

Abstract
Disease Overview
Chronic myelomonocytic leukemia (CMML) is a clonal hematopoietic stem cell disorder with overlapping features of myelodysplastic syndromes and myeloproliferative neoplasms, with an inherent risk for leukemic transformation (~15–20% over 3–5 years).
Diagnosis
Diagnosis is based on the presence of sustained (>3 months) peripheral blood monocytosis (≥1 x 109/L; monocytes ≥10%), along with bone marrow dysplasia. Clonal cytogenetic abnormalities occur in ~ 30% of patients, while >90% have gene mutations. Mutations involving TET2 (~60%), SRSF2 (~50%), ASXL1 (~40%) and the oncogenic RAS pathway (~30%) are frequent; while the presence of ASXL1 and DNMT3A mutations and the absence of TET2 mutations negatively impact over-all survival.
Risk stratification
Molecularly integrated prognostic models include; the Groupe Français des Myélodysplasies (GFM), Mayo Molecular Model (MMM) and the CMML specific prognostic model (CPSS-Mol). Risk factors incorporated into the MMM include presence of nonsense or frameshift ASXL1 mutations, absolute monocyte count>10 × 109/L, hemoglobin <10 gm/dl, platelet count <100 × 109/L and the presence of circulating immature myeloid cells. The MMM stratifies CMML patients into 4 groups; high (≥3 risk factors), intermediate-2 (2 risk factors), intermediate-1 (1 risk factor) and low (no risk factors), with median survivals of 16, 31, 59 and 97 months, respectively.
Risk-adapted therapy
Hypomethylating agents such as 5-azacitidine and decitabine are commonly used, with overall response rates of ~30–40% and complete remission rates of ~7–17%; with no impact on mutational allele burdens. Allogeneic stem cell transplant is the only potentially curative option, but is associated with significant morbidity and mortality.
Keywords: myelodysplastic syndrome, myeloproliferative neoplasm, chronic myelomonocytic leukemia, ASXL1, TET2
DISEASE OVERVIEW
The 2016 iteration of the World Health Organization (WHO) classification of myeloid neoplasms defines chronic myelomonocytic leukemia (CMML) as a clonal hematopoietic stem cell disorder characterized by the presence of sustained (>3 months) peripheral blood (PB) monocytosis (≥1 x 109/L; monocytes ≥10% of white blood cell count) along with dysplastic features in the bone marrow (BM).[1] Secondary to the overlapping features of both, myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPN), the classification of CMML as a unique myeloid neoplasm has undergone several changes dating back to the original French-American-British (FAB) co-operative group effort in 1982.[2] Due to renewed evidence demonstrating clinical, morphological and molecular differences, the 2016 WHO classification has once again recommended categorization of CMML into “proliferative” (MPN-CMML) and “dysplastic” (MDS-CMML) sub-types; based on a white blood cell count of ≥13 x 109/L for MPN-CMML.[1, 3] In addition, based on PB and BM blast %, CMML can be sub-classified into three categories; a) CMML-0 (<2% PB blasts including promonocytes and <5% BM blasts), b) CMML-1 (2–4% PB blasts including promonocytes and 5–9 % BM blasts), and c) CMML-2 (>5% PB blasts including promonocytes and 10–19% BM blasts and/or when any Auer rods are present).[1]
The median age at CMML diagnosis is ~71–74 years, with a male preponderance (1.5–3:1).[4–6] The exact incidence of CMML remains unknown, but is estimated at 4 cases per 100,000 persons per year.[7, 8] Therapy related CMML (t-CMML) cases have been described (~10% of all CMML), and like their MDS counterparts are associated with poor clinical outcomes.[9–11] Patients with t-CMML, in comparison to their de novo counterparts, are more likely to have cytogenetic abnormalities with higher risk karyotypic stratifications and shorter median over-all survivals (OS).[11] The presentation of patients with CMML is variable and the clinical heterogeneity is effectively captured by the current categorization into MDS-CMML and MPN-CMML.[12] Those with a MDS phenotype tend to present with peripheral blood cytopenias, effort intolerance, easy bruising, recurrent infections and transfusion dependance.[13] Those with a MPN phenotype tend to present with leukocytosis, monocytosis, hepatomegaly, splenomegaly and features of myeloproliferation such as; fatigue, night sweats, symptoms from organomegaly, bone pains, weight loss and cachexia.[13] Patients with MPN-CMML have a higher frequency of oncogenic RAS pathway mutations (NRAS, KRAS, CBL and PTPN11) and unique gene expression profiles.[3] Approximately 30% of CMML patients can present with antecedent or concomitant autoimmune diseases (rheumatoid arthritis, psoriasis, etc) and poorly defined systemic inflammatory syndromes.[14, 15] Rarely, CMML can present with leukemia cutis as an initial manifestation,[16] or directly present with blast phase disease.[17]
DIAGNOSIS
General Principles
An approach to patients with monocytosis is shown in Figure 1. It is important to exclude reactive causes of monocytosis before embarking on a workup of CMML. Monocytosis could be attributable to a number of non-malignant causes – infectious etiologies such as tuberculosis, chronic fungal infections, subacute bacterial endocarditis, viral and protozoal infections (leishmaniasis); connective tissue disorders such as systemic lupus erythematosus and sarcoidosis, and lipid storage disorders. The recovery phase of an acute infection (usually viral) or bone marrow regeneration post chemotherapy is commonly associated with monocytosis.[18]
Figure 1.

A Schematic approach to the differential diagnosis of peripheral blood monocytosis.
*: Peripheral blood abnormalities include unexplained anemia, thrombocytopenia, thrombocytosis, leukocytosis, eosinophilia, granulocytic dysplasia (pseudo Pelger Huët cells), circulating immature myeloid cells such as myelocytes, metamyelocytes and promyelocytes, promonocytes and blasts.
**: FISH – fluorescence in-situ hybridization, PDGFRA and PDGFRB: Platelet-derived growth factor – A and Platelet-derived growth factor – B.
FISH testing for PDGFRA and PDGFRB rearrangements is highly recommended if the peripheral blood monocytosis is associated with concomitant eosinophilia. The ETV6-PDGFRB fusion oncogene can give rise to clonal monocytosis mimicking CMML, but is in fact a unique molecularly defined myeloid neoplasm (not to be diagnosed as CMML). Similarly PDGFRA fusions are commonly associated with eosinophilia, but rarely can have associated monocytosis. Most PDGFRA fusions occur due to the karyotypically occult CHIC2 deletion (not detectable by metaphase cytogenetics) resulting in the FIP1L1-PDGFRA fusion oncogene. The World Health Organization also mandates FISH testing for FGFR1 rearrangements and the PCM1-JAK2 fusion, however, these abnormalities are very uncommonly associated with monocytosis.
*** While estimating peripheral blood blasts in a patient with CMML, the blasts have to be summated with circulating promonocytes.
Once these etiologies have been ruled out, molecularly defined clonal hematopoietic disorders need to be considered. Firstly, chronic myeloid leukemia (CML) with the distinctive Philadelphia chromosome and the BCR-ABL1 fusion oncogene must be evaluated and excluded.[19] Rearrangement of the platelet-derived growth factor receptors A (PDGFRA) and B (PDGFRB) should then be evaluated for. PDGFRA (chromosome 4q12) and PDGFRB (chromosome 5q31–q32) are type III receptor tyrosine kinases. Chromosomal translocations involving PDGFRA/B have been associated with myeloid neoplasms characterized by prominent eosinophilia and responsiveness to imatinib.[20, 21] At times, PDGFR rearranged myeloid neoplasms can present with monocytosis and BM dysplasia, but given their unique responsiveness to imatinib, these are no longer classified as CMML.[22] Patients presenting with a clinical phenotype of CMML with eosinophilia, should be assessed for t(5;12)(q31–q32;p13), giving rise to the ETV6(TEL)-PDGFRB fusion oncogene.[22] The association between monocytosis and PDGFRA rearrangements is uncommon.[23, 24] Additional molecular markers that should be assessed for, in the context of monocytosis and eosinophilia include FGFR1 rearrangements and the PCM1-JAK2 fusion.[25] Monocytosis can be associated with MPN such as primary myelofibrosis and polycythemia vera, where its presence adversely impacts survival.[26, 27] The presence of a prior well documented diagnosis of a MPN, or MPN-associated driver mutations such as MPL and CALR, make the diagnosis of CMML less likely.[1] Finally, the presence of bone marrow dysplasia in at least one hematopoietic lineage should be established. If myelodysplasia is absent or minimal, a diagnosis of CMML can still be made if clonal cytogenetic or molecular abnormalities are present (discussed below). Table 1 lists the 2016 WHO recommended diagnostic criteria for CMML.
Table 1.
2016 World Health Organization (WHO) recommended diagnostic criteria for chronic myelomonocytic leukemia (CMML)
|
Myeloproliferative neoplasms (MPN) such as primary myelofibrosis and polycythemia vera can present with concurrent monocytosis. A previous documented history of MPN excludes a diagnosis of CMML. In addition, the presence of MPN like features in the bone marrow, or the presence of MPN-associated driver mutations, especially MPL and CALR make the diagnosis of CMML unlikely.
PDGFRA abnormalities most often involve the cryptic CHIC2 deletion at chromosome 4q12, resulting in the FIP1L1-PDGFRA fusion, commonly associated with peripheral blood eosinophilia and increased bone marrow mast cells.
PDGFRB abnormalities most often involve the ETV6-PDGFRB gene fusion with ~25 additional reported partners. This fusion is associated with peripheral blood monocytosis and concomitant eosinophilia.
FGFR1 rearrangements often result in an aggressive stem cell leukemia/lymphoma syndrome characterized by MPN, eosinophilia and the development of T cell-acute lymphoblastic leukemia (ALL).
The PCM1-JAK2 fusion usually results in eosinophilia with T-ALL or B-ALL.
While gene mutations involving TET2 (~60%), SRSF2 (~50%), ASXL1 (~40%) and SETBP1 (~15%) are common in CMML, they are not specific for the disease. TET2 and ASXL1 mutations can also be detected in patients with normal blood counts as a part of age related clonal hematopoiesis (clonal hematopoiesis of indeterminate potential).
Flow cytometry
Peripheral blood flow cytometry is a recent measure that has been used to help diagnose CMML.[28] Human monocytes can be divided into three subsets; CD14+/CD16− (classical), CD14+/CD16+ (intermediate) and CD14low/CD16+ (non- classical), with different gene expression profiles, chemokine receptor expressions and phagocytic activities.[28, 29] The classical monocytes constitute majority of the human monocytes (~85%) in healthy conditions.[29] Compared to healthy donors and patients with reactive monocytosis, CMML patients demonstrate an increase in the fraction of classical monocytes (CD14+/CD16−) [cut off value 94%].[28] In the abovementioned French study, the associated specificity and sensitivity values were reported at 95.1% and 91.9% respectively.[28] Importantly, this repartition was noted to be independent of CMML mutational status and this increment corrected in CMML patients that responded to hypomethylating agents (HMA).[28] This technique has also been used to effectively distinguish monocytosis associated with CMML from monocytosis seen in patients with MPN,[30] and in identifying MDS patients with monocyte counts <1 x 109/L who eventually develop CMML.[31] False negatives with this technique have been encountered in CMML patients with autoimmune diseases, where the M02 fraction increases, resulting in a false decrease in the M01 fraction.[31] We hope that by using additional monocyte markers such as CCR2, CD36, HLA-DR and CD11c and better assessment techniques such as mass cytometry, we can improve upon the sensitivity and specificity of this assay.[32]
Histopathology and Immunohistochemistry
There is no single finding pathognomonic of the diagnosis of CMML. Bone marrow biopsies are often hypercellular with granulocytic hyperplasia and dysplasia. Monocytic proliferation can be present, but is often difficult to appreciate and immunohistochemical studies that aid in the identification of monocytes and their precursors is recommended.[33] Almost 80% of patients will demonstrate micro-megakaryocytes with abnormal nuclear contours and lobations, and 30% of patients can have an increase in BM reticulin fibrosis.[33] Twenty percent of patients can demonstrate nodules composed of mature plasmacytoid dendritic cells. The identification of promonocytes requires expertise and these cells are to be summated with blasts while estimating the blast count.[34]. Promonocytes are described as monocytic precursors that have a delicately convoluted, folded or grooved nucleus with finely dispersed chromatin, a small indistinct or absent nucleolus, and finely granulated cytoplasm (Figures two and three). [34, 35] On immunophenotyping the abnormal BM cells often express myelomonocytic antigens such as, CD13, CD33, with variable expression of CD14, CD68 and CD64. Markers of aberrant expression include CD2, CD15, CD56 or decreased expression of CD14, CD13, HLA-DR, CD64 or CD36. The presence of myeloblasts can be detected by expression of CD34. The most reliable markers on immunohistochemistry include CD68R and CD163. The monocytic cells are often positive for non-specific esterases and lysozyme, while the granulocytic precursors are often positive for lysozyme and chloroacetate esterase. This process can help differentiate CMML from other MPN such as CML and atypical CML, where BM monocytosis is uncommon.
Figure Two.
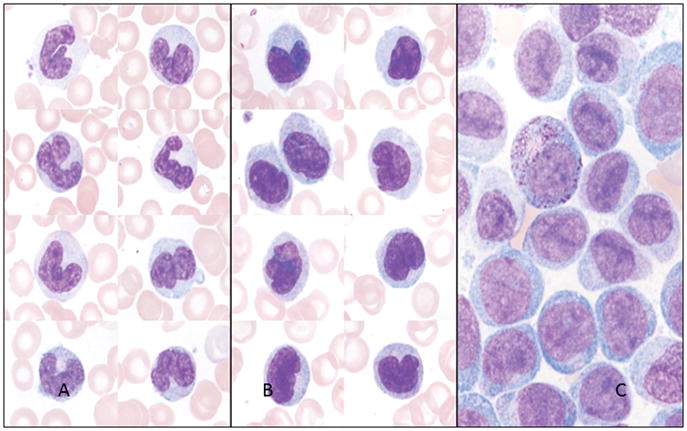
A. Peripheral blood smear demonstrating monocytes with well-defined nuclear lobes and mature chromatin. Wright-Giemsa 1000 X magnification.
B. Peripheral blood smear demonstrating promonocytes with open chromatin, nuclear folds and less defined nuclear lobes. Wright-Giemsa 1000 X magnification.
C. Bone marrow aspirate demonstrating monoblasts with open chromatin, lack of nuclear lobes and variable nucleoli. Wright-Giemsa 1000 X magnification.
Figure Three.
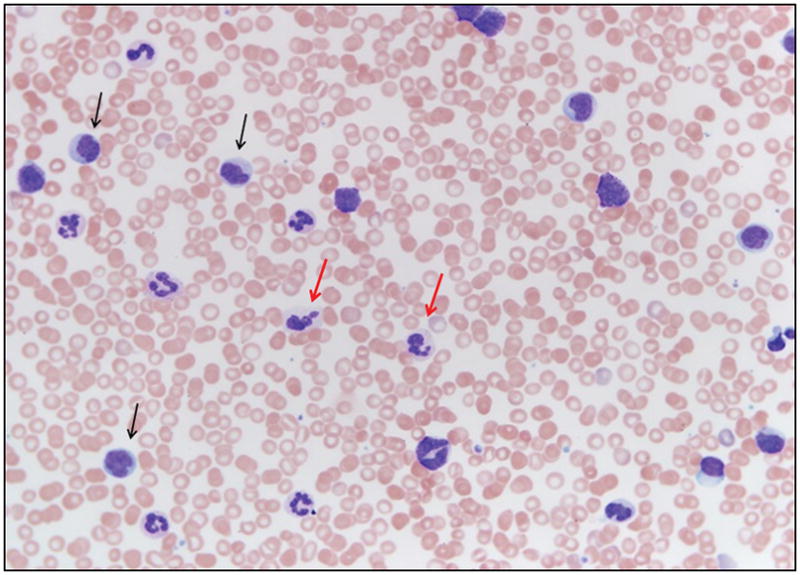
Peripheral blood smear of a patient with chronic myelomonocytic leukemia demonstrating circulating promonocytes (black arrows) and dysplastic granulocytes (red arrows). Wright-Giemsa 100 X magnification.
The diagnostic criteria for CMML place a heavy onus on the presence of PB monocytosis (Figure three). As discussed, monocytosis is associated with a variety of reactive and clonal causes. Persistent reactive monocytosis with marrow dysplasia can wrongly be labelled as CMML. Similarly, CMML patients with progressive dysplasia or splenomegaly might develop peripheral blood cytopenias, and in spite of having monocytosis, fail to meet the diagnostic criteria for CMML. Bone marrow monocytosis can be seen in patients with underlying dysplasia and while these patients may eventually progress to CMML, at this point, BM monocytosis is not incorporated into the diagnostic algorithm.
Cytogenetic abnormalities in CMML
Clonal cytogenetic abnormalities are seen in ~20–30% of CMML patients.[5, 36–38] Common alterations include; trisomy 8, - Y, abnormalities of chromosome 7 (monosomy 7 and del7q), trisomy 21, and complex karyotypes.[37] The Spanish CMML specific cytogenetic risk stratification (CPSS) system categorizes patients in to three groups; high risk (trisomy 8, chromosome 7 abnormalities, or complex karyotype), intermediate risk (all chromosomal abnormalities, except for those in the high and low risk categories), and low risk (normal karyotype or –Y), with 5-year OS of 4%, 26% and 35%, respectively.[37] Recently, in a large international study, 409 patients with CMML were analyzed for cytogenetic and molecular abnormalities.[39] Thirty percent displayed an abnormal karyotype; with common abnormalities being, +8 (23%), −Y (20%), −7/7q-(14%), 20q- (8%), +21 (8%) and der(3q) (8%).[39] A step-wise survival analysis resulted in three distinct cytogenetic risk categories: high (complex and monosomal karyotypes), intermediate (all abnormalities not in the high or low risk groups) and low (normal, sole -Y and sole der (3q)) with median survivals of 3 (HR 8.1, 95% CI 4.6–14.2), 21 (HR 1.7, 95% CI 1.2–2.3) and 41 months, respectively (Mayo-French cytogenetic risk stratification system).[39]
Molecular abnormalities in CMML
There has been an exponential discovery of several molecular abnormalities in patients with CMML. On an average, patients with CMML demonstrate ~10–15 mutations per kilobase of coding DNA regions, similar to patients with acute myeloid leukemia (AML), but several folds lower than other malignancies such as melanoma and lung cancer.[40, 41] These mutations can broadly be divided into the following categories: (a) mutations in epigenetic control of transcription,[42–47] such as histone modification (EZH2, ASXL1, UTX), and DNA methylation (TET2, DNMT3A, IDH1 and IDH2); (b) mutations in the spliceosome machinery (SF3B1, SRSF2, U2AF1, ZRSR2, PRPF8);[5] (c) mutations in genes that regulate cell signaling (JAK2, KRAS, NRAS, CBL, PTPN11 and FLT3);[48–52] (d) mutations in transcription factors and nucleosome assembly (RUNX1, SETBP1);[49, 53, 54] and (e) mutations in DNA damage response genes such as TP53 and PHF6.[55] The relative frequency of these mutations in individuals with CMML is shown in Table 2. Of these, mutations involving TET2 (~60%), SRSF2 (~50%), ASXL1 (~40%) and the oncogenic RAS pathway (~30%) are most frequent, with only frame-shift and non-sense ASXL1 mutations independently and adversely impacting OS.[44, 56]
Table 2.
Relative frequencies of somatic mutations in patients with chronic myelomonocytic leukemia
| Major class of genetic mutation | Gene | Frequency of mutation | |
|---|---|---|---|
| Epigenetic Control | Histone modification | ASXL1* | 40% |
| EZH2 | 5% | ||
| DNA methylation | TET2 | 60% | |
| DNMT3A* | 5% | ||
| Dual effect | IDH1 | 1% | |
| IDH2 | 5–10% | ||
| Cell signaling | JAK2V617F | 5–10% | |
| CBL | 15% | ||
| NRAS* | 15% | ||
| KRAS | 10% | ||
| PTPN11 | 5% | ||
| FLT3 | <5% | ||
| Pre-mRNA splicing | SRSF2 | 50% | |
| SF3B1 | 5–10% | ||
| U2AF1 | 5–10% | ||
| ZRSR2 | 5% | ||
| Transcription and nucleosome assembly | RUNX1* | 15% | |
| SETBP1* | 15% | ||
| DNA damage | TP53 | 1% | |
| PHF6 | 5% | ||
Annotates genes that have been shown in various studies to have an independent and adverse prognostic impact on survival outcomes.
The ASXL1 gene (chromosome 20q11) regulates chromatin by interacting with the polycomb- group repressive complex proteins (PRC1 and PRC2).[42] In a seminal paper, Abdel-Wahab et al. demonstrated that ASXL1 mutations resulted in loss of PRC2-mediated H3K27 (histone 3 lysine 27) tri-methylation.[57] In addition, Balasubramani et al. demonstrated that ASXL1 truncations conferred enhanced activity on the ASXL1-BAP1 (BRCA associated protein 1) complex.[58] These interactions result in a global erasure of H2AK119Ub and depletion of H327Kme3, promoting dysregulated transcription and oncogenesis. EZH2 mutations (chromosome 7q36.1) occur in <5% of CMML patients, and unlike in epithelial malignancies and lymphoproliferative disorders are loss-of-function mutations.[59] EZH2 mutations in CMML almost always co-occur with ASXL1 mutations, are frequently associated with a MPN-CMML phenotype, and while they themselves do not impact either OS or LFS, ASXL1/EZH2 co-mutated patients have a shorter OS, in comparison to ASXL1mt patients alone.[59]
The TET2 gene located on chromosome 4q24 is a member of the TET family of proteins.[60] TET2 has a dioxygenase enzymatic activity and converts 5-methyl-cytosine to 5-hydroxymethyl-cytosine (5hmC). 5hmC, represents a new base in genomic DNA, which may have a specific effect on transcription.[61, 62] Although TET2 mutations are widely prevalent in CMML (~60%), they have not been shown to independently impact either OS or LFS.[44, 63] In a recent study, the presence of clonal TET2 mutations, in the absence of clonal ASXL1 mutations (ASXL1wt/TET2mut), had a favorable impact on OS.[64] The reason for this association is unclear. In MDS and younger patients with CMML (age <65years), the presence of clonal TET2 mutations, in the absence of clonal ASXL1 mutations, has been associated with response to HMA.[65, 66]. Mutations involving TET1, TET3 and ASXL2 are extremely uncommon in CMML.[67]
DNA methylation is mediated by a family of DNA methyltransferase enzymes (DNMT), including DNMT1, DNMT3A (chromosome 2p23), and DNMT3B.[68] DNMT1 primarily maintains pre-existing DNA methylation patterns, whereas DNMT3A and DNMT3B carry out de novo DNA methylation.[68] DNMT3A mutations are seen in ~5% of CMML patients and independently and adversely impact both OS and LFS.[69] Of note, a recurrent Arginine882 (R882) hot spot accounts for 40–60% of DNMT3A mutations, with limited data suggesting loss of methyltransferase activity in in vitro assays.
Spliceosome component mutations (SRSF2, SF3B1, U2AF1, PRPF8 and ZRSR2) affect pre-mRNA splicing.[5] SRSF2 mutations are common in CMML (~50%) and are associated with increasing age, less pronounced anemia and a diploid karyotype.[5] Thus far, SRSF2 mutations have not demonstrated an independent prognostic impact on both, OS and LFS.[5, 44, 70] SF3B1 mutations have a high prevalence (~80%) in patients with MDS and ring sideroblasts (RS)[71] and can also be seen in patients with CMML and RS (~10%).[5] These mutations do not influence either the OS or LFS.[72, 73] Similarly, U2AF1 and ZRSR2 mutations are seen in ~10% of CMML patients and have thus far lacked an independent prognostic effect.[74]
Common signal pathway mutations in CMML include; oncogenic RAS pathway mutations (~30%, NRAS, KRAS, CBL and PTPN11), and JAK2V617F (~10%).[44, 49] RAS pathway mutations are associated with a MPN-like phenotype.[75] Although univariate analysis studies with RAS mutations have demonstrated inferior outcomes in CMML, these findings have not been substantiated in multivariable models.[36, 44] The CBL gene codes for an E3 ubiquitin ligase involved in degradation of activated receptor tyrosine kinases. RING finger domain (RFD) mutations of CBL are frequently associated with UPD11q (uniparental disomy) and have been reported in 10–20% of patients with CMML.[44, 49] RUNX1 is essential for normal hematopoiesis and mutations can be seen in ~10–15% of patients with CMML.[44, 49] Although these mutations do not impact OS, there is a trend towards a higher risk for AML progression.[76]
The sequence of genetic events leading to the clinical phenotype of CMML remains under investigation. It is thought that the initial driver mutation is likely to be a mutation in TET2 or ASXL1. This assumption is based on the high frequency of these mutations (~40–60%) in CMML,[77, 78] and results of single-cell tracking experiments.[79] Secondary mutations in the spliceosome machinery (such as in SRSF2) and cytokine signaling (NRAS or CBL) may allow a subset of these clones to progress, resulting in the typical phenotype associated with this disease.[55, 80] A recent paper has demonstrated that concurrent Tet2 loss and NrasG12D expression in hematopoietic cells induced myeloid transformation, with a fully penetrant CMML phenotype in mice.[81]
RISK STRATIFICATION
Numerous prognostic models have been developed for CMML. In this regard, the value of Bournemouth, Lille, International Prognostic Scoring Systems (IPSS) and the revised-IPSSS is limited, as they were designed primarily for patients with MDS, excluding CMML patients with a MPN phenotype.[82, 83] The MD Anderson prognostic system (MDAPS) is CMML specific and identified a hemoglobin (HB) level <12 gm/dl, presence of PB immature myeloid cells (IMC), absolute lymphocyte count (ALC) >2.5 x 109/L and ≥ 10% BM blasts as independent predictors for inferior OS.[36] The MDAPS was subsequently applied to 212 CMML patients in the Dusseldorf registry[84]; in a univariate analysis circulating IMC had no prognostic impact, while on multivariable analysis, elevated lactate dehydrogenase, BM blast count >10%, male gender, HB <12 gm/dl and ALC >2.5 x 109/L were independently prognostic.[84]
The Global MDAPS (2008) was developed for patients with de novo MDS, secondary MDS and CMML (n=1,915).[85] Independent prognostic factors included; older age, poor performance status, thrombocytopenia, anemia, increased BM blasts, leukocytosis (>20 x 109/L), chromosome 7 or complex cytogenetic abnormalities and a prior history of red blood cell transfusions.[85] This model identified 4 prognostic groups with median survivals of 54 (low), 25 (intermediate-1), 14 (intermediate-2) and 6 months (high), respectively.[85] The CMML-specific prognostic scoring system (CPSS) identified 4 variables as being prognostic for both OS and LFS; FAB and WHO CMML-subtypes, red blood cell transfusion dependency, and the Spanish cytogenetic risk stratification system. [6, 37] One point was accorded for each variable, with the exception of high risk cytogenetics which earned 2 points, and four risk categories were determined: low (0 points), intermediate-1 (1), intermediate-2 (2–3), and high risk (4–5).[6]
The discovery of gene mutations in CMML has resulted in the development of molecular prognostic models. A Mayo Clinic study (n=226) analyzed several parameters, including ASXL1 mutations; on multivariable analysis, risk factors for survival included HB <10 gm/dl, platelet count <100 × 109/L, AMC>10 × 109/L and circulating IMC.[86] ASXL1 mutations did not impact either the OS or the LFS. The study resulted in the development of the Mayo prognostic model, with three risk categories, low (0 risk factor), intermediate (1 risk factor) and high (≥2 risk factors), with median survivals of 32, 18.5 and 10 months, respectively.[86] The GFM demonstrated an adverse prognostic effect for ASXL1 mutations in 312 patients with CMML; additional risk factors on multivariable analysis included age >65 years, WBC >15 × 109/L, platelet count <100 × 109/L and HB <10 gm/dl in females and <11 gm/dl in males.[44] The GFM model assigns 3 adverse points for WBC >15 × 109/L and 2 adverse points for each one of the remaining risk factors, resulting in a three-tiered risk stratification; low (0–4 points), intermediate (5–7) and high (8–12), with respective median survivals of 56, 27.4 and 9.2 months.[44] It should be noted that all nucleotide variations (missense, nonsense and frameshift) were regarded as ASXL1 mutations in the Mayo study,[86] whereas only nonsense and frameshift ASXL1 mutations were considered in the French study[44].
To further clarify the prognostic relevance of ASXL1 mutations, an international collaborative cohort of 466 CMML patients was analyzed.[39] In univariate analysis, survival was adversely affected by ASXL1 (nonsense and frameshift) mutations. In multivariable analysis, ASXL1 mutations, AMC >10 × 109/L, HB <10 gm/dl, platelets <100 × 109/L and circulating IMC were independently predictive of shortened OS. A regression coefficient-based prognostic model based on these five risk factors delineated high (≥3 risk factors; HR 6.2, 95% CI 3.7–10.4) intermediate-2 (2 risk factors; HR 3.4, 95% CI 2.0–5.6) intermediate-1 (one risk factor; HR 1.9, 95% CI 1.1–3.3) and low (no risk factors) risk categories with median survivals of 16, 31, 59 and 97 months, respectively.[56] This model is referred to as the Mayo Molecular Model. Recently the CPSS model was updated to include molecular abnormalities including ASXL1, RUNX1, NRAS and SETBP1 mutations (CPSS-Mol).[87] These mutations, in addition to the prior CPSS cytogenetic scores were used to calculate the CPSS genetic score. One point each was assigned for an intermediate-1 genetic score, WBC ≥ 13 x 109/L, BM blasts≥ 5% and red blood cell transfusion dependancy, 2 points for intermediate-2 genetic score and 3 points for a high risk genetic score.[87] The CPSS-Mol stratified CMML patients into four risk categories, low (0 risk factors), intermediate-1(1 risk factor), intermediate-2 (2–3 risk factors) and high (≥4 risk factors) risk, with median OS of not reached, 64, 37 and 18 months; with respective 4-year leukemic transformation rates of 0%,3%, 21% and 48%.[87] Table 3 highlights the CMML specific prognostic models along with their relevant components.
Table 3.
Prognostic scoring systems for chronic myelomonocytic leukemia
| Prognostic Score | Year | Number of Patients | External Validation | Variables included in the final model | Median Survival in months | Transformation into AML | |||
|---|---|---|---|---|---|---|---|---|---|
| Low risk | Intermediate-1 risk | Intermediate-2 risk | High risk | ||||||
| Onida et al5 (MDAPS) | 2002 | 213 | No | 1. Hemoglobin <12 gm/dL 2. Circulating immature myeloid cells 3. Absolute lymphocyte count >2.5 x 109/L 4. Bone marrow blasts >10% |
24 | 15 | 8 | 5 | 19% developed AML after a median time of 7 months |
| Germing et al4 (Dusseldorf score for CMML) | 2004 | 288 | No | 1. Bone marrow blasts ≥5% 2. LDH >200 U/L 3. Hemoglobin ≤9 gm/dl 4. Platelets ≤100 x 109/L |
93 | 26 | 11 | 8%, 23% and 23% at 5 years, respectively | |
| Such et al42 (CPSS Model) | 2013 | 578 | Yes, in 274 patients | 1. CMML FAB type 2. CMML WHO type 3. CMML-specific cytogenetics 4. RBC transfusion dependence |
72 | 31 | 13 | 5 | Probability of AML evolution at 5 years, 13%, 29%, 60%, and 73%, respectively |
| Itzykson et al22 (GFM Model) | 2013 | 312 | Yes, 165 patients | 1. Age >65 years 2. WBC >15x109/L 3. Anemia 4. Platelets <100 x109/L 5. ASXL1 mutation |
Not reached | 38.5 | 14.4 | AML-free survival was 56.0, 27.4, and 9.2 months, respectively | |
| Patnaik et al34 (Mayo Model) | 2013 | 226 | Yes, 268 patients | 1. Increased absolute monocyte count >10×109/L 2. Presence of circulating blasts 3. Hemoglobin <10 gm/dL 4. Platelet count <100 ×109/L |
32 | 18.5 | 10 | NR | |
| Patnaik et al (Mayo Molecular Model) | 2014 | 466 | No | 1. Increased absolute monocyte count >10×109/L 2. Presence of circulating blasts 3. Hemoglobin <10 gm/dL 4. Platelet count <100 ×109/L 5. Frameshift and nonsense ASXL1 mutations |
97 | 59 | 31 | 16 | At a median follow up of 23 months, 75 (16%) leukemic transformations occurred. |
| Elena et al (CPSS-Mol) | 2016 | 214 | 260 | 1. Genetic risk groups as defined by *CPSS cytogenetic risk stratification and gene mutations involving ASXL1, NRAS, SETBP1 and RUNX1. 2. Bone marrow blasts ≥ 5%. 3. WBC count ≥ 13x109/L 4. Red blood cell transfusion dependancy |
Not reached | 64 | 37 | 18 | 48 months cumulative incidence of AML evolution; 0%, 3%, 21% and 48%, respectively. |
Key: MDAPS- MD Anderson Prognostic Scoring System, CPSS- CMML specific prognostic scoring system, CMML- chronic myelomonocytic leukemia, GFM- Groupe Francophone des Myélodysplasies, LDH- lactate dehydrogenase, FAB- French American British, WHO – World Health Organization, WBC- white blood cell count.
The CPSS-Mol used a genetic risk group stratification that assigned a score of 0 for low risk cytogenetics and absence of ASXL1/NRAS/SETBP1/RUNX1 mutations, a score of 1 for intermediate risk cytogenetics and mutations involving ASXL1/SETBP1 and NRAS, and a score of 2 for high risk cytogenetics and RUNX1 mutations
Seven clinical prognostic models, not incorporating ASXL1 mutational status (IPSS, R-IPSS, MDAPS, Global MDAPS, Dusseldorf, CPSS and Mayo model) were statistically compared in a large dataset of CMML patients (n=1832).[82] All seven models were found to be valid with comparable performance, but were vulnerable to upstaging.[82]
Rates of leukemic transformation vary among different series of CMML patients reported in the literature. However, most studies quote an incidence of 15–20%.[88–90] In a study of 274 CMML patients followed for a median of 17.1 months, blast transformation (BT) occurred in 36 (13%).[17] On multivariable analysis, risk factors for BT were presence of PB blasts (HR 5.7; 95% CI 2.8–11.9) and female gender (HR 2.6; 95% CI 1.3–5.1); and the results remained unchanged when analysis was restricted to CMML-1. ASXL1/SRSF2/SETBP1 mutational frequencies were not significantly different between time of CMML diagnosis and BT. Median survival post-BT was 4.7 months (5-year survival 6%) and was better with allogeneic stem cell transplant (HCT) (14.3 months vs. 4.3 months for chemotherapy vs. 0.9 months for supportive care; P = 0.03).[17]
RISK ADAPTED THERAPY
After its inclusion as a specific category of myeloid neoplasms in the 2008 WHO classification, treatment options for CMML have evolved. In the late 1990’s, major treatment options consisted of chemotherapy such as etoposide, cytarabine, all-trans retinoic acid,[91–93] topotecan,[94],[95] 9-nitro-campothecin (topoisomerase inhibitor),[96] and lonafarnib (farnesyltransferase inhibitor).[97] Collectively, response rates in these trials were disappointing and therapy was associated with significant toxicities.
The United States Food and Drug Administration (FDA) approved two HMA, 5-azacitidine and decitabine, for treatment of patients with MDS. Two pivotal randomized studies that established the efficacy and safety of these drugs included a total of 361 patients with MDS.[98, 99] However, these studies only had 14 patients with CMML each, and the response rates for patients with CMML were not reported separately. Since the publication of these studies, several Phase II studies have reported the outcomes of patients with CMML who were treated with HMA.[73, 100–107] A complete list of the studies, including the dose and schedule of the drugs used, toxicities, response rates and survival are shown in Table 4. The overall response rate ranged from 25–70% (~30–40%), and median OS ranged from 12 to 37 months. Braun et al showed that mutations in ASXL1, NRAS, KRAS, CBL, FLT3, and JAK2 genes, and hypermethylation of the promoter of the tumor suppressor gene - transcription-intermediary factor-1 gene (TIF1γ) did not predict response or survival in 39 CMML patients treated with decitabine. However, lower CJUN and CMYB gene expression levels independently predicted improved OS. There was a trend towards higher response rate in patients with a TET2 mutation (when not associated with an ASXL1 mutation), although it did not reach statistical significance.[101] On multivariable analysis, Ades et al showed that bone marrow blasts >10% and WBC>13 x 109/L had a prognostic impact on OS among 76 patients treated with azacitidine.[100] Fianchi et al showed that improved OS was associated with an absolute monocyte count <10 x 109/L, and PB blasts <5% at the start of therapy with HMA.[103] Pleyer et al conducted a matched-pair analysis of CMML patients treated with azacitidine (n=42) versus those who were treated with best supportive care (BSC, n=42) or with hydroxyurea (n=22). Although there was an improvement in median OS in the azacitidine arm (31 months) compared to BSC (17 months), these results were not statistically different (p=0.25), possibly due to the small sample size. Similarly, there was no difference in median OS between the azacitidine and hydroxyurea treatment arms (7.5 vs. 6.2 months, respectively, p=0.22). Next generation sequencing studies of CMML patients treated with HMA have shown that these agents do not alter the mutational allele burdens, even in responding patients.[40] Hematological responses obtained are often not durable, and are associated with significant changes in DNA methylation arguing for an epigenetic restoration of normal hematopoiesis, without significantly altering disease biology or progression to AML.[40]
Table 4.
Use of hypomethylating agents in chronic myelomonocytic leukemia (CMML)
| Reference | Number of patients |
Median Age (years, range) |
Phase of Study |
Treatment Regimen | Response Rates | Toxicity | Median Survival |
Progression to Acute Myeloid Leukemia |
|---|---|---|---|---|---|---|---|---|
| Aribi (2007)59 | 19 | 66 (44–82) | II | Decitabine 100 mg/m2 per course in three different schedules, repeated every 4 weeks | CR: 58% HI: 11% |
Myelosuppression associated complications: 8% | 19 months | NR |
| Wijermans (2008)65 | 31 | 71 (53–81) | II | Decitabine 15 mg/m2 over 4 hours IV three times per day on three consecutive days, with a total dose of 135 mg/m2 per course, every 6 weeks | CR: 10% PR: 16% HI: 19% |
Nausea, vomiting, pneumonia, mortality due to sepsis: 3% | 15 months | NR |
| Costa (2010)61 | 38 | 70 (36–83) | II | Azacitidine 75 mg/m2/day for 7 days or 100 mg/m2/day for 5 days every 4 weeks | CR: 11% PR: 3% HI: 25% |
Pneumonia, mortality due to sepsis: 3% | 12 months | NR |
| Garcia-Manero (2011)63 | 41 (4 with CMML) | 70 (31–91) | I | 1 cycle of subcutaneous azacitidine 75 mg/m2 on the first 7 days of cycle 1, followed by oral azacitidine daily,120 to 600 mg, on the first 7 days of each additional 28-day cycle | ORR: 35% in previously treated patients and 73% in previously untreated patients | diarrhea, nausea, vomiting, febrile neutropenia, fatigue | NR | NR |
| Braun (2011)60 | 39 | 71 (54–88) | II | Decitabine 20 mg/m2 per day intravenously for 5 days every 28 days | CR: 10% PR: 20% HI: 8% ORR: 38% |
Neutropenia and thrombocytopenia (36%), severe infection (20%) | 18 months | NR |
| Thorpe (2012)64 | 10 | 66 (41–76) | II | Azacitidine 75 mg/m2 for 7 days or azacitidine 100 mg/m2 for 5 days every 28 days | CR: 20% HI: 40% ORR: 60% |
Thrombocytopenia, pneumonia (20%) | 29 months | NR |
| Ades (2013)58 | 76 | 70 (33–85) | II | Azacitidine 75 mg/m2 for 5–7 days every 28 days | CR: 17% PR: 1% Marrow CR: 8% HI: 17% ORR: 43% |
NR | 29 months | 31% after 1.2 years from azacitidine initiation |
| Wong (2013)66 | 11 | 65 (42–80) | II | Azacitidine 75 mg/m2 for 7 days every 28 days | CR: 9% Marrow CR: 27% PR: 9% HI: 9% ORR: 55% |
Local skin reactions (55%), nausea (36%), infection (73%) | 17 months | 18% |
| Fianchi (2013)62 | 31 | 69 (53–84) | II | Azacitidine 50–75 mg/m2 for 7 days in 22 patients, and 100 mg flat dose for 5–7 days in 9 patients | CR: 45% PR: 3% HI: 6% ORR: 54% |
Grade 4 thrombocytopenia (6%), grade 4 anemia (6%) | 37 months | 16% after 12.7 months |
| Santini (2013) | 44 | 71 (42–84) | II | Decitabine 20 mg/m2 for 5 days, every 28 days | CR: 14% PR: 2% Marrow CR: 17% ORR: 33% |
Severe infections (17%) | 19 months | NR |
| Pleyer (2014) | 48 | 71 (38–87) | II | Azacitidine 75 mg/m2 for 7 days in 42 patients, and 100 mg flat dose for 5–7 days in 6 patients | CR/marrow CR: 13% HI: 50% ORR: 54% |
Grade 3–4 cardiac events (21%) | 12.6 months | 4% after 9 months |
| Drummond (2014) | 32 | 70 (57–85) | II | Azacitidine 75 mg/m2 for 7 days, every 28 days | CR: 7% PR: 0 Marrow CR: 7% HI:3% ORR: 17% |
NR | 16 months | 33% after 13 months |
| Sekeres (2017) | 53 with CMML | 70 (28–93) | Randomized phase II | Azacitidine (75 mg/m2/day on days 1 to 7 of a 28–day cycle); Azacitidine plus lenalidomide (10 mg/day on days 1 to 21); or Azacitidine plus vorinostat (300 mg twice daily on days 3 to 9). |
ORR: 38% (68% in the azacitidine and lenalidomide arm) | Azacitidine plus lenalidomide associated with higher incidence of skin rashes, while azacitidine and vorinostat with a higher incidence of Gastrointestinal side effects | Not reached | NR |
| Santini (2018) | 43 | 71.5 (42–84) | II | Decitabine 20 mg/m2 for 5 days, every 28 days | CR: 16% PR: 2.4% Marrow CR: 19% HI: 9.5% ORR: 47.6% |
Thrombocytopenia (64%) Anemia (52%) Gastrointestinal side effects (23.8%) |
17 months | 57.5%after 51.5 months |
Abbreviations Used:
CR: complete remission; PR: partial remission; HI: hematologic improvement; ORR: overall response rate; NR: not reported; CMML: chronic myelomonocytic leukemia.
A recent phase I clinical trial (n=20) has demonstrated safety and potential efficacy (35% MDS international working group (IWG) and spleen response) with ruxolitinib (JAK inhibitor), in patients with CMML.[108] This trial has currently been expanded to a phase II design and is currently accruing. Additional JAK/STAT inhibitors being preclinically assessed include momelotinib and pacritinib.[109] Given the inherent, demonstrable, GM-CSF (granulocyte-macrophage colony stimulating factor) dependant pSTAT5 (phosphorylated Signal Transducer and Activator of Transcription 5) sensitivity in CMML patients, targeted anti-GM-CSF monoclonal antibody therapy (lenzilumab) is being developed (NCT02546284-www.clinicaltrials.gov).[110] Tipifarnib a farnesyl transferase inhibitor (NCT02807272) and SL-401 a recombinant fusion protein composed of the catalytic and translocation domains of diphtheria toxin (DT) fused via a Met-His linker to IL3.11 (NCT02268253), are also undergoing clinical trial assessments in patients with CMML.
Allogeneic Stem Cell Transplantation
Allogeneic stem cell transplantation remains the only curative option for patients with CMML. This modality is however fraught with complications including, acute and chronic graft versus host disease (GVHD), non-relapse mortality and post-transplant disease relapse. There unfortunately exists no prospective data analyzing the risks and benefits for HCT in CMML. The response rates in retrospective studies have ranged from 17% to 50%, with corresponding treatment related mortality rates ranging from 12% to 52% (Table 5).[111–118] The ten-year OS of 85 patients who underwent HCT at Fred Hutchinson Cancer Center was 40%. A multivariable model identified increasing age, higher HCT comorbidity index and poor-risk cytogenetics to be associated with increased mortality and reduced relapse-free survival (RFS).[111] The European Group for Blood and Marrow Transplantation reported an OS of 42% for 283 patients with CMML that underwent HCT. None of the baseline factors including the conditioning regimen, age, disease status at transplant, cytogenetics, donor-recipient gender match, HLA-type of donor, stem cell source, T-cell depletion or the development of GVHD affected the RFS or OS.[117] A recent application of the CPSS in the HCT setting, assessed 209 adult patients from 2001 to 2012 with a median age of 57 years and followed for a median of 51 months.[7] On multivariate analysis, CPSS score, Karnofsky performance status and graft source were significant predictors of OS.
Table 5.
Summary of select allogeneic stem cell transplant studies for chronic myelomonocytic leukemia (CMML)
| Reference | N | Age, yrs (median) | Disease Stage | Cytogenetics | Donor Type and Stem Cell Source | Conditioning (Myeloablative, reduced intensity) | Relapse rate and Treatment-related mortality | Outcomes |
|---|---|---|---|---|---|---|---|---|
| Kroger (2002) | 50 | 44 (19–61) | CMML-1: 28 CMML-2: 17 Unknown: 5 |
Diploid: 18 Abnormal: 11 Unknown: 21 |
MRD: 43 MUD: 7 BM: 40 PBSC: 9 |
MAC: 50 RIC: 0 |
RR: 28% TRM: 52% |
5-year OS: 21% 5-year DFS: 18% |
| Mittal (2004) | 8 | 51 (20–64) | NR | Diploid: 3 Abnormal: 4 Unknown: 1 |
MRD: 6 MUD: 2 BM: 4 PBSC: 4 |
MAC: 4 RIC: 4 |
RR: 50% TRM: 12% |
18 month OS: 35% 18 month DFS: 31% |
| Elliott (2006) | 17 | 50 (20–60) | NR | Diploid: 9 Abnormal: 8 |
MRD:14 MUD:3 BM: 8 PBSC: 7 |
MAC: 16 RIC: 1 |
RR: 41% TRM: 41% |
3 year OS: 18% 3-year RFS: 18% |
| Ocheni (2009) | 12 | 56 (38–67) | CMML-1: 7 CMML-2: 3 Unknown: 2 |
Diploid: 7 Abnormal: 4 Unknown: 1 |
MUD: 11 MRD: 1 BM: 0 PBSC: 12 |
MAC: 7 RIC: 6 |
RR: 17% TRM: 25% |
2-year OS: 75% 2-year DFS: 67% |
| Krishnamurthy (2010) | 18 | 54 (38–66) | CMML-1: 8 CMML-2: 10 |
Diploid: 7 Abnormal: 11 |
MRD: 10 MUD: 8 BM: 6 PBSC: 12 |
MAC: 1 RIC: 17 |
RR: 44% TRM: 31% |
3-year OS: 31% 3-year DFS: 47% (< 5% blasts) 3-year DFS: 20% (>5 % blasts) |
| Symeonidis (2010) | 283 | 50 (NR) | CMML-MDS:45 CMML-MPN:60 Unknown: 178 |
NR | MRD: 160 MUD: 85 Unknown: 38 BM: 108 PBSC: 175 |
MAC: 152 RIC: 87 |
RR: 25% TRM: 37% |
OS: 42% DFS: 38% (time interval not specified) |
| Eissa (2011) | 85 | 51 (1–69) | CMML-1: 57 CMML-2: 26 |
Good: 45 Intermediate: 14 Poor: 22 |
MRD: 38 MUD: 47 BM: 32 PBSC: 53 |
MAC: 58 RIC: 27 |
RR (10 yrs): 27% TRM (10 yrs): 35% |
10-year OS: 40% 10-year DFS: 40% |
| Park (2013) | 73 | 53 (27–66) | CMML-1: 40 CMML-2: 29 |
Good: 48 Intermediate: 13 Poor: 9 |
MRD: 41 MUD: 32 BM: 27 PBSC: 46 |
MAC: 30 RIC: 43 |
RR (3 yrs): 35% | 3-year OS: 32% 3-year DFS: 29% |
| Itonaga (2013) | 141 | 49 (NR) | NR | NR | MRD: 68 MUD: 53 Cord: 10 |
MAC: 101 RIC: 40 |
NR | 3-year OS: 47% |
| Duong (2015) | 209 | 57 (23–74) | CPSS low/intermediate-1– 88 (42%) Intermediate-2/high- 79 (38%) Missing- 42 |
CPSS Cytogenetic groups Low- 50% Intermediate-19% High- 17% Missing- 14% |
MRD: 35% MUD: 45% MMUD: 19% BM: 16% PBSC: 84% |
MAC: 51% RIC: 41% NMA: 5% |
NR | OS at 1, 3 and 5 years for CPSS low /intermediate-1: 61%, 48%, 41%. Intermediate-2/high- 38%, 32%, 19% |
| Symeonidis (2015) | 513 | 53 (18.5–75.4) | CMML-MDS:73 CMML-MPN: 110 CMML-1: 87 CMML-2: 32 Secondary AML- 95 |
Normal- 104 Abnormal- 60 |
MRD: 285 MUD: 228 BM: 119 PBSC: 394 |
MAC- 249 RIC- 226 |
RR (4yrs): 32% NRM (4yrs): 41% |
4-year OS: 33% 4-year DFS: 27% |
| Liu HD (2017) | 209 | 57 (23–74) | CMML-1: 140 (67%) CMML-2: 52 (25%) Missing: 17 (8) |
NR | MRD: 73 MUD: 95 MMUD: 36 |
MAC: 105 RIC: 99 Missing: 5 |
RR (1,3 and 5 yrs.): 46%, 50% and 52% TRM (1,3 and 5 years): 19%, 23% and 28% |
OS at 1,3 and 5 years: 50%, 38% and 30%. |
Abbreviations Used:
N: total number of patients, yrs: years, CMML-1: chronic myelomonocytic leukemia-1, CMML-2: chronic myelomonocytic leukemia-2, AML- acute myeloid leukemia, CMML-MDS: chronic myelomonocytic leukemia-myelodysplasia type, CMML-MPN: chronic myelomonocytic leukemia- myeloproliferative type, MRD: matched related donor, MUD: matched unrelated donor, MMUD: mismatched unrelated donor, BM: bone marrow donor, PBSC: peripheral blood stem cell donor, MAC: myeloablative conditioning, RIC: reduced intensity conditioning, NMA: non myeloablative, RR: relapse rate, TRM: treatment-related mortality, NRM: non relapse mortality, OS: overall survival, DFS: disease-free survival; NR: not reported, CPSS- CMML specific prognostic scoring system.
In general, for younger patients with higher risk disease and an acceptable co-morbidity index, allogeneic HCT is the preferred treatment modality.[65] With the advent of reduced intensity conditioning and alternate donor sources (haploidentical HCT and double umbilical cord blood units), an increasing number of patients have access to HCT. While reduced intensity conditioning is associated with lower non-relapse mortality, disease relapse rates are higher in comparison to myeloablative regimens.[119, 120] Similar to MDS, cytoreductive therapy or HMA are often considered prior to HCT in patients with increased BM blasts (CMML-2) or prior to a reduced intensity conditioning.[121] A recent retrospective study (n=83) demonstrated prior therapy with HMA followed by allogeneic HCT was associated with a lower cumulative incidence of relapse (22% versus 35%; p=0.03), without a significant increase in the one-year transplant related mortality.[122] This finding needs prospective validation.
Recommendations: Hydroxyurea remains the cornerstone of therapy for patients with myeloproliferative features. Guidelines for supportive care measures such as the use of erythropoietin analogs for the treatment of anemia, prophylactic antibiotics for isolated neutropenia and iron chelation therapy for patients with a heavy transfusion burden are in general similar to patients with MDS, and data for their use specifically in patients with CMML do not exist. Standard induction chemotherapy should be considered for all eligible patients who develop blast transformation. Hypomethylating agents remain the most commonly used therapeutic intervention for patients with CMML. The presence of an elevated WBC count (>13x109/L), palpable splenomegaly and increased bone marrow blast percentage (>10%) are all associated with a worse survival while on therapy with hypomethylating agents. Although several novel mutations (such as ASXL1, RUNX1, NRAS and SETBP1) have been described to adversely affect survival of untreated CMML patients, their impact on patients undergoing therapy with hypomethylating agents is unclear at this time. Unfortunately, the response rates and survival following therapy is suboptimal, and therefore clinical trial participation is strongly encouraged.
The role of allogeneic HCT in CMML remains controversial. Similar to MDS, younger patients with an adverse survival, as determined by newer prognostic models incorporating molecular aberrations, should be considered for HCT. Older patients with a high HCT comorbidity index do not benefit from HCT, and are best suited for clinical trials.
CONCLUSION
CMML, a myeloid neoplasm with features of MDS and MPN, often presents with PB monocytosis and has an inherent risk for transformation to AML. Clonal cytogenetic changes are seen in ~30% of patients and the CPSS and Mayo French Cytogenetic systems effectively risk stratify CMML patients based on cytogenetic abnormalities. Gene mutations are seen in >90% of patients, with common abnormalities involving; epigenetic regulators (TET2~60% and ASXL1~40%), spliceosome components (SRSF2~50%) and cell signaling (oncogenic RAS~30%). Of these, only frame-shift and nonsense ASXL1 mutations have universally been shown to negatively impact OS. Lower risk, CMML patients that present with MPN-like features are effectively managed with hydroxyurea. Hypomethylating agents are associated with overall response rates of ~ 30–40%, with complete remission rates of ~ 15%. These responses are generally not sustained, do not alter mutational allele burdens, and survival after loss of response is often dismal. Allogeneic HCT remains the treatment of choice for younger patients with higher risk disease. Complications of HCT including, non-relapse mortality, acute and chronic graft versus host disease, limit generalized applicability of this treatment strategy. The development of CMML specific disease response criteria and clinical trials exploiting the genetic and epigenetic abnormalities in CMML, are important milestones to look forward to.[123]
Acknowledgments
Current publication is supported in part by grants from the “The Gerstner Family Career Development Award” and the Mayo Clinic Center for Individualized Medicine, Mayo Clinic, Rochester, MN, USA”.
This publication was supported by CTSA Grant Number KL2 TR000136 from the National Center for Advancing Translational Science (NCATS). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
The authors would like to thank Dr. Matthew Howard from the department of laboratory medicine and pathology and Dr. Sameer A. Parekh from the division of hematology, Mayo Clinic, for their contribution in the preparation of this manuscript.
Footnotes
Conflict of interest statement: None of the authors have any conflict of interest to disclose in regards to the current manuscript
References
- 1.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 2.Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51:189–199. [PubMed] [Google Scholar]
- 3.Ricci C, Fermo E, Corti S, et al. RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res. 2010;16:2246–2256. doi: 10.1158/1078-0432.CCR-09-2112. [DOI] [PubMed] [Google Scholar]
- 4.Ades L, Sekeres MA, Wolfromm A, et al. Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk Res. 2013;37:609–613. doi: 10.1016/j.leukres.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Patnaik MM, Lasho TL, Finke CM, et al. Spliceosome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: prevalence, clinical correlates, and prognostic relevance. Am J Hematol. 2013;88:201–206. doi: 10.1002/ajh.23373. [DOI] [PubMed] [Google Scholar]
- 6.Such E, Germing U, Malcovati L, et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood. 2013;121:3005–3015. doi: 10.1182/blood-2012-08-452938. [DOI] [PubMed] [Google Scholar]
- 7.Williamson PJ, Kruger AR, Reynolds PJ, et al. Establishing the incidence of myelodysplastic syndrome. Br J Haematol. 1994;87:743–745. doi: 10.1111/j.1365-2141.1994.tb06733.x. [DOI] [PubMed] [Google Scholar]
- 8.Rollison DE, Howlader N, Smith MT, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001–2004, using data from the NAACCR and SEER programs. Blood. 2008;112:45–52. doi: 10.1182/blood-2008-01-134858. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi K, Pemmaraju N, Strati P, et al. Clinical characteristics and outcomes of therapy-related chronic myelomonocytic leukemia. Blood. 2013;122:2807–2811. doi: 10.1182/blood-2013-03-491399. quiz 2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subari S, Patnaik M, Alfakara D, et al. Patients With Therapy-Related CMML Have Shorter Median Overall Survival Than Those With De Novo CMML: Mayo Clinic Long-Term Follow-Up Experience. Clin Lymphoma Myeloma Leuk. 2015;15:546–549. doi: 10.1016/j.clml.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Patnaik MM, Vallapureddy R, Yalniz FF, et al. Therapy related-chronic myelomonocytic leukemia (CMML): Molecular, cytogenetic, and clinical distinctions from de novo CMML. Am J Hematol. 2017 doi: 10.1002/ajh.24939. [DOI] [PubMed] [Google Scholar]
- 12.Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451–458. doi: 10.1111/j.1365-2141.1976.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 13.Patnaik MM, Parikh SA, Hanson CA, et al. Chronic myelomonocytic leukaemia: a concise clinical and pathophysiological review. Br J Haematol. 2014;165:273–286. doi: 10.1111/bjh.12756. [DOI] [PubMed] [Google Scholar]
- 14.Zahid MF, Barraco D, Lasho TL, et al. Spectrum of autoimmune diseases and systemic inflammatory syndromes in patients with chronic myelomonocytic leukemia. Leuk Lymphoma. 2017;58:1488–1493. doi: 10.1080/10428194.2016.1243681. [DOI] [PubMed] [Google Scholar]
- 15.Peker D, Padron E, Bennett JM, et al. A close association of autoimmune-mediated processes and autoimmune disorders with chronic myelomonocytic leukemia: observation from a single institution. Acta Haematol. 2015;133:249–256. doi: 10.1159/000365877. [DOI] [PubMed] [Google Scholar]
- 16.Mathew RA, Bennett JM, Liu JJ, et al. Cutaneous manifestations in CMML: Indication of disease acceleration or transformation to AML and review of the literature. Leuk Res. 2012;36:72–80. doi: 10.1016/j.leukres.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Patnaik MM, Wassie EA, Lasho TL, et al. Blast transformation in chronic myelomonocytic leukemia: Risk factors, genetic features, survival, and treatment outcome. Am J Hematol. 2015;90:411–416. doi: 10.1002/ajh.23962. [DOI] [PubMed] [Google Scholar]
- 18.Patnaik MM, Parikh SA, Hanson CA, et al. Chronic myelomonocytic leukaemia: a concise clinical and pathophysiological review. Br J Haematol. 2014;165:273–286. doi: 10.1111/bjh.12756. [DOI] [PubMed] [Google Scholar]
- 19.Pophali PA, Patnaik MM. The Role of New Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Cancer J. 2016;22:40–50. doi: 10.1097/PPO.0000000000000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Apperley JF, Gardembas M, Melo JV, et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med. 2002;347:481–487. doi: 10.1056/NEJMoa020150. [DOI] [PubMed] [Google Scholar]
- 21.Pardanani A, Ketterling RP, Li CY, et al. FIP1L1-PDGFRA in eosinophilic disorders: prevalence in routine clinical practice, long-term experience with imatinib therapy, and a critical review of the literature. Leuk Res. 2006;30:965–970. doi: 10.1016/j.leukres.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 22.Patnaik MM, Lasho TL, Finke CM, et al. Targeted next generation sequencing of PDGFRB rearranged myeloid neoplasms with monocytosis. Am J Hematol. 2016;91:E12–14. doi: 10.1002/ajh.24267. [DOI] [PubMed] [Google Scholar]
- 23.Tefferi A, Gilliland DG. Oncogenes in myeloproliferative disorders. Cell Cycle. 2007;6:550–566. doi: 10.4161/cc.6.5.3919. [DOI] [PubMed] [Google Scholar]
- 24.Pardanani A, Lasho T, Barraco D, et al. Next generation sequencing of myeloid neoplasms with eosinophilia harboring the FIP1L1-PDGFRA mutation. Am J Hematol. 2016;91:E10–11. doi: 10.1002/ajh.24273. [DOI] [PubMed] [Google Scholar]
- 25.Patnaik MM, Ketterling RP, Tefferi A. FGFR1 rearranged hematological neoplasms - molecularly defined and clinically heterogeneous. Leuk Lymphoma. 2018:1–3. doi: 10.1080/10428194.2018.1429607. [DOI] [PubMed] [Google Scholar]
- 26.Barraco D, Cerquozzi S, Gangat N, et al. Monocytosis in polycythemia vera: Clinical and molecular correlates. Am J Hematol. 2017;92:640–645. doi: 10.1002/ajh.24740. [DOI] [PubMed] [Google Scholar]
- 27.Tefferi A, Shah S, Mudireddy M, et al. Monocytosis is a powerful and independent predictor of inferior survival in primary myelofibrosis. Br J Haematol. 2017 doi: 10.1111/bjh.15061. [DOI] [PubMed] [Google Scholar]
- 28.Selimoglu-Buet D, Wagner-Ballon O, Saada V, et al. Characteristic repartition of monocyte subsets as a diagnostic signature of chronic myelomonocytic leukemia. Blood. 2015;125:3618–3626. doi: 10.1182/blood-2015-01-620781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:e74–80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 30.Patnaik MM, Timm MM, Vallapureddy R, et al. Flow cytometry based monocyte subset analysis accurately distinguishes chronic myelomonocytic leukemia from myeloproliferative neoplasms with associated monocytosis. Blood Cancer J. 2017;7:e584. doi: 10.1038/bcj.2017.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talati C, Zhang L, Shaheen G, et al. Monocyte subset analysis accurately distinguishes CMML from MDS and is associated with a favorable MDS prognosis. Blood. 2017;129:1881–1883. doi: 10.1182/blood-2016-12-753210. [DOI] [PubMed] [Google Scholar]
- 32.Thomas GD, Hamers AAJ, Nakao C, et al. Human Blood Monocyte Subsets: A New Gating Strategy Defined Using Cell Surface Markers Identified by Mass Cytometry. Arterioscler Thromb Vasc Biol. 2017;37:1548–1558. doi: 10.1161/ATVBAHA.117.309145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swerdlow S, Camp E, Harris NL, Jaffe ES, Stefano PA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon: International Agency for Research on Cancer; 2008. [Google Scholar]
- 34.Swederlow S, Camp E, Harris NL, Jaffe ES, Stefano PA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumors of the Haematopoietic and Lymphoid Tissue. Lyon: International Agency for Research on Cancer (IARC); 2008. [Google Scholar]
- 35.Bain BJ. What is a promonocyte? Am J Hematol. 2013 doi: 10.1002/ajh.23548. [DOI] [PubMed] [Google Scholar]
- 36.Onida F, Kantarjian HM, Smith TL, et al. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood. 2002;99:840–849. doi: 10.1182/blood.v99.3.840. [DOI] [PubMed] [Google Scholar]
- 37.Such E, Cervera J, Costa D, et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica. 2011;96:375–383. doi: 10.3324/haematol.2010.030957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang G, Zhang L, Fu B, et al. Cytogenetic risk stratification of 417 patients with chronic myelomonocytic leukemia from a single institution. Am J Hematol. 2014;89:813–818. doi: 10.1002/ajh.23751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wassie EA, Itzykson R, Lasho TL, et al. Molecular and prognostic correlates of cytogenetic abnormalities in chronic myelomonocytic leukemia: a Mayo Clinic-French Consortium Study. Am J Hematol. 2014;89:1111–1115. doi: 10.1002/ajh.23846. [DOI] [PubMed] [Google Scholar]
- 40.Merlevede J, Droin N, Qin T, et al. Mutation allele burden remains unchanged in chronic myelomonocytic leukaemia responding to hypomethylating agents. Nat Commun. 2016;7:10767. doi: 10.1038/ncomms10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ball M, List AF, Padron E. When clinical heterogeneity exceeds genetic heterogeneity: thinking outside the genomic box in chronic myelomonocytic leukemia. Blood. 2016;128:2381–2387. doi: 10.1182/blood-2016-07-692988. [DOI] [PubMed] [Google Scholar]
- 42.Abdel-Wahab O, Pardanani A, Patel J, et al. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia. 2011;25:1200–1202. doi: 10.1038/leu.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ernst T, Chase A, Zoi K, et al. Transcription factor mutations in myelodysplastic/myeloproliferative neoplasms. Haematologica. 2010;95:1473–1480. doi: 10.3324/haematol.2010.021808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Itzykson R, Kosmider O, Renneville A, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31:2428–2436. doi: 10.1200/JCO.2012.47.3314. [DOI] [PubMed] [Google Scholar]
- 45.Gelsi-Boyer V, Trouplin V, Roquain J, et al. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br J Haematol. 2010;151:365–375. doi: 10.1111/j.1365-2141.2010.08381.x. [DOI] [PubMed] [Google Scholar]
- 46.Grossmann V, Kohlmann A, Eder C, et al. Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80% of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2011;25:877–879. doi: 10.1038/leu.2011.10. [DOI] [PubMed] [Google Scholar]
- 47.Tefferi A, Lim KH, Abdel-Wahab O, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2009;23:1343–1345. doi: 10.1038/leu.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gelsi-Boyer V, Trouplin V, Adelaide J, et al. Genome profiling of chronic myelomonocytic leukemia: frequent alterations of RAS and RUNX1 genes. BMC cancer. 2008;8:299. doi: 10.1186/1471-2407-8-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohlmann A, Grossmann V, Klein HU, et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72. 8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010;28:3858–3865. doi: 10.1200/JCO.2009.27.1361. [DOI] [PubMed] [Google Scholar]
- 50.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 51.Makishima H, Cazzolli H, Szpurka H, et al. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol. 2009;27:6109–6116. doi: 10.1200/JCO.2009.23.7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daver N, Strati P, Jabbour E, et al. FLT3 mutations in myelodysplastic syndrome and chronic myelomonocytic leukemia. Am J Hematol. 2013;88:56–59. doi: 10.1002/ajh.23345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laborde RR, Patnaik MM, Lasho TL, et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia. 2013 doi: 10.1038/leu.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Damm F, Itzykson R, Kosmider O, et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 2013;27:1401–1403. doi: 10.1038/leu.2013.35. [DOI] [PubMed] [Google Scholar]
- 55.Itzykson R, Solary E. An evolutionary perspective on chronic myelomonocytic leukemia. Leukemia. 2013;27:1441–1450. doi: 10.1038/leu.2013.100. [DOI] [PubMed] [Google Scholar]
- 56.Patnaik MM, Itzykson R, Lasho TL, et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia. 2014;28:2206–2212. doi: 10.1038/leu.2014.125. [DOI] [PubMed] [Google Scholar]
- 57.Abdel-Wahab O, Adli M, LaFave LM, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22:180–193. doi: 10.1016/j.ccr.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Balasubramani A, Larjo A, Bassein JA, et al. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat Commun. 2015;6:7307. doi: 10.1038/ncomms8307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patnaik MM, Vallapureddy R, Lasho TL, et al. EZH2 mutations in chronic myelomonocytic leukemia cluster with ASXL1 mutations and their co-occurrence is prognostically detrimental. Blood Cancer J. 2018;8:12. doi: 10.1038/s41408-017-0045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamazaki J, Taby R, Vasanthakumar A, et al. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics. 2012;7:201–207. doi: 10.4161/epi.7.2.19015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abdel-Wahab O, Levine RL. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood. 2013;121:3563–3572. doi: 10.1182/blood-2013-01-451781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–147. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patnaik MM, Zahid MF, Lasho TL, et al. Number and type of TET2 mutations in chronic myelomonocytic leukemia and their clinical relevance. Blood Cancer J. 2016;6:e472. doi: 10.1038/bcj.2016.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patnaik MM, Lasho TL, Vijayvargiya P, et al. Prognostic interaction between ASXL1 and TET2 mutations in chronic myelomonocytic leukemia. Blood Cancer J. 2016;6:e385. doi: 10.1038/bcj.2015.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patnaik MM, Wassie EA, Padron E, et al. Chronic myelomonocytic leukemia in younger patients: molecular and cytogenetic predictors of survival and treatment outcome. Blood Cancer J. 2015;5:e280. doi: 10.1038/bcj.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bejar R, Lord A, Stevenson K, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014 doi: 10.1182/blood-2014-06-582809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lasho TL, Vallapureddy R, Finke CM, et al. Infrequent occurrence of TET1, TET3, and ASXL2 mutations in myelodysplastic/myeloproliferative neoplasms. Blood Cancer J. 2018;8:32. doi: 10.1038/s41408-018-0057-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15:152–165. doi: 10.1038/nrc3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patnaik MM, Barraco D, Lasho TL, et al. DNMT3A mutations are associated with inferior overall and leukemia-free survival in chronic myelomonocytic leukemia. Am J Hematol. 2017;92:56–61. doi: 10.1002/ajh.24581. [DOI] [PubMed] [Google Scholar]
- 70.Meggendorfer M, Roller A, Haferlach T, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML) Blood. 2012;120:3080–3088. doi: 10.1182/blood-2012-01-404863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patnaik MM, Lasho TL, Hodnefield JM, et al. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood. 2012;119:569–572. doi: 10.1182/blood-2011-09-377994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patnaik MM, Hanson CA, Sulai NH, et al. Prognostic irrelevance of ring sideroblast percentage in World Health Organization defined myelodysplastic syndromes without excess blasts. Blood. 2012 doi: 10.1182/blood-2012-03-415356. [DOI] [PubMed] [Google Scholar]
- 73.Aribi A, Borthakur G, Ravandi F, et al. Activity of decitabine, a hypomethylating agent, in chronic myelomonocytic leukemia. Cancer. 2007;109:713–717. doi: 10.1002/cncr.22457. [DOI] [PubMed] [Google Scholar]
- 74.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 75.Ricci C, Fermo E, Corti S, et al. RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res. 2010;16:2246–2256. doi: 10.1158/1078-0432.CCR-09-2112. [DOI] [PubMed] [Google Scholar]
- 76.Kuo MC, Liang DC, Huang CF, et al. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia. 2009;23:1426–1431. doi: 10.1038/leu.2009.48. [DOI] [PubMed] [Google Scholar]
- 77.Jankowska AM, Makishima H, Tiu RV, et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood. 2011;118:3932–3941. doi: 10.1182/blood-2010-10-311019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith AE, Mohamedali AM, Kulasekararaj A, et al. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood. 2010;116:3923–3932. doi: 10.1182/blood-2010-03-274704. [DOI] [PubMed] [Google Scholar]
- 79.Itzykson R, Kosmider O, Renneville A, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121:2186–2198. doi: 10.1182/blood-2012-06-440347. [DOI] [PubMed] [Google Scholar]
- 80.Patnaik MM, Tefferi A. Cytogenetic and molecular abnormalities in chronic myelomonocytic leukemia. Blood Cancer J. 2016;6:e393. doi: 10.1038/bcj.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kunimoto H, Meydan C, Nazir A, et al. Cooperative Epigenetic Remodeling by TET2 Loss and NRAS Mutation Drives Myeloid Transformation and MEK Inhibitor Sensitivity. Cancer Cell. 2018;33:44–59. e48. doi: 10.1016/j.ccell.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Padron E, Garcia-Manero G, Patnaik MM, et al. An international data set for CMML validates prognostic scoring systems and demonstrates a need for novel prognostication strategies. Blood Cancer J. 2015;5:e333. doi: 10.1038/bcj.2015.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Patnaik MM, Tefferi A. Chronic Myelomonocytic Leukemia: Focus on Clinical Practice. Mayo Clinic proceedings. 2016;91:259–272. doi: 10.1016/j.mayocp.2015.11.011. [DOI] [PubMed] [Google Scholar]
- 84.Germing U, Strupp C, Aivado M, et al. New prognostic parameters for chronic myelomonocytic leukemia. Blood. 2002;100:731–732. doi: 10.1182/blood-2002-01-0330. author reply 732–733. [DOI] [PubMed] [Google Scholar]
- 85.Kantarjian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113:1351–1361. doi: 10.1002/cncr.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Patnaik MM, Padron E, LaBorde RR, et al. Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. Leukemia. 2013;27:1504–1510. doi: 10.1038/leu.2013.88. [DOI] [PubMed] [Google Scholar]
- 87.Elena C, Galli A, Such E, et al. Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood. 2016;128:1408–1417. doi: 10.1182/blood-2016-05-714030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Germing U, Gattermann N, Minning H, et al. Problems in the classification of CMML--dysplastic versus proliferative type. Leukemia research. 1998;22:871–878. doi: 10.1016/s0145-2126(97)00192-6. [DOI] [PubMed] [Google Scholar]
- 89.Nosslinger T, Reisner R, Gruner H, et al. Dysplastic versus proliferative CMML--a retrospective analysis of 91 patients from a single institution. Leukemia research. 2001;25:741–747. doi: 10.1016/s0145-2126(01)00014-5. [DOI] [PubMed] [Google Scholar]
- 90.Breccia M, Latagliata R, Mengarelli A, et al. Prognostic factors in myelodysplastic and myeloproliferative types of chronic myelomonocytic leukemia: a retrospective analysis of 83 patients from a single institution. Haematologica. 2004;89:866–868. [PubMed] [Google Scholar]
- 91.Cambier N, Wattel E, Menot ML, et al. All-trans retinoic acid in adult chronic myelomonocytic leukemia: results of a pilot study. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 1996;10:1164–1167. [PubMed] [Google Scholar]
- 92.Gerhartz HH, Marcus R, Delmer A, et al. A randomized phase II study of low-dose cytosine arabinoside (LD-AraC) plus granulocyte-macrophage colony-stimulating factor (rhGM-CSF) in myelodysplastic syndromes (MDS) with a high risk of developing leukemia. EORTC Leukemia Cooperative Group Leukemia. 1994;8:16–23. [PubMed] [Google Scholar]
- 93.Venditti A, Tamburini A, Buccisano F, et al. A phase-II trial of all trans retinoic acid and low-dose cytosine arabinoside for the treatment of high-risk myelodysplastic syndromes. Annals of hematology. 2000;79:138–142. doi: 10.1007/s002770050569. [DOI] [PubMed] [Google Scholar]
- 94.Beran M, Estey E, O’Brien SM, et al. Results of topotecan single-agent therapy in patients with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk Lymphoma. 1998;31:521–531. doi: 10.3109/10428199809057611. [DOI] [PubMed] [Google Scholar]
- 95.Beran M, Estey E, O’Brien S, et al. Topotecan and cytarabine is an active combination regimen in myelodysplastic syndromes and chronic myelomonocytic leukemia. J Clin Oncol. 1999;17:2819–2830. doi: 10.1200/JCO.1999.17.9.2819. [DOI] [PubMed] [Google Scholar]
- 96.Quintas-Cardama A, Kantarjian H, O’Brien S, et al. Activity of 9-nitro-camptothecin, an oral topoisomerase I inhibitor, in myelodysplastic syndrome and chronic myelomonocytic leukemia. Cancer. 2006;107:1525–1529. doi: 10.1002/cncr.22186. [DOI] [PubMed] [Google Scholar]
- 97.Feldman EJ, Cortes J, DeAngelo DJ, et al. On the use of lonafarnib in myelodysplastic syndrome and chronic myelomonocytic leukemia. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2008;22:1707–1711. doi: 10.1038/leu.2008.156. [DOI] [PubMed] [Google Scholar]
- 98.Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 99.Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 100.Ades L, Sekeres MA, Wolfromm A, et al. Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk Res. 2013;37:609–613. doi: 10.1016/j.leukres.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 101.Braun T, Itzykson R, Renneville A, et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: a phase 2 trial. Blood. 2011;118:3824–3831. doi: 10.1182/blood-2011-05-352039. [DOI] [PubMed] [Google Scholar]
- 102.Costa R, Abdulhaq H, Haq B, et al. Activity of azacitidine in chronic myelomonocytic leukemia. Cancer. 2011;117:2690–2696. doi: 10.1002/cncr.25759. [DOI] [PubMed] [Google Scholar]
- 103.Fianchi L, Criscuolo M, Breccia M, et al. High rate of remissions in chronic myelomonocytic leukemia treated with 5-azacytidine: results of an Italian retrospective study. Leuk Lymphoma. 2013;54:658–661. doi: 10.3109/10428194.2012.719617. [DOI] [PubMed] [Google Scholar]
- 104.Garcia-Manero G, Gore SD, Cogle C, et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol. 2011;29:2521–2527. doi: 10.1200/JCO.2010.34.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thorpe M, Montalvao A, Pierdomenico F, et al. Treatment of chronic myelomonocytic leukemia with 5-Azacitidine: a case series and literature review. Leuk Res. 2012;36:1071–1073. doi: 10.1016/j.leukres.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 106.Wijermans PW, Ruter B, Baer MR, et al. Efficacy of decitabine in the treatment of patients with chronic myelomonocytic leukemia (CMML) Leuk Res. 2008;32:587–591. doi: 10.1016/j.leukres.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 107.Wong E, Seymour JF, Kenealy M, et al. Treatment of chronic myelomonocytic leukemia with azacitidine. Leuk Lymphoma. 2013;54:878–880. doi: 10.3109/10428194.2012.730615. [DOI] [PubMed] [Google Scholar]
- 108.Padron E, Dezern A, Andrade-Campos M, et al. A Multi-Institution Phase 1 Trial of Ruxolitinib in Patients with Chronic Myelomonocytic Leukemia (CMML) Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yoshimi A, Balasis ME, Vedder A, et al. Robust patient-derived xenografts of MDS/MPN overlap syndromes capture the unique characteristics of CMML and JMML. Blood. 2017;130:397–407. doi: 10.1182/blood-2017-01-763219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Padron E, Painter JS, Kunigal S, et al. GM-CSF-dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood. 2013;121:5068–5077. doi: 10.1182/blood-2012-10-460170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Eissa H, Gooley TA, Sorror ML, et al. Allogeneic hematopoietic cell transplantation for chronic myelomonocytic leukemia: relapse-free survival is determined by karyotype and comorbidities. Biol Blood Marrow Transplant. 2011;17:908–915. doi: 10.1016/j.bbmt.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Elliott MA, Tefferi A, Hogan WJ, et al. Allogeneic stem cell transplantation and donor lymphocyte infusions for chronic myelomonocytic leukemia. Bone Marrow Transplant. 2006;37:1003–1008. doi: 10.1038/sj.bmt.1705369. [DOI] [PubMed] [Google Scholar]
- 113.Krishnamurthy P, Lim ZY, Nagi W, et al. Allogeneic haematopoietic SCT for chronic myelomonocytic leukaemia: a single-centre experience. Bone Marrow Transplant. 2010;45:1502–1507. doi: 10.1038/bmt.2009.375. [DOI] [PubMed] [Google Scholar]
- 114.Kroger N, Zabelina T, Guardiola P, et al. Allogeneic stem cell transplantation of adult chronic myelomonocytic leukaemia. A report on behalf of the Chronic Leukaemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT) Br J Haematol. 2002;118:67–73. doi: 10.1046/j.1365-2141.2002.03552.x. [DOI] [PubMed] [Google Scholar]
- 115.Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009;114:1545–1552. doi: 10.1182/blood-2009-03-206672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ocheni S, Kroger N, Zabelina T, et al. Outcome of allo-SCT for chronic myelomonocytic leukemia. Bone Marrow Transplant. 2009;43:659–661. doi: 10.1038/bmt.2008.366. [DOI] [PubMed] [Google Scholar]
- 117.Perez Botero J, Oliveira JL, Chen D, et al. ASXL1 mutated chronic myelomonocytic leukemia in a patient with familial thrombocytopenia secondary to germline mutation in ANKRD26. Blood Cancer J. 2015;5:e315. doi: 10.1038/bcj.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Park S, Labopin M, Yakoub-Agha I, et al. Allogeneic stem cell transplantation for chronic myelomonocytic leukemia: a report from the Societe Francaise de Greffe de Moelle et de Therapie Cellulaire. Eur J Haematol. 2013;90:355–364. doi: 10.1111/ejh.12073. [DOI] [PubMed] [Google Scholar]
- 119.Cutler CS, Lee SJ, Greenberg P, et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood. 2004;104:579–585. doi: 10.1182/blood-2004-01-0338. [DOI] [PubMed] [Google Scholar]
- 120.Koreth J, Pidala J, Perez WS, et al. Role of reduced-intensity conditioning allogeneic hematopoietic stem-cell transplantation in older patients with de novo myelodysplastic syndromes: an international collaborative decision analysis. J Clin Oncol. 2013;31:2662–2670. doi: 10.1200/JCO.2012.46.8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Damaj G, Duhamel A, Robin M, et al. Impact of azacitidine before allogeneic stem-cell transplantation for myelodysplastic syndromes: a study by the Societe Francaise de Greffe de Moelle et de Therapie-Cellulaire and the Groupe-Francophone des Myelodysplasies. J Clin Oncol. 2012;30:4533–4540. doi: 10.1200/JCO.2012.44.3499. [DOI] [PubMed] [Google Scholar]
- 122.Kongtim P, Popat U, Jimenez A, et al. Treatment with Hypomethylating Agents before Allogeneic Stem Cell Transplant Improves Progression-Free Survival for Patients with Chronic Myelomonocytic Leukemia. Biol Blood Marrow Transplant. 2016;22:47–53. doi: 10.1016/j.bbmt.2015.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Savona MR, Malcovati L, Komrokji R, et al. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood. 2015;125:1857–1865. doi: 10.1182/blood-2014-10-607341. [DOI] [PMC free article] [PubMed] [Google Scholar]