

Abstract
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) are carcinogenic in animal models and are believed to play an important role in human lung carcinogenesis in cigarette smokers. Cytochrome P450-mediated metabolism of these tobacco-specific nitrosamines produces reactive species that alkylate DNA in the form of pyridyloxobutyl (POB)- or pyridylhydroxybutyl (PHB)-DNA adducts. Understanding the formation mechanism and overall levels of these adducts can potentially enhance cancer prevention methods through the identification of particularly susceptible smokers. Previous studies have identified and measured a panel of POB- and PHB-DNA base adducts of dGuo, dCyd, and Thd; however, dAdo adducts have yet to be determined. In this study, we complete this DNA adduct panel by identifying and quantifying levels of NNK- and NNAL-derived dAdo adducts in vitro and in vivo. To accomplish this, we synthesized standards for expected dAdo-derived DNA adducts and used isotope-dilution LC-ESI+-MS/MS to identify POB adducts formed in vitro from the reaction of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone (NNKOAc) with calf thymus DNA. Adduct levels were then quantified in lung and liver DNA of rats chronically treated with NNK or NNAL for 50 weeks using similar LC-MS detection methods. The in vitro studies identified N6-POB-dAdo and N1-POB-dIno as products of the reaction of NNKOAc with DNA, which supports our proposed mechanism of formation. Though both N6-dAdo and N1-dIno adducts were found in vitro, only N6-dAdo adducts were found in vivo, implying possible intervention by DNA repair mechanisms. Analogous to previous studies, levels of N6-POB-dAdo and N6-PHB-dAdo varied both with tissue and treatment type. Despite the adduct levels being relatively modest compared to most other POB- and PHB-DNA adducts, they may play a biological role and could be used in future studies as NNK- and NNAL-specific DNA damage biomarkers.
Graphical Abstract
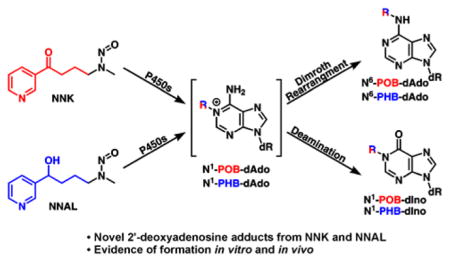
Introduction
Tobacco use remains an important global health crisis by claiming nearly 6 million lives per year, a figure which is expected to rise to 8 million annually by 2030.1 In the United States alone, 15% of the adult population are cigarette smokers,2 resulting in ~480,000 preventable deaths annually.3 Of these, 160,000 are due to cancer which accounts for 30% of all annual U.S. cancer deaths.4 Though tobacco use causes a variety of cancers, lung cancer is of particular concern as ~70–80% of all lung cancers are tobacco-related,3,4 and each year, 1.5 million lung cancer deaths occur worldwide.
Understanding the mechanisms of tobacco-induced carcinogenesis can potentially lead to new approaches to cancer prevention by identifying cigarette smokers particularly susceptible to lung cancer and other tobacco-related cancers. One important carcinogen in tobacco and tobacco smoke is 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK, 2), a well-established genotoxic agent evaluated, along with the related carcinogen N′-nitrosonornicotine, as carcinogenic to humans by the International Agency for Research on Cancer.5–7 NNK is found solely in tobacco products and readily causes adenocarcinoma of the lung in rats, mice, hamsters, and ferrets regardless of the route of administration and at relatively low doses.5 Additionally, epidemiological studies have correlated NNK metabolites in smokers’ urine to future lung cancer onset.8–10
An overview of NNK metabolism leading to DNA adduct formation is presented in Scheme 1. NNK is converted in humans to both enantiomers of the carcinogenic metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL, 3) by enzymatic carbonyl reduction.5,11 Studies suggest that this process may be partially reversible as (S)-NNAL can be enzymatically oxidized to NNK in vivo while (R)-NNAL commonly remains reduced.12 Both enantiomers are O- or N-glucuronidated and excreted in the urine.13 These and unmodified NNAL serve as useful biomarkers and were the basis of the epidemiology studies noted above.8–10
Scheme 1.

Overview of NNK (2) and NNAL (3) metabolism and DNA adduct formation in vivo. NNK is in enzymatic equilibrium with NNAL, which is commonly glucuronidated. Both NNK and NNAL can alternatively be oxidized to α-hydroxynitrosamines 4 or 5. These further decompose to diazonium ions 8 or 9 and ultimately hydrolyze to 10 or 11, or form DNA adducts.
Both NNK and NNAL are metabolized by P450s, activating them to DNA-alkylating agents.5 Initial hydroxylation of the terminal methyl group of NNK or NNAL by either P450 2A6 or P450 2A1314 produces the unstable α-hydroxy species 4 or 5. A small fraction of these may be converted further to nitrosamides,15 but most will rapidly decompose to diazohydroxides 6 and 7, before losing H2O and forming highly reactive diazonium ions 8 and 9. These intermediates hydrolyze to form metabolites 10 and 11, which are excreted in the urine after further enzymatic processing. However, 8 and 9 can also react with DNA to form a variety of products referred to as pyridyloxobutyl- (POB) or pyridylhydroxybutyl- (PHB) DNA adducts, respectively. The POB-DNA adducts can also be formed in vitro from NNKOAc, a precursor to 4 that does not require P450 metabolism. Analogously, the α-methylene group can be hydroxylated to ultimately form methyl DNA adducts. DNA adducts left unrepaired can cause mutations and initiate tumor development.5,6 To date, only four POB- and three PHB- base adducts have been detected and quantified in animal models (Figure 1).12,16–18 In addition to these base adducts, a large family of DNA phosphate adducts have recently been uncovered and quantified in vivo (Figure 1).19,20
Figure 1.

Structures of previously measured POB- and PHB-DNA adducts in vivo.
Notably missing from this panel of adducts are those formed with 2′-deoxyadenosine (dAdo). Therefore, the goal of this study was to determine the structure and levels of dAdo-derived POB- and PHB-adducts in NNK- and NNAL-treated rats. We hypothesized that initial alkylation of dAdo would occur at the N1-position (Scheme 2). This results in an unstable cationic intermediate (19), which can deprotonate and undergo spontaneous Dimroth rearrangement to give N6-dAdo adducts 20 and 21.21–23 Alternatively, deamination via an addition-elimination mechanism can occur yielding N1-dIno adducts 22 and 23.23–26 In support of this general mechanism, N6-dAdo and N1-dIno type adducts have been found after in vitro treatment of DNA with styrene oxide and butadiene monoxide.27,28 Along with these adduct types, previous studies have identified N3- and N7-dAdo adducts, but these are known to be unstable and generate abasic sites,29,30 thus were not considered in this study. N1-dIno adducts are thought to be potentially important as they can cause A:G mutations through Hoogstein base pairing,31 while N6-dAdo adducts may interfere with base pairing due to their steric bulk. Regardless of their potential mutagenicity, these adducts also may serve as DNA damage biomarkers.32 Therefore, to meet the goals of this study, we synthesized standards for all four proposed dAdo-derived adducts, detected POB adduct formation in vitro, and quantified dAdo adduct formation in the livers and lungs of rats treated chronically with NNK and NNAL.
Scheme 2.

Proposed mechanism for dAdo adduct formation. After initial N1-alkylation, the resulting cationic intermediate either undergoes Dimroth rearrangement or deamination to yield N6-dAdo (20–21) or N1-dIno (22–23) DNA adducts, respectively.
Experimental Procedures
Caution: NNK and NNAL are carcinogenic in animal models. NNKOAc, an activated form of NNK, is mutagenic and is presumed to be carcinogenic. Handle these compounds in a well-ventilated fume hood while wearing proper personal protective equipment.
Chemicals and Enzymes
NNK and NNKOAc were purchased from Toronto Research Chemicals. (S)-NNAL, (R)-NNAL, N6-PHB-dAdo, 2-(3-pyridyl)-1,3-dithiane, tert-butyl 3-(2-(3-pyridyl)-1,3-dithianyl)-1-propylcarbamate, and [pyridine-D4]tert-butyl 3-(2-(3-pyridyl)-1,3-dithianyl)-1-propylcarbamate were synthesized as previously described.15,33,34 [15N5]2′-deoxyadenosine (>99% 15N5 incorporation) was obtained from Cambridge Isotope Laboratories (Tewksbury, MA). 6-Chloropurine-2′-deoxyriboside was obtained from Carbosynth (Compton, United Kingdom). Reagents for DNA isolation were purchased from Qiagen (Hilden, Germany). Calf thymus DNA, phosphodiesterase II, and micrococcal nuclease were obtained from Worthington Biochemical Co. (Lakewood, NJ). Alkaline phosphatase was procured from Roche Diagnostics GmbH (Mannheim, Germany). Porcine liver esterase and all other chemicals and solvents were obtained from either Sigma Aldrich (Milwaukee, WI) or Thermo Scientific (Waltham, MA) in reagent grade or higher and used without further purification.
General Synthetic Procedures
NMR spectra were recorded on a Bruker 500 MHz spectrometer. Chemical shifts are reported as parts per million (ppm). Residual solvent peaks were used as an internal reference for 1H-NMR (7.26 ppm CDCl3; 2.50 ppm D6-DMSO) and 13C-NMR (77.2 ppm CDCl3; 39.5 ppm D6-DMSO). Peak splitting was abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, dt = doublet of triplets, ddd = doublet of doublet of doublets, bs = broad singlet, and m = multiplet. High resolution mass spectrometry (HRMS) of selected compounds was performed on an LTQ Orbitrap Velos (Thermo Scientific, Waltham, MA) and reported as m/z. Thin-layer chromatography (TLC) utilized Polygram pre-coated silica gel TLC plate (40 × 80 mm, 0.2 mm thick) with 254 nm fluorescent indicator. TLC plates were visualized with permanganate stain when necessary, otherwise UV lamp irradiation sufficed. Flash chromatography was performed on SiliCycle 60 (70–150) mesh silica gel. Reactions were performed with oven-dried glassware and under an atmosphere of N2 unless specified otherwise.
[15N5]6-N-(4-(3-pyridyl)-4-hydroxy-1-butyl)-2′-deoxyadenosine ([15N5]N6-PHB-dAdo, 21)
This was prepared as previously described34 for N6-PHB-dAdo except using [15N5]2′-deoxyadenosine as starting material.
1H-NMR (500 MHz; DMSO-d6): δ 8.51 (d, J = 10.4 Hz, 1H, 2-Pyr), 8.42 (d, J = 4.7 Hz, 1H, 6-Pyr), 8.33-8.29 (m, 1H, C8), 8.18 (s, 1H, C2), 7.87 (bs, 1H, NH), 7.71-7.69 (m, 1H, 4-Pyr), 7.32 (dd, J = 7.8, 4.8 Hz, 1H, 5-Pyr), 6.34 (t, J = 7.0 Hz, 1H, C1′), 5.31 (m, 2H, 3′-OH/CHOH), 5.24 (s, 1H, 5′-OH), 4.62 (m, 1H, CH), 4.41-4.40 (m, 1H, C3′), 3.88 (d, J = 2.2 Hz, 1H, C4′), 3.62 (m, 1H, C5′), 3.50 (m, 3H, C5′/NHCH2), 2.72 (dt, J = 12.0, 5.5 Hz, 1H, C2′), 2.25 (dd, J = 12.4, 4.4 Hz, 1H, C2′), 1.69-1.54 (m, 4H, CH2CH2).
13C-NMR: (126 MHz; DMSO-d6): δ 152.6 (C2), 148.4 (6-Pyr), 148.1 (2-Pyr), 139.5 (C8), 133.9 (4-Pyr), 123.5 (5-Pyr), 88.5 (C4′), 84.3 (C1′), 71.4 (C3′), 70.4 (CH), 62.4 (C5′), 39.9 (C2′), 39.8 (NCH2), 36.8 (CHCH2)), 25.9 (NCH2CH2). 3-Pyr, C4, C5, and C6 not observed.
HRMS: calc’d: 406.17835; found: 406.17756
3-(2-(3-Pyridyl)-1,3-dithianyl)-1-propylamine (26)
A solution of 25 (87.6 mg, 0.247 mmol) in CH2Cl2 (3 mL) was treated with trifluoroacetic acid (TFA, 1,49 g, 13.1 mmol, 1 mL) and stirred at room temperature. After 3h, the solution was concentrated by rotary evaporation to remove excess TFA. The resulting oil was dissolved in CH2Cl2 and washed with sat’d NaHCO3 solution and brine. The organic layer was then dried over MgSO4, filtered, and concentrated in vacuo to give a yellow oil. Purification by column chromatography (CH2Cl2/MeOH/Et3N 85:15:1) yielded the product as a light, yellow oil (41.4 mg, 66%).
1H-NMR (500 MHz; CDCl3): δ 9.11 (d, J = 1.8 Hz, 1H), 8.49-8.48 (m, 1H), 8.21 (d, J = 8.1 Hz, 1H), 7.31 (dd, J = 8.0, 4.8 Hz, 1H), 2.72-2.64 (m, 6H), 2.10-2.06 (m, 2H), 1.95 (dd, J = 8.9, 4.7 Hz, 2H), 1.53-1.49 (m, 2H). 13C-NMR (126 MHz; CDCl3): δ 150.5, 148.1, 137.6, 136.9, 123.3, 56.3, 42.1, 41.2, 27.5, 26.2, 24.9
6-N-(4-(3-pyridyl)-4-oxo-1-butyl)-2′deoxyadenosine (N6-POB-dAdo, 20)
A solution of 26 (6.35 mg, 0.0250 mmol), 6-chloropurine-9-2′-deoxyriboside (10.4 mg, 0.0384 mmol), and iPr2EtN (7.42 mg, 0.0574 mmol,10 μL) in DMSO (300 μL) was heated to 60 °C for 16 h. The compound was crudely isolated by solid-phase extraction using a Strata-X 33 μm polymeric reversed phase cartridge (30 mg/mL; Phenomenex, Torrance, CA). The cartridge was preconditioned with 1 mL of MeOH, followed by 1 mL of H2O. After sample addition, the cartridge was washed with 2 mL of H2O, 1 mL of 5% (v/v), 10%, and 50% MeOH, and eluted with 1 mL of 80% MeOH. The eluent was evaporated to dryness in a Speedvac.
The isolated product was dissolved in 4:1 MeCN/H2O (1 mL) and added to a suspension of N-chlorosuccinimide (NCS, 11.9 mg, 0.0891 mmol) and AgNO3 (20.2 mg, 0.1188) in 4:1 MeCN/H2O (1 mL) at 0 °C. The reaction was stirred at 0 °C for 30 min before quenching with sat’d Na2SO3, sat’d NaHCO3, and brine solutions in succession (1 mL each). The product was isolated by HPLC using a 250 mm × 10 mm, 5 μm, Luna C-18 column (Phenomenex) with H2O and MeCN as mobile phases. The gradient was 30% to 45% MeCN over 5 min at 3 mL/min. The product eluted at 4.1 min. The product was a light yellow solid (2.37 mg, 24% over two steps).
1H-NMR (500 MHz; DMSO-d6): δ 9.09 (d, J = 1.1 Hz, 1H, 2-Pyr), 8.77 (dd, J = 4.7, 0.8 Hz, 1H, 6-Pyr), 8.32 (s, 1H, C-8), 8.25 (d, J = 8.0 Hz, 1H, 4-Pyr), 8.12 (s, 1H, C-2), 7.94-7.93 (bs, 1H, NH), 7.54 (dd, J = 7.9, 4.8 Hz, 1H, 5-Pyr), 6.34 (t, J = 7.0 Hz, 1H, C1′), 5.30 (d, J = 3.9 Hz, 1H, 3′-OH), 5.22 (t, J = 5.7 Hz, 1H, 5′-OH), 4.41-4.40 (m, 1H, C3′), 3.88 (d, J = 2.5 Hz, 1H, C4′), 3.64-3.50 (m, 4H, C5′+ NHCH2), 3.15 (t, J = 6.9 Hz, 2H, COCH2), 2.74-2.69 (m, 1H, C2′), 2.27-2.23 (m, 1H, C2′), 1.96 (quintet, J = 6.8 Hz, 2H, COCH2CH2). 13C-NMR (126 MHz; DMSO-d6): δ 199.5 (CO), 155.1 (C6), 153.7 (6-Pyr), 152.7 (C2), 149.6 (2-Pyr), 148.5 (C4), 139.8 (C8), 135.7 (4-Pyr), 132.4 (3-Pyr), 124.3 (5-Pyr), 120.1 (C5), 88.5 (C4′), 84.4 (C1′), 71.4 (C3′), 62.4 (C5′), 39.9 (C2′), 39.3 (NHCH2), 36.0 (COCH2), 21.6 (COCH2CH2).
HRMS: calc’d: 399.17753; found: 399.17742
[Pyridine-D4]3-(2-(3-Pyridyl)-1,3-dithianyl)-1-propylamine
This compound was prepared analogously to 26 except [pyridine-D4]25 (>98% D4-incorporation) was used as starting material. The product was a light yellow oil (7.67 mg, 35%).
1H-NMR (500 MHz; CDCl3): δ 2.71-2.64 (m, 4H), 2.61 (t, J = 7.0 Hz, 2H), 2.05 (dt, J = 7.9, 4.2 Hz, 2H), 1.98-1.93 (m, 2H), 1.47-1.41 (m, 2H).
13C-NMR (126 MHz; CDCl3): δ 56.5, 42.5, 41.8, 27.52, 27.42, 25.0
[Pyridine-D4]6-N-(4-(3-pyridyl)-4-oxo-1-butyl)2′-deoxyadenosine ([pyridine-D4]N6-POB-dAdo)
This compound was prepared analogously to N6-POB-dAdo except [pyridine-D4]26 (7.67mg, 0.0297 mmol) was used as the starting material. The isolated product was a light yellow solid (2.08 mg, 18% over two steps).
1H-NMR (500 MHz; DMSO-d6): δ 8.32 (s, 1H, C8), 8.11 (s, 1H, C2), 7.94 (bs, 1H, NH), 6.33 (dd, J = 7.7, 6.2 Hz, 1H, C1′), 5.33 (d, J = 0.3 Hz, 1H, 3′-OH), 5.24 (m, 1H, 5′-OH), 4.41-4.40 (m, 1H, C3′), 3.88 (q, J = 3.4 Hz, 1H, C4′), 3.62-3.50 (m, 4H, C5′/COCH2), 3.15 (dd, J = 10.6, 5.9 Hz, 2H, NHCH2), 2.71-2.69 (m, 1H, C2′), 2.25 (ddd, J = 13.1, 6.1, 2.8 Hz, 1H, C2′), 1.96 (t, J = 6.9 Hz, 2H, CH2).
13C-NMR (126 MHz; DMSO-d6): δ 199.6 (CO), 155.1 (C6), 152.7 (C2), 147.0 (C4), 139.7 (C8), 132.3 (3-Pyr), 120.4 (C5), 88.5 (C4′), 84.4 (C1′), 71.4 (C3′), 62.3 (C5′), 39.6 (C2′), 39.2 (NHCH2), 36.1 (COCH2), 23.8 (COCH2CH2).
HRMS: calc’d: 403.20264; found: 403.20173
3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyinosine (29)
2′-Deoxyinosine (463 mg, 1.84 mmol) and imidazole (554.5 mg, 8.14 mmol) were suspended in anhydrous pyridine (5 mL) and treated with tert-butyldimethylsilyl chloride (TBS-Cl, 585 mg, 3.88 mmol). The mixture was stirred at room temperature. After 4 h, the mixture became completely homogenous, and the reaction was quenched with H2O (10 mL). The solution was extracted twice with CH2Cl2 (15 mL). The pooled organics were dried over MgSO4, filtered, and concentrated. The resulting product was evaporated from toluene (5 mL) multiple times to remove residual pyridine. NMR indicated that purity was sufficient to carry forward without further purification.
1H-NMR (500 MHz; CDCl3): δ 12.61 (s, 1H), 8.19 (s, 1H), 8.08 (s, 1H), 6.40 (t, J = 6.4 Hz, 1H), 4.60 (dt, J = 5.6, 3.6 Hz, 1H), 4.01 (q, J = 3.5 Hz, 1H), 3.85-3.75 (m, 2H), 2.58-2.42 (m, 2H), 0.90 (d, J = 2.6 Hz, 18H), 0.10-0.08 (m, 12H).
13C NMR (126 MHz; CDCl3): δ 158.7, 148.3, 144.6, 138.4, 125.0, 88.0, 84.5, 71.7, 62.7, 41.6, 26.0, 25.7, 18.4, 18.0, −4.67, −5.40.
1-N-(2,4-dinitrophenyl)-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyinosine (30)
A mixture of 29 (1384 mg, 2.14 mmol), K2CO3 (608.6 mg, 4.40 mmol), and 1-chloro-2,4-dinitrobenzene (499.5 mg, 2.22 mmol) in DMF (10 mL) was heated to 80 °C for 2.5 h. The bright red solution turned orange during the reaction. After 2.5 h, the reaction mixture was cooled to room temperature and diluted with EtOAc (10 mL). The mixture was filtered over Celite and concentrated to a thick, red oil. Purification by column chromatography (EtOAc/hexanes 1:2 →1:1) yielded pure product as a yellow solid (1.26 g, 91% over 2 steps).
1H-NMR (500 MHz; CDCl3): δ 9.05 (d, J = 1.2 Hz, 1H), 8.68-8.66 (m, 1H), 8.21 (d, J = 10.3 Hz, 1H), 8.00 (s, 1H), 7.72-7.68 (m, 1H), 6.47-6.41 (m, 1H), 4.64-4.61 (m, 1H), 4.06-4.04 (m, 1H), 3.89-3.79 (m, 2H), 2.60-2.45 (m, 2H), 0.93 (s, 18H), 0.13-0.11 (m, 12H). 13C-NMR (126 MHz; CDCl3): δ 155.2, 148.2, 147.1, 146.50, 146.48, 144.72, 144.61, 139.2, 138.9, 135.9, 131.96, 131.83, 128.75, 128.70, 124.37, 124.17, 121.43, 121.38, 88.32, 88.18, 84.64, 84.44, 71.87, 71.71, 62.77, 62.63, 42.1, 41.8, 26.0, 25.7, 18.4, 18.0, −4.63, −4.66, −5.37, −5.48
1-N-(3-hydroxypropyl)-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyinosine (31)
A solution of 30 (701.2 mg, 1.08 mmol) was dissolved in DMF (10 mL) and treated with 3-amino-1-propanol (810.2 mg, 10.8 mmol, 820 μL). The yellow solution immediately became red. The solution was brought to 80 °C for 2 h, then back to room temperature. It was concentrated to a dark red oil. Purification by column chromatography (EtOAc/hexanes 2:1 → 1:0) yielded pure product as a yellow oil that solidified upon refrigeration (522.9 mg, 90%).
1H-NMR (500 MHz; CDCl3): δ 8.08 (s, 1H), 8.03 (s, 1H), 6.35 (t, J = 6.4 Hz, 1H), 4.58 (dt, J = 5.7, 3.1 Hz, 1H), 4.24 (t, J = 6.4 Hz, 2H), 3.99 (d, J = 3.4 Hz, 1H), 3.82-3.73 (m, 2H), 3.59 (t, J = 5.5 Hz, 2H), 2.52 (dt, J = 13.0, 6.4 Hz, 1H), 2.40 (ddd, J = 13.0, 5.9, 4.0 Hz, 1H), 1.97 (quintet, J = 5.9 Hz, 2H), 0.89 (d, J = 3.9 Hz, 18H), 0.09-0.06 (m, 12H).
13C-NMR (126 MHz; CDCl3): δ 157.4, 147.29, 147.18, 138.5, 124.6, 88.0, 84.2, 71.8, 62.7, 57.9, 43.1, 41.5, 32.6, 25.9, 18.4, 18.0, −4.7, −5.4
1-N-(3-iodopropyl)-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyinosine (32)
Imidazole (69.0 mg, 1.01 mmol) and triphenylphosphine (222.7 mg, 0.849 mmol) were dissolved in CH2Cl2 (7 mL) and brought to 0 °C. To this was added I2 (218.8 mg, 0.862 mmol). After 15 min, the color of the mixture changed from yellow to dark orange. A CH2Cl2-diluted solution of 31 (415.9 mg, 0.772 mmol, 7 mL) was then added. After 10 min, the reaction was warmed to room temperature and stirred for 2 h. Then the mixture was filtered over Celite and washed with sat’d Na2S2O3 solution. The organic layer was separated and further washed with brine, dried over MgSO4, filtered, and concentrated to a yellow solid. Purification by column chromatography (EtOAc/hexanes 3:1) yielded pure product as a yellow oil (461.4 mg, 95%).
1H-NMR (500 MHz; CDCl3): δ 8.26 (s, 1H), 8.08 (s, 1H), 6.39 (t, J = 6.4 Hz, 1H), 4.60 (dt, J = 5.9, 3.2 Hz, 1H), 4.19 (t, J = 6.6 Hz, 2H), 4.03 (q, J = 3.5 Hz, 1H), 3.85-3.76 (m, 2H), 3.17 (t, J = 6.4 Hz, 2H), 2.59-2.43 (m, 2H), 2.33 (quintet, J = 6.5 Hz, 2H), 0.91 (d, J = 4.7 Hz, 18H), 0.11-0.08 (m, 12H). 13C-NMR (126 MHz; CDCl3): δ 155.5, 147.5, 146.9, 138.3, 123.8, 88.3, 84.7, 71.8, 62.7, 47.5, 41.6, 31.9, 26.0, 25.7, 18.4, 18.0, 2.3, −4.65, −4.78, −5.35, −5.45.
1-N-(3-(2-(3-Pyridyl)-1,3-dithianyl)propyl)-3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyinosine (34)
A solution of 3315 (78.1 mg, 0.396 mmol) and tetramethylethylenediamine (TMEDA, 46.5 mg, 0.400 mmol, 60 μL) in anhydrous THF (4 mL) was cooled to −78 °C and treated with n-BuLi in hexanes dropwise (0.480 mmol, 300 μL). The resulting red solution was stirred for 10 min. A THF-diluted solution of 32 (228.5 mg, 0.352 mmol, 3 mL) was then added dropwise. The mixture was stirred for 4 h and the color slowly transitioned from red to yellow. The reaction was quenched with H2O (5 mL) and warmed to room temperature. The mixture was diluted with EtOAc and washed with brine. The aqueous phase was extracted twice with EtOAc. The pooled organics were dried over MgSO4, filtered, and concentrated to an orange oil. Purification by column chromatography (100% EtOAc) yielded the product as a yellow solid (179.4 mg, 71%).
1H-NMR (500 MHz; CDCl3): δ 9.12 (d, J = 1.9 Hz, 1H), 8.53 (d, J = 3.9 Hz, 1H), 8.30 (m, 1H), 8.05 (s, 1H), 7.85 (s, 1H), 7.42 (dd, J = 8.0, 4.9 Hz, 1H), 6.38-6.33 (m, 1H), 4.59-4.55 (m, 1H), 4.27 (s, 1H), 3.97 (m, 4H), 3.82-3.59 (m, 2H), 2.77-2.49 (m, 6H), 2.06 (dd, J = 9.8, 6.4 Hz, 2H), 1.98-1.76 (m, 4H), 0.78 (s, 18H), 0.07 (m, 12H).
13C-NMR (126 MHz; CDCl3): δ 156.5, 149.0, 147.2, 146.86, 146.69, 139.5, 138.6, 138.3, 124.9, 89.9, 84.8, 73.2, 63.5, 56.1, 46.2, 41.9, 41.7, 27.6, 26.1, 24.7, 18.6, 18.1, −4.53, −4.67, −5.35, −5.39
1-N-(4-oxo-4-(3-pyridyl)-butyl)-2′-deoxyinosine (N1-POB-dIno, 22)
N-Chlorosuccinimide (NCS, 11.5 mg, 0.0861) and AgNO3 (15.5 mg, 0.0912) were suspended in 1:1 MeCN/H2O (400 μL) and cooled to 0 °C. To this was added a solution of 34 in MeCN (11.1 mg, 0.0155 mmol, 600 μL). The reaction mixture was stirred at 0 °C for 30 min before quenching with sat’d Na2SO3, sat’d NaHCO3, and brine solutions in succession (1 mL each). The mixture was diluted with CH2Cl2 (5 mL) and filtered. The organic layer was collected and the aqueous phase further extracted once with CH2Cl2. The pooled organics were dried over MgSO4, filtered, and concentrated to a yellow oil. The crude product was then transferred and concentrated into a 2-mL polypropylene tube and reconstituted in THF (200 μL). To this was added 5 μL of a K2HPO4-buffered solution of tetra-n-butylammonium fluoride (TBAF, 3.4M, 25mM 2:1 K2PO4, pH 7.1/THF). The solution immediately turned orange and was shaken for 24 h. The solvent was evaporated to give an orange solid, which was reconstituted in H2O (2 mL) and purified by HPLC using a 250 mm × 10 mm, 5 μm, Luna C-18 column (Phenomenex) with H2O and MeCN as mobile phases. The gradient was 30% to 45% MeCN over 5 min at 3 mL/min. The product eluted at 10.7 min. The product was a white solid (2.37 mg, 24% over two steps).
1H-NMR (500 MHz; DMSO-d6): δ 9.09 (d, J = 1.7 Hz, 1H, 2-Pyr), 8.78 (dd, J = 4.8, 1.6 Hz, 1H, 6-Pyr), 8.42 (s, 1H, C2), 8.31 (s, 1H, C8), 8.25 (dt, J = 8.0, 1.9 Hz, 1H, 4-Pyr), 7.56 (dd, J = 7.9, 4.8 Hz, 1H, 5-Pyr), 6.30 (t, J = 6.9 Hz, 1H, C1′), 5.32 (d, J = 3.7 Hz, 1H, 3′-OH), 4.97 (t, J = 5.5 Hz, 1H, 5′-OH), 4.39-4.37 (m, 1H, C3′), 4.09 (t, J = 7.0 Hz, 2H, NCH2), 3.86 (q, J = 3.7 Hz, 1H, C4′), 3.61-3.49 (m, 2H, C5′), 3.16 (t, J = 7.0 Hz, 2H, COCH2), 2.62 (ddd, J = 13.3, 7.2, 6.1 Hz, 1H, C2′), 2.31-2.27 (m, 1H, C2′), 2.05 (t, J = 7.0 Hz, 2H, COCH2CH2).
13C-NMR (126 MHz; DMSO-d6): δ 198.6 (CO), 156.1 (C6), 153.3 (6-Pyr), 149.1 (2-Pyr), 148.4 (C2), 147.1 (C4), 139.3 (C8), 135.3 (4-Pyr), 131.8 (3-Pyr), 123.8 (5-Pyr), 123.4 (C5), 88.6 (C4′), 83.6 (C1′), 70.6 (C3′), 61.5 (C5′), 45.2 (NCH2), 40.4 (C2′), 35.2 (COCH2), 23.5 (COCH2CH2)
HRMS: calc’d: 400.16155; found: 400.16162
1-N-(4-hydroxy-4-(3-pyridyl)-butyl)- 3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyinosine (35)
N-Chlorosuccinimide (NCS, 12.9 mg, 0.0966) and AgNO3 (20.6 mg, 0.121) were suspended in 1:1 MeCN/H2O (200 μL) and cooled to 0 °C. To this was added a solution of 34 in 1:1 MeCN:H2O (15.6 mg, 0.0217 mmol, 800 μL). The reaction mixture was stirred at 0 °C for 30 min before quenching with sat’d Na2SO3, sat’d NaHCO3, and brine solutions in succession (1 mL each). The mixture was diluted with CH2Cl2 (5 mL) and filtered. The organic layer was collected and the aqueous phase extracted once with CH2Cl2. The pooled organics were dried over MgSO4, filtered, and concentrated to a yellow oil. The crude product was then dissolved in MeOH (800 μL) and treated with NaBH4 (7.0 mg, 0.185 mmol) at room temperature. After 1 h, the reaction mixture was quenched with sat’d NH4Cl, diluted with CH2Cl2 and washed with sat’d NaHCO3. The aqueous phase was further extracted three times with CH2Cl2. The pooled organics were dried over MgSO4, filtered, and concentrated to a white solid (6.15 mg). The crude material was carried forward without further purification.
1-N-(4-hydroxy-4-(3-pyridyl)-butyl)-2′-deoxyinosine (N1-PHB-dIno, 23)
A solution of 35 (3.4 mg, 0.00540 mmol) in THF (450 μL) was treated with TBAF in THF (0.0150 mmol, 15 μL) at room temperature. After 1 h, the reaction mixture was diluted with H2O and filtered through a 4 mm Acrodisc syringe filter (Sigma Aldrich). The resulting solution was purified by HPLC using a 250 mm × 10 mm, 5 μm, Luna C-18 column (Phenomenex) with H2O and MeCN as mobile phases. The gradient was 30% to 45% MeCN over 5 min at 3 mL/min. The product eluted at 7.1 min. The product was a white solid (1.61 mg, 34% over 3 steps).
1H-NMR (500 MHz; DMSO-d6): δ 8.51-8.51 (m, 1H, 2-Pyr), 8.43 (dd, J = 4.7, 1.5 Hz, 1H, 6-Pyr), 8.40 (s, 1H, C2), 8.33 (s, 1H, C8), 7.70 (dd, J = 7.8, 1.6 Hz, 1H, 4-Pyr), 7.33 (dd, J = 7.7, 4.7 Hz, 1H, 5-Pyr), 6.29 (dd, J = 7.7, 2.8 Hz, 1H, C1′), 5.43 (m, 2H, 3′-OH/5′-OH), 4.85 (s, 1H, OH), 4.63-4.61 (m, 1H, CH), 4.32-4.29 (m, 1H, C3′), 4.14-4.11 (m, 1H, C4′), 4.03-4.00 (m, 2H, NCH2), 3.48-3.41 (m, 2H, C5′), 2.76-2.70 (m, 1H, C2′), 2.33-2.29 (m, 1H, C2′), 1.79-1.56 (m, 4H, CH2CH2).
13C-NMR (126 MHz; DMSO-d6): δ 156.4 (C6), 148.8 (C2), 148.5 (6-Pyr), 148.0 (2-Pyr), 146.8 (C4), 139.8 (C8), 136.5 (3-Pyr), 133.8 (4-Pyr), 125.5 (C5), 123.7 (5-Pyr), 89.5 (C4′), 84.1 (C1′), 71.0 (C3′), 70.2 (CH), 62.0 (C5′), 46.1 (NCH2), 36.3 (CHCH2), 26.1 (CHCH2CH2).
HRMS: calc’d: 402.17720; found: 402.17661
In vitro detection of POB-DNA adducts using NNKOAc
Calf thymus DNA (0.99 mg) was dissolved in 1.0 mL of 25 mM phosphate buffer, pH 7.4. To this was added porcine liver esterase (1.33 mg, 24U) and NNKOAc (1 mg, 3.7 μmol in 20 μL of MeOH) and the mixture was placed in a 37 °C water bath with shaking for 24 h. Then, the solution was diluted with 1 mL of H2O and extracted twice with 2 mL of CHCl3/isoamyl alcohol (24:1). The DNA was precipitated by addition of 0.4 mL of 5M NaCl and 2 mL of ice-cold isopropyl alcohol. The DNA was further washed with 1 mL of 70% EtOH, followed by 1 mL of 100% EtOH and dried under a stream of N2. DNA was stored at −20 °C.
Isolated DNA was dissolved in 0.5 mL of 10 mM sodium succinate, 5 mM CaCl2 buffer and incubated at 37 °C with micrococcal nuclease (30 U) and phosphodiesterase II (0.5 U) for 5 h, followed by 37 °C incubation with alkaline phosphatase (150 U) for 16 h. [Pyridine-D4]N6-POB-dA (100 fmol) was added as an internal standard. Samples were placed in 0.45 μm Nylon SpinX centrifugal tube filters and centrifuged at 12,000 g for 10 min. A 20 μL aliquot was removed from each sample and diluted as necessary for dGuo quantiation by HPLC.18,35 The remaining sample was purified by solid-phase extraction using a Strata-X 33 μm polymeric reversed phase cartridge (30 mg/mL; Phenomenex, Torrance, CA). The cartridge was preconditioned with 1 mL of MeOH, followed by 1 mL of H2O. After sample addition, the cartridge was washed with 2 mL of H2O and 1 mL of 10% (v/v) MeOH, and eluted with 1 mL of 100% MeOH. The eluent was evaporated to dryness in a Speedvac. The residue was dissolved in 25 μL of H2O and analyzed by liquid chromatography-positive electrospray ionization-tandem mass spectrometry (LC-ESI+-MS/MS).
A previously reported LC-ESI+-MS/MS method was performed with modifications.18 Briefly, a 0.5 × 150 mm Zorbax SB-C18, 5 μm column (Agilent, Santa Clara, CA) was eluted with a multi-step gradient and flow rate of 10 μL/min. After a 5-min hold at 5% B, the eluent was brought to 65% B over 25 min, followed by a wash at 85% B and re-equilibration, where solvent A was 15 mM NH4OAc and solvent B was methanol. MS was performed on a TSQ Vantage triple quadrupole mass analyzer (Thermo Scientific). The selected reaction monitoring (SRM) transitions were m/z 400.1 → m/z 284.1 for N1-POB-dIno, m/z 399.1 → m/z 265.1 for N6-POB-dAdo, and m/z 403.1 → m/z 269.1 for [pyridine-D4]N6-POB-dAdo using collision energies of 10, 20, and 20 eV, respectively, and a 0.4 amu scan width for all transitions.
In vivo detection and quantitation of POB- and PHB-DNA adducts in rat liver and lung tissues
The lung and liver tissues were obtained from a previous study in which male F-344 rats were treated with 5 ppm of NNK, (S)-NNAL, or (R)-NNAL in drinking water for 50 weeks.18,35 Control rats were given unmodified tap water. DNA was isolated from these tissues (n = 3) using a modified Qiagen protocol that has been applied in previous studies.17,35,36 Isolated DNA was stored at −20 °C until further analysis.
Isolated DNA samples were hydrolyzed and spiked with [pyridine-D4]N6-POB-dAdo and [15N5]N6-PHB-dAdo as described above for our in vitro experiment, then analyzed for adducts as described previously.17,18,36,37 LC-ESI+-MS/MS conditions were the same as described above for our in vitro experiment except SRM transitions were added for N6-PHB-dAdo (m/z 401.1 → m/z 132.1), N1-PHB-dIno (m/z 402.1 → m/z 286.1), and [15N5]N6-PHB-dAdo (m/z 406.1 → m/z 132.1) at collision energies of 26, 10, 26 eV, respectively. Corresponding fragmentation patterns are shown in Figure S2.
Samples were further analyzed by an optimized liquid chromatography-positive nanoelectrospray ionization-high resolution tandem mass spectrometry (LC-NSI+-HRMS/MS) method. A New Objective (Woburn, MA) emitter (75 μm ID, 10 μm orifice) was hand-packed with Luna (Phenomenex) C18, 5 μm stationary phase to create a 200 mm capillary column and a multistep gradient was used for chromatographic separation. With 5 mM NH4OAc and MeCN as solvents A and B, respectively, the sample was loaded onto the column by running 2% B at 1 μL/min for 5 min. Then the flow rate was decreased to 0.3 μL/min over 1 min, the injection valve was removed from the flow path, and a linear gradient was started that ramped from 2% B to 40% B over 20 min. This was followed by an increase to 98% B over 1 min, in which the column was washed for 2 min at 1 μL/min before a 5 min re-equilibration at 2% B. All dAdo and dIno adducts were monitored by MS2 fragmentation on an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific, Waltham, MA). The spray voltage was 2.2 kV, the capillary temperature was 300 °C, and the S-Lens RF level was 60%. Each parent ion was isolated (1.5 amu isolation width) and fragmented by higher-energy collision dissociation (HCD) at a normalized energy of 22% with a resolution of 60,000 and mass range of m/z 100 – 425. Accurate product ion masses from characteristic transitions for N1-POB-dIno (m/z 400 → 284.1140, 148.0757), N1-PHB-dIno (m/z 402 → 286.1296, 268.1191), N6-POB-dAdo (m/z 399 → 283.1300, 265.1194), N6-PHB-dAdo (m/z 401 → 287.1550, 269.1445), [D4]N6-POB-dAdo (m/z 403 → 285.1456, 267.1350), and [15N5]N6-PHB-dAdo (m/z 406 → 290.1307, 272.1202) were extracted at a mass tolerance of 5 ppm (Figure S2).
Quantitation of other DNA base adducts (12, 14, and 15) for comparison was performed as previously described using similar DNA isolation and LC-ESI+-MS/MS or LC-NSI+-HRMS/MS techniques.18,19,38
Results
Synthesis of DNA adduct standards
N6-PHB-dAdo was previously synthesized by a 2-step procedure34 (Scheme 3A) which was applied to produce a 15N5-labelled standard starting from [15N5]2′-deoxyadenosine.
Scheme 3.

Synthetic route for N6-PHB-dAdo and N6-POB-dAdo. dAdo = 2′-deoxyadenosine, TFA = trifluoroacetic acid, NCS = N-chlorosuccinimide
Initial attempts to oxidize N6-PHB-dAdo to N6-POB-dAdo were unsuccessful. Therefore, we devised a new route that utilized previously prepared compound 25 as starting material15 (Scheme 3B). After Boc deprotection by TFA, primary amine 26 was coupled to 6-chloropurine-2′-deoxyribose. The synthesis was completed by a final oxidative dithiane deprotection39 and HPLC purification resulting in a 17% overall yield. A [pyridine-D4]-labeled standard was synthesized analogously using [pyridine-D4]25, which was prepared in 2 steps15 from commercially available [pyridine-D4]3-pyridylcarboxaldehyde.
The synthesis of N1-POB-dIno (22, Scheme 4A) began with protection of both hydroxyl groups of 2′-deoxyinosine with tert-butyldimethylsilyl chloride and N1-arylation with 1-chloro-2,4-dinitrobenzene to produce 30 as a 1:1 mixture of atropisomers in excellent yield. Compound 30 was heated with 3-amino-1-propanol to give 31 in good to excellent yield through a transamidation-like mechanism.40 This was converted into iodide 3241 and further coupled to dithiane 34 under Seebach’s conditions42 with good yields for both steps. Lastly, the keto and hydroxyl groups were deprotected by N-chlorosuccinimide oxidation and a pH-neutral, catalytic TBAF procedure,43 respectively, to produce N1-POB-dIno (22) in modest yield over these two steps.
Scheme 4.

Synthetic Scheme for (A) N1-POB-dIno and (B) N1-PHB-dI. TBSCl = tert-butyldimethylsilyl chloride, Imid = imidazole, Pyr = pyridine, DMF = N,N-dimethylformamide, DCM = dichloromethane, TMEDA = N,N,N,N-tetramethylethylenediamine, TBAF = tetra-n-butylammonium fluoride.
N1-PHB-dIno (23, Scheme 4B) was obtained by reducing the ketone after deprotection of 34 and removing the TBS groups with an unbuffered TBAF solution with moderate yield. All final DNA adduct structures were confirmed by one- and two-dimensional NMR experiments (1H, 13C, HSQC, HMBC) and high-resolution mass spectrometry.
In vitro detection of 2′-deoxyadenosine-derived POB adducts
Mixtures of calf thymus DNA, NNKOAc, and porcine liver esterase were incubated at 37 °C and pH 7.4 for 24 h. After DNA hydrolysis and enrichment, samples were analyzed by LC-ESI+-MS/MS using [pyridine-D4]N6-POB-dAdo as an internal standard. As shown in Figure 2, peaks corresponding to both N6-POB-dAdo and N1-POB-dIno were observed. Based on relative quantitation, N1-POB-dIno was 25 times more abundant than N6-POB-dAdo. These peaks were not detected in control incubations that either contained esterase alone or lacked NNKOAc. This indicates that both deamination and Dimroth rearrangement occur under these conditions and implicates the N1-position as the initial site of alkylation.
Figure 2.

Representative LC-ESI+-MS/MS chromatograms for in vitro formation of N6-POB-dA and N1-POB-dI. Samples containing 25 mM K2PO3 buffer with esterase, esterase + DNA, or esterase + DNA + NNKOAc were incubated at 37 °C for 24 h. Samples were analyzed after DNA hydrolysis and purification by solid-phase extraction (SPE). In each case, the top two channels are monitoring separate transitions for the N1-dIno adducts (POB: m/z 400 → m/z 284, 148; PHB: m/z 402 → m/z 286, 132). The bottom two channels are monitoring the N6-dAdo adducts (POB: m/z 399 → m/z 265; PHB: m/z 401 → m/z 132) and its isotopically-labeled internal standard (POB: m/z 403 → m/z 269; PHB: m/z 406 → m/z 132).
In vivo detection and quantification of 2′-dAdo adducts
DNA was extracted from the livers and lungs of rats chronically treated with 5 ppm NNK, (S)-NNAL, or (R)-NNAL in the drinking water for 50 weeks.18 Hydrolysis and analysis by LC-ESI+-MS/MS of these samples were performed as in our in vitro experiment. Representative chromatograms for lung samples are shown in Figure 3. N6-POB-dAdo and N6-PHB-dAdo were detected, while both N1-POB-dIno and N1-PHB-dIno remained unobserved. Though a characteristic peak at 20.9 min, possibly corresponding to N1-POB-dIno, was found in the m/z 400 → m/z 148 SRM channel, the corresponding peak in the m/z 400 → m/z 284 SRM channel was not observed. The 20.9 min peak was detected in all control samples indicating this signal is ubiquitous to the sample matrix. This same trend was also seen for N1-PHB-dIno.
Figure 3.

Representative chromatograms obtained from the LC-ESI+-MS/MS analyses of (A) POB- and (B) PHB-DNA adducts in the lungs of rats chronically treated with 5 ppm NNK for 50 weeks. In each case, the top two channels are monitoring separate transitions for the N1-dIno adducts (POB: m/z 400 → m/z 284, 148; PHB: m/z 402 → m/z 286, 132). The bottom two channels are monitoring the N6-dAdo adducts (POB: m/z 399 → m/z 265; PHB: m/z 401 → m/z 132) and its isotopically-labeled internal standard (POB: m/z 403 → m/z 269; PHB: m/z 406 → m/z 132).
In the liver, levels of N6-POB-dAdo were higher in the NNK and (S)-NNAL treatment groups (27–32 fmol/mg DNA or 9–10.7 adducts/109 nts) as compared to the (R)-NNAL group (4 fmol/mg DNA or 1.3 adduct/109 nts). The trend was reversed for N6-PHB-dAdo in that levels were lower in the NNK and (S)-NNAL treatment groups (4–6 fmol/mg DNA or 1–2 adducts/109 nts) than in the (R)-NNAL group (21–25 fmol/mg DNA or 7–8.3 adducts/109 nts) (Figure 4). Analysis of rat lung DNA revealed the same trend, however, at higher overall levels. N6-POB-dAdo was relatively abundant in the NNK and (S)-NNAL groups at 99 fmol/mg DNA (33 adducts/109 nts) and 130 fmol/mg DNA (43.3 adducts/109 nts), respectively, while N6-PHB-dAdo was relatively high in the (R)-NNAL group at 125 fmol/mg DNA (41.7 adducts/109 nts). Similar to the liver, N6-POB-dAdo was a minor product after (R)-NNAL treatment (16 fmol/mg DNA or 5.3 adducts/109 nts) and N6-PHB-dAdo was less abundant in both NNK and (S)-NNAL treatment groups (35 fmol/mg DNA or 11.7 adducts/109 nts). Control rats given unmodified tap water showed no adduct formation. To evaluate if the lack of dIno adducts was due to insufficient sensitivity, these samples were reanalyzed by a LC-NSI+-HRMS/MS method utilizing accurate mass detection. After extensive method optimization, the dIno adducts were still undetectable, while dAdo adducts were easily observed (Figure S1).
Figure 4.
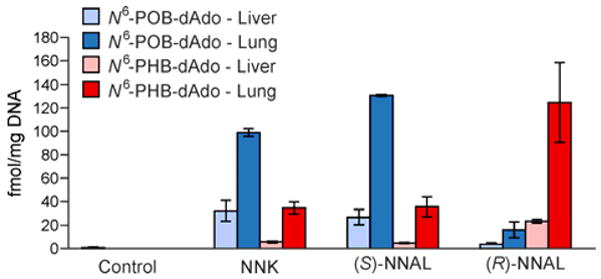
Levels of N6-POB-dA and N6-PHB-dA in the liver and lung DNA of rats treated with NNK-, (S)-NNAL, and (R)-NNAL at a dose of 5 ppm in their drinking water for 50 weeks. Values are the average of three replicates and error bars denote their standard deviation. POB- and PHB-adducts are represented in blue and red, respectively. Liver and lung tissue is differentiated by light and dark hues, respectively.
Discussion
We describe the first characterization of dAdo base adducts formed in the metabolism of NNK by α-hydroxylation. Synthesis of the standards N6-POB-dAdo, N6-PHB-dAdo, N1-POB-dIno, and N1-PHB-dIno as well as stable isotope-labelled analogues allowed us to interrogate their presence and abundance in vitro and in vivo. Detection of both N6-dAdo and N1-dIno type adducts in vitro, from reactions of NNKOAc with calf thymus DNA, supports our hypothesized mechanism of formation involving either a spontaneous Dimroth rearrangement or deamination, respectively. Likewise, quantitation of the N6-dAdo adducts in liver and lung DNA of NNK and NNAL-treated rats supports the occurrence of this mechanism in vivo.
Levels of the dAdo adducts in liver and lung of rats treated chronically with NNK or enantiomers of NNAL in their drinking water for 50 weeks are compared to those of some other characterized NNK-DNA adducts19 in Table 1. dAdo adduct levels were relatively low in both tissues, exceeding only those of O6-POB-dGuo when measurable. Levels were otherwise considerably lower than the 7-Gua and O2-Thd POB- and PHB-DNA adducts. Levels of the dAdo adducts were higher in lung than in liver of the NNK treated rats, consistent with the comparative tissue levels of the other DNA adducts.18 This reflects extensive metabolic activation of NNK and NNAL in rat lung upon treatment with relatively low doses of these carcinogens. Consistent with previous studies, levels of N6-POB-dAdo were higher in rats treated with NNK or (S)-NNAL than those found in rats treated with (R)-NNAL. In comparison, N6-PHB-dAdo amounts were highest in lung and liver DNA of (R)-NNAL-treated rats. These consistent findings presumably result from the facile conversion of (S)-NNAL to NNK in vivo and slow conversion of (R)-NNAL to NNK as compared to direct metabolic activation by P450s.12
Table 1.
Levels of DNA adducts in lung and liver DNA from rats chronically treated with NNK, (S)-NNAL, or (R)-NNAL (5 ppm in their drinking water) for 50 weeks. Values are the average of three replicates and their standard deviations.
| Tissue | Treatment | N6-POB-dAdo (fmol/mg DNA) | O6-POB-dGuo (fmol/mg DNA) | 7-POB-Gua (fmol/mg DNA) | O2-POB-Thd (fmol/mg DNA) | O2-POB-Cyt (fmol/mg DNA) |
|---|---|---|---|---|---|---|
| Liver | NNK | 32 ± 9 | < LOQa | 257 ± 32a | 1531 ± 161a | < LOQ |
| (S)-NNAL | 27 ± 6 | < LOQ | 356 ± 18 | 1680 ± 100 | < LOQ | |
| (R)-NNAL | 4 ± 1 | < LOQ | 28 ± 5 | 130 ± 27 | < LOQ | |
| Lungb | NNK | 99 ± 3 | 9 ± 2 | 688 ± 65 | 4409 ± 320 | < LOQ |
| (S)-NNAL | 130 ± 1 | 10 ± 5 | 568 ± 68 | 3951 ± 328 | < LOQ | |
| (R)-NNAL | 16 ± 7 | < LOQ | 136 ± 10 | 584 ± 45 | < LOQ | |
|
| ||||||
| N6-PHB-dAdo (fmol/mg DNA) | O6-PHB-dGuo (fmol/mg DNA) | 7-PHB-Gua (fmol/mg DNA) | O2-PHB-Thd (fmol/mg DNA) | |||
|
|
||||||
| Liver | NNK | 6 ± 1 | < LOQ | 295 ± 42 | 658 ± 107 | |
| (S)-NNAL | 5 ± 1 | < LOQ | 133 ± 15 | 422 ± 36 | ||
| (R)-NNAL | 24 ± 2 | < LOQ | 436 ± 23 | 2995 ± 121 | ||
| Lungb | NNK | 35 ± 5 | < LOQ | 129 ± 34 | 1259 ± 66 | |
| (S)-NNAL | 36 ± 8 | < LOQ | 100 ± 25 | 1152 ± 41 | ||
| (R)-NNAL | 125 ± 34 | < LOQ | 379 ± 19 | 6291 ± 885 | ||
In contrast to the results of our in vitro studies, in which NNKOAc was reacted with calf thymus DNA, we found no evidence for the presence of either N1-dIno adduct in lung and liver DNA of rats treated with NNK or enantiomers of NNAL. This was in spite of the use of a highly sensitive and specific LC-NSI+-HRMS/MS method. The accurate mass detection feature of this method, when coupled with nano-spray ionization, guarantees both high specificity and sensitivity in detection of analytes. Even after rigorous application of this method, the dIno adducts remained undetected, perhaps due to their efficient repair in vivo. Since other N1-dIno adducts form Hoogstein base pairs and flip outside the DNA helix, it is possible that our POB- and PHB-N1-dIno adducts are prime targets for base excision repair or other DNA repair mechanisms.31,44,45
In our preliminary studies of the reaction of NNKOAc with calf thymus DNA, we observed an HPLC peak which decreased relatively rapidly with time, as peaks corresponding to N6-POB-dAdo and N1-POB-dIno increased (data not shown). We presume that this peak was N1-POB-dA. Ultimately, the concentration of N1-POB-dIno was about 25 times greater than that of N6-POB-dAdo in these in vitro reactions. This preference is consistent with previous studies where deamination dominated under neutral pH and when the N1-alkyl group possessed a nucleophilic oxygen group.23–26 In the case of styrene oxide, the latent alcohol was shown to facilitate the addition-elimination mechanism of deamination,24 however, it is unclear how these results translate to the POB- and PHB-adducts. While Dimroth rearrangement is the most plausible explanation for the presence of N6-POB-dAdo and N6-PHB-dAdo, direct N6-alkylation cannot be excluded as there is some evidence for this in reactions of acrolein46 and 1,2,3,4-diepoxybutane47 with DNA.
Synthesis of the adduct standards proceeded as expected with the exception of two major findings. First, the transformation of 30 (Scheme 4) to N1-alkyl structures showed a dependence on steric features. It was our original plan to directly produce 34 by using 26 (Scheme 3 & 4) as the nucleophile, but this reaction gave little to no yield. We believe that steric repulsion between the dithiane and TBS-groups blocked the amino group from engaging the C2-position of the nucleoside. Therefore, we simplified the attacking group to 3-amino-1-propanol and obtained good yields of 31, which was further elaborated to the final product. Next, we found TBS-deprotection of 34 to be very sensitive to pH. After dithiane removal, unbuffered TBAF removed not only the TBS groups, but also the entire POB moiety, perhaps due to the basicity of fluoride. We surmise that N1-POB cleavage occurred via an enolate-ring closure that released dIno and presumably a cyclopropyl byproduct.48 When recently developed conditions using catalytic fluoride and neutral pH buffer were applied,43 selective TBS cleavage was observed. In agreement with the enolate-ring closure hypothesis, unbuffered TBAF could be used to deprotect compound 35 with no cleavage of the PHB group.
Though adduct levels were relatively low in vivo, they may play a biological role. As discussed earlier, O6-POB-dGuo is found at very low or undetectable levels in vivo (Table 1); however, this adduct is thought to contribute to tumorigenesis as it is strongly mutagenic in a variety of cell models.44 Though efficient repair tempers this mutagenicity in vivo, it may increase the persistence of other adducts such as O6-Me-dGuo, since previous work identified GC to AT transitions as a major NNK-induced mutation.49 The same study also found moderate levels of AT to GC mutations, perhaps due to the persistence of N6-dAdo adducts. This is reasonable as the bulky N6-linkage could block base pairing with Thd. In studies with 1,3-butadiene, particular N6-dAdo adducts were bypassed by several DNA polymerases and in some cases caused mutations in vitro.50 However, in a follow-up study,51 the same adducts were found to be far less mutagenic, possibly due to base-excision repair. In studies with polycyclic aromatic hydrocarbons (PAHs), N6-dAdo adducts were highly mutagenic, and sometimes tumorigenic, depending on the PAH structure and location on the DNA strand.52,53 When adducts significantly distorted the DNA helix, cells were found to halt during DNA replication, possibly alluding to DNA polymerases stalling near the adduct site.52 Together, these findings seem to indicate that effective N6-dAdo adducts require steric balance; adducts should be large enough to disrupt base pairing, but small enough to evade detection by cellular machinery. It is currently unknown how POB- and PHB- adducts fit into this context and thus requires future study. Along with possible biological effects, N6-POB-dAdo and N6-PHB-dAdo have potential as biomarkers. These products directly link to the DNA damaging properties of NNK and NNAL and could be used to study differences between demographic groups.32 With analytical power continuing to increase, measurement of low levels of DNA adducts in humans is starting to become an achievable feat.54
In summary, this work has identified and quantified the first dAdo-derived DNA adducts of NNK and its enantiomers NNAL formed in vitro and in vivo. We found general support for our proposed adduct formation mechanism by quantifying both N1-POB-dIno and N6-POB-dAdo in vitro. Though N1-dIno type adducts were the major ones formed in vitro, they were undetectable in vivo. N6-dAdo adduct levels were relatively modest in vivo when compared to most DNA adducts of NNK and NNAL previously detected. This research allows and encourages future studies on the biological properties of these adducts and their potential utility as specific DNA damage biomarkers.
Supplementary Material
Acknowledgments
We thank Bob Carlson for editorial assistance, Xun Ming and Makenzie Pillsbury for mass spectrometry assistance in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, and Dr. Adam T. Zarth for valuable discussions and NMR assistance.
Funding Information:
This study was supported by grant no. CA-81301 from the U.S. National Cancer Institute. Salary support for P.W.V. was provided by the U.S. National Cancer Institute [Grant R50-CA211256]. The Analytical Biochemistry Shared Resource is partially supported by National Cancer Institute Cancer Center Support Grant CA-77598.
Abbreviation List
- 7-PHB-Gua
N7-(4-hydroxy-4-(3-pyridyl)-1-butyl)-guanine
- 7-POB-Gua
N7-(4-oxo-4-(3-pyridyl)-1-butyl)-guanine
- Boc
tert-butylcarbamate
- B1p(POB)B2
B1p(4-oxo-4-(3-pyridyl)-1-butyl)B2
- B1p(PHB)sB2
B1p(4-oxo-4-(3-pyridyl)-1-butyl)B2
- B1p(PHB)bB2
B1p(4-oxo-4-(3-pyridyl)-2-butyl)B2
- DIPEA
N,N′-diisopropylethylamine
- DMF
N,N′-dimethylformamide
- DMSO
dimethylsulfoxide
- HCD
Higher-energy collisional dissociation
- Imid
imidazole
- LC-ESI+-MS/MS
liquid chromatography-positive electrospray ionization-tandem mass spectrometry
- LC-NSI+-HRMS/MS
liquid chromatography-positive nanoelectrospray ionization-high resolution tandem mass spectrometry
- [15N5]N6-PHB-dAdo
[15N5]N6-(4-hydroxy-4-(3-pyridyl)-1-butyl)-2′-deoxyadenosine
- N6-PHB-dAdo
N6-(4-hydroxy-4-(3-pyridyl)-1-butyl)-2′-deoxyadenosine
- N6-POB-dAdo
N6-(4-oxo-4-(3-pyridyl)-1-butyl)-2′-deoxyadenosine
- N1-PHB-dIno
N1-(4-hydroxy-4-(3-pyridyl)-1-butyl)-2′-deoxyinosine
- N1-POB-dIno
N1-(4-oxo-4-(3-pyridyl)-1-butyl)-2′-deoxyinosine
- NCS
N′-chlorosuccinimide
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNAL
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol
- NNKOAc
4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone
- O6-Me-dGuo
O6-methyl-2′-deoxyguanosine
- O2-PHB-dThd
O2-(4-hydroxy-4-(3-pyridyl)-1-butyl)-thymidine
- O2-POB-dThd
O2-(4-oxo-4-(3-pyridyl)-1-butyl)-thymidine
- O2-POB-Cyt
O2-(4-oxo-4-(3-pyridyl)-1-butyl)-cytosine
- O6-PHB-dG
O6-(4-hydroxy-4-(3-pyridyl)-1-butyl)-2′-deoxyguanosine
- O6-POB-dG
O6-(4-oxo-4-(3-pyridyl)-1-butyl)-2′-deoxyguanosine
- POB
pyridyloxobutyl
- PHB
pyridylhydroxybutyl
- [Pyridine-D4]N6-POB-dAdo
[Pyridine-D4]N6-(4-oxo-4-(3-pyridyl)-1-butyl)-2′-deoxyadenosine
- SRM
selected reaction monitoring
- TBAF
tetra-n-butylammonium fluoride
- TBS-Cl
tert-butyldimethylsilyl chloride
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TMEDA
tetramethylethylenediamine
Footnotes
LC-NSI+-HRMS/MS chromatograms from the analysis of POB- and PHB-DNA adducts in the lungs of rats chronically treated with NNK, (S)-NNAL, and (R)-NNAL, and fragmentation patterns for all N6-dAdo and N1-dIno adducts. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.World Health Organization, T. WHO report on the global tobacco epidemic, 2011: warning about the dangers of tobacco. Geneva: 2011. [Google Scholar]
- 2.Jamal A, King BA, Neff LJ, Whitmill J, Babb SD, Graffunder CM, Jamal A. Current cigarette smoking among adults — United States, 2005–2015. Morb Mortal Wkly Rep. 2016;65:1205–1211. doi: 10.15585/mmwr.mm6544a2. [DOI] [PubMed] [Google Scholar]
- 3.United States Department of Health and Human Services, Center for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health. The Health Consequences of Smoking—50 Years of Progress A Report of the Surgeon General. Atlanta: 2014. [Google Scholar]
- 4.American Cancer Society. Cancer Facts Fig. Atlanta: 2014. Cancer Facts & Figures 2014. [Google Scholar]
- 5.Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 6.Rom WN, Markowitz SB, editors. Environmental and Occupational Medicine. 4. Lippincott Williams & Wilkins; Philadelphia, PA: 2007. N-Nitrosamines; pp. 1226–1239. [Google Scholar]
- 7.International Agency for Research on Cancer. Smokeless Tob Some Tobacco-specific N-Nitrosamines. Lyon: 2007. Monographs on the Evaluation of Carcinogenic Risks to Humans - Volume 89. [Google Scholar]
- 8.Yuan JM, Gao YT, Murphy SE, Carmella SG, Wang R, Zhong Y, Moy KA, Davis AB, Tao L, Chen M, Han S, Nelson HH, Yu MC, Hecht SS. Urinary levels of cigarette smoke constituent metabolites are prospectively associated with lung cancer development in smokers. Cancer Res. 2011;71:6749–6757. doi: 10.1158/0008-5472.CAN-11-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan JM, Koh WP, Murphy JSE, Fan Y, Wang R, Carmella SG, Han S, Wickham K, Gao YT, Yu MC, Hecht SS. Urinary levels of tobacco-specific nitrosamine metabolites in relation to lung cancer development in two prospective cohorts of cigarette smokers. Cancer Res. 2009;69:2990–2995. doi: 10.1158/0008-5472.CAN-08-4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan JM, Nelson HH, Carmella SG, Wang R, Kuriger-Laber J, Jin A, Adams-Haduch J, Hecht SS, Koh WP, Murphy SE. CYP2A6 genetic polymorphisms and biomarkers of tobacco smoke constituents in relation to risk of lung cancer in the Singapore Chinese Health Study. Carcinogenesis. 2017;38:411–418. doi: 10.1093/carcin/bgx012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finckh C, Atalla A, Nagel G, Stinner B, Maser E. Expression and NNK reducing activities of carbonyl reductase and 11beta-hydroxysteroid dehydrogenase type 1 in human lung. Chem Biol Interact. 2001;130–132:761–773. doi: 10.1016/s0009-2797(00)00306-9. [DOI] [PubMed] [Google Scholar]
- 12.Zhang S, Wang M, Villalta PW, Lindgren BR, Upadhyaya P, Lao Y, Hecht SS. Analysis of pyridyloxobutyl and pyridylhydroxybutyl DNA adducts in extrahepatic tissues of F344 rats treated chronically with 4-(methylnitrosamino) -1-(3-pyridyl)-1-butanone and enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl) -1-butanol. Chem Res Toxicol. 2009;22:926–936. doi: 10.1021/tx900015d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy SE, Spina DA, Nunes MG, Pullo DA. Glucuronidation of 4-((hydroxymethyl)nitrosamino)-1-(3-pyridyl)-1-butanone, a metabolically activated form of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, by phenobarbital-treated rats. Chem Res Toxicol. 1995;8:772–9. doi: 10.1021/tx00047a018. [DOI] [PubMed] [Google Scholar]
- 14.Jalas JR, Murphy SE, Hecht SS. Cytochrome P450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a tobacco-specific carcinogen. Chem Res Toxicol. 2005;18:95–110. doi: 10.1021/tx049847p. [DOI] [PubMed] [Google Scholar]
- 15.Carlson ES, Upadhyaya P, Hecht SS. Evaluation of nitrosamide formation in the cytochrome P450-mediated metabolism of tobacco-specific nitrosamines. Chem Res Toxicol. 2016;29:2194–2205. doi: 10.1021/acs.chemrestox.6b00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lao Y, Yu N, Kassie F, Villalta PW, Hecht SS. Analysis of pyridyloxobutyl DNA adducts in F344 rats chronically treated with (R)-and (S)-N′-nitrosonornicotine. Chem Res Toxicol. 2007;20:246–256. doi: 10.1021/tx060208j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Upadhyaya P, Kalscheuer S, Hochalter JB, Villalta PW, Hecht SS. Quantitation of pyridylhydroxybutyl-DNA adducts in liver and lung of F-344 rats treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem Res Toxicol. 2008;21:1468–1476. doi: 10.1021/tx8001109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balbo S, Johnson CS, Kovi RC, James-Yi SA, O’Sullivan MG, Wang M, Le CT, Khariwala SS, Upadhyaya P, Hecht SS. Carcinogenicity and DNA adduct formation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F-344 rats. Carcinogenesis. 2014;35:2798–2806. doi: 10.1093/carcin/bgu204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma B, Villalta PW, Zarth AT, Kotandeniya D, Upadhyaya P, Stepanov I, Hecht SS. Comprehensive high-resolution mass spectrometric analysis of DNA phosphate adducts formed by the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem Res Toxicol. 2015;28:2151–2159. doi: 10.1021/acs.chemrestox.5b00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma B, Zarth AT, Carlson ES, Villalta PW, Upadhyaya P, Stepanov I, Hecht SS. Identification of more than one hundred structurally unique DNA-phosphate adducts formed during rat lung carcinogenesis by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 2018;39:232–241. doi: 10.1093/carcin/bgx135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engel JD. Mechanism of the dimroth rearrangement in adenosine. Biochem Biophys Res Commun. 1975;64:581–586. doi: 10.1016/0006-291x(75)90361-7. [DOI] [PubMed] [Google Scholar]
- 22.Fujii T, Saito T. Purines. XXVI The dimroth rearrangement of 9-substituted 1-methyladenines: accelerating effect of a b-D-ribofuranosyl group at the 9-position. Chem Pharm Bull. 1985;33:3635–3644. [PubMed] [Google Scholar]
- 23.Fujii T, Saito T, Terahara N. Purines XXVII. Hydrolytic deamination versus dimroth rearrangement in the 9-substituted adenine ring: effect of an omega-hydroxyalkyl group at the 1-position. Chem Pharm Bull. 1986;34:1094–1107. [Google Scholar]
- 24.Barlow T, Ding J, Vouros P, Dipple A. Investigation of hydrolytic deamination of 1-(2-hydroxy-1-phenylethyl)adenosine. Chem Res Toxicol. 1997;10:1247–1249. doi: 10.1021/tx970091m. [DOI] [PubMed] [Google Scholar]
- 25.Fujii T, Saito T, Hisata H, Shinbo K. Purines. XLVII Dimroth rearrangement versus hydrolytic deamination of 1-ethyladenine. Chem Pharm Bull. 1990;38:3326–3330. [Google Scholar]
- 26.Saito T, Murakami M, Inada T, Hayashibara H, Fujii T. Purines. LIII Deamination of 1-(omega-hydroxyalkyl)adenine derivatives by nucleophiles. Chem Pharm Bull(Tokyo) 1992;40:3201–3205. [Google Scholar]
- 27.Selzer RR, Elfarra AA. Characterization of N1- and N6-adenosine adducts and N1-inosine adducts formed by the reaction of butadiene monoxide with adenosine: evidence for the N1-adenosine adducts as major initial products. Chem Res Toxicol. 1996;9:875–881. doi: 10.1021/tx960039a. [DOI] [PubMed] [Google Scholar]
- 28.Barlow T, Takeshita J, Dipple A. Deamination and dimroth rearrangement of deoxyadenosine-styrene oxide adducts in DNA. Chem Res Toxicol. 1998;11:838–845. doi: 10.1021/tx980038d. [DOI] [PubMed] [Google Scholar]
- 29.Fujii T, Saito T, Nakasaka T. Purines. XXXIV 3-Methyladenosine and 3-methyl-2′-deoxyadenosine: their synthesis, glycosidic hydrolysis, and ring fission. Chem Pharm Bull. 1989;37:2601–2609. [Google Scholar]
- 30.Fujii T, Saito T, Iguchi K. Purines. LXI An attempted synthesis of 2′-deoxy-7-methyladenosine: glycosidic hydrolyses of the N6-methoxy derivative and 2′-deoxy-Nx-methyladenosines. Chem Pharm Bull. 1994;42:495–499. [Google Scholar]
- 31.Scholdberg TA, Merritt WK, Dean SM, Kowalcyzk A, Harris CM, Harris TM, Rizzo CJ, Lloyd RS, Stone MP. Structure of an oligodeoxynucleotide containing a butadiene oxide-derived N1 beta-hydroxyalkyl deoxyinosine adduct in the human N-ras codon 61 sequence. Biochemistry. 2005;44:3327–3337. doi: 10.1021/bi0482452. [DOI] [PubMed] [Google Scholar]
- 32.Hecht SS. Oral cell DNA adducts as potential biomarkers for lung cancer susceptibility in cigarette smokers. Chem Res Toxicol. 2017;30:367–375. doi: 10.1021/acs.chemrestox.6b00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hecht SS, Spratt TE, Trushin N. Absolute configuration of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol formed metabolically from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 1997;18:1851–1854. doi: 10.1093/carcin/18.9.1851. [DOI] [PubMed] [Google Scholar]
- 34.Upadhyaya P, Hecht SS. Identification of adducts formed in the reactions of 5′-acetoxy- N′-nitrosonornicotine with deoxyadenosine, thymidine, and DNA. Chem Res Toxicol. 2008;21:2164–2171. doi: 10.1021/tx8002559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lao Y, Villalta PW, Sturla SJ, Wang M, Hecht SS. Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high-performance liquid chromatography- electrospray ionization-tandem mass spectrometry. Chem Res Toxicol. 2006;19:674–682. doi: 10.1021/tx050351x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lao Y, Yu N, Kassie F, Villalta PW, Hecht SS. Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino )-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem Res Toxicol. 2007;20:235–245. doi: 10.1021/tx060207r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Upadhyaya P, Lindgren BR, Hecht S. Comparative levels of O6-methylguanine, pyridyloxobutyl-, and pyridylhydroxybutyl-DNA adducts in lung and liver of rats treated chronically with the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Drug Metab Dispos. 2009;37:1147–1151. doi: 10.1124/dmd.109.027078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma B, Zarth AT, Carlson ES, Villalta PW, Stepanov I, Hecht SS. Pyridylhydroxybutyl and pyridyloxobutyl DNA phosphate adduct formation in rats treated chronically with enantiomers of the tobacco-specific nitrosamine metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Mutagenesis. 2017;32:561–570. doi: 10.1093/mutage/gex031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corey EJ, Erickson BW. Oxidative hydrolysis of 1, 3-dithiane derivatives to carbonyl compounds using N-halosuccinimide reagents. J Org Chem. 1971;36:3553–3560. [Google Scholar]
- 40.Napoli L, De Messere A, Montesarchio D, Piccialli G, Varra M. 1-Substituted 2′-deoxyinosine analogues. J Chem Soc Perkin Trans 1. 1997:2079–2082. [Google Scholar]
- 41.Appel R. Tertiary phosphane/tetrachloromethane, a versatile reagent for chlorination, dehydration, and P-N linkage. Angew Chemie Int Ed English. 1975;14:801–811. [Google Scholar]
- 42.Corey EJ, Seebach D. Carbanions of 1,3-dithianes. Reagents for C-C bond formation by nucleophilic displacement and carbonyl addition. Angew Chemie Int Ed English. 1965;4:1075–1077. [Google Scholar]
- 43.Dilauro AM, Seo W, Phillips ST. Use of catalytic fluoride under neutral conditions for cleaving silicon-oxygen bonds. J Org Chem. 2011;76:7352–7358. doi: 10.1021/jo200848j. [DOI] [PubMed] [Google Scholar]
- 44.Peterson LA. Context matters: contribution of specific DNA adducts to the genotoxic properties of the tobacco-specific nitrosamine NNK. Chem Res Toxicol. 2017;30:420–433. doi: 10.1021/acs.chemrestox.6b00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wickramaratne S, Banda DM, Ji S, Manlove AH, Malayappan B, Nuñez NN, Samson L, Campbell C, David SS, Tretyakova N. Base excision repair of N6-deoxyadenosine adducts of 1,3-butadiene. Biochemistry. 2016;55:6070–6081. doi: 10.1021/acs.biochem.6b00553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pawłowicz AJ, Munter T, Zhao Y, Kronberg L. Formation of acrolein adducts with 2′-deoxyadenosine in calf thymus DNA. Chem Res Toxicol. 2006;19:571–576. doi: 10.1021/tx0503496. [DOI] [PubMed] [Google Scholar]
- 47.Seneviratne U, Antsypovich S, Goggin M, Dorr DQ, Guza R, Moser A, Thompson C, York DM, Tretyakova N. Exocyclic deoxyadenosine adducts of 1,2,3,4-diepoxybutane: synthesis, structural elucidation, and mechanistic studies. Chem Res Toxicol. 2010;23:118–133. doi: 10.1021/tx900312e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spratt TE, Peterson LA, Confer WL, Hecht SS. Solvolysis of model compounds for α-hydroxylation of N′-nitrosonornlcotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: evidence for a cyclic oxonium ion intermediate in the alkylation of nucleophiles. Chem Res Toxicol. 1990;3:350–356. doi: 10.1021/tx00016a013. [DOI] [PubMed] [Google Scholar]
- 49.Belinsky SA, Devereux TR, Anderson MW, Maronpot RR, Stoner GD. Relationship between the formation of promutagenic adducts and the activation of the k-ras protooncogene in lung tumors from A/J mice treated with nitrosamines. Cancer Res. 1989;49:5305–5311. [PubMed] [Google Scholar]
- 50.Kotapati S, Wickramaratne S, Esades A, Boldry EJ, Quirk Dorr D, Pence MG, Guengerich FP, Tretyakova NY. Polymerase bypass of N6-deoxyadenosine adducts derived from epoxide metabolites of 1,3-butadiene. Chem Res Toxicol. 2015;28:1496–1507. doi: 10.1021/acs.chemrestox.5b00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang SC, Seneviratne UI, Wu J, Tretyakova N, Essigmann JM. 1,3-Butadiene-induced adenine DNA adducts are genotoxic but only weakly mutagenic when replicated in escherichia coli of various repair and replication backgrounds. Chem Res Toxicol. 2017;30:1230–1239. doi: 10.1021/acs.chemrestox.7b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dipple A. DNA reactions, mutagenic action and stealth properties of polycyclic aromatic hydrocarbon carcinogens (Review) Int J Oncol. 1999;14:103–111. doi: 10.3892/ijo.14.1.103. [DOI] [PubMed] [Google Scholar]
- 53.Bigger CAH, Pontén I, Page JE, Dipple A. Mutational spectra for polycyclic aromatic hydrocarbons in the supF target gene. Mutat Res - Fundam Mol Mech Mutagen. 2000;450:75–93. doi: 10.1016/s0027-5107(00)00017-8. [DOI] [PubMed] [Google Scholar]
- 54.Villalta PW, Hochalter JB, Hecht SS. Ultrasensitive high-resolution mass spectrometric analysis of a DNA adduct of the carcinogen benzo[a]pyrene in human lung. Anal Chem. 2017;89:12735–12742. doi: 10.1021/acs.analchem.7b02856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.