

Abstract
Determining the fitness of viral genotypes has become a standard practice in virology as it is essential to evaluate their evolutionary potential. Darwinian fitness, defined as the advantage of a given genotype with respect to a reference one, is a complex property that captures, in a single figure, differences in performance at every stage of viral infection. To what extent does viral fitness result from specific molecular interactions with host factors and regulatory networks during infection? Can we identify host genes in functional classes whose expression depends on viral fitness? Here, we compared the transcriptomes of tobacco plants infected with seven genotypes of tobacco etch potyvirus that differ in fitness. We found that the larger the fitness differences among genotypes, the more dissimilar the transcriptomic profiles are. Consistently, two different mutations, one in the viral RNA polymerase and another in the viral suppressor of RNA silencing, resulted in significantly similar gene expression profiles. Moreover, we identified host genes whose expression showed a significant correlation, positive or negative, with the virus' fitness. Differentially expressed genes which were positively correlated with viral fitness activate hormone- and RNA silencing-mediated pathways of plant defense. In contrast, those that were negatively correlated with fitness affect metabolism, reducing growth, and development. Overall, these results reveal the high information content of viral fitness and suggest its potential use to predict differences in genomic profiles of infected hosts.
Keywords: host–virus interaction; Nicotiana tabacum, Potyvirus; response to infection; systems biology; TEV; transcriptomics; virus evolution; viral fitness
Introduction
Fitness is a complex parameter often used by evolutionary biologists and ecologists to quantitatively describe the reproductive ability and evolutionary potential of an organism in a particular environment (Linnen and Hoekstra 2009; Orr 2009). Despite this apparently simple definition, measuring fitness is difficult and most studies only measure one or more fitness components (e.g., survival to maturity, fecundity, number of mates, or number of offspring produced) as proxies to total fitness (Linnen and Hoekstra 2009; Orr 2009). In the field of virology, it has become standard to measure fitness by growth-competition experiments in mixed infections with a reference strain (Holland et al. 1991; Wargo and Kurath 2012). With this experimental set up, fitness is just the relative ability of a viral strain to produce stable infectious progeny in a given host (cell type, organ, individual, or species) when resources have to be shared with a competitor (Domingo and Holland 1997). Regardless its limitations, this approach provides a metric for ranking viral strains according to their performance in a particular environment/host. Such a fitness measure has been pivotal for quantitatively understanding many virus evolution processes: the effect of genetic bottlenecks and accumulation of deleterious mutations (Chao 1990; Duarte et al. 1992; De la Iglesia and Elena 2007), the rates and dynamics of adaptive evolution into novel hosts (Novella, Duarte, et al. 1995), the pleiotropic cost of host range expansion (Novella, Clarke, et al. 1995; Turner and Elena 2000; Lalić et al. 2011), the cost of genome complexity (Pesko et al. 2015; Willemsen et al. 2016), the cost of antiviral escape mutations (Novella et al. 2005; Westerhout et al. 2005; Martínez-Picado and Martínez 2008), the topography of adaptive fitness landscapes (Da Silva and Wyatt 2014; Lalić and Elena 2015; Cervera et al. 2016), and the role of robustness in virus evolution (Codoñer et al. 2006; Sanjuán et al. 2007; Novella et al. 2013).
But differences in viral fitness should also matter in genome-wide studies seeking to understand the mode of action of the viruses (i.e., the precise way they interact with their hosts). It has been argued that an integrative systems biology approach to viral pathogenesis would result in a better understanding of pathogenesis and in the identification of common targets for different viruses, therefore serving as a guide to a more rational design of therapeutic drugs (Tan et al. 2007; Viswanathan and Früh 2007; Bailer and Haas 2009; Barabási et al. 2011; Friedel and Haas 2011; Elena and Rodrigo 2012; Finzer 2017). Pioneering studies have ignored the high genetic variability of viruses in fitness and in mode of action. Experimental evidence supports that even single nucleotide substitutions have significant effects on viral fitness regardless of whether they are synonymous or nonsynonymous, or they affect coding or noncoding genomic regions (Sanjuán et al. 2004; Carrasco, De la Iglesia, et al. 2007; Domingo-Calap et al. 2009; Peris et al. 2010; Acevedo et al. 2014; Bernet and Elena 2015; Visher et al. 2016). A common trend among all these studies is that, whenever fitness is evaluated in the standard host, the distribution of mutational effects is highly skewed toward deleterious effects, with a large fraction of mutations being lethal. Furthermore, increasing evidence suggests that the distribution of fitness effects increases as the genetic divergence among two hosts (e.g., a tested host and the natural one) increases (Lalić et al. 2011; Vale et al. 2012; Lalić and Elena 2013; Cervera et al. 2016). Together, all these observations suggest that the fitness of a given viral genotype depends not only on its own genetic background but also on the host where fitness is evaluated. Arguably, differences in viral fitness reflect differences in the virus–host interaction. As cellular parasites, viruses need to utilize all sort of cellular factors and resources, reprogram gene expression patterns into their own benefit, and block and interfere with cellular defenses. All these processes take place in the host complex network of intertwined interactions and regulations. Interacting in suboptimal ways with any of the elements of the host network may have profound effects in the progression of a successful infection and therefore in viral fitness; inefficient interactions may result in attenuated or even abortive infections.
Little is known about how viral fitness informs about the underlying changes occurring in host gene expression and protein function at a genome-wide scale. In this work, we have investigated the potential association between viral fitness and host transcriptional regulation upon infection as a first step into this direction. We have characterized the transcriptomic profiles of Nicotiana tabacum L. var Xanthi NN plants inoculated with a collection of genotypes of Tobacco etch virus (TEV; genus Potyvirus, family Potyviridae) that differ in their fitness in this natural host. Analyses of gene expression data allowed us to characterize differential gene expression upon infection with different TEV genotypes, as well as to identify sets of candidate genes whose expressions positively or negatively correlated with the magnitude of TEV fitness. Differences in expression for representative genes from these two categories were experimentally validated by an alternative method.
Results and Discussion
Differences in Viral Fitness and Host Symptomatology
Figure 1 shows relevant information about the seven TEV genotypes used for this study. The mutant genotypes differ from the wild-type (WT) genotype in a rather limited number of nonsynonymous mutations (1 or 2). However, their fitness values and the severity of symptoms induced differ widely. In this study, the fitness of each mutant genotype was estimated as the ratio of Malthusian growth rates of the mutant and the WT (see Materials and Methods for details). Significant differences existed among the fitness values of the seven selected viral genotypes (fig. 1B; likelihood ratio test: χ2 = 373.006, 6 df, P < 0.001) and among plants inoculated with the same viral genotype (χ2 = 2927.885, 14 df, P < 0.001). As a measure of the quality of data, the percentage of total variance for relative fitness explained by true genetic differences among genotypes was 70.56%, whereas differences among plants inoculated with the same viral genotype accounted for 29.16% of the observed variance. The remaining 0.28% of the observed variability was assigned to error measurements. A Bonferroni post hoc test classifies the seven genotypes into five groups (labeled as a to f in fig. 1B). Interestingly, the three genotypes with the lowest fitness values (AS13, CLA2, and CLA11) contain mutations in the multifunctional protein HC-Pro, whose most relevant role during infection is to serve as suppressor of the RNA-silencing (VSR) defense (Revers and García 2015). These mutants were originally described as hyposupressors (Torres-Barceló et al. 2008), meaning that the suppressor activity of their HC-Pro was significantly reduced compared with the WT protein. The two genotypes with mutations in the CI protein (PC55 and PC48), a helicase also involved in cell-to-cell movement (Revers and García 2015), have the mildest deleterious fitness effects. Mutant PC95, which contains a point mutation in the replicase NIb protein (Revers and García 2015) occupies an intermediate position in the fitness scale (fig. 1B).
Fig. 1.
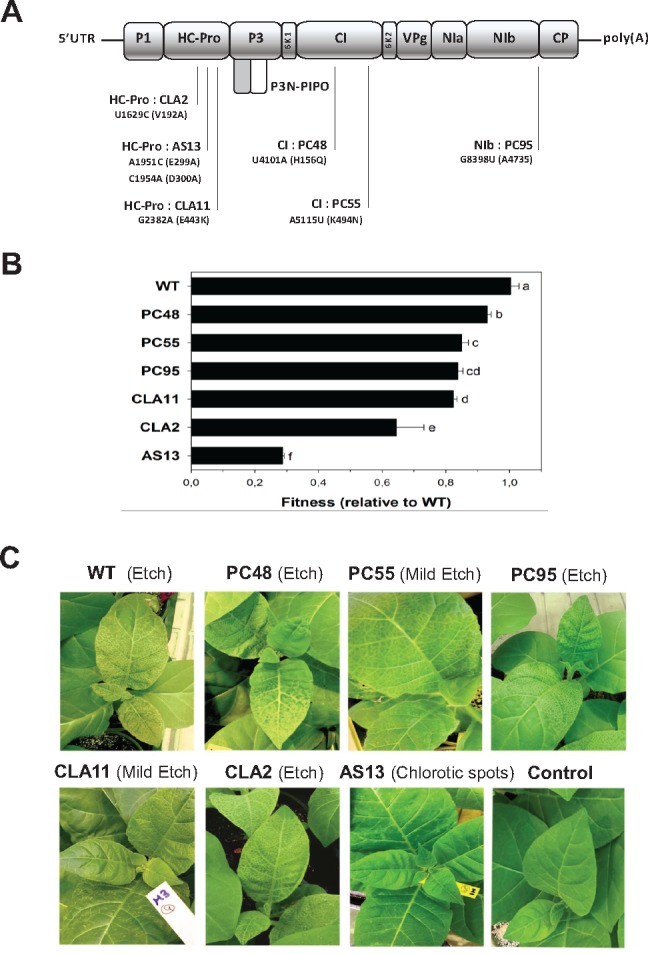
Properties of the TEV mutants used in this study. (A) Schematic representation of the TEV genome together with the location of the mutations used in this study. The corresponding amino acid replacement(s) are indicated within parenthesis. (B) Fitness values estimated for each genotype in the natural host Nicotiana tabacum. Error bars represent ±1 SEM. Letters (a–f) next to the bars define the homogenous groups identified using a post hoc Bonferroni test. (C) Representative pictures of the symptomatology induced by each genotype in tobacco plants. Picture labeled as “Control” shows a mock-inoculated plant. For CLA11 and AS13 mutants, arrows point to symptoms.
Figure 1C illustrates the differences in symptoms induced by each one of the seven genotypes. Symptoms ranged from the asymptomatic infection or local chlorotic spots characteristic of mutant AS13, the mild etching of mutant CLA11 and the severe etching induced by the WT and the other mutants. No correlation exists between virus fitness and symptoms, a finding previously reported for this experimental system (Carrasco, De la Iglesia, et al. 2007).
Differences in Viral Fitness Are Associated with Differences in the Magnitude of the Change in the Host Transcriptome
First, we sought to test whether differences in TEV fitness might be associated with differences in the gene expression profiles of infected plants. We hypothesized that viral fitness results from a particular interaction between virus and host factors and assumed that the outcome of infection of a WT virus in its natural host results from an optimal (from the virus perspective) modulation of the host’s gene expression profile. As viral fitness is reduced, interactions are less optimal and, consequently, the gene expression profile of the plant will be increasingly different from that resulting from the infection with the WT virus. To test this hypothesis, we infected N. tabacum plants with each one of the seven TEV genotypes described earlier. Eight-day postinoculation (dpi) symptomatic tissues were collected for all mutants except for the very low fitness mutant AS13, for which tissues were collected 15 dpi because the delay in symptoms appearance and severity (fig. 1C). Total RNAs were extracted, normalized, and used to hybridize N. tabacum Gene Expression 4 × 44 K Microarrays (Agilent). Slides were handled as described in the Materials and Methods section; intensity signals were normalized using tools in BABELOMICS (Alonso et al. 2015). Normalized expression data are contained in supplementary file S1, Supplementary Material online. Figure 2A shows the clustering (unweighted average distance method; UPGMA) of average expression data for those genes that significantly changed expression (±2-fold) among plants infected with the seven viral genotypes (1-way ANOVAs with false discovery rate (FDR) correction; overall P < 0.05) relative to the mock-inoculated plants.
Fig. 2.

Analyses of the transcriptomic data from tobacco plants infected with each TEV genotype. (A) Heat-map representation of gene expression values (infected vs. mock-inoculated control plants) for all viral genotypes. Only genes that show significant differences among all infections (one-way ANOVA with FDR, adjusted P < 0.05) are included in the heat-map. Hierarchical clustering of genes done with UPGMA by using the correlations between all pairs of mean profiles as distance metric. Genes down-regulated (in blue) mainly correspond to metabolic and developmental processes, whereas genes up-regulated (in red) mainly correspond to stress responses. (B) TEV genotypes clustered (UPGMA) according to the similarity of the mean expression profiles of plants infected with each one of them. The heat-map represents the value of the Pearson’s correlation coefficient between pairs of mean profiles. (C) Representation of the three major principal components from the data shown in panel (B). The three first PCs explain up to 93% of the total observed variance. Lines link each genotype with the centroid of the 3D space. The arrow represents a putative trajectory of increasing viral fitness. (D) Association between viral fitness and the magnitude of the perturbation (vs. mock-inoculated control plants) both relative to WT (P = 0.005). (E) Association between viral fitness and the distance of each genotype to the WT (P = 0.003), from the dendrogram shown in panel (B).
Regarding individual genes, two major clusters can be distinguished, one corresponding to the overexpression of genes related to stress response and a second one corresponding to the underexpression of genes involved with metabolism and plant development. To further explore the similarity in the perturbation induced by each viral genotype into the plants’ transcriptome, we computed all pairwise Pearson product-moment correlation coefficients (r) between the mean expression values for all genes in the microarray. These correlations were used as a measure of similarity to build a UPGMA dendrogram. The rationale for this analysis is as follows: the more correlated two expression profiles are, the more similar the effects induced in infected plants. When comparing expression profiles from a pair of infected plants, a high correlation may indicate that genes that changed expression relative to the mock-inoculated plants, are exactly the same in both samples, showing a similar expression pattern. Conversely, if genes with differential expression do not match in the two profiles being compared, then the correlation will be low. Figure 2B shows both the heat-map of the correlation coefficients and the resulting dendrogram. Three clusters result from this analysis (fig. 2B). The first cluster is constituted by the three viral genotypes with the higher fitness values, that is, WT, PC55, and PC48. Genotypes of intermediate fitness CLA11, PC95, and CLA2 constitute a second cluster. Finally, plants infected with AS13 show the most dissimilar gene expression profile. The heat-map is shown with viral genotypes ordered according to the UPGMA clustering. Correlations decreased as the distance in the cladogram increases. Within clusters, r > 0.85, whereas between clusters the correlations ranged between 0.65 < r < 0.75, except for plants infected with AS13, whose similarity with other infected plants was r < 0.65.
Next, to further investigate the similarity between expression profiles of plants infected with different TEV genotypes, we performed a principal components (pc) analysis of all the gene expression data. The percentage of total observed variance explained by the first three components was ∼93% (the first pc itself explained 81%). Figure 2C shows the distribution of values in the space defined by the three first principal components. Results are equivalent to those obtained with the two previous clustering methods, where genotypes are classified into three groups. WT, PC55, and PC48 are closer in the space and characterized by positive values of first pc but negative values of the second and third pcs. CLA11, PC95, and CLA2 form a second group, with positive values of first and second pcs but negative values of the third. As before, AS13 effect on host transcriptome is clearly different, and has negative values of the first and second pcs but positive of the third. Interestingly, figure 2C shows that genotypes are located in this principal component space following a trajectory of increasing fitness values (indicated by the arrow in fig. 2C). Along this trajectory, pcs switch sign in different directions. This transition suggests that the over- or under-expression of a set of genes is associated with particular levels of viral fitness: low fitness AS13 is characterized by a positive third pc and a negative first pc while high fitness viruses are characterized by the opposite sign. That is, over- or under-expressed genes are not progressively accumulated as long as viral fitness changes. These genes will be evaluated in the following sections.
Following from our working hypothesis, if the WT virus has evolved to optimize its interaction with the host, it is logical that small departures in viral fitness will be associated with small deviations between the transcriptomes of plants infected with the WT virus and with viruses whose fitness is close to the WT. Conversely, the less similar fitness between the WT and mutant viruses, the more dissimilar would be the transcriptional profiles of infected plants. To test this prediction, we have explored the following: 1) the correlation between the similarity of transcriptional profiles of plants infected with the WT TEV and with each mutant (again using Pearson’s r) and fitness and 2) the correlation between the distance from WT in the cladogram shown in figure 2B and fitness. The results of these analyses are shown in figure 3D and E. As expected, both correlations were significant (r = 0.826, P = 0.005 and r = −0.857, P = 0.003, respectively; in both cases 5 df) and of the expected sign.
Fig. 3.

Comparative analysis of differential gene expression in plants infected with the distinct TEV genotypes. (A) Distribution of the number of DEGs (up- and down-regulated vs. mock-inoculated control plants) for each TEV genotype. (B) Heat-map of the degree of overlapping between two lists of DEGs. Three different groups are highlighted (dashed squares). (C) Distribution of the number of DEGs (up- and down-regulated vs. plants infected with the WT TEV) for each TEV genotype. (D) Correlation plot between the total number of DEGs relative to WT TEV infection and the fitness of each TEV genotype (P < 0.001).
The Number of Altered Genes Depends on Viral Fitness
Figure 3A (data contained in supplementary file S2, Supplementary Material online) shows the number of differentially expressed genes (DEG), both up- and down-expressed, relative to the transcriptome of mock-inoculated plants. The number of down-expressed DEGs ranges between 531 (for AS13) and 2,809 (for CLA11), while in the case of up-expressed DEGs the range is slightly narrower: from 781 (AS13) to 2,696 (CLA11). Figure 3B illustrates the number of DEGs in common between all pairs of transcriptomes from infected plants. The heat-map shows a pattern of modularity, with three well-defined modules. The first module contains the three viruses with highest fitness (WT, PC48, and PC55), the second module contains the three viruses with intermediate fitness (PC95, CLA11, and CLA2), and the very low fitness genotype AS13 is the only member of the third module. The number of shared DEGs within each of these modules is > 75% of total. The number of shared DEGs between modules drops <60%.
Next, following the same rationale as the previous section, we sought to determine whether the number of DEGs also depends on the difference in fitness from WT. In this case, we hypothesized that the overlap in the lists of DEGs must be similar for WT and viruses of equivalent fitness (e.g., PC48 or PC55), whereas the magnitude of the overlap between DEG lists would decrease as differences in fitness became larger. Figure 3C shows the counts of DEGs that are differentially expressed (for up- and down-expressed genes) between WT and the other six viral genotypes. As expected, PC48, PC55, and PC95 alter the same genes, though in a different magnitude (as shown in the previous section). The number of genes that are not in common with WT increases from CLA11 (502), CLA2 (969), and AS13 (2001). A highly significant negative correlation exists between the number of differences in DEGs list (adding up the number of up- and down-expressed DEGs in fig. 3C) and differences in viral fitness (fig. 3D:r = −0.937, 5 df, P < 0.001).
Of particular interest is the similarity between PC95, a mutant of the replicase NIb gene, and CLA11, a mutant of the VSR HC-Pro gene. These two mutations led to close fitness values (fig. 3A), but also resulted in significantly similar gene expression profiles (fig. 3B). At first sight, one may argue that their impact in transcriptomic profiles should be different since these mutations affect virus proteins that are functionally unrelated. However, our results suggest that the effects on the overall virus–host interaction of each mutant are canalized in the same way. This clearly shows that viral fitness contains high information about the virus–host interaction.
Viral Fitness Conditions the Functional Categories That Are Altered
Lists of genes are difficult to interpret and functional analyses provide a good tool to cluster genes into groups with related functions. To this end, we performed an analysis of enriched functional categories (GO terms) for each viral genotype. Figure 4 illustrates the way that plants infected with each one of the seven TEV genotypes differ in the functional categories significantly overrepresented relative to the mock-inoculated plants. Figure 4 shows the GO terms ordered from the highest to the lowest viral fitness. The upper plane shows the functional categories altered in plants infected with WT TEV, with metabolic process (GO: 0008152) containing the largest number and photosynthesis (GO: 0015979) the smallest. Regulation of response to biotic stimulus (GO: 0002831), defense response (GO: 006952), immune system process (GO: 0002376), protein modification process (GO: 0036211), hormone-mediated signaling (GO: 0009755), and cell death (GO: 008219) are all enriched in up-expressed DEGs, while photosynthesis, lipid metabolic process (GO: 0006629), and regulation of nitrogen compound metabolic process (GO: 0051171) are categories significantly enriched in down-expressed DEGs. Categories such as metabolic process, unspecific response to stress (GO: 0006950), response to stimulus (GO: 0050896), response to abiotic stimulus (GO: 0009628), localization (GO: 0051641), or transport (GO: 0008150) are enriched in both types of DEGs. Therefore, overall speaking, genes involved in different aspects of plant defense pathways and response to infection are up-expressed, whereas genes involved in metabolism and photosynthesis are down-expressed.
Fig. 4.
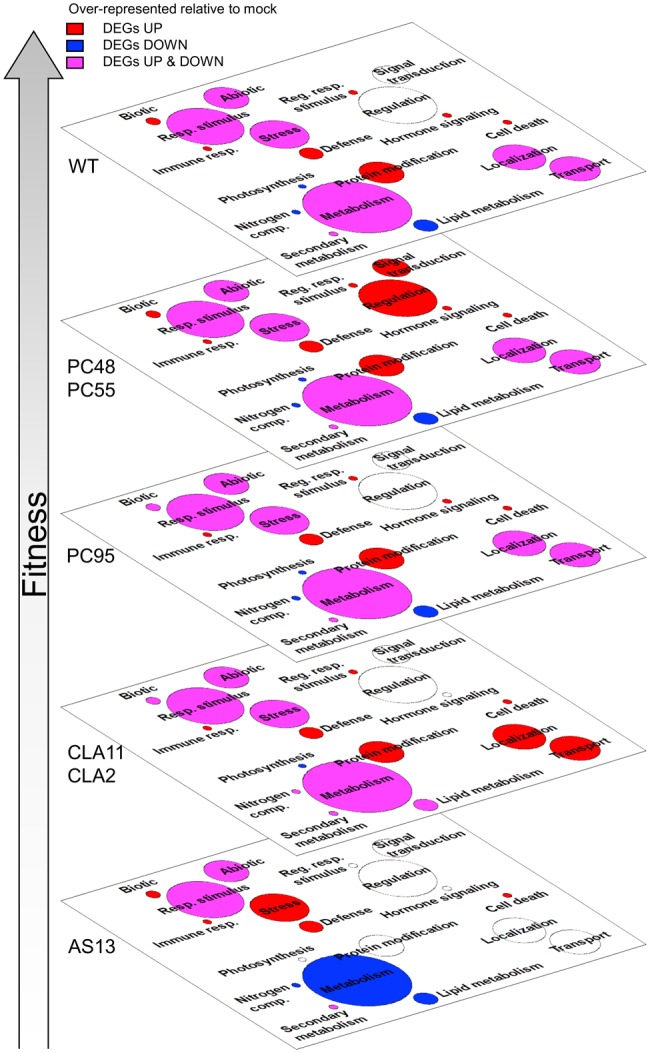
Functional analysis associated to differential gene expression. Artwork of meaningful biological processes (in a plane). Categories that are overrepresented in the two lists of DEGs for each TEV genotype (up- and down-regulated), either in one of them or in both, are indicated with different colors. In red, we represent categories that are significantly enriched by up-expressed DEGs, in blue categories that are significantly enriched by down-expressed DEGs, and in pink categories enriched in both types of DEGs; the surface of each circle is proportional to the number of DEGs included in each category. Enrichments were evaluated by Fisher’s exact tests with FDR (adjusted P < 0.05). The different planes are organized according to viral fitness. We considered infected versus mock-inoculated control plants (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.).
The second fitness plane corresponds to genotypes PC48 and PC55, both of mild effect and carrying mutation in the CI gene. The most remarkable difference between these two genotypes and the rest of genotypes is the significant enrichment in up-expressed DEGs related to signal transduction (GO: 0007165) and regulation of gene expression (GO: 0010468). Drifting down in the virus fitness scale, the next plane in figure 4 corresponds to genotype PC95, which shows a similar distribution of GOs as the WT except for a lack of enrichment in up-expressed DEGs in the regulation of response to biotic stress category. Next plane corresponds to genotypes CLA2 and CLA11 hyposuppressors of moderate fitness and carrying point mutations in the HC-Pro gene. Plants infected with these two viral genotypes differ from plants infected with the WT virus in three main functional categories: the loss of significant enrichment in the hormone-mediated signaling category, and a significant enrichment in up-expressed DEGs into the localization and transport categories. Finally, the bottom plane in the fitness scale corresponds to genotype AS13 (fig. 4), which has a very week VSR activity, very low fitness and induces no symptoms or very mild symptoms. These differences in fitness and severity of symptoms have a direct translate into the enrichment of the different functional categories. Compared with WT, metabolic process is enriched with down-expressed DEGs while nonspecific response to stress is very much enriched now in up-expressed DEGs, and response to stimulus, protein modification process, localization, and transport are not enriched in any particular type of DEGs. Moreover, no significant enrichment in the hormone-mediated signaling module was found in plants infected with AS13, or for the other HC-Pro mutant genotypes CLA2 and CLA11.
Correlation between Gene Expression and Viral Fitness
Taken together, the results shown in the previous sections suggest that the transcriptomic response of plants to infection varies with the fitness of the virus being inoculated. This observation motivated us to identify genes whose expression significantly correlates with viral fitness; that is, systematic changes in virus fitness are associated with an increase or decrease in the expression level of a particular gene. This is a correlation analysis and as such does not assume a functional dependence between viral fitness and the expression of individual host genes. Yet, it may provide a list of candidate genes to be considered as determinants of viral fitness. We computed a nonparametric Spearman’s correlation coefficient between viral fitness and the normalized degree of expression (z-score) for each one of the previously characterized DEGs (fig. 4A). A total of 326 DEGs show a significant positive correlation while 154 show a significant negative one (fig. 5A; red dots for positively correlated genes, blue dots for negatively correlated ones; data in supplementary file S4, Supplementary Material online).
Fig. 5.

Association between TEV fitness and plant gene expression. (A) Correlation plots between host gene expression and viral fitness for those genes that significantly vary across all viral infections (one-way ANOVA with FDR, adjusted P < 0.05), and that exhibit a significant positive (upper panel; red dots) or negative (lower panel; blue dots) trend (Spearman’s correlation test, P < 0.05). Expression data represented as z-scores. (B) Pie charts of biological and molecular functions. On the top, for genes whose expression increases with TEV fitness (red dots in panel A); on the bottom, for genes whose expression decreases with fitness (blue dots in panel A). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.).
Next, we sought to explore which functional categories and molecular functions, if any, were enriched among these two subsets of DEGs. Results are shown in figure 5B and functional annotations are all reported in the supplementary file S4, Supplementary Material online. There are significant differences in the distribution of positively and negatively correlated DEGs into different functional categories (fig. 5B, left column; homogeneity test: χ2 = 29.225, 6 df, P < 0.001), although the difference in magnitude is moderate (Cramér’s V = 0.304). Among DEGs whose expression is positively correlated with viral fitness, biological regulation (GO: 0065008) and developmental processes (GO: 0032502) are strongly enriched compared with the negatively correlated DEGs. By contrast, negatively correlated DEGs disproportionally contribute more than positively correlated ones to the categories response to stimulus, localization, metabolic processes, and cell death. Focusing on molecular functions (fig. 5B, right column), a significant difference also exists among DEGs whose expression is positively- and negatively correlated with viral fitness (homogeneity test: χ2 = 36.720, 6 df, P < 0.001), with a magnitude of the difference in the moderate to large magnitude range (Cramér’s V = 0.341). On the one hand, among positively correlated DEGs, nucleic acid binding (GO: 003676) shows the largest departure from negatively correlated ones. On the other hand, catalytic activity (GO: 003824) and transporter activity (GO: 0005215) are the two molecular functions that appear to be enriched among negatively correlated DEGs. Together, these results suggest that positively correlated DEGs play a role in the transcriptional regulation of host defenses. By contrast, DEGs with negative correlation between expression and TEV fitness participate more in catalytic and transport activities than genes with positive correlation, suggesting a redirection of resources by the host that is not independent of viral fitness.
Validation of Microarray Data by RT-qPCR for Nine Representative Genes
Normalized expression data used in figure 5 were estimated from changes in spot intensity in the N. tabacum Gene Expression 4 × 44 K Microarrays (Agilent). To validate these results with RT-qPCR, we selected four positively correlated and five negatively correlated DEGs that cover the entire range of observed significant Spearman’s correlation coefficients (supplementary file S4, Supplementary Material online). They represent different biological functions and are expressed at different developmental stages and under different environmental situations (see below). The four positively correlated DEGs selected were (ordered according to the observed rS values): dicer-like 2 gene (DCL2; rS = 0.893), the gene encoding for the VQ motif-containing protein 29 (VQ29; rS = 0.893), the gene encoding for the GAST1 protein homolog 1 (GASA1; rS = 0.857), and a gene encoding for a member of the lipase/lipoxygenase PLAT/LH2 family (PLAT1; rS = 0.786). DCL2 is involved in defense response to viruses, maintenance of DNA methylation and production of ta-siRNAs involved in RNA interference (Parent et al. 2015). VQ29 is a negative transcriptional regulator of light-mediated inhibition of hypocotyl elongation that likely promotes the transcriptional activation of phytochrome interacting factor 1 (PIF1) during early seedling development, and participates in the jasmonic acid-mediated (JA) plant basal defense, as the VQ proteins interact with WRKY transcription factors (Li et al. 2014). GASA1 encodes for a gibberellin- and brassinosteroid-regulated protein possibly involved in cell elongation (Bouquin et al. 2001), also reported to be involved in resistance to abiotic stress through ROS signaling (O’Brien et al. 2012). PLAT1 encodes for a lipase/lipoxygenase that promotes abiotic stress tolerance (Hyun et al. 2015), is a positive regulator of plant growth, and regulates the abiotic-biotic stress cross-talk. The negatively correlated DEGs selected for validation are the adenosine kinase 2 gene (ADK2; rS = −0.857), the gene encoding for the small 3B chain of the Rubisco (RBCS3B; rS = −0.857), the AGAMOUS-like 20 gene (AGL20; rS = −0.786), the factor of DNA methylation 1 gene, (FDM1; rS = −0.786), and the granule-bound starch synthase 1 gene (GBSS1; rS = −0.821). ADK2 encodes for an adenosine kinase involved in adenosine metabolism, including the homeostasis of cytokinines (Schoor et al. 2011), controls methyl cycle flux in a S-adenosyl methionine-dependent manner and plays a role in RNA silencing by methylation. RBCS3B is involved in carbon fixation during photosynthesis and in yielding sufficient Rubisco content (Zhan et al. 2014). AGL20 is a DNA-binding MADS-box transcription activator modulating the expression of homeotic genes involved in flower development and maintenance of inflorescence meristem identity, transitions between vegetative stages of plant development and in tolerance to cold (Lee et al. 2000). FDM1 is an SGS3-like protein that acts in RNA-directed DNA methylation participating in the RNA silencing defense pathway (Xie et al. 2012). GBSS1 is involved in glucan biosynthesis and responsible of amylase synthesis that is essential for plant growth and other developmental processes (Denyer et al. 1996).
RT-qPCR-based relative expression data were calculated using the ΔΔCT method normalized by each one of the two reference genes and then averaged (see Materials and Methods). To make expression data by microarray readings and RT-qPCR readily comparable, they were both transformed into z-scores. Figure 6A shows the comparison of the two expression measures for the four DEGs with positive correlation with TEV fitness and figure 6B for the five DEGs with negative correlation with TEV fitness. Two different plots are presented for each gene. In all cases, the left plot illustrates the relationship between the expression z-scores obtained with the microarray method (x-axis) and with RT-qPCR method (y-axis) for each one of the seven TEV genotypes; the solid lines indicate the best linear fit between these two data sets. In this representation, a regression line of slope 1 is expected if both quantification methods provide identical z-scores. In all nine cases, both expression z-scores are highly and significantly correlated; Pearson’s r values ranged from 0.696 (VQ29) to 0.970 (GASA1) (in all cases 5 df, 1-tailed P ≤ 0.041). If a more stringent Holm-Bonferroni correction of the overall significance level is taken, then VQ29 would not remain significant.
Fig. 6.

Validation of the transcriptomic data by RT-qPCR for a set of selected genes. For each gene, two different scatter plots are presented. The left panel shows the relationship between the normalized (z-score) expression levels measured by transcriptomics with microarrays (x-axis) and by RT-qPCR (y-axis). The solid line represents the null hypothesis of equal expression values (i.e., a perfect match between both quantifications). The Pearson’s correlation coefficient (r) is shown on the top of each panel. The right panel shows the correlation plots between gene expression (in red measured by microarray and in blue by RT-qPCR) and viral fitness. The solid lines represent linear models; the closer the slopes of both lines, the more similarity between microarray and RT-qPCR expression data. (A) Genes whose expression increases with viral fitness (cases from fig. 5A, red dots). (B) Genes whose expression decreases with viral fitness (cases from fig. 5B, blue dots). Bidimensional error bars represent ±1 SD. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
For each gene, the right plot shows both expression z-scores as a function of TEV fitness; solid lines represent the best linear fit between normalized expressions and TEV fitness. In this representation, the more overlap between the two regression lines, the better the agreement between both quantitative methods. In this representation, VQ29 and ADK2 show the largest departure between both regression lines, though even in these extreme cases, the difference was not large enough as to be significant in a nonparametric Wilcoxon’s signed ranks test (P ≥ 0.499 in all nine cases) or in a Student’s t-test for the comparison of regression coefficients (P ≥ 0.285). Thus, we conclude that, at least for the sample of genes analyzed, the observed correlations between host’s gene expression and viral fitness are consistent for both experimental methods used to evaluate levels of gene expression.
A Model of TEV Infection Incorporating the Effect of Viral Fitness
We further delineated a picture of virus–plant interaction reflected in precise alterations of transcriptomic profiles and regulatory networks. Plant–virus interactions result from the confrontation of two players with opposed strategies and interests. From the plant perspective, activation of basal defenses, immunity, hormone-regulated pathways, and RNA-silencing (some of which are not virus-specific) will result in an immediate benefit to control virus replication and spread. We found that some plant defense responses are expressed upon infection regardless the fitness of the virus, whereas other defenses are induced progressively as viral fitness increases. Consistent with the first mode, we observed the activation of the genes EDS1 and PAD4, components of R gene-mediated disease resistance with homology to lipases, in every infection (fig. 7A). These are master regulators of plant defenses that connect pathogen signals with salicylic acid signaling (Cui et al. 2017). Salicylic acid is involved in resistance to a broad spectrum of pathogens, and in particular viruses (Alamillo et al. 2006; Conti et al. 2017). Consistent with the second mode, we observed the activation of many genes involved in defenses in proportion to TEV fitness (fig. 7A). For example, the DCL2 and AGO1 genes—key for the RNA silencing response-, genes modulating resistance to pathogens such as the subtilisin-like protease (SBT1.9), or genes expressing proteins involved in hormone-regulated defenses such as GASA1 and VQ29, brassinosteroids (e.g., brassinosteroid enhanced expression 2 BEE2, brassionosteroid-signaling kinases 2 and 7, brassinosteroid BAK1 BRI1-associated receptor kinase), ethylene response factors (e.g., ERF1B, ERF71, or cytokine response factor 1 CRF1), and members of abscisic acid perception pathway (e.g., PYL4-RCAR10, a regulatory component of the abscisic acid receptor family). Likewise, genes involved in methylation-mediated stress responses, such ADK2, FDM1 or the methionine adenosyltransferase MAT3 reduce their expression as virus replication is more efficient, thus resulting in less methylation and increased expression of genes that participate in apoptosis and posttranscriptional gene silencing (Schoor et al. 2011). In this way, the overexpression of genes that modulate histone acetylation or chromatin organization, such as the histone acetyltransferase HAC1 and the chromatin remodeling factor R17 (CHR17) would regulate differentiation, apoptosis, transcriptional activation, or ethylene response just as viral fitness increases.
Fig. 7.

Cartoon regulatory model between TEV and plant components. (A) Transcriptional regulatory scheme underlying the virus–host interaction. In general, genes participating in host defenses are activated, genes participating in host metabolism are repressed, and genes required by the virus are either activated or unaffected. (B) Protein–protein interactome of HC-Pro, the multifunctional protein with VSR activity, contextualizing gene expression data over multiple TEV infections. Different biological processes are indicated.
However, these activations have a cost, mainly in terms of resources that can be invested into secondary metabolism and development. Consistent with this idea is the fact that many genes participating in metabolic processes (e.g., CYSC1, a cysteine synthase) are highly repressed upon infection (fig. 7A). There are also central genes for the plant metabolism whose repression correlates with viral fitness, such as GBSS1, photosystem components, or assembly factors (e.g., LHCB1.3 and HCF136), Rubisco subunits and ATPases, catalases, transketolases, nucleotide and phosphate transporters, synthases involved in flavonoid, isoprenoid, ascorbate, or tryptophan biosynthesis, and GAPDH (fig. 6).
Diverting host cell resources and reprograming the metabolic machinery to support RNA metabolism and ATP production is a general strategy both of plant (Rodrigo et al. 2012) and animal viruses (Tang et al. 2005; Tiwari et al. 2017). TEV achieves this reprogramming by altering the expression of a series of genes to its own benefit. For example, we found the expression of genes involved in actin cytoskeleton organization such as ADF4 and PFN3 to be negatively correlated with TEV fitness. The profilin PNF3 is an actin-binding protein and ADF4 participates in the depolymerization of actin filaments that results from microbial-associated molecular patterns being recognized by the corresponding pattern-recognition receptors (Henty-Ridilla et al. 2014). Therefore, by downregulating this function, longer and more stable actin filaments are produced that virions can use to move around the cell from the ER-associated replication factories to plasmodesmata. Another example is the repression observed for the UBP1B gene, a negative regulator of potyvirus translation, that would allow for a more optimal virus accumulation (Hafrén et al. 2015). Genes involved in nonsense-mediated decay (NMD) defenses (Wachter and Hartmann 2014), such as the ATP-dependent RNA helicase UPF1, also show reduction in levels of expression. Other group of proteins that show alteration during viral infection are those involved in protein degradation, via ubiquitination and downstream into the proteasome pathway (e.g., ubiquitin-protein ligase 1, UPL1; ubiquitin-conjugating enzyme 2, UBC2; ubiquitin E2 variant 1B, MMZ2; EIN3-binding F box protein 1, EBF) or via autophagy (e.g., the ATP-driven chaperone CDC48C and the plant autophagy adaptor NBR1). Moreover, TEV activates in a fitness-dependent manner the expression of genes RH8, an RNA helicase, and PCaP1, a membrane-associated cation-binding protein, also required by potyviruses for cell-to-cell movement (Vijayapalani et al. 2012) (fig. 7A), and of a diversity of transcription factors including global (e.g., GRA2, GTE8), sequence-specific (e.g., SACL1 and SPL12), GATA/NAC family members (e.g., GATA1, NAC083, NAC029), bZIP G-box finding factors (e.g., GBF1 and BZIP63), and involved in homeotic gene expression (e.g., AGL20 and homeobox-1). We also found genes related to genome integrity, (e.g., the cohesins SYN2 and SMC3, and the chromatin protein SPT2), DNA replication and nucleosome assembly, alternative splicing (e.g., SF1, an homolog nuclear splicing factor), chromatin transition (e.g., SPT16, an histone chaperone involved in transcription elongation from RNApolII promoters and regulation of chromatin transitions; or the histone acetyltransferase HAC1, a coactivator of gene transcription with a major role in controlling flowering time and also essential for resistance to bacterial infections), DNA replication, and cell division (e.g., the mitotic cohesin RAD21 and the cyclin-dependent protein kinase CYCH1). However, not all host factors recruited by the virus present alterations in their expression. According to our data, the translation initiation factor eIF4E, known to be exploited by TEV for its own translation (Robaglia and Caranta 2006), was found to be unperturbed (fig. 7A) while eIF3A and eIF4G expression is positively correlated with TEV fitness.
In essence, there are genes that are significantly altered (up or down) upon infection irrespective of the ability of the virus to replicate, genes whose expression correlates with this ability (positively or negatively), and genes that remain unaltered. Nevertheless, this picture of virus–plant interaction may be biased by the limited number of viral genotypes analyzed in this work. Three out of six genotypes correspond to HC-Pro mutants. As a multifunctional protein, it is not surprising that different fitness levels can be reached by introducing mutations in different functional domains. But, certainly, more mutants should be analyzed in future work to provide a comprehensive picture and avoid bias toward certain virus proteins. In addition, we here focused on the transcription regulation, but other interlinked networks exist in the cell (e.g., metabolism, protein–protein interactions, …). To provide an insight on these other networks, we constructed the interactome (fig. 7B) of HC-Pro with the host proteins known to interact with this virus protein (Revers and García 2015). We then contextualized our gene expression data over multiple TEV infections. Many of the cellular functions in which HC-Pro participate (protein degradation, translation, redox processes, and cation signaling) are not regulated transcriptionally upon infection (or regulated marginally). Presumably, the virus exploits these processes for its own benefit (mainly to enhance replication and movement within a cell), and the normal expression of the corresponding genes is sufficient for such subversion. By contrast, RNA silencing and methylation are functions involved in defense against pathogens that are quantitatively regulated, as a sort of control strategy exerted by the plant, as long as they are needed, that is, according to viral fitness.
Some Concluding Remarks of Relevance for the Evolutionary Biology of RNA Viruses
Biological systems and processes can be analyzed and modeled at every scale of complexity. It is expected that components of each level of complexity may contribute to determine the behavior of processes at other levels. The complexity at the molecular level (i.e., the lowest level of biological organization) is astonishing both in terms of possible elements (genes, functional RNAs, proteins, and metabolites) and of interactions among them (Barabási et al. 2011). Thus, if the components at lower scales of complexity, presumed to be more accessible experimentally, are informative enough about the underlying processes, they result in excellent proxies to understand biological systems. In the case of a disease (in plants or animals), the symptoms exhibited by the organism have been traditionally used as macroscopic indicators of what occurred within the organism. This allows diagnosing diseases without the need to perform further analyses. However, symptoms are generally uncoupled from the magnitude of the perturbation at the molecular level in the host (with respect to a healthy state) (Barabási et al. 2011; Finzer 2017). This is particularly true in the case of a virus-induced disease, a paradigmatic example of a system-wide perturbation (Tan et al. 2007; Viswanathan and Früh 2007; Bailer and Haas 2009; Friedel and Haas 2011; Elena and Rodrigo 2012).
Here, we have studied for the first time the use of viral fitness as an indicator of the molecular changes occurring in the host upon infection. After all, the progress of a viral infection depends on the fitness of the virus mutant swarm. Classically, viral fitness has been evaluated by means of parameters describing the absolute growth and accumulation, by competition experiments (Holland et al. 1991; Wargo and Kurath 2012), or even by correlating it with the development of host’s symptoms (Carrasco, De la Iglesia, et al. 2007; Wargo and Kurath 2012). We focused on the infections exerted by different genotypes of a given virus in the same host. Fitness differences among genotypes are due to several causes. First, they may be a direct consequence of the effect of mutations on viral proteins, perhaps even resulting in altered folding, and thus jeopardizing their functions. Second, in the case of mutations affecting regulatory regions (e.g., RNA stems and loops), the effect may be due to altered structural configurations that impede the binding of virus own proteins or of cellular factors. Plenty of examples illustrate the effect of mutations via these two mechanisms (Bernet and Elena 2015). A third, more intriguing, yet poorly explored possibility is that mutated viral components (i.e., RNAs and proteins) may interact in nonoptimal ways with the complex network of genetic and biochemical interactions of the cell as a whole. Interacting in nonoptimal ways with any of the elements of the host regulatory and biochemical networks may have profound effects in the progression of a successful infection and therefore of viral fitness. In this work, we considered mutations affecting the CI protein (with RNA helicase, ATPase, and membrane activities), the viral replicase NIb, and the HC-Pro protein (VSR, protease, and helper-component during transmission by aphids). Our results point out that fitness, irrespective of what type of mutation is introduced, is a good indicator of how a given mutant reprograms gene expression patterns, to its own benefit or as a consequence of cellular defenses (e.g., fig. 2C).
Despite the interest of this hypothesis, none of the early studies tackled the relationship between genotype and fitness of the virus and transcriptomic profiles of the host in a systematic manner, but rather focused on comparing two viral genotypes. Evolution experiments simulating the spillover of TEV from its natural host N. tabacum into a novel, poorly susceptible one, Arabidopsis thaliana, have shown that adaptation of TEV to the novel host (i.e., concomitant to large increases in fitness) was associated with a profound change in the way the ancestral and evolved viruses interacted with the plant’s transcriptome, with genes involved in the response to biotic stresses, including signal transduction and innate immunity pathways, being significantly underexpressed in plants infected with the evolved virus than in plants infected with the ancestral one (Agudelo-Romero et al. 2008). Further evolution experiments into different ecotypes of A. thaliana that differed in their susceptibility to infection illustrated a pattern of adaptive radiation in which viruses were better adapted to their local host ecotype than to any alternative one, but with viruses evolved into more restrictive ecotypes being more generalists than viruses evolved in the more permissive ones (Hillung et al. 2014). Interestingly, these differences in fitness had a parallelism with differences in the transcriptomic profiles of plants from different ecotypes; the more generalist viruses altering similar genes in every ecotype, whereas the more specialist viruses altered different genes in different ecotypes (Hillung et al. 2016). Similarly, A. thaliana plants infected either with a mild or a virulent isolate of turnip mosaic potyvirus also showed profound differences in the genes and functional categories altered (Sánchez et al. 2015). In this case, the more virulent strain mainly altered stress responses and transport functions compared with the mild one (Sánchez et al. 2015). In a recent study, the transcriptomic alterations induced in Nicotiana benthamiana plants infected either with a WT tobacco vein banding mosaic potyvirus or a genotype deficient in the VSR were compared (Geng et al. 2017). Both transcriptomes differed in many aspects, including repression of photosynthesis-related genes, genes involved in the RNA silencing pathway, the jasmonic acid signaling pathway, and the auxin signaling transduction (Geng et al. 2017).
Altogether, the results reported in this study illustrate the complex interaction between viruses and their native host plants, and how the outcome of this interaction, in terms of viral replication and accumulation, correlates with the expression of host genes (fig. 7A). Our observation that viral fitness correlates positively or negatively with the expression of certain genes is of particular interest. By simply measuring the fitness of the virus infecting a given host, we may predict the whole genomic profile of the host cell to characterize its state (molecular impact of infection). Moreover, by specifically targeting host genes that are essential for high fitness virus variants but not for milder ones, we may prevent the spreading of the former variants, whereas still allowing mild variants to replicate and, perhaps, act as attenuated vaccines that enhance the antiviral response of the plant.
Materials and Methods
Virus Genotypes and Plant Inoculations
The infectious clone pMTEV contains a full copy of the genome of a WT TEV strain isolated from tobacco (fig. 1A; GenBank accession DQ986288) (Bedoya and Daròs 2010). Six TEV mutant genotypes were constructed by site-directed mutagenesis starting from template plasmid pMTEV as described in Torres-Barceló et al. (2008) (mutants AS13, CLA2, and CLA11) and in Carrasco, De la Iglesia, et al. (2007) (mutants PC48, PC55, and PC95). Figure 1A shows the characteristics of the seven genotypes used in the study.
The pMTEV-derived plasmids contain a unique BglII restriction site. After linearization with BglII, each plasmid was transcribed with mMESSAGE mMACHINE SP6 kit (Ambion), following the manufacturer’s instructions, to obtain infectious 5′-capped RNAs. Transcripts were precipitated (1.5 volumes of diethyl pyrocarbonate [DEPC]-treated water, 1.5 volumes of 7.5 M LiCl, 50 mM EDTA), collected, and resuspended in DEPC-treated water (Carrasco, Daròs, et al. 2007). RNA integrity and quantity were assessed by gel electrophoresis. In addition, each transcript was confirmed by sequencing of a ca. 800-bp fragment circumventing the mutation site as described elsewhere (Lalić et al. 2010). In short, reverse transcription (RT) was performed using M-MuLV reverse transcriptase (Thermo Scientific) and a reverse primer outside the region of interest to be PCR-amplified for sequencing. PCR was then performed with Phusion DNA polymerase (Thermo Scientific) and appropriate sets of primers for each transcript. Sequencing was performed at IBMCP Sequencing Service. Templates were labelled with Big Dyes v3.1 and resolved in an ABI 3130 XL machine (Life Technologies).
Nicotiana tabacum L. cv. Xanthi NN plants were used for production of virus particles of each of the seven genotypes (fig. 1A). The 5′-capped RNA transcripts were mixed with a 1:10 volume of inoculation buffer (0.5 M K2HPO4, 100 mg/ml Carborundum). Batches of 8-week-old N. tabacum plants were inoculated with ∼5 µg of RNA of each viral genotype by abrasion of the third true leaf. Inoculations were done in two experimental blocks, the first one including AS13, CLA2, CLA11, PC95, and their controls, and the second one including PC48, PC55, and their corresponding controls. All plants were at similar growth stages. Afterward, plants were maintained in a Biosafety Level-2 greenhouse chamber at 25°C under a 16-h natural sunlight (supplemented with 400 W high-pressure sodium lamps as needed to ensure a minimum light intensity of PAR 50 μmol/m2/s) and 8 h dark photoperiod. All infected plants showed symptoms 5–8 dpi, except the AS13 infected plants, which remained asymptomatic and only showed erratic chlorotic spots. At 8 dpi virus-infected leafs and apexes from each plant were collected individually in plastic bags (after removing the inoculated leaf), with the exception of the AS13 infected plants that were collected at 15 dpi. Next, plant tissue was frozen with liquid N2, homogenized using a Mixer Mill MM 400 (Retsch), and aliquoted in 1.5 ml tubes (100 mg each). These aliquots of TEV-infected tissue were stored at −80°C.
RNA Preparations
RNA extraction from 100 mg of fresh tissue per plant was performed using Agilent Plant RNA Isolation Mini Kit (Agilent Technologies) following the manufacturer’s instructions. The concentration of total plant RNA extract was adjusted to 50 ng/µl for each sample. Each RNA sample was resequenced again at this stage to ensure the constancy of the genotypes as described earlier.
Viral loads were measured by absolute real-time RT-quantitative PCR (RT-qPCR), using standard curves (Lalić et al. 2010). Standard curves were constructed using ten serial dilutions of the WT TEV genome, synthetized in vitro as described earlier, in total plant RNA obtained from healthy tobacco plants treated like all other plants in the experiment. Quantification amplifications were done in a 20-µl volume, using a GoTaq 1-Step RT-qPCR system (Promega) following the manufacturer’s instructions. The forward (q-TEV-F 5′-TTGGTCTTGATGGCAACGTG-3′) and reverse (q-TEV-R 5′-TGTGCCGTTCAGTGTCTTCCT-3′) primers were chosen to amplify a 71-nt fragment in the 3′ end of TEV genome and would only amplify complete genomes (Lalić et al. 2011). Amplifications were done using an ABI StepOne Plus Real-time PCR System (Applied Biosystems) and the following cycling conditions: the RT phase consisted of 15 min at 37°C and 10 min at 95°C; the PCR phase consisted of 40 cycles of 10 s at 95°C, 34 s at 60°C, and 30 s at 72°C; and the final phase consisted of 15 s at 95°C, 1 min at 60°C, and 15 s at 95°C. Amplifications were performed in a 96-well plate containing the corresponding standard curve. Three technical replicates per infected plant were done. Quantification results were examined using StepOne software version 2.2.2 (Applied Biosystems).
Fitness Evaluations
Total RNA was extracted and virus accumulation quantified by RT-qPCR as described earlier and detailed previously (Lalić et al. 2010). Virus accumulation (expressed as genomes/ng of total RNA) was quantified 8 dpi for all genotypes with the exception of AS13, that was quantified 15 dpi. These sampling times assure that viral populations were at a quasi-stationary plateau in N. tabacum (Carrasco, Daròs, et al. 2007). These values were then used to compute the fitness of the mutant genotypes relative to that of the WT genotype using the expression W = (Rt⁄/R0)1/t, where R0 and Rt are the ratios of accumulations estimated for the mutant and WT viruses at inoculation and after t days of growth, respectively (Carrasco, De la Iglesia, et al. 2007).
Fitness (W) data were fitted to a generalized linear model with a Normal distribution and an identity link function. The model incorporates two random factors, the TEV genotype (G) and the replicate plants (P), with the second nested within the first: , where μ is the grand mean value and ɛijk is the error associated with individual measure k (estimated from the three technical replicates of the RT-qPCR reaction). The statistical significance of each factor was evaluated using a likelihood ratio test that asymptotically follows a χ2 probability distribution. This statistical analysis was performed with IBM SPSS version 23.
Microarray Hybridizations
Total RNA was isolated as described earlier and its integrity assessed using a BioAnalyzer 2100 (Agilent) before and after hybridization. The RNA samples were hybridized onto a genotypic designed N. tabacum Gene Expression 4 × 44 K Microarray (AMADID: 021113), which contained 43,803 probes (60-mer oligonucleotides) and was used in a one-color experimental design according to Minimum Information About a Microarray Experiment guidelines (Brazma et al. 2001). Three biological replicates for each of the six TEV mutant genotypes, four replicates for the WT TEV, plus four mock-inoculated negative control plants were analyzed. Sample RNAs (200 ng) were amplified and labeled with the Low Input Quick Amp Labeling Kit (Agilent). The One-color Spike-in Kit (Agilent) was used to assess the labeling and hybridization efficiencies. Hybridization and slide washing were performed with the Gene Expression Hybridization Kit (Agilent) and Gene Expression Wash Buffers (Agilent) as detailed in the manufacturer’s instructions kits. After washing and drying, slides were scanned with a GenePix 4000B (Axon) microarray scanner, at 5 µm resolution. Image files were extracted with the Feature Extraction software version 9.5.1 (Agilent). Microarray hybridizations were performed at IBMCP Genomics Service.
Differential Gene Expression Analyses
Interarray analyses were performed with tools implemented in the BABELOMICS 5 webserver (Alonso et al. 2015). Firstly, all Agilent files were uploaded together to standardize the expression-related signals using quantile normalization (Bolstad et al. 2003). This process resulted in a matrix of normalized expression with genes in rows and samples (TEV genotypes, controls, and their replicates) in columns, provided as supplementary file S1, Supplementary Material online. To compare the expression profiles of two TEV genotypes, the expression level corresponding to mock-inoculated plants (control) was first subtracted.
Secondly, differential expression was carried out by comparing two different samples, including replicates (against mock-inoculated or WT TEV-infected plants), by using the Limma test (Smyth 2004) with FDR according to Benjamini and Hochberg (1995) (adjusted P < 0.05). An additional criterion of at least 2-fold change in mean expression, that is |log2(fold change)| > 1, was imposed to discard genes presenting minimal increases or decreases. Lists of DEGs, up- or down-regulated, provided in supplementary file S2, Supplementary Material online.
Thirdly, one-way ANOVA tests were performed to identify genes that vary across all conditions (with FDR as above, adjusted P < 0.05). To identify the genes shown in figure 2A, the test was done over all samples, including the control. By contrast, to identify the genes shown in figure 5A, the test was done over all samples corresponding to infections with distinct TEV genotypes. An additional criterion of significant Spearman’s correlation between mean fitness and mean expression (P < 0.05) was imposed in this latter case. Lists of genes whose expressions correlate with viral fitness, either positive or negative, provided in supplementary file S4, Supplementary Material online.
The similarity between expression profiles of plants infected with different TEV genotypes was evaluated with a principal components (pc) analysis with MATLAB version R2014b (MathWorks) with default parameters (singular value decomposition).
Functional Analyses from Gene Lists
The annotation of the individual probes in the Agilent’s tobacco microarray (files 021113_D_AA_20130122.txt and 021113_D_GeneList_20130122.txt provided by Agilent) was updated by BLASTing the oligo sequence file (021113_D_Fasta_20130122.txt) against the most recent version of the N. tabacum mRNA database (Ntab-BX_AWOK-SS_Basma.mrna.annot.fna) available at the Sol Genomics Network (Fernández-Pozo et al. 2015). Sequences that did not return a significant BLAST hit were removed from the output. A total of 40,430 annotated probes were generated. In 2,673 cases, more than one probe pointed to the same N. tabacum gene (e.g., probes A_95_P217927 and A_95_P046006 were both complementary to gene EB438730), and in those cases the target appeared twice in the output. Each one of the hits could be associated with an alternatively spliced mature mRNA in the Sol Genomics Network database. We then proceeded to generate the list of N. tabacum orthologous genes in the A. thaliana genome. To do so, we used BLAST against the TAIR version ten database of A. thaliana cDNAs (Lamesch et al. 2012), with a cutoff e-value < 10−4. The resulting mapping between N. tabacum and A. thaliana orthologues is listed in supplementary file S3, Supplementary Material online.
The determination of the gene ontology (GO) categories overrepresented within the lists of DEGs was carried out in the AgriGO webserver (Tian et al. 2017) by using the Fisher’s exact test (with FDR adjusted P < 0.05 according to Benjamini and Yekutieli [2001] criterion). For the graphical representation, we constructed a plane involving the most relevant categories, depicted as circles with size proportional to the total number of host genes belonging to that category (in log scale). In addition, with the lists of genes whose expression correlates with viral fitness, we calculated the pie charts associated to the following: 1) biological function and 2) molecular function in the PANTHER webserver (Mi et al. 2017).
Validation of Gene Expression through RT-qPCR
Total RNA was extracted from 100 mg of fresh tissue of plants infected with each one of the seven TEV genotypes as described earlier. The concentration of total plant RNA was adjusted to 50 ng/µl.
Nine candidate genes were selected to validate the effect of each TEV genotype on expression. Specific primers were designed for each gene that amplified the matured version of their corresponding mRNAs. Primers were designed using OLIGO Primer Analysis Software version 7 (www.oligo.net).
Gene expression was quantified by RT-qPCR relative to the expression of two housekeeping genes (Schmidt and Delaney 2010). The first housekeeping gen encodes for the L25 ribosomal protein (GenBank accession L18908). Forward NtL25-F (5′-CCCCTCACCACAGAGTCTGC-3′) and reverse NtL25-R (5′-AAGGGTGTTGTTGTCCTCAATCTT-3′) primers were chosen to amplify a 51-nt long fragment. The second housekeeping gen encodes for the elongation factor 1α (GenBank accession AF120093). For this second gene, forward NtEF1α-F (5′-TGAGATGCACCACGAAGCTC-3′) and reverse NtEF1α-R (5′-CCAACATTGTCACCAGGAAGTG-3′) primers were chosen to produce a 51-nt long amplicon.
Amplifications were done in 20 µl volume, using GoTaq 1-Step RT-qPCR System (Promega) following the manufacturer’s instructions. The forward and reverse primers for each target gene were chosen to amplify a 68–137 nt fragments in the corresponding tobacco mature mRNA. Amplifications were done using an ABI StepOne Plus Real-time PCR System (Applied Biosystems) and the following cycling conditions: the RT phase consisted of 15 min at 37°C and 10 min at 95°C; the PCR phase consisted of 40 cycles of 10 s at 95°C, 34 s at 60°C, and 30 s at 72°C; and the final phase consisted of 15 s at 95°C, 1 min at 60°C, and 15 s at 95°C. Amplifications were performed individually for each target gene (with the corresponding set of primers) in a 96-well plate containing three biological replicates and two technical replicates per infected plant. In addition, each plate incorporates the two housekeeping genes. Since each plate served for the quantification of a single mature mRNA together with the two housekeeping reference genes, a baseline value of 0.1056, resulting from averaging the threshold baselines of all plates analyzed, was used as default threshold. Quantification results were examined using the StepOne version 2.2.2 software (Applied Biosystems).
Details on the primers used for amplifications, the size of the amplicons, the GenBank identification IDs, and RT-qPCR threshold crossing (CT) values for the nine DEGs and the corresponding internal reference genes from the same samples are all reported in supplementary file S5, Supplementary Material online.
Data Availability
The microarray data that support the findings of this study have been deposited at NCBI GEO with accession number GSE99838. Processed data are presented in the Supplementary Material online. All other relevant data are available from the corresponding author on request.
Supplementary Material
Supplementary data are available at LabArchives under doi: 10.6070/H4NP22XX.
Acknowledgments
We thank Francisca de la Iglesia and Paula Agudo for excellent technical assistance, the EvolSysVir lab members for help, comments and discussions, Rachel Whitaker for English proofreading, and Lorena Latorre (IBMCP Genomics Service) and Javier Forment (IBMCP Bioinformatics Service) for their assistance. This research was supported by grants from Spain’s Agencia Estatal de Investigación—FEDER (BFU2012-30805 and BFU2015-65037-P to S.F.E. and BFU2015-66894-P to G.R.) and Generalitat Valenciana (PROMETEOII/2014/021).
References
- Acevedo A, Brodsky L, Andino R.. 2014. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 5057485:686–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agudelo-Romero P, Carbonell P, Pérez-Amador MA, Elena SF.. 2008. Virus adaptation by manipulation of host’s gene expression. PLoS One 36:e2397.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamillo JM, Saénz P, García JA.. 2006. Salicylic acid-mediated and RNA-silencing defense mechanisms cooperate in the restriction of systemic spread of Plum pox virus in tobacco. Plant J. 482:217–227. [DOI] [PubMed] [Google Scholar]
- Alonso R, Salavert F, García-García F, Carbonell-Caballero J, Bleda M, García-Alonso L, Sanchís-Juan A, Pérez-Gil D, Marín-García P, Sánchez R et al. , . 2015. BABELOMICS 5.0: functional interpretation for new generations of genomic data. Nucleic Acids Res. 43(W1):W117–W121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailer SM, Haas J.. 2009. Connecting viral with cellular interactomes. Curr Opin Microbiol. 124:453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási AL, Gulbahce N, Loscalzo J.. 2011. Network medicine: a network-based approach to human disease. Nat Rev Genet. 121:56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedoya L, Daròs JA.. 2010. Stability of Tobacco etch virus infectious clones in plasmid vectors. Virus Res. 1492:234–240. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y.. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300. [Google Scholar]
- Benjamini Y, Yekutieli D.. 2001. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 294:1165–1188. [Google Scholar]
- Bernet GP, Elena SF.. 2015. Distribution of mutational fitness effects and of epistasis in the 5’ untranslated region of a plant RNA virus. BMC Evol Biol. 15:274.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP.. 2003. A comparison of normalization methods for high density oligonucleotide array data base don variance and bias. Bioinformatics 192:185–193. [DOI] [PubMed] [Google Scholar]
- Bouquin T, Meier C, Foster R, Nielsen ME, Mundy J.. 2001. Control of specific gene expression by gibberellin and brassinosteroid. Plant Physiol. 1272:450–458. [PMC free article] [PubMed] [Google Scholar]
- Brazma A, Hingamp P, Quackensbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC et al. , . 2001. Minimum information about a microarray experiment (MIAME). Toward standards for microarray data. Nat Genet. 29:365–371. [DOI] [PubMed] [Google Scholar]
- Carrasco P, Daròs JA, Agudelo-Romero P, Elena SF.. 2007. A real-time RT-PCR assay for quantifying the fitness of Tobacco etch virus in competition experiments. J Virol Methods 139:181–188. [DOI] [PubMed] [Google Scholar]
- Carrasco P, De la Iglesia F, Elena SF.. 2007. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco etch virus. J Virol. 81:12979–12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervera H, Lalić J, Elena SF.. 2016. Effect of host species on topography of the fitness landscape for a plant RNA virus. J Virol. 9022:10160–10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao L. 1990. Fitness of RNA virus decreased by Muller’s ratchet. Nature 3486300:454–455. [DOI] [PubMed] [Google Scholar]
- Codoñer FM, Daròs JA, Solé RV, Elena SF.. 2006. The fittest versus the flattest: experimental confirmation of the quasispecies effect with subviral pathogens. PLoS Pathog. 212:e136–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti G, Rodríguez MC, Venturuzzi AL, Asurmendi S.. 2017. Modulation of host plant immunity by tobamovirus proteins. Ann Bot. 1195:737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Gobbato E, Kracher B, Qiu J, Bautor J, Parker JE.. 2017. A core function of EDS1 and PAD4 is to protect the salicylic acid defense sector in Arabidopsis immunity. New Phytol. 2134:1802–1817. [DOI] [PubMed] [Google Scholar]
- Da Silva J, Wyatt SK.. 2014. Fitness valleys constrain HIV-1’s adaptation to its secondary chemokine coreceptor. J Evol Biol. 273:604–615. [DOI] [PubMed] [Google Scholar]
- De la Iglesia F, Elena SF.. 2007. Fitness declines in Tobacco etch virus upon serial bottleneck transfers. J Virol. 8110:4941–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denyer K, Clarke B, Hylton C, Tatge H, Smith AM.. 1996. The elongation of amylose and amylopectin chains in isolated starch granules. Plant J. 106:1135–1143. [Google Scholar]
- Domingo E, Holland JJ.. 1997. RNA virus mutations and fitness for survival. Annu Rev Microbiol. 51:151–178. [DOI] [PubMed] [Google Scholar]
- Domingo-Calap P, Cuevas JM, Sanjuán R.. 2009. The fitness effects of random mutations in single-stranded DNA and RNA bacteriophages. PLoS Genet. 511:e1000742.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte EA, Clarke DK, Duarte EA, Moya A, Domingo E, Holland JJ.. 1992. Rapid fitness losses in mammalian RNA virus clones due to Muller’s ratchet. Proc Natl Acad Sci U S A. 8913:6015–6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena SF, Rodrigo G.. 2012. Towards an integrated molecular model of plant-virus interactions. Curr Opin Virol. 26:719–718. [DOI] [PubMed] [Google Scholar]
- Fernández-Pozo N, Menda N, Edwards JD, Saha S, Tecle IY, Strickler SR, Bombarely A, Fisher-York T, Pujar A, Foerster H, Yan A, Mueller LA.. 2015. The Sol Genomics network (SGN) – from genotype to phenotype to breeding. Nucleic Acids Res. 43:D1036–D1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzer P. 2017. How we become ill. EMBO Rep. 184:515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel CC, Haas J.. 2011. Virus-host interactomes and global models of virus-infected cells. Trends Microbiol. 1910:501–508. [DOI] [PubMed] [Google Scholar]
- Geng C, Wang HY, Liu J, Yan ZY, Tian YP, Yuan XF, Gao R, Li XD.. 2017. Transcriptomic changes in Nicotiana benthamiana plants inoculated with the wild-type or an attenuated mutant of Tobacco vein banding mosaic virus. Mol Plant Pathol. 188:1175–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafrén A, Löhmus A, Mäkinen K.. 2015. Formation of Potato virus A-induced RNA granules and viral translation are interrelated processes required for optimal virus accumulation. PLoS Pathog. 1112:e1005314.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henty-Ridilla JL, Li J, Day B, Staiger CJ.. 2014. Acting depolymerization factor 4 regulates actin dynamics during innate immune signaling in Arabidopsis. Plant Cell 261:340–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillung J, Cuevas JM, Valverde S, Elena SF.. 2014. Experimental evolution of an emerging plant virus in host genotypes that differ in their susceptibility to infection. Evolution 689:2467–2480. [DOI] [PubMed] [Google Scholar]
- Hillung J, García-García F, Dopazo J, Cuevas JM, Elena SF.. 2016. The transcriptomics of an experimentally evolved plant-virus interaction. Sci Rep. 6:24901.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland JJ, de la Torre JC, Clarke DK, Duarte EA.. 1991. Quantitation of relative fitness and great adaptability of clonal populations of RNA viruses. J Virol. 65:2960–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun TK, Albacete A, Van der Graaff E, Eom SH, Großkinsky DK, Böhm H, Janschek U, Rim Y, Ali WW, Kim SY, Roitsch T.. 2015. The Arabidopsis PLAT domain protein 1 promotes abiotic stress tolerance and growth in tobacco. Transgenic Res. 244:651–663. [DOI] [PubMed] [Google Scholar]
- Lalić J, Agudelo-Romero P, Carrasco P, Elena SF.. 2010. Adaptation of tobacco etch potyvirus to a susceptible ecotype of Arabidopsis thaliana capacitates it for systemic infection of resistant ecotypes. Philos Trans R Soc B 3651548:1997–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalić J, Cuevas JM, Elena SF.. 2011. Effect of host species on the distribution of mutational fitness effects for an RNA virus. PLoS Genet. 711:e1002378.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalić J, Elena SF.. 2013. Epistasis between mutations is host-dependent for an RNA virus. Biol Lett. 91:20120396.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalić J, Elena SF.. 2015. The impact of higher-order epistasis in the within-host fitness of a positive-sense plant RNA virus. J Evol Biol. 2812:2236–2247. [DOI] [PubMed] [Google Scholar]
- Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, Muller R, Dreher K, Alexander DL, García-Hernández M et al. , . 2012. The Arabidopsis information resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40(D1):D1202–D1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Suh SS, Park E, Cho E, Ahn JH, Kim SG, Lee JS, Kwon YM, Lee I.. 2000. The Agamous-like 20 MADS domain protein integrates floral inductive pathways in Arabidopsis. Genes Dev. 1418:2366–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jing Y, Li J, Xu G, Lin R.. 2014. Arabidopsis VQ MOTIF-CONTAINING PROTEIN 29 represses seedling deetiolation by interacting with phytochrome-interacting factor 1. Plant Physiol. 1644:2068–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnen CR, Hoekstra HE.. 2009. Measuring natural selection on genotypes and phenotypes in the wild. Cold Spring Harb Symp Quant Biol. 74:155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Picado J, Martínez MA.. 2008. HIV-1 reverse transcriptase inhibitor resistance mutations and fitness: a view from the clinic and ex vivo. Virus Res. 134(1–2):104–123. [DOI] [PubMed] [Google Scholar]
- Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD.. 2017. PANTHER version 11: expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45(D1):D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novella IS, Clarke DK, Quer J, Duarte EA, Lee CH, Weaver SC, Elena SF, Moya A, Domingo E, Holland JJ.. 1995. Extreme fitness differences in mammalian and insect hosts after continuous replication of Vesicular stomatitis virus in sandfly cells. J Virol. 69:6805–6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novella IS, Duarte EA, Elena SF, Moya A, Domingo E, Holland JJ.. 1995. Exponential increases of RNA virus fitness during large populations transmissions. Proc Natl Acad Sci U S A. 92:5841–5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novella IS, Gilbertson DL, Borrego B, Domingo E, Holland JJ.. 2005. Adaptability costs in immune escape variants of Vesicular stomatitis virus. Virus Res. 1071:27–34. [DOI] [PubMed] [Google Scholar]
- Novella IS, Presloid JB, Beech C, Wilke CO.. 2013. Congruent evolution of fitness and genetic robustness in Vesicular stomatitis virus. J Virol. 879:4923–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JA, Daudi A, Finch P, Butt VS, Whitelegge JP, Souda P, Ausubel FM, Bolwell GP.. 2012. A peroxidase-dependent apoplastic oxidative burst in cultured Arabidopsis cells functions in MAMP-elicited defense. Plant Physiol. 158:2013–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. 2009. Fitness and its role in evolutionary genetics. Nat Rev Genet. 108:531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JS, Bouteiller N, Elmayan T, Vaucheret H.. 2015. Respective contributions of Arabidopsis DCL2 and DCL4 to RNA silencing. Plant J. 812:223–232. [DOI] [PubMed] [Google Scholar]
- Peris JB, Davis P, Cuevas JM, Nebot MR, Sanjuán R.. 2010. Distribution of fitness effects caused by single-nucleotide substitutions in bacteriophage f1. Genetics 1852:603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesko K, Voigt EA, Swick A, Morley VJ, Timm C, Yin J, Turner PE.. 2015. Genome rearrangements affects RNA virus adaptability on prostate cancer cells. Front Genet. 6:121.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revers F, García JA.. 2015. Molecular biology of potyviruses. Adv Virus Res. 92:101–199. [DOI] [PubMed] [Google Scholar]
- Robaglia C, Caranta C.. 2006. Translation initiation factors: a weak link in plant RNA virus infection. Trends Plant Sci. 111:40–45. [DOI] [PubMed] [Google Scholar]
- Rodrigo G, Carrera J, Ruiz-Ferrer V, Del Toro FJ, Llave C, Voinnet O, Elena SF.. 2012. A meta-analysis reveals the commonalities and differences in Arabidopsis thaliana response to different viral pathogens. PLoS One 77:e40526.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez F, Manrique P, Mansilla C, Lunello P, Wang X, Rodrigo G, López-González S, Jenner CE, González-Melendi P, Elena SF et al. , . 2015. Viral strain-specific differential alterations in Arabidopsis developmental patterns. Mol Plant Microbe Interact. 2812:1304–1315. [DOI] [PubMed] [Google Scholar]
- Sanjuán R, Cuevas JM, Furió V, Holmes EC, Moya A.. 2007. Selection for robustness in mutagenized RNA viruses. PLoS Genet. 36:e93–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R, Moya A, Elena SF.. 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc Natl Acad Sci U S A. 10122:8396–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt GW, Delaney SK.. 2010. Stable internal reference genes for normalization of real-time RT-PCR in tobacco (Nicotiana tabacum) during development and abiotic stress. Mol Genet Genomics 2833:233–241. [DOI] [PubMed] [Google Scholar]
- Schoor S, Farrow S, Blaschke H, Lee S, Perry G, Von Schwartzenberg K, Emery N, Moffatt B.. 2011. Adenosine kinase contributes to cytokinin interconversion in Arabidopsis. Plant Physiol. 1572:659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 3:3. [DOI] [PubMed] [Google Scholar]
- Tan SL, Ganji G, Paeper B, Proll S, Katze MG.. 2007. Systems biology and the host response to viral infection. Nat Biotechnol. 2512:1383–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang BSF, Chan KH, Cheng VCC, Woo PCY, Lau SKP, Lam CCK, Chan TL, Wu AKL, Hung IFN, Leung SY, Yuen KY.. 2005. Comparative host gene transcription by microarray analysis early after infection of the Huh7 cell line with severe acute respiratory syndrome coronavirus and human coronavirus 229E. J Virol. 7910:6180–6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, Xu W, Su Z.. 2017. AgriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 45(W1):W122–W129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari SK, Dang J, Qin Y, Lichinchi G, Bansal V, Rana TM.. 2017. Zika virus infection reprograms global transcription of host cells to allow sustained infection. Emerg Microb Infect. 64:e24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Barceló C, Martín S, Daròs JA, Elena SF.. 2008. From hypo- to hypersuppression: effect of amino acid substitutions on the RNA-silencing suppressor activity of the tobacco etch potyvirus HC-Pro. Genetics 1802:1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PE, Elena SF.. 2000. Cost of host radiation in an RNA virus. Genetics 156:1465–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale PF, Choisy M, Froissar R, Sanjuán R, Gandon S.. 2012. The distribution of mutational fitness effects of phage φX174 on different hosts. Evolution 6611:3495–3507. [DOI] [PubMed] [Google Scholar]
- Vijayapalani P, Maeshima M, Nagasaki-Takekuchi N, Miller WA.. 2012. Interaction of the trans-frame potyvirus protein P3N-PIPO with host protein PCaP1 facilitates potyvirus movement. PLoS Pathog. 84:e1002639.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visher E, Whitefield SE, McCrone JT, Fitzsimmons W, Lauring AS.. 2016. The mutational robustness of Influenza A virus. PLoS Pathog. 128:e1005856.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan K, Früh K.. 2007. Viral proteomics: global evaluation of viruses and their interaction with the host. Exp Rev Proteomics 46:815–829. [DOI] [PubMed] [Google Scholar]
- Wachter A, Hartmann L.. 2014. NMD: nonsense-mediated defense. Cell Host Microbe 163:273–275. [DOI] [PubMed] [Google Scholar]
- Wargo AR, Kurath G.. 2012. Viral fitness: definitions, measurements, and current insights. Curr Opin Virol. 25:538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhout EM, Ooms M, Vink M, Das AT, Berkhout B.. 2005. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 332:796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen A, Zwart MP, Higueras P, Sardanyés J, Elena SF.. 2016. Predicting the stability of homologous gene duplications in a plant RNA virus. Genome Biol Evol. 89:3065–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Ren G, Zhang C, Yu B.. 2012. The DNA- and RNA-binding protein factor of DNA methylation 1 requires XH domain-mediated complex formation for its function in RNA-directed methylation. Plant J. 723:491–500. [DOI] [PubMed] [Google Scholar]
- Zhan GM, Li RJ, Hu ZY, Liu J, Deng LB, Lu SY, Hua W.. 2014. Cosupression of RBCS3B in Arabidopsis leads to severe photoinhibition caused by ROS accumulation. Plant Cell Rep. 337:1091–1108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The microarray data that support the findings of this study have been deposited at NCBI GEO with accession number GSE99838. Processed data are presented in the Supplementary Material online. All other relevant data are available from the corresponding author on request.