

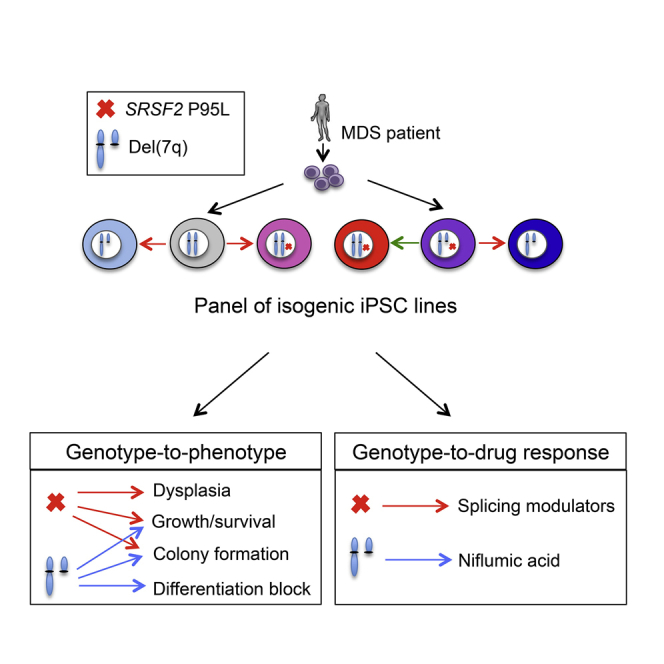
Summary
Connecting specific cancer genotypes with phenotypes and drug responses constitutes the central premise of precision oncology but is hindered by the genetic complexity and heterogeneity of primary cancer cells. Here, we use patient-derived induced pluripotent stem cells (iPSCs) and CRISPR/Cas9 genome editing to dissect the individual contributions of two recurrent genetic lesions, the splicing factor SRSF2 P95L mutation and the chromosome 7q deletion, to the development of myeloid malignancy. Using a comprehensive panel of isogenic iPSCs—with none, one, or both genetic lesions—we characterize their relative phenotypic contributions and identify drug sensitivities specific to each one through a candidate drug approach and an unbiased large-scale small-molecule screen. To facilitate drug testing and discovery, we also derive SRSF2-mutant and isogenic normal expandable hematopoietic progenitor cells. We thus describe here an approach to dissect the individual effects of two cooperating mutations to clinically relevant features of malignant diseases.
Keywords: induced pluripotent stem cells, myelodysplastic syndrome, CRISPR/Cas9, gene editing, mutational cooperation, splicing factor mutations, spliceosomal mutations, SRSF2, chr7q deletion
Graphical Abstract
Highlights
-
•
A panel of isogenic iPSCs with SRSF2 P95L mutation and del(7q) was generated
-
•
The contributions of each genetic lesion to phenotype were separated
-
•
Each genetic lesion could be linked to specific therapeutic vulnerabilities
-
•
Expandable hematopoietic progenitor cell lines facilitate drug testing
Papapetrou and colleagues develop a comprehensive panel of isogenic iPSC lines with SRSF2 P95L mutation and chr7q deletion. They use these cells to identify cellular phenotypes contributed by each genetic lesion and therapeutic vulnerabilities specific to each one and develop expandable hematopoietic progenitor cell lines to facilitate drug discovery.
Introduction
Precision oncology aims to match patients with drugs that target the specific genetic makeup of their malignant cells to enhance their chances of response. Pairing specific mutations with drug responses is central to this goal, but hindered by the immense genetic diversity and complexity of both hematologic malignancies and solid tumors. The vast majority of human malignancies contain more than one recurrent genetic lesion that cooperatively confer their phenotypic characteristics. In addition to associations of specific mutations to drug responses, associations of mutations to cellular phenotypes would also be valuable to help determine the most useful functional assays to guide drug testing. Furthermore, understanding the relative contribution of each genetic lesion to the malignant phenotype can aid the understanding of mechanisms of oncogenesis and the importance of the order of mutation acquisition.
Current experimental methods afford limited opportunities for drawing genotype-to-phenotype and genotype-to-drug response associations. Very few associations of gene mutations to drug responses can be made directly in the clinic due to the complexity and heterogeneity of cancer genomes, the relative rarity of some cancers and genotypes and the constrains imposed by currently administered therapies. Patient-derived xenograft models have been used to predict therapeutic responses but variable clonal dynamics and genetic drifts hamper systematic connections of specific mutations with drug responses (Gao et al., 2015, Papapetrou, 2016). Genetically engineered mouse models offer precise genetic models amenable to studies of mutational cooperation, but can be limited by species-determined differences in disease phenotypes, downstream mechanisms, and drug specificities (Day et al., 2015, Gould et al., 2015, Kronke et al., 2015).
Myelodysplastic syndrome (MDS) genomes typically harbor two or more genetic lesions, including gene mutations and larger-scale genetic abnormalities, mainly deletions of the long arm of chromosomes 5, 7, and 20 (Papaemmanuil et al., 2013). Mutations in genes encoding splicing factors have recently been discovered as the most frequent class of mutations in MDS (Graubert et al., 2012, Papaemmanuil et al., 2011, Yoshida et al., 2011). Serine/arginine-rich splicing factor 2 (SRSF2) is a member of the serine/arginine-rich (SR) protein family that contributes to both constitutive and alternative splicing by binding to RNA sequence motifs that are enriched in exons, called exonic splicing enhancer (ESE) sequences. Mutations of SRSF2 are found in 20%–30% of MDS patients and, less frequently, in other hematologic malignancies and solid tumors and are almost always heterozygous missense substitutions at codon P95 (P95 L/R/H) (Dvinge et al., 2016, Papaemmanuil et al., 2013, Yoshida et al., 2011). Somatic loss of one copy of the long arm of chromosome 7 (del(7q)) is a characteristic cytogenetic abnormality in MDS and other myeloid malignancies, associated with unfavorable prognosis and can co-occur with the SRSF2 P95 mutation in patients with MDS and acute myeloid leukemia (AML) (Papaemmanuil et al., 2013, Papaemmanuil et al., 2016).
Here we combined patient-derived induced pluripotent stem cells (iPSCs) with the CRISPR/Cas9 system to interrogate the contributions of the SRSF2 P95 mutation and of the del(7q) to cellular phenotype and drug responses. We find that the SRSF2 P95 mutation confers dysplastic morphology and other phenotypic characteristics to iPSC-derived hematopoietic progenitor cells (iPSC-HPCs) in support of a role early in the transformation process, while del(7q)-iPSC-HPCs exhibit a more severe differentiation block, concomitant with disease progression—findings consistent with clinical observations and population genetics analyses. We show that SRSF2 mutant iPSC-HPCs are preferentially sensitive to splicing modulator drugs and identify candidate compounds preferentially targeting del(7q) cells through an unbiased large-scale small-molecule screen. To facilitate drug testing and screening, we report the derivation of iPSC-derived expandable HPCs (eHPCs) that can be grown like conventional cell lines while maintaining specific drug sensitivities. These results demonstrate the power of patient-derived iPSCs and genome editing in dissecting the individual contributions of cooperating genetic lesions to clinically relevant cancer features.
Results
Introduction of the SRSF2 P95L Mutation in Normal Patient-Derived iPSCs
We previously derived normal and MDS iPSC lines from a patient with MDS harboring SRSF2 P95L mutation and del(7q) (Kotini et al., 2015, Kotini et al., 2017). The MDS-2.13 line was derived from the MDS clone of this patient and harbors the SRSF2 P95L mutation and a deletion of chr(7q), and contains no additional mutations recurrently found in myeloid malignancies, as determined by whole-exome sequencing of the iPSC line and of the starting patient cells (Kotini et al., 2015). The N-2.12 line originated from normal bone marrow (BM) hematopoietic cells of the same patient, as it was not found to share any common somatic variants with the patient's MDS clone by whole-exome sequencing (Kotini et al., 2015). To study the effects of the SRSF2 P95L mutation in isolation, we first introduced the mutation into the iPSC line N-2.12 (Figure 1A) (Kotini et al., 2015). We designed four guide RNAs (gRNAs) targeting the first intron of the SRSF2 gene and a donor plasmid containing a selection cassette (Figure 1B). We selected two gRNAs, which we co-transfected with the donor DNA (Figures S1A–S1C). Cells with targeted integration (TI) of the donor DNA were detected by PCR, but no puromycin-resistant colonies could be retrieved, presumably because expression of the puromycin resistance gene from the SRSF2 locus was not sufficient for successful selection. We therefore attempted to obtain targeted clones by first selecting pools of transfected cells enriched for targeting events and subsequent screening of single-cell clones (Figure S1D). TI of the donor could be detected in all 48 pools of approximately 20,000 transfected cells. Two pools (no. 2 and no. 5) with the strongest signal were selected. Two out of 48 and 4 out of 48 targeted clones were found after single-cell subcloning of the two pools, respectively (Figures S1E–S1G). These six clones were tested with a second set of TI-specific primers, DNA sequencing of the introduced 284C > T mutation, as well as detection and sequencing of the untargeted allele (Figures S1H, S1I, and S2A–S2C). All six clones contained indels in the untargeted allele, which were restricted to intronic sequences (Figure S2C). Out of 4 clones with confirmed TI of the intact donor (Figure S1H), clone 5-16, harboring the smallest indel, a deletion of 16 nt at a distance of 125 and 193 bp from the splice donor and splice acceptor sites, respectively, was selected and confirmed to maintain a normal karyotype (Figures S2C and S2D). Two subclones, 5-16Cre5 and 5-16Cre20, were selected after Cre-mediated excision (Figures S2E and S2F)(Papapetrou et al., 2011, Papapetrou and Sadelain, 2011). Off-target insertions of the donor cassette were excluded and excision of the selection cassette was confirmed by Southern blotting (Figure 1C). The two clones were confirmed to harbor the 284C > T mutation, to maintain a normal karyotype and to express SRSF2 at levels similar to the parental line (Figures 1D–1F).
Figure 1.

Introduction of the SRSF2 P95L Mutation in Normal iPSCs and Correction of the SRSF2 P95L Mutation in MDS Patient-Derived iPSCs
(A and B) Schematic representation of the gene-editing strategy for the generation of SRSF2 P95L iPSCs. Depicted from top to bottom in (B): the SRSF2 locus with the position of the 284C > T mutation and the position targeted by the gRNAs (scissors) shown; the donor DNA with 5′ and 3′ homology arms; scheme of the targeted allele with the 284T mutation introduced and the selection cassette integrated; scheme of the targeted allele following Cre recombinase-mediated excision of the floxed selection cassette. Triangles depict loxP sites. E1, exon 1; E2, exon 2; hPGK, human phosphoglycerate kinase promoter; PuroR, puromycin resistance gene; pA, poly-adenylation signal.
(C) Southern blot analysis of CRISPR/Cas9-edited clones before and after targeting and excision of the selection cassette with probes shown in the upper panel. H depicts HindIII restriction sites.
(D) Sanger sequencing of the parental line, the targeted clone 5-16 and two excised derivative clones 5-16 Cre5 and 5-16 Cre20, harboring heterozygous 284T SRSF2 mutation. The arrow shows the position of the 284C (WT) or 284T (mutant) nucleotide.
(E) Karyotypic analysis of the two targeted and excised clones showing normal male karyotype.
(F) Total SRSF2 expression by qRT-PCR, showing reduced expression by approximately half in the intermediate targeted clone 5-16, relative to the parental N-2.12 line, presumably because the pA in the selection cassette, shown in (B), results in premature transcription termination and inactivation of the targeted allele. Expression of total SRSF2 is restored to normal levels in the two clones (5-16 Cre5 and 5-16 Cre20) after excision of the cassette. Error bars represent standard deviation.
(G) Schematic representation of the gene-editing strategy for the correction of the SRSF2 P95L iPSC line MDS-2.13, harboring a chr7q deletion in addition to the SRSF2 P95L mutation.
(H) Identification of monoallelically targeted clones with primers shown in the right panel. The 760 bp band (lower) corresponds to the untargeted allele. The 2,105 bp band (upper) corresponds to the targeted allele.
(I) Sanger sequencing of the parental SRSF2 mutant MDS-2.13 line and five targeted clones found positive by PCR screening for targeted integration of the donor, as shown in (H). The arrow shows the position of the 284C (WT) or 284T (mutant) nucleotide. Clones 8, 18, and 24 are targeted in the mutant allele and therefore corrected (homozygous for 284C), whereas clones 7 and 21 are targeted in the WT allele and remain mutant (heterozygous 284C/T).
(J) Sanger sequencing of the PCR band corresponding to the untargeted allele, shown in (H), to test for indels generated through non-homologous end joining. Clone 8 harbors a 19-nt deletion, clone 18 a 1-nt (G) insertion, whereas clone 24 has an intact untargeted allele and was therefore selected for further experiments.
Correction of the SRSF2 P95L Mutation in MDS Patient-Derived iPSCs
To further probe the contribution of the SRSF2 P95L mutation in MDS with cooperating genetic events, we corrected this mutation in the iPSC line MDS-2.13, derived from the MDS clone of the same patient from whom the N-2.12 line was derived and harboring a del(7q) in addition to the SRSF2 P95L mutation (Figure 1G) (Kotini et al., 2015, Kotini et al., 2017). We used a strategy similar to the one used for introducing the mutation, this time using only gRNA-1, in order to minimize indels in the untargeted allele, a donor plasmid carrying a normal SRSF2 284C allele and a modified selection strategy (Figures S3A and S3B). Ten out of 46 clones were found to have TI of the donor cassette in one allele (Figure 1H). In 5 out of 10 the donor was integrated in the mutant allele reverting it to wild-type (WT) (Figure 1I). Clone 24 (C24), found to have an intact untargeted allele, was selected (Figure 1J). Two excised clones (C24 Cre6 and C24 Cre7) were confirmed to be homozygous for the WT SRSF2 284C allele (Figures S3C and S3D).
The SRSF2 P95L Mutation Imparts Splicing Alterations
To assess gene expression and splicing in SRSF2 mutant iPSCs, we compared the two CRISPR/Cas9-engineered SRSF2 P95L mutant clones 5-16Cre 5 and 5-16Cre20 with the parental normal N-2.12 line by RNA sequencing of undifferentiated iPSCs, CD34+ cells isolated on day 9 of differentiation and CD45+ cells isolated on day 12 of differentiation (when almost all CD45+ cells are also CD34+) (Figure S4). The SRSF2 mutant iPSC expressed SRSF2 at levels similar to the normal N-2.12 line and expressed the mutant allele at approximately 50% of total levels (Figures S5A and S5B). Global gene expression changes were found that varied with cell type (i.e., undifferentiated pluripotent stem cells, CD34+ hemogenic endothelium, and CD45+ hematopoietic progenitors) (Figures S5C and S5D). Differentially expressed genes included predominantly cell-type-specific genes (Figure S5E). Cell proliferation, differentiation, apoptosis, and regulation of hematopoiesis were among the predominant gene ontology categories (Figure S5F).
SRSF2 mutant cells exhibited genome-wide alterations in ESE motif recognition in all three cell states. C-rich variants of the SSNG (S = C or G) motif recognized by WT SRSF2 were enriched within exons promoted by SRSF2 P95L, while G-rich variants were enriched within exons repressed by SRSF2 P95L (Figures 2A and 2B). These findings are consistent with findings previously reported by us and others in a knockin mouse model and in K562 cells expressing mutant SRSF2 ectopically or through gene editing (Kim et al., 2015, Zhang et al., 2015). Cell-type-specific differences in differentially spliced genes were less prominent than those found in differentially expressed genes (Figure 2C). Mis-spliced genes included genes of potential disease relevance with isoforms previously reported to be promoted by mutant SRSF2, including EZH2 and FYN (Figures 2D and 2E) (Kim et al., 2015, Zhang et al., 2015). Importantly, iPSC-derived CD34+ and CD45+ hematopoietic progenitors recapitulated a higher fraction of the mis-spliced events that were observed in patient cells than either the knockin mouse or the K562 SRSF2 mutant models (Figures 2F and 2G), suggesting that iPSCs capture disease-relevant splicing alterations more faithfully than other existing models.
Figure 2.

Splicing Alterations in SRSF2 Mutant iPSCs and iPSC-HPCs
(A) Mean enrichment of all variants of the SSNG (S = C or G) motif in cassette exons that are promoted versus repressed in SRSF2 mutant 5-16 Cre5 cells (upper panels) or 5-16 Cre20 cells (lower panels) compared with the isogenic SRSF2 WT N-2.12 cells. Error bars indicate 95% confidence intervals estimated by bootstrapping.
(B) Relative occurrence of CCNG and GGNG motifs within and adjacent to cassette exons that are differentially spliced in association with the SRSF2 mutation in undifferentiated iPSCs (top), CD34+ cells (middle), and CD45+ cells (lower). Shading indicates 95% confidence interval by bootstrapping. The schematic illustrates a portion of a metagene centered on the differentially spliced cassette exon. From left to right, the features are the upstream exon (gray box) and intron (black line), the cassette exon (black box, vertical dashed lines), and the downstream intron (black line) and exon (gray box). Horizontal axis, genomic coordinates defined with respect to the 5′ and 3′ splice sites where 0 is the splice site itself. Vertical axis, relative frequency of the indicated motifs over genomic loci containing cassette exons promoted versus repressed by SRSF2 mutations (log scale).
(C) Venn diagram depicting the overlap between genes that are differentially spliced in SRSF2 mutant (in either 5-16 Cre5 or 5-16 Cre20) versus SRSF2 WT (N-2.12) cells at the undifferentiated (iPSC) state, CD34+ or CD45+ state.
(D) Percentage of EZH2 transcripts harboring the poison cassette exon in the indicated cell lines and cell types, as measured by RNA sequencing (RNA-seq) data. Error bars indicate 95% confidence intervals. SRSF2 mutant cells exhibit preferential inclusion of a “poison” cassette exon of EZH2, giving rise to an isoform of EZH2 that contains a premature termination codon, resulting in degradation by nonsense-mediated RNA decay.
(E) Percentage of FYN transcripts corresponding to the FYN-B isoform in the indicated cell lines and cell types, as measured by RNA-seq data. Error bars indicate 95% confidence intervals. SRSF2 mutant cells exhibit preferential inclusion of the upstream exon 7 (exon 7A) of FYN, a gene encoding a tyrosine kinase, giving rise to an isoform (FYN-B) with altered kinase activity.
(F) Fraction of mis-spliced genes observed in patients that are also mis-spliced in each indicated model system. The overlap between genes exhibiting differential splicing in association with SRSF2 mutational status in either an AML or CMML cohort (Kim et al., 2015) and genes exhibiting differential splicing in each system was computed. All analyses were restricted to human genes with orthologous mouse genes to avoid potential biases resulting from incomplete orthology assignments.
(G) Venn diagram depicting the overlap between genes that are differentially spliced in SRSF2 mutant versus SRSF2 WT cells, comparing data from mice (Lineage− Sca1+ c-Kit+ [LSK] cells), K562 cells (Kim et al., 2015) and iPSC-derived CD45+ cells.
Hematopoietic Phenotypes Exclusively Produced by either SRSF2 P95L or del(7q)
To connect each of the two genetic lesions—SRSF2 P95L mutation and chr7q deletion—with specific hematopoietic phenotypes, we assembled a comprehensive panel of isogenic iPSC lines with both, one or none of the two genetic lesions (Figure 3A; Table S1). The patient-derived lines N-2.12 and MDS-2.13, as well as the lines derived from them through CRISPR/Cas9 (5-16Cre5, 5-16Cre20, and C24Cre6), were described above. We also analyzed a line, MDS-2.A3C, derived from MDS-2.13 after spontaneous correction of the del(7q), as well as two lines, Cre10 and 8Cre21, derived from N-2.12 by engineered del(7q) through a previously described inverted Cre-loxP genetic engineering strategy (Kotini et al., 2015). This panel was subjected to hematopoietic differentiation and detailed phenotypic characterization (Figure 3B). Both normal and SRSF2 P95L mutant lines without del(7q) gave rise to comparable percentages of CD45+ HPCs (80%–90% on day 14 of differentiation) (Figures 3C and 3D). In contrast, all del(7q) lines generated very few CD45+ cells (<10%), and these levels remained unchanged even after correction of the SRSF2 P95L mutation (Figures 3C and 3D, compare MDS-2.13 with C24 Cre6). These results suggest that the SRSF2 P95L mutation does not affect the numbers of CD45+ myeloid progenitors. Hematopoietic progenitors and mature cells from SRSF2 P95L mutant iPSCs displayed dysplastic morphological features, whereas cells with isolated del(7q) showed no apparent morphological changes of dysplasia (Figure 3E; Table S2).
Figure 3.

Hematopoietic Phenotypes Exclusively Contributed by SRSF2 P95L or del(7q)
(A) All isogenic iPSC lines used in this study. Lines N-2.12 (WT SRSF2) and MDS-2.13 (mutant SRSF2) were derived from BM mononuclear cells (BMMCs) of an MDS patient in parallel in the same reprogramming experiment (Kotini et al., 2015, Kotini et al., 2017). The latter also contains a chr7q deletion. Lines 5-16 Cre5 and 5-16 Cre20 were derived from N-2.12 through CRISPR/Cas9-mediated editing to introduce the SRSF2 P95L mutation (Figure 1). C24 Cre6 was derived from MDS-2.13 through CRISPR/Cas9-mediated correction of the SRSF2 P95L mutation (Figure 1). MDS-2.A3C was derived from MDS-2.13 through spontaneous correction of the chr7q deletion and maintains the SRSF2 P95L mutation (Kotini et al., 2015). Cre10 and 8Cre21 were derived from N-2.12 through engineering of del(7q) with a modified Cre/loxP strategy (Kotini et al., 2015). The red arrows depict CRISPR/Cas9-mediated gene editing. The green arrow depicts spontaneous conversion.
(B) Hematopoietic differentiation scheme.
(C) CD45 expression on day 14 of hematopoietic differentiation. Mean and SEM from independent differentiation experiments are shown for each line. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, not significant.
(D) CD34 and CD45 expression on day 14 of hematopoietic differentiation in representative experiments.
(E) May-Giemsa staining of cytospin preparations of hematopoietic cells derived from the indicated iPSC lines after 14 days of hematopoietic differentiation culture. Upper and middle left panels: arrows indicate normal myeloid cells. Upper right panel: arrow indicates giant multinucleated myeloid progenitor cells. Remaining panels: arrows indicate hypogranular and/or hypolobular myeloid cells. Scale bars, 10 μm; ns, not significant.
Hematopoietic Phenotypes Cooperatively Produced by SRSF2 P95L and del(7q)
We previously identified reduced growth and viability as a disease-relevant phenotype of MDS-iPSC-derived HPCs (Kotini et al., 2015, Kotini et al., 2017). HPCs derived from both SRSF2 P95L mutant and del(7q)- iPSCs grew at a slower rate and exhibited increased cell death, compared with the normal isogenic cells (Figures 4A and 4B). Viability was even lower in HPCs derived from iPSCs harboring both the SRSF2 P95L mutation and the chr7q deletion. This defect was partially restored by correcting either the SRSF2 P95L mutation (C24 Cre6 line) or the chr7q deletion (MDS-2.A3C line), albeit without reaching normal levels (Figures 4A and 4B). SRSF2-mutant HPCs consistently gave rise to lower numbers of methylcellulose colonies than normal cells, whereas del(7q) cells gave rise to very few if any colonies (Figure 4C). The patient-derived line MDS-2.13 did not give rise to any colonies before or after correction of the SRSF2 mutation, whereas correction of the chr7q deletion (MDS-2.A3C line) partially restored colony formation, albeit not to normal levels (Figure 4C). These results suggest that the impaired viability and clonogenicity phenotypes are cooperatively generated by both the SRSF2 P95L mutation and the chr7q deletion.
Figure 4.

Cooperative Hematopoietic Phenotypes of SRSF2 P95L and del(7q)
(A) Cell viability measured by DAPI staining on day 14 of hematopoietic differentiation. Mean and SEM from independent differentiation experiments are shown for each line. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001; ns, not significant.
(B) Growth competition assay. The cells were mixed 1:1 with the N-2.12 line stably expressing GFP at the beginning of hematopoietic differentiation and followed for 12 days by flow cytometry. The relative population size was calculated as the percentage of GFP− cells at each time point relative to the population size at day 2. Error bars represent SEM from one to four independent differentiation experiments.
(C) Methylcellulose assays on day 14 of hematopoietic differentiation. The number of colonies from 5,000 seeded cells is shown. CFU-GEMM, colony-forming unit-granulocyte, erythrocyte, monocyte, megakaryocyte; CFU-GM, CFU-granulocyte, monocyte; CFU-G, colony-forming unit-granulocyte; CFU-M, CFU-monocyte; BFU-E, burst-forming unit-erythrocyte.
(D) Summary of phenotypic analyses.
(E) Scheme of phenotypes contributed exclusively by each genetic lesion or cooperatively by both.
Taken together, our phenotypic analysis of an expansive panel of isogenic iPSCs allowed us to determine phenotypes that are specific to each isolated genetic lesion—morphologic dysplasia to the SRSF2 P95L mutation and loss of myeloid progenitors to the del(7q)—as well as phenotypes that are cooperatively produced by both lesions—impaired clonogenicity, viability, and growth (Figures 4D and 4E).
The SRSF2 P95L Mutation Confers Selective Susceptibility to Splicing Inhibitors
We recently reported that SRSF2-mutant primary human leukemias preferentially respond to treatment with the splicing inhibitor E7107 (Lee et al., 2016). To precisely assess the selectivity of this growth inhibitory effect for the SRSF2 P95L mutation, we tested the growth of iPSC-derived HPCs with isolated SRSF2 mutation (5-16Cre20) and isolated chr7q deletion (Cre10) in the presence of the E7107 drug (Figure 5A). These experiments showed selective growth inhibition of SRSF2 mutant, but not of isogenic normal or of cells with isolated del(7q) (Figure 5A). We then sought to test other classes of splicing-modulating drugs for selectivity against SRSF2-mutant cells. Cpd-1, Cpd-2, and Cpd-3 are small-molecule inhibitors of CDC-like kinases and serine-arginine protein kinases, which modulate splicing (Araki et al., 2015). All three compounds could inhibit the SRSF2-mutant line, whereas they had no effect in the growth of the isogenic normal or del(7q) HPCs (Figure 5A). Quantification of un-spliced and spliced mRNA levels of two genes very sensitive to inhibition of splicing, DNAJB1 and EIF4A1 (Eskens et al., 2013), confirmed that the effect of these compounds was mediated through splicing inhibition (Figure 5B).
Figure 5.

The SRSF2 P95L Mutation Confers Selective Sensitivity to Splicing Inhibitor Drugs
(A) The normal N-2.12, SRSF2-mutant 5-16 Cre20 and del(7q) Cre10 lines were mixed 1:1 with a GFP-marked clone derived from the parental normal N-2.12 line to allow easy tracking of the relative rations of the two cell populations by flow cytometry. The cells were treated with E7107 0.1 nM, Cpd-1 5 μM, Cpd-2 0.5 μM, or Cpd-3 0.5 μM, as indicated, starting on day 8 of hematopoietic differentiation. The relative population size was calculated as the percentage of GFP− cells in the treated cells relative to the percentage of GFP− cells in the untreated cells at each time point. Error bars represent SEM from three independent experiments.
(B) Modulation of un-spliced pre-mRNA and mature mRNA of genes DNAJB1 and EIF4A1, (chosen on the basis of their documented sensitivity to splicing inhibition), in hematopoietic cells differentiated from the SRSF2 mutant 5-16 Cre20 line and treated with the indicated drugs starting at day 8 of differentiation. Fold-change overexpression levels of day 8 untreated cells is shown. The un-spliced precursor pre-mRNA levels of both genes is markedly increased, whereas the spliced (mature) mRNA levels are largely unchanged, consistent with splicing inhibition by the compounds.
A Small-Molecule Screen Identifies a Hit Compound against del(7q) Cells
To identify putative new compounds with selective growth-inhibitory activity against SRSF2-mutant and/or del(7q) cells, we screened a library of 2,000 compounds containing Food and Drink Administration-approved drugs, natural products, and other bioactive compounds at four different concentrations using a luminescence-based viability assay in the MDS-2.13 and isogenic normal N-2.12 cells. This experimental design allowed us to take advantage of parallel screening in normal isogenic control cells to filter hits with unspecific effects in cell growth inhibition. Primary hits were defined as compounds that inhibited the growth of the MDS-iPSC line, but not of the control normal iPSC line in a compound dose-dependent manner. A total of 66 compounds had a dose-dependent inhibitory effect on MDS cells. Thirty-four of these also inhibited the normal cells, whereas the remaining 32 appeared selective for the MDS cells (Figure 6A; Table S3. Results of Small-Molecule Screen in MDS-2.13 Line, Related to Figure 6, Table S4. Results of Small-Molecule Screen in N-2.12 Line, Related to Figure 6, Table S5. Hits of the Small-Molecule Screen, Related to Figure 6). These were evaluated based on physical-chemical properties, propensity for promiscuous reactivity, overall medicinal chemistry viability, and drug-like properties (Arrowsmith et al., 2015, Dahlin et al., 2015, Dahlin and Walters, 2014). Three compounds—niflumic acid, a cyclooxygenase-2 inhibitor; albendazole, and fenbendazole, two broad-spectrum benzimidazole-derivative antihelminthic agents (Figure S6A)—were further tested with a competitive growth assay (Figure 6B). Only niflumic acid showed detectable selectivity against the MDS line (Figure 6C). We further validated this result in a larger set of isogenic iPSC lines following hematopoietic differentiation (Figure 6D). Niflumic acid selectively inhibited the growth of HPCs from two del(7q) iPSC clones Cre10 and 8Cre21, but not that of HPCs derived from the two SRSF2-edited clones. Moreover, it inhibited an additional del(7q) iPSC line derived from a different MDS patient (MDS-3.1) (Kotini et al., 2015, Kotini et al., 2017), but had no effect on the growth of the del(7q)-corrected MDS-2A.3C clone.
Figure 6.

Identification and Validation of Hit Compounds Selective against del(7q) Cells
(A) General scheme of the small-molecule screening strategy, yielding 138 and 66 hits for the N-2.12 and the MDS-2.13 lines, respectively. Thirty-four of these were shared by the two lines, whereas 32 were specific for the MDS-2.13 cells.
(B and C) Competitive growth assay for hit validation. MDS-2.13 iPSCs were mixed at an equal ratio with the normal GFP-marked N-2.12 line to allow easy tracking of the ratio of the MDS to the normal cells. The cells were treated with the indicated drugs at the indicated concentrations (C). The relative population size was calculated as the percentage of GFP− cells in the treated cells relative to the percentage of GFP− cells in the untreated cells at each time point.
(D) Competitive growth assay in HPCs derived from the indicated iPSC lines. The cells were differentiated to hematopoietic cells for 12 days, mixed at an equal ratio with the normal GFP-marked N-2.12 line, differentiated in parallel, and treated with niflumic acid at 100–500 μM for 2 days. The relative population size was calculated as the percentage of GFP− cells in the treated cells relative to the percentage of GFP− cells in the untreated cells at each time point.
(E) Treatment of primary cells from patients with MDS and secondary AML with (lower panels) or without (upper panels) chromosome 7q loss (see Table S6 for details on patient characteristics) and of cord blood CD34+ cells (CB) with niflumic acid. The cells were thawed on day −2 and treated with niflumic acid on days 0, 1, and 2. The relative population was calculated from cell numbers counted on a hemocytometer on days 1, 3, and 5, relative to the DMSO-treated (untreated) cells.
We next treated primary cells from BM or peripheral blood of patients with MDS and secondary AML with or without chr7/7q abnormalities (Figures 6E and S6B; Table S6). Treatment with niflumic acid suppressed the growth of cells with monosomy 7 or del(7q), both in liquid and methylcellulose-based culture, while it had little effect in cells from patients without del7/7q or in cord blood CD34+ cells. These results further support a selective effect of niflumic acid in suppressing the growth of del(7q)-MDS cells.
Generation of Expandable iPSC-Derived HPCs
A major limiting step in the use of iPSC models in drug testing is the requirement to generate the appropriate cell types in sufficient cell numbers through in vitro differentiation procedures, which generally require specialized expertise and are often difficult to scale up. We thus sought to derive from our iPSCs HPCs that could be expanded and maintained as conventional hematopoietic cell lines. We assembled a lentiviral library of 22 factors (22F) that may confer enhanced self-renewal of HPCs in various contexts (Table S7) (Doulatov et al., 2013, Lis et al., 2017, Pereira et al., 2013, Riddell et al., 2014, Sandler et al., 2014, Sugimura et al., 2017). Day 8 embryoid bodies (EBs) from normal and SRSF2-mutant iPSCs transduced with the library gave rise to hematopoietic cells that could be expanded in culture for at least several weeks and serially replated in methylcellulose-based media (Figures 7A–7D and S7A). In contrast, control untransduced HPCs from both normal and SRSF2-mutant iPSCs did not serially replate or expand past day 28 of hematopoietic differentiation culture (Figure 7D). The eHPCs exhibited morphology and immunophenotype of myeloid progenitors and required interleukin-3 (IL-3) for growth (Figures 7E, 7F, and S7B). They could be frozen and thawed without loss of phenotypic properties (Figures S7C and S7D).
Figure 7.

Establishment of Expandable SRSF2 P95L and Isogenic Normal HPCs
(A) Experimental scheme. Normal and SRSF2-mutant iPSCs were transduced with the lentiviral library (Table S7) at day 8 of hematopoietic differentiation to generate expandable HPCs (eHPCs).
(B) Representative bright-field images of eHPC cultures 49 days after the beginning of differentiation. Scale bars, 25 μm.
(C) Serial replating of eHPCs derived from normal and SRSF2-mutant iPSCs in methylcellulose culture. The first plating was performed on day 14 from the beginning of differentiation and cells were replated every 14 days thereafter. CFU-GEMM, colony-forming unit-granulocyte, erythrocyte, monocyte, megakaryocyte; CFU-GM, CFU-granulocyte, monocyte; CFU-G, colony-forming unit-granulocyte; CFU-M, CFU-monocyte; BFU-E, burst-forming unit-erythrocyte.
(D) Expansion of eHPCs in liquid culture estimated by cell counts. TR, transduced with library; UT, untransduced.
(E) eHPCs are immunophenotypically stable for several weeks.
(F) May-Giemsa staining of cytospin preparations of day 49 eHPCs derived from the indicated iPSC lines. Scale bars, 100 μm.
(G) eHPCs derived from the N-2.12 and the 5-16 Cre20 iPSC lines were treated every 2 days with 0.1 nM E7107. The relative population size was calculated as the number of the treated cells relative to the number of untreated cells at each time point. Error bars represent SEM from three independent experiments.
To determine the factors essential for the maintenance of eHPCs, we performed barcode enrichment analysis in cells maintained in culture for 42–130 days or serially replated in methocult compared with the initial cells 6–8 days after transduction in 14 comparisons from 11 independent experiments (Figure S7E). Of the 22F, SOX17, MYB, HMGA2, GFI1b, ETV6, FOS, SCL/TAL1, LMO2, and RORA were dispensable (Figures S7F and S7G). BMI1, GATA2, ERG, c-MYC, RUNX1c, BCL-XL, HOXA9, SOX4, and MDH2 were among the most highly enriched in the majority of independent experiments, whereas GFI1, HOXB8, HOXB4, and PU1/SPI1 were also found enriched, albeit at a lower magnitude and frequency (Figures S7F and S7G). We therefore transduced these 9 factors (BMI1, GATA2, ERG, c-MYC, RUNX1c, BCL-XL, HOXA9, SOX4, and MDH2) with and without the additional 4 factors ([9+4F], BMI1, GATA2, ERG, c-MYC, RUNX1c, BCL-XL, HOXA9, SOX4, MDH2, GFI1, HOXB8, HOXB4, and PU1/SPI1) in SRSF2-mutant HPCs. The 9+4F combination, but not the 9 factors alone, generated IL-3-dependent eHPCs that could be expanded for more than 100 days (Figures S7H and S7I). Notably, the 9+4F eHPCs were capable of higher expansion than the 22F cells in two independent experiments and upregulated CD34 expression (Figure S7J). Importantly, the selective inhibition of SRSF2-mutant, but not normal cells, by the splicing inhibitor E7107 was maintained in eHPCs (Figure 7G). These results demonstrate that expandable tissue-specific progenitor cell lines can be derived from isogenic mutant and normal iPSCs and used to model therapeutic responses.
Discussion
Here, we used somatic cell reprogramming and genome editing to study the individual contributions of two cooperating genetic lesions to cellular phenotypes and therapeutic vulnerabilities. SRSF2-mutant iPSCs recapitulated several hallmarks of SRSF2-mutant MDS, including impaired hematopoiesis, splicing defects, and sensitivity to splicing inhibition. The SRSF2 mutation in isolation conferred relatively mild perturbation of hematopoiesis, compared with the profound phenotypic impairment mediated by the chr7q deletion (Kotini et al., 2015, Kotini et al., 2017). This is consistent with the SRSF2 mutation being an early, potentially initiating, event, supported by its frequent presence in the dominant clone and in individuals with clonal hematopoiesis of indeterminate potential (CHIP) (Genovese et al., 2014, Jaiswal et al., 2014, Papaemmanuil et al., 2013, Xie et al., 2014). Furthermore, the morphologic findings in our model support a role for the SRSF2 mutation in driving overt dysplasia, which might explain its lower relative frequency in CHIP compared with MDS (Malcovati et al., 2017, Sperling et al., 2017). In contrast, del(7q) resulted in a profound differentiation block in our model, consistent with a role in advanced disease and its typical association with progression to high-risk disease.
By comparing SRSF2 WT and mutant cells at different differentiation stages, we could find cell-type-specific gene expression and splicing changes. Although these may be simply due to diverse sets of genes being expressed at different differentiation stages, cell-type-specific expression of other splicing factors or global changes in splicing efficiency may also contribute to these stage-specific effects (Pimentel et al., 2016, Wong et al., 2013), and may conceivably modify disease phenotypes depending on patient BM cell composition. Our model captured a greater percentage of the genes characterized as mis-spliced in patients than previous models (Figure 2F). This suggests that the iPSC lines may be a more faithful model of mutant SRSF2, particularly for molecular studies into disease mechanisms. These results are consistent with the observation that, while splicing regulation itself is highly conserved between human and mouse, most specific isoforms are not (Barbosa-Morais et al., 2012, Merkin et al., 2012, Yeo et al., 2005). Therefore, disease-relevant targets of mutant SRSF2 may be missed in the mouse. In contrast to K562 or other immortalized cell lines, iPSCs enable the study of the SRSF2 P95L mutation in the context of a normal diploid genome both in isolation and together with cooperating mutations. All isogenic iPSC lines generated here harbor two SRSF2 alleles, maintaining correct stoichiometry of mutant to WT protein. In contrast, K562 cells typically contain three copies of the SRSF2 gene (Zhang et al., 2015). This is likely crucial in view of recent evidence on the importance of the relative expression levels of mutant and WT splicing factor genes (Fei et al., 2016, Lee et al., 2016). Furthermore, while SRSF2 mutations are early, potentially initiating events in MDS and are also found in patients with CHIP, they have so far mostly been investigated in the context of full blown leukemia. The iPSC model provides the opportunity to study their effects in isolation as they would present in pre-leukemic hematopoietic stem/progenitor cells before the onset of overt disease. Thus, the mutant iPSC lines we describe here can greatly facilitate future investigations into the mechanisms by which splicing gene mutations drive malignancy and the identification of relevant targets for therapeutic intervention.
We provide evidence for a potential therapeutic value of inhibiting kinases regulating splicing, supporting splicing modulation as a broader strategy for therapeutic intervention in MDS with splicing gene mutations. Our screening approach highlights new opportunities for drug discovery and drug repurposing that iPSC models present, such as phenotype-based screening in the absence of predetermined targets (Eggert, 2013, Engle and Puppala, 2013) and the ability to use broad phenotypes such as growth and viability as readouts with isogenic controls. Although del(7q) has been known as a recurrent chromosomal abnormality in MDS for decades, the genetic and molecular mechanisms by which it drives MDS remain incompletely understood prohibiting rational development of targeted therapies. While follow-up and translation of our findings with niflumic acid is not straightforward, as the drug exerted its effect at a very high concentration in our study, this study provides proof of principle that our approach can lead to the identification of lead compounds targeting del(7q) and other intractable genetic lesions for future drug development. Parallel testing in isogenic controls, as we show here, can enhance sensitivity and specificity and obviate the need for multiple normal controls to account for genetic background differences.
A major limiting step in the use of iPSC-derived HPCs in disease modeling and drug testing is the derivation of the desired cell types through directed differentiation. This step introduces variability, increases experimentation time and cost, and limits scalability. Here we report the generation of expandable tissue-specific progenitor cells that can be grown similarly to conventional cell lines from iPSCs. These “secondary” cell lines can overcome some of the limitations of iPSC disease models, while maintaining disease-relevant features. Such cell lines can be widely accessible and particularly valuable for precise genetic studies of the effects of driver mutations in the context of a normal diploid genome, as most established immortalized cell lines harbor complex karyotypes with frequent gene copy-number gains or losses. Indeed, no human hematopoietic cell lines harboring splicing factor mutations in the context of a normal diploid genome currently exist. Our results suggest that different factor combinations can potentially sustain expandable cultures. Future experiments could define a minimal factor combination required to generate such expandable cells, which can, further, be expressed in an inducible system.
Experimental Procedures
CRISPR/Cas9 Genome Editing
A plasmid co-expressing Cas9 linked to mCitrine by a P2A peptide driven by the cytomegalovirus promoter and a gRNA driven by the U6 promoter was constructed and gRNAs targeting the SRSF2 locus within the first intron were designed and inserted. A donor plasmid containing a floxed cassette consisting of a human phosphoglycerate kinase promoter-driven puromycin resistance gene and 5′ and 3′ homology arms was constructed. One million iPSCs were nucleofected with 5 μg of CRISPR/Cas9 plasmid(s) and 10 μg of donor plasmid. After 7–10 days, single colonies were picked, allowed to grow for approximately 3–6 days and screened by PCR.
Human iPSC Culture and Hematopoietic Differentiation
Culture of human iPSCs on mouse embryonic fibroblasts or in feeder-free conditions was performed as described previously (Papapetrou and Sadelain, 2011). For hematopoietic differentiation, spin EBs were prepared and cultured in APEL medium, as described by Ng et al. (2008).
RNA Sequencing
Poly(A)-tailed mRNA was selected with beads using the NEBNext Poly(A) mRNA Magnetic Isolation Module. cDNAs were generated using random hexamers and ligated to barcoded Illumina adaptors with the NEXTflex Rapid Directional RNA-Seq Library Prep Kit (BioScientific). Sequencing of 75-nt-long single-end reads was performed in a NextSeq 500 (Illumina).
Small-Molecule Screen
The MicroSource Discovery Systems “Spectrum Collection” library was used. Using robotic equipment, 384-well plates were first coated with 20 μL of Matrigel (BD Biosciences) diluted 1:20 in DMEM/F12 media for 30 min at room temperature. The iPSCs were subsequently plated as a single-cell suspension at a density of 1,500 per well in 50 μL TeSR medium (STEMCELL Technologies) with 10 μM Rock inhibitor Y-27632. The next day, the cells were washed and the compounds were added at a final concentration range from 10, 1.0, 0.1 to 0.01 μM. On day 5, luminescence was measured using Promega's CellTiter-Glo assay.
Author Contributions
C.-J.C. performed the experiments, analyzed the data, and assisted with manuscript preparation. A.G.K., M.O., and M.G. performed the experiments. J.T.-F. performed cytological analyses. R.K.B. analyzed and interpreted RNA sequencing data and contributed to discussion of the results and manuscript preparation. O.A.-W. provided the splicing inhibitor drugs and contributed to discussion of the results and critical reading of the manuscript. H.S. analyzed barcode enrichment data. T.J.M. performed and analyzed the small-molecule screen. R.S. and R.D. evaluated the small-molecule screen results. E.P.P. conceived, designed, and supervised the study, analyzed data, and prepared the manuscript with assistance from C.J.C. and R.K.B.
Acknowledgments
We thank Chrystel Husser for excellent technical assistance. We thank H3 Biomedicine for kindly providing E7107 and Elli Papaemmanuil for helpful comments. This work was supported by NIH grants R01HL121570 and R01HL137219, by a Damon Runyon-Rachleff Innovation Award from the Damon Runyon Cancer Research Foundation, by a Pershing Square Sohn Prize from the Pershing Square Sohn Cancer Research Alliance, and by grants from the Edward P. Evans Foundation, the Ellison Medical Foundation, and the Henry and Marilyn Taub Foundation, all to E.P.P. R.K.B. was supported by NIH grants R01HL128239 and R01DK103854, the Edward P. Evans Foundation, the Ellison Medical Foundation (AG-NS-1030-13), and the Department of Defense Bone Marrow Failure Research Program (BM150092). The small-molecule screen was funded by a Quellos Research Acceleration Award by the Institute for Stem Cell and Regenerative Medicine, University of Washington, Seattle, WA.
Published: April 19, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.03.020.
Accession Numbers
The accession number for the data reported in this paper is GEO: SRP108379.
Supplemental Information
References
- Araki S., Dairiki R., Nakayama Y., Murai A., Miyashita R., Iwatani M., Nomura T., Nakanishi O. Inhibitors of CLK protein kinases suppress cell growth and induce apoptosis by modulating pre-mRNA splicing. PLoS One. 2015;10:e0116929. doi: 10.1371/journal.pone.0116929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith C.H., Audia J.E., Austin C., Baell J., Bennett J., Blagg J., Bountra C., Brennan P.E., Brown P.J., Bunnage M.E. The promise and peril of chemical probes. Nat. Chem. Biol. 2015;11:536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais N.L., Irimia M., Pan Q., Xiong H.Y., Gueroussov S., Lee L.J., Slobodeniuc V., Kutter C., Watt S., Colak R. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338:1587–1593. doi: 10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- Dahlin J.L., Inglese J., Walters M.A. Mitigating risk in academic preclinical drug discovery. Nat. Rev. Drug Discov. 2015;14:279–294. doi: 10.1038/nrd4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin J.L., Walters M.A. The essential roles of chemistry in high-throughput screening triage. Future Med. Chem. 2014;6:1265–1290. doi: 10.4155/fmc.14.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day C.P., Merlino G., Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell. 2015;163:39–53. doi: 10.1016/j.cell.2015.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S., Vo L.T., Chou S.S., Kim P.G., Arora N., Li H., Hadland B.K., Bernstein I.D., Collins J.J., Zon L.I. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell. 2013;13:459–470. doi: 10.1016/j.stem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H., Kim E., Abdel-Wahab O., Bradley R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer. 2016;16:413–430. doi: 10.1038/nrc.2016.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert U.S. The why and how of phenotypic small-molecule screens. Nat. Chem. Biol. 2013;9:206–209. doi: 10.1038/nchembio.1206. [DOI] [PubMed] [Google Scholar]
- Engle S.J., Puppala D. Integrating human pluripotent stem cells into drug development. Cell Stem Cell. 2013;12:669–677. doi: 10.1016/j.stem.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Eskens F.A., Ramos F.J., Burger H., O'Brien J.P., Piera A., de Jonge M.J., Mizui Y., Wiemer E.A., Carreras M.J., Baselga J. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013;19:6296–6304. doi: 10.1158/1078-0432.CCR-13-0485. [DOI] [PubMed] [Google Scholar]
- Fei D.L., Motowski H., Chatrikhi R., Prasad S., Yu J., Gao S., Kielkopf C.L., Bradley R.K., Varmus H. Wild-type U2AF1 antagonizes the splicing program characteristic of U2AF1-mutant tumors and is required for cell survival. PLoS Genet. 2016;12:e1006384. doi: 10.1371/journal.pgen.1006384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H., Korn J.M., Ferretti S., Monahan J.E., Wang Y., Singh M., Zhang C., Schnell C., Yang G., Zhang Y. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015;21:1318–1325. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- Genovese G., Kahler A.K., Handsaker R.E., Lindberg J., Rose S.A., Bakhoum S.F., Chambert K., Mick E., Neale B.M., Fromer M. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould S.E., Junttila M.R., de Sauvage F.J. Translational value of mouse models in oncology drug development. Nat. Med. 2015;21:431–439. doi: 10.1038/nm.3853. [DOI] [PubMed] [Google Scholar]
- Graubert T.A., Shen D., Ding L., Okeyo-Owuor T., Lunn C.L., Shao J., Krysiak K., Harris C.C., Koboldt D.C., Larson D.E. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2012;44:53–57. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S., Fontanillas P., Flannick J., Manning A., Grauman P.V., Mar B.G., Lindsley R.C., Mermel C.H., Burtt N., Chavez A. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E., Ilagan J.O., Liang Y., Daubner G.M., Lee S.C., Ramakrishnan A., Li Y., Chung Y.R., Micol J.B., Murphy M.E. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell. 2015;27:617–630. doi: 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotini A.G., Chang C.J., Boussaad I., Delrow J.J., Dolezal E.K., Nagulapally A.B., Perna F., Fishbein G.A., Klimek V.M., Hawkins R.D. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat. Biotechnol. 2015;33:646–655. doi: 10.1038/nbt.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotini A.G., Chang C.J., Chow A., Yuan H., Ho T.C., Wang T., Vora S., Solovyov A., Husser C., Olszewska M. Stage-specific human induced pluripotent stem cells map the progression of myeloid transformation to transplantable leukemia. Cell Stem Cell. 2017;20:315–328 e317. doi: 10.1016/j.stem.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke J., Fink E.C., Hollenbach P.W., MacBeth K.J., Hurst S.N., Udeshi N.D., Chamberlain P.P., Mani D.R., Man H.W., Gandhi A.K. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature. 2015;523:183–188. doi: 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.C., Dvinge H., Kim E., Cho H., Micol J.B., Chung Y.R., Durham B.H., Yoshimi A., Kim Y.J., Thomas M. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016;22:672–678. doi: 10.1038/nm.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis R., Karrasch C.C., Poulos M.G., Kunar B., Redmond D., Duran J.G.B., Badwe C.R., Schachterle W., Ginsberg M., Xiang J. Conversion of adult endothelium to immunocompetent haematopoietic stem cells. Nature. 2017;545:439–445. doi: 10.1038/nature22326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcovati L., Galli A., Travaglino E., Ambaglio I., Rizzo E., Molteni E., Elena C., Ferretti V.V., Catricala S., Bono E. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129:3371–3378. doi: 10.1182/blood-2017-01-763425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J., Russell C., Chen P., Burge C.B. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science. 2012;338:1593–1599. doi: 10.1126/science.1228186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng E.S., Davis R., Stanley E.G., Elefanty A.G. A protocol describing the use of a recombinant protein-based, animal product-free medium (APEL) for human embryonic stem cell differentiation as spin embryoid bodies. Nat. Protoc. 2008;3:768–776. doi: 10.1038/nprot.2008.42. [DOI] [PubMed] [Google Scholar]
- Papaemmanuil E., Cazzola M., Boultwood J., Malcovati L., Vyas P., Bowen D., Pellagatti A., Wainscoat J.S., Hellstrom-Lindberg E., Gambacorti-Passerini C. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011;365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E., Gerstung M., Bullinger L., Gaidzik V.I., Paschka P., Roberts N.D., Potter N.E., Heuser M., Thol F., Bolli N. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E., Gerstung M., Malcovati L., Tauro S., Gundem G., Van Loo P., Yoon C.J., Ellis P., Wedge D.C., Pellagatti A. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–3627. doi: 10.1182/blood-2013-08-518886. quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetrou E.P. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat. Med. 2016;22:1392–1401. doi: 10.1038/nm.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetrou E.P., Lee G., Malani N., Setty M., Riviere I., Tirunagari L.M., Kadota K., Roth S.L., Giardina P., Viale A. Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat. Biotechnol. 2011;29:73–78. doi: 10.1038/nbt.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetrou E.P., Sadelain M. Generation of transgene-free human induced pluripotent stem cells with an excisable single polycistronic vector. Nat. Protoc. 2011;6:1251–1273. doi: 10.1038/nprot.2011.374. [DOI] [PubMed] [Google Scholar]
- Pereira C.F., Chang B., Qiu J., Niu X., Papatsenko D., Hendry C.E., Clark N.R., Nomura-Kitabayashi A., Kovacic J.C., Ma'ayan A. Induction of a hemogenic program in mouse fibroblasts. Cell stem cell. 2013;13:205–218. doi: 10.1016/j.stem.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel H., Parra M., Gee S.L., Mohandas N., Pachter L., Conboy J.G. A dynamic intron retention program enriched in RNA processing genes regulates gene expression during terminal erythropoiesis. Nucleic Acids Res. 2016;44:838–851. doi: 10.1093/nar/gkv1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell J., Gazit R., Garrison B.S., Guo G., Saadatpour A., Mandal P.K., Ebina W., Volchkov P., Yuan G.C., Orkin S.H. Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell. 2014;157:549–564. doi: 10.1016/j.cell.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler V.M., Lis R., Liu Y., Kedem A., James D., Elemento O., Butler J.M., Scandura J.M., Rafii S. Reprogramming human endothelial cells to haematopoietic cells requires vascular induction. Nature. 2014;511:312–318. doi: 10.1038/nature13547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling A.S., Gibson C.J., Ebert B.L. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer. 2017;17:5–19. doi: 10.1038/nrc.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura R., Jha D.K., Han A., Soria-Valles C., da Rocha E.L., Lu Y.F., Goettel J.A., Serrao E., Rowe R.G., Malleshaiah M. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature. 2017;545:432–438. doi: 10.1038/nature22370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J.J., Ritchie W., Ebner O.A., Selbach M., Wong J.W., Huang Y., Gao D., Pinello N., Gonzalez M., Baidya K. Orchestrated intron retention regulates normal granulocyte differentiation. Cell. 2013;154:583–595. doi: 10.1016/j.cell.2013.06.052. [DOI] [PubMed] [Google Scholar]
- Xie M., Lu C., Wang J., McLellan M.D., Johnson K.J., Wendl M.C., McMichael J.F., Schmidt H.K., Yellapantula V., Miller C.A. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G.W., Van Nostrand E., Holste D., Poggio T., Burge C.B. Identification and analysis of alternative splicing events conserved in human and mouse. Proc. Natl. Acad. Sci. USA. 2005;102:2850–2855. doi: 10.1073/pnas.0409742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K., Sanada M., Shiraishi Y., Nowak D., Nagata Y., Yamamoto R., Sato Y., Sato-Otsubo A., Kon A., Nagasaki M. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- Zhang J., Lieu Y.K., Ali A.M., Penson A., Reggio K.S., Rabadan R., Raza A., Mukherjee S., Manley J.L. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. USA. 2015;112:E4726–E4734. doi: 10.1073/pnas.1514105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.