conditions to other commercially available amines, the pH 8 buffer could be omitted when using the free base instead of the hydrochloride salt (15–26; Figure 1). The use of linear aliphatic amines afforded the products 16a and 16b in good yields, despite the presence of a methylene adjacent to the nitrogen center, which is prone to oxidative degradation. Halogenated amine coupling partners also provided the aminated products 17a and 17b in moderate yield. Allylamine afforded moderate to high yields of 18a and 18b, and benzylamine derivatives were also competent (19 and 20). Enantiopure (S)-α-methylbenzylamine did not racemize over the course of the reaction, thus indicating that even chiral benzylic amines retain stereo-chemical fidelity throughout the transformation. This reaction was performed on 2.5 mmol scale using a flow apparatus, affording the desired product in 61% yield. Amines bearing increased steric bulk also participated in the reaction, however in diminished yield, with tert-butylamine providing the aniline 21 in 34% yield.
The use of more highly substituted amines was demonstrated with the addition of adamantylamine and memantine, anti-Parkinson and anti-Alzheimer pharmaceuticals, respectively, affording the corresponding anilines 22 and 23 in modest to good yields and high ortho selectivities with anisole (Figure 1).[22] Furthermore, the antiviral rimantidine[23] produced the aminated arene 24 as a single regioisomer in 45% yield. Gabapentin methyl ester, a pharmaceutical used to treat seizures,[24] afforded the aryl amine 25 in 34% yield, while anti-arrhythmic pharmaceutical mexiletine[25] provided 26 in 41% yield as a single regioisomer. The coupling of L-valyl-L-phenylalanine ethyl ester with anisole in 58% yield (27) highlights the application of this methodology to peptidic compounds.
With a wide variety of primary amines shown as competent coupling partners, the arene scope was explored using valine methyl ester hydrochloride as the amine component (Figure 2). As discussed above, the regioselectivity of the amination could be altered by changing the substituent on the phenolic ether. Exposure of aliphatic (Me, Et, tBu) and silyl-protected (TES, TBS, TBDPS) phenolic ethers to the reaction conditions demonstrated that increased steric demand leads to higher amounts of the para adduct (3a/b, 28–31). Diphenyl ether, diphenyl sulfide, and biphenyl underwent amination in modest to good yields, also showing a preference for formation of the para isomer (32–34). Furthermore, halogens were tolerated on the arene to afford 35–38, using modified reaction conditions employing substoichiometric amounts of TEMPO as an additive, similar to our prior work.[20] For these substrates, the major product resulted from addition ortho to the methoxy substituent. Importantly, the presence of a halogen in these products allows subsequent derivatization through cross-coupling reactions, thus highlighting the utility of the present methodology in the synthesis of complex benzenoids. 4-Alkyl substituents were also tolerated, with amination of 4-isopropyl anisole proceeding in 43% yield (39) in high selectivity for the C2 isomer. Additionally, 1,3-disubstituted benzenes reacted well, with 1,3-dimethoxybenzene and meta-xylene providing the aminated products 40 and 41 in moderate to good yields and excellent regioselectivity. 1,2-Disubstituted arenes such as 2-chloroanisole also participated in the amination, leading to 42 in 63% yield. More complex substrates bearing an additional aryl ring afforded the anilines 43 and 44 in good yields, with functionalization only occurring on the more-electron-rich ring. Heterocycles also served as efficient arene coupling partners, with 2,6-dimethoxypyridine and N-methylindazole providing 45 and 46, respectively, as single regioisomers in good yields. Moreover, amination of halogenated heterocycles such as 7-bromo-N-methylindazole afforded a single regioisomer (47) in good yield.
Figure 2.
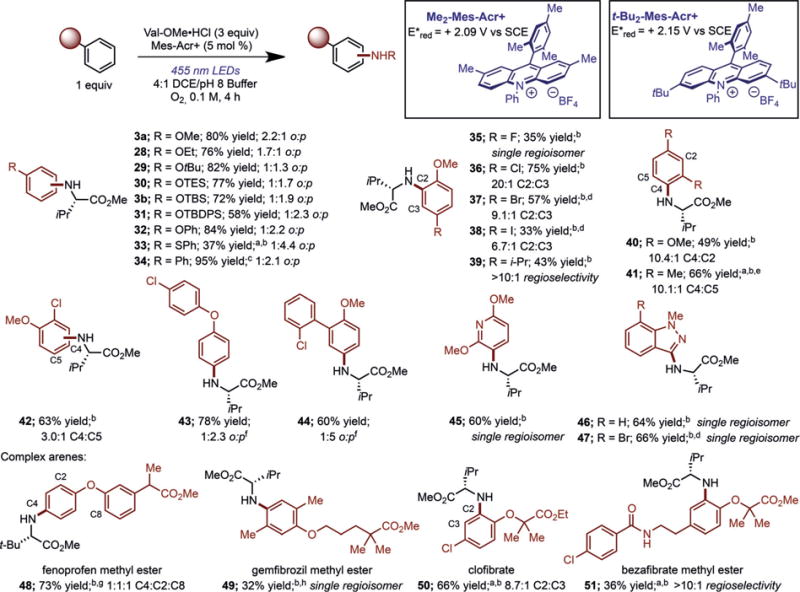
Product of C–H amination illustrating arene scope. Ratios refer to ortho/para (o:p) selectivity for the given isomer unless otherwise specified. Reactions were run using Me2-Mes-Acr+ in 4:1 DCE/pH 8 phosphate buffer at 0.1M concentration with respect to the arene limiting reagent. [a] Yield of adduct as determined by 1H NMR spectroscopy. [b] Reactions run with 40 mol% TEMPO and 5 mol% tBu2-Mes-Acr+ for 15– 26 h. [c] Reaction run for 13 h. [d] Reactions were run in 4:1 trifluorotoluene/pH 8 phosphate buffer. [e] 10 equiv of meta-xylene was used with the limiting amine. [f] The o:p ratio determined after isolation. [g] Reaction was run with 3 equiv of tert-leucine methyl ester hydrochloride. [h] 5 equiv of amine used. TBDPS=tert-butyldiphenysilyl.
To highlight the derivatization of complex arenes using this methodology, fenoprofen, a nonsteroidal anti-inflammatory drug, was subjected to amination with tert-leucine methyl ester, affording 48 in 73% yield (Figure 2). Gemfibrozil, a lipid-lowering drug,[26] also participated in the amination reaction with valine methyl ester to give 49 as a single regioisomer in 32% yield. Clofibrate and bezafibrate methyl ester, pharmaceuticals that are also used to lower LDL cholesterol,[27] were both functionalized in moderate yields and good selectivity (50 and 51), further illustrating the utility for late-stage pharmaceutical derivatization.
In our previous work on arene functionalization, we exploited electrophilic arene cation radicals generated by SET to the acridinium photoredox catalyst. Since the present system allows the primary amine to be oxidized to the amine cation radical, we questioned whether arenes with oxidation potentials above the reduction potential of the excited-state acridinium could undergo amination. Under slightly modified reaction conditions, benzene (E1/2ox =+ 2.75 V vs. SCE) afforded 52 in 40% yield, thus indicating a pathway for C– H amination, which is not accessible through an arene cation radical generated by the acridinium catalyst (Figure 3). Toluene (E1/2ox =+ 2.42 V vs. SCE) also underwent amination, affording 53 as a mixture of regioisomers in 50% combined yield.
Figure 3.
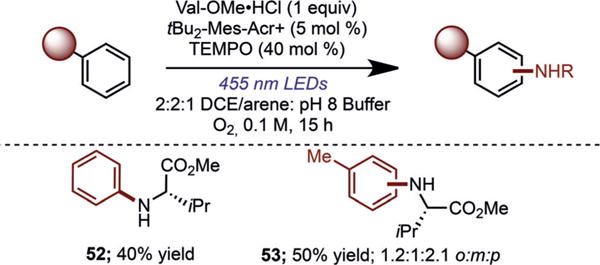
Amination of benzene and toluene. Reactions were run in 2:2:1 DCE/arene/pH 8 phosphate buffer at 0.1M concentration with respect to the amine limiting reagent. TEMPO=(2,2,6,6-tetramethylpiperidin-1-yl)oxyl.
We hypothesize that the reaction begins with excitation of Mes-Acr + with 455 nm LEDs to Mes-Acr + * (Figure 4). The excited state of the catalyst oxidizes the amine to the cation radical to generate Mes-Acr·. The addition of the arene forms a cyclohexadienyl radical intermediate that can be rearomatized using molecular oxygen as originally proposed by Fukuzumi and co-workers.[28] However, since several arenes in Figure 2 possess similar oxidation potentials to those of the amine coupling partner, an arene cation radical could also account for the observed reactivity.[20] As determined by Stern–Volmer fluorescence quenching analysis (see Figure S1), amines and electron-rich arenes both quench the acridinium excited state, but insufficiently electron-rich arenes, such as toluene, do not. Based on these results, insufficiently electron-rich arenes cannot undergo amination through an arene cation radical and instead react by an amine cation radical pathway. For electron-rich arenes, neither reactive intermediate can be excluded since both the amine and the arene quench the acridinium excited state.
Figure 4.
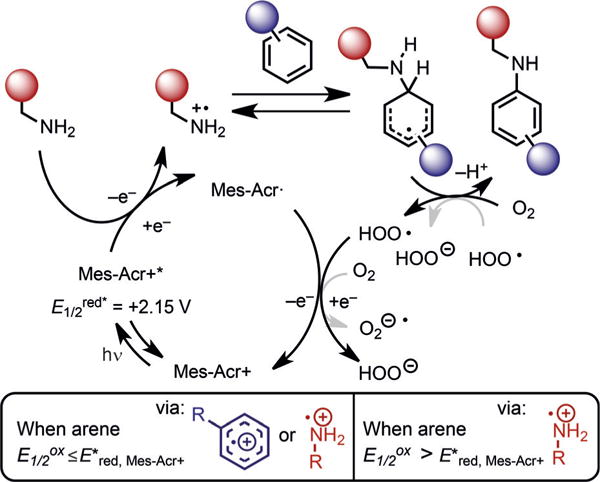
Proposed mechanism for direct C–H amination by photoredox catalysis using primary amines.
In conclusion, we have developed a direct aryl C–H amination using primary amines and organic photoredox catalysis. Further work will be required to extend this protocol to secondary amines. This methodology is mild and compatible with a variety of functional groups on both the arene and the amine, and enables extension of the arene coupling partners to non-activated aromatics such as benzene.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS; Award No. R01 GM120186) and an Eli Lilly Grantee Award (D.A.N.). A.L. thanks the American Australian Association for a Chevron Fellowship. This research made use of an Edinburgh FLS920 emission spectrometer funded by the UNC EFRC: Center for Solar Fuels, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001011. We thank Cole Cruz for his assistance with spectroscopic measurements. We thank the University of North Carolina’s Department of Chemistry Mass Spectrometry Core Laboratory for its assistance with mass spectrometry analysis supported by the National Institute of General Medical Sciences of the National Institutes of Health (R35GM118055).
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201709523.
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Kaila A. Margrey, Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290 (USA)
Alison Levens, Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290 (USA).
David A. Nicewicz, Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290 (USA).
References
- 1.Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW. Chem Soc Rev. 2016;45:546–576. doi: 10.1039/c5cs00628g. [DOI] [PubMed] [Google Scholar]
- 2.Vitaku E, Smith DT, Njardarson JT. J Med Chem. 2014;57:10257–10274. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 3.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies HML, Morton D. J Org Chem. 2016;81:343–350. doi: 10.1021/acs.joc.5b02818. [DOI] [PubMed] [Google Scholar]
- 5.Gensch T, Hopkinson MN, Glorius F, Wencel-Delord J. Chem Soc Rev. 2016;45:2900–2936. doi: 10.1039/c6cs00075d. [DOI] [PubMed] [Google Scholar]
- 6.Wolfe JP, Wagaw S, Marcoux JF, Buchwald SL. Acc Chem Res. 1998;31:805–818. [Google Scholar]
- 7.Hartwig JF. Acc Chem Res. 1998;31:852–860. [Google Scholar]
- 8.Rao KS, Wu TS. Tetrahedron. 2012;68:7735–7754. [Google Scholar]
- 9.Boursalian GB, Ngai MY, Hojczyk KN, Ritter T. J Am Chem Soc. 2013;135:13278–13281. doi: 10.1021/ja4064926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foo K, Sella E, ThomØ I, Eastgate MD, Baran PS. J Am Chem Soc. 2014;136:5279–5282. doi: 10.1021/ja501879c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawakami T, Murakami K, Itami K. J Am Chem Soc. 2015;137:2460–2463. doi: 10.1021/ja5130012. [DOI] [PubMed] [Google Scholar]
- 12.Jiao J, Murakami K, Itami K. ACS Catal. 2016;6:610–633. [Google Scholar]
- 13.Liu Y, Ge H. Nat Chem. 2016;9:26. [Google Scholar]
- 14.Xu Y, Young MC, Wang C, Magness DM, Dong G. Angew Chem Int Ed. 2016;55:9084–9087. doi: 10.1002/anie.201604268. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2016;128:9230–9233. [Google Scholar]
- 15.Chen G, Shigenari T, Jain P, Zhang Z, Jin Z, He J, Li S, Mapelli C, Miller MM, Poss MA, Scola PM, Yeung KS, Yu JK. J Am Chem Soc. 2015;137:3338–3351. doi: 10.1021/ja512690x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen G, Zhuang Z, Li GC, Saint-Denis TG, Hsiao Y, Joe CL, Yu JQ. Angew Chem Int Ed. 2017;56:1506–1509. doi: 10.1002/anie.201610580. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2017;129:1528–1531. [Google Scholar]
- 17.Boursalian GB, Ham WS, Mazzotti AR, Ritter T. Nat Chem. 2016;8:810–815. doi: 10.1038/nchem.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chow YL, Danen WC, Nelsen SF, Rosenblatt DH. Chem Rev. 1978;78:243–274. [Google Scholar]
- 19.Minisci F. Synthesis. 1973:1–24. [Google Scholar]
- 20.Romero NA, Margrey KA, Tay NE, Nicewicz DA. Science. 2015;349:1326–1330. doi: 10.1126/science.aac9895. [DOI] [PubMed] [Google Scholar]
- 21.Roth H, Romero N, Nicewicz D. Synlett. 2016;27:714–723. [Google Scholar]
- 22.Brown PD, Pugh S, Laack NN, Wefel JS, Khuntia D, Meyers C, Choucair A, Fox S, Suh JH, Roberge D, Kavadi V, Bentzen SM, Mehta MP, Watkins-Bruner D. Neuro-Oncology. 2013;15:1429–1437. doi: 10.1093/neuonc/not114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donath E, Herrmann A, Coakley WT, Groth T, Egger M, Taeger M. Biochem Pharmacol. 1987;36:481–487. doi: 10.1016/0006-2952(87)90355-8. [DOI] [PubMed] [Google Scholar]
- 24.Traa BS, Mulholland JD, Kadam SD, Johnston MV, Comi AM. Pediatr Res. 2008;64:81–85. doi: 10.1203/PDR.0b013e318174e70e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Logigian EL, Martens WB, Moxley RT, McDermott MP, Dilek N, Wiegner AW, Pearson AT, Barbieri CA, Annis CL, Thornton CA, Moxley RT. Neurology. 2010;74:1441–1448. doi: 10.1212/WNL.0b013e3181dc1a3a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corbett GT, Roy A, Pahan K. J Immunol. 2012;189:1002–1013. doi: 10.4049/jimmunol.1102624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beggs PW, Clark DWJ, Williams SM, Coulter DM. Br J Clin Pharmacol. 2001;47:99–104. doi: 10.1046/j.1365-2125.1999.00846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohkubo K, Mizushima K, Iwata R, Fukuzumi S. Chem Sci. 2011;2:715. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.