

Summary
Human pluripotent stem cells (hPSCs) generate hematopoietic progenitor cells (HPCs) but fail to engraft xenograft models used to detect adult/somatic hematopoietic stem cells (HSCs) from donors. Recent progress to derive hPSC-derived HSCs has relied on cell-autonomous forced expression of transcription factors; however, the relationship of bone marrow to transplanted cells remains unknown. Here, we quantified a failure of hPSC-HPCs to survive even 24 hr post transplantation. Across several hPSC-HPC differentiation methodologies, we identified the lack of CXCR4 expression and function. Ectopic CXCR4 conferred CXCL12 ligand-dependent signaling of hPSC-HPCs in biochemical assays and increased migration/chemotaxis, hematopoietic progenitor capacity, and survival and proliferation following in vivo transplantation. This was accompanied by a transcriptional shift of hPSC-HPCs toward somatic/adult sources, but this approach failed to produce long-term HSC xenograft reconstitution. Our results reveal that networks involving CXCR4 should be targeted to generate putative HSCs with in vivo function from hPSCs.
Keywords: human pluripotent, hematopoietic stem cells, progenitors, bone marrow, transplantation, chemokine receptor, cell signaling
Graphical Abstract
Highlights
-
•
Transplant kinetics indicate human PSC-HPCs fail in the first 24 hr in bone marrow
-
•
hPSC-HPCs aberrantly express chemokine receptors, specifically lacking CXCR4
-
•
Ectopic CXCR4 enhances hPSC-HPC function in vitro and transplantation in vivo
-
•
CXCR4 linked with global transcriptional shift of hPSC-HPCs toward somatic HPCs
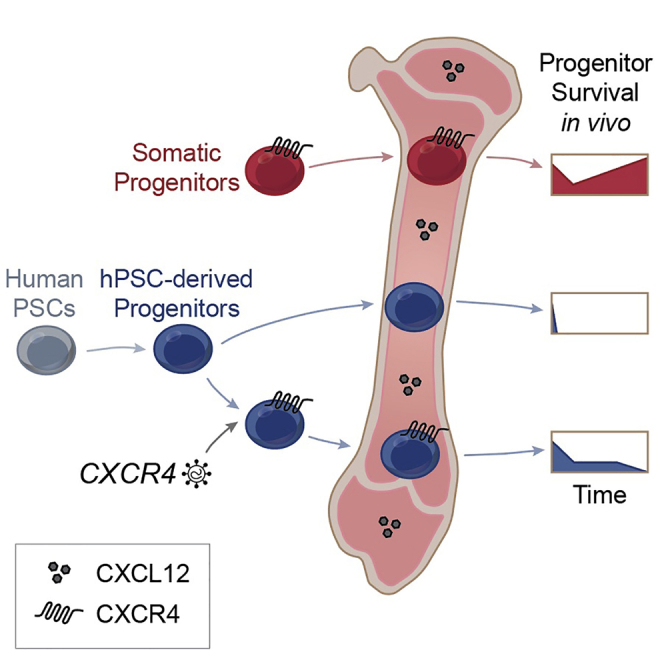
Bhatia and colleagues reveal that human PSC-derived hematopoietic progenitor cells fail to survive even 24 hr following in vivo bone marrow transplantation, while these same progenitors survive and proliferate for weeks in vitro. They link these observations to deficiencies in CXCR4 signaling, which, when rectified, lead to enhanced progenitor function and survival in the bone marrow and a transcriptional shift toward somatic hematopoietic stem cells gene profiles.
Introduction
Hematopoietic stem cell (HSC) transplants are the only globally adopted stem cell therapy for patients and have been shown to be curative for hematological malignancies and diseases along with certain solid tumors (Copeland, 2006). However, given the scarcity of compatible donors against the number of patients in need (Gratwohl et al., 2015), developing alternative sources of HSCs is paramount. While hematopoietic progenitor cells (HPCs) can readily be generated by human pluripotent stem cells (hPSCs) in vitro, they lack robust engraftment potential (Gori et al., 2015, Lee et al., 2017, Ng et al., 2016, Risueño et al., 2012, Wang et al., 2005). Ectopic transcription factor (TF) expression has been used in attempts to induce bone marrow (BM) engraftment of hPSC-HPCs (Doulatov et al., 2013, Doulatov et al., 2017, Ramos-Mejia et al., 2014, Ran et al., 2013, Wang et al., 2005). Recently, expression of seven TFs in hPSC-derived hemogenic endothelium generated HSC/HPCs, but only after BM transplantation (Sugimura et al., 2017). Despite this progress, the in-vivo-dependent approach did not produce an abundance of HSCs and these cells remain molecularly unrelated to somatic HSCs—factors that require modification for successful clinical translation.
Unlike solid organ transplants, injected HSCs must migrate to and reside in specialized niches in the BM, the primary site of adult hematopoiesis (Boyd and Bhatia, 2014). Adult HSCs receive complex and dynamic cues from the BM for survival, quiescence, homeostasis, and proliferation. Likewise, using co-cultures of BM stroma cells or embryonic niche cells improves hPSC-HPC derivation (Ledran et al., 2008, Tian et al., 2006, Vodyanik et al., 2006, Weisel et al., 2006), suggesting these cells too require specific niche cues. BM secreted CXCL12 (formerly known as SDF1) is a powerful regulator of HSC function and binds its cognate receptor, CXCR4, expressed by HSC/HPCs (Lapidot and Kollet, 2002, Nagasawa et al., 1996, Sugiyama et al., 2006). CXCR4 represents the sole chemokine receptor utilized by HSCs for migration/chemotaxis (Wright et al., 2002) and regulates the proliferation of somatic HSCs (Kahn et al., 2004). This is sustained through an auto-regulatory loop that is dynamically regulated from cell surface to intracellular stores (Lapidot and Kollet, 2002). CXCR4 is regulated by BM factors, some of which include hypoxia (Scheurer et al., 2004), Notch (Wang et al., 2017), glucocorticoid (Guo et al., 2017), and prostaglandin E2 (PGE2) (Goessling et al., 2011) signaling pathways. However, the functional capacity of hPSC-HPCs to respond to BM regulatory cues remains largely unknown.
Previous studies assessing hPSC-HPC engraftment potential have reported low levels of human hematopoietic microchimerism in immunocompromised mouse BM 4 weeks or more post transplant (Doulatov et al., 2013, Doulatov et al., 2017, Gori et al., 2015, Lee et al., 2017, Ramos-Mejia et al., 2014, Ran et al., 2013, Risueño et al., 2012, Wang et al., 2005). Here, we reveal the previously unappreciated early transplantation failure of hPSC-HPCs in vivo that occurs within the first 24 hr, despite robust hematopoietic progenitor capacity detected for weeks in vitro. Across a broad range of differentiation methodologies, global transcriptional analysis identified the chemokine receptor CXCR4 as aberrantly regulated in hPSC-HPCs. Molecular and biochemical characterization of ectopic CXCR4 in hPSC-HPCs demonstrated that CXCR4 enhances survival and progenitor retention following BM transplantation. We propose the CXCR4 network is essential for physiological responsiveness toward generating bona fide HSCs from hPSCs.
Results
Defective In Vivo Retention of hPSC-HPCs
Early properties of hPSC-HPC integration into the BM in vivo have not been explored by direct side by side comparisons with human adult/somatic HPC sources. Cord blood (CB) is readily available for experimentation as a somatic source of HSCs that establish long-term multilineage hematopoietic engraftment in xenograft models (Boyd et al., 2017). Furthermore, transplantation of CB cells has been used clinically for long-term reconstitution of donor-derived healthy hematopoiesis in patients (Cutler et al., 2013). As such, we used CB as a source of transplantable cells to analyze early HPC behavior and compare this directly with HPCs derived from hPSCs. hPSC-derived HPCs were derived using embryoid body (EB) formation and differentiated with hematopoietic cytokines and BMP4 (Chadwick et al., 2003), and were utilized on EB day 15 for analysis and transplantation. Somatic and hPSC-HPCs do not share equivalent frequencies of phenotypic or functional progenitors, as quantified by human specific CD34+CD45+ cell surface expression (versus mouse mCD45; Figure 1A) and colony forming unit (CFU) composition (Figure S1A), respectively. These results are consistent with previous reports across a broad range of methodologies to produce phenotypic or functional progenitors from hPSCs (Doulatov et al., 2013, Lee et al., 2017, Ramos-Mejia et al., 2014, Risueño et al., 2012, Saxena et al., 2016, Tian et al., 2006, Vodyanik et al., 2006), as well as non-human primate Macaca nemestrina-induced PSC-derived HPCs (Gori et al., 2015). To accurately control for the number of input human CD34+CD45+ cells and functional progenitors injected from hPSC-HPC cultures, we therefore selected two doses of CB for transplantation (Figure 1B). The higher CB dose, CBCD34, closely approximates the number of total phenotypic hPSC-HPCs transplanted (dark red, 5 × 104 CD34+ cells). The lower dose, CBPRO, closely approximates the total number of functional hPSC-HPC progenitors, as well as the clinical dose of 2 × 105 CD34+ cells/kg (Gluckman, 2009) when scaled by mass to a 25 g mouse (light red, 5 × 103 CD34+ cells). Total phenotypic and functional HPCs prior to injection were measured at day 0 (Figures 1A and 1B).
Figure 1.

Transplantation Kinetics of Somatic and hPSC-Derived HPCs
(A) Phenotype of CB and hPSC-derived HPCs in injection fraction (human CD34+ human CD45+ mouse CD45–), analyzed on the day of transplant. Mouse CD45 antibody use validated to exclude mouse cells upon BM transplantation.
(B) Total viable cells, and total phenotypic (CD34+CD45+) and functional (CFU) CB and hPSC-derived HPCs in injection fraction, analyzed on the day of transplant. CBCD34 and CBPRO cell doses control for input phenotypic and functional hPSC-HPCs, respectively. Data points represent the transplanted cells of three independent transplants. Data are represented as means ± SEM.
(C) Tissue allocation for phenotypic and functional assays.
(D) n numbers represent transplanted mice, pooled from three independently performed experiments with six harvest analyses.
(E) Phenotype of CB and hPSC-derived HPCs from harvested BM.
(F) Total mCD45–hCD45+CD34+ cells retained in the BM of injected (IF) and contralateral (CF) femurs. To assess BM retention separately from proliferation, only 24 and 48 hr data for CB shown. Data points represent n transplanted mice, Ø is zero. Two-way ANOVA, ∗∗∗∗p < 0.0001. Data are represented as means ± SEM.
(G and H) Total mCD45–hCD45+CD34+ cells per injected femur. Same hPSC-HPC data in both panels.
(I) CFU from CB-transplanted BM, harvested at day 5. Arrowheads: red, burst-forming unit-erythroid; gray, CFU-granulocyte and/or -monocyte.
(J) Total human CFU per harvested IF and CF BM. To assess BM retention of progenitors separately from cellular proliferation and expansion, only 24 and 48 hr retention data for CB shown; day 3 and 5 data omitted. Data points represent n transplanted mice, Ø is zero. One-way ANOVA, ∗∗p < 0.01. Data are represented as means ± SEM.
(K and L) Total human CFU per IF. Same hPSC-HPC data in both panels.
(M) Linear regression of total CB phenotypic versus functional HPCs quantified per IF. Data points represent n transplanted mice.
Using this carefully quantitated approach to phenotypically and functionally enumerate equivalency of transplanted cells, human CB versus hPSC-derived HPCs were injected into the femurs of murine recipients, where the BM was assessed for human chimerism at the functional and phenotypic level at multiple time points within the first week. At the same time points as injected femur assessment, we determined migration capacity in vivo by analysis of contralateral femur BM, spleen, and lungs (Figure 1C). The number of individual mice from four transplant groups were compared at 24 hr and 2, 3, and 5 days as indicated (Figure 1D) to address the classical time of homing, within 24 hr (Jetmore et al., 2002), while also being inclusive of longer periods of homing, up to 4 days (Foster et al., 2015). The frequency of human hematopoietic cell chimerism was rare, but could be captured by flow cytometric analysis for human HPCs (mCD45–hCD45+CD34+, Figures 1E, S1B, and S1C). Phenotypic CB HPC expansion was evident within the injected femur BM well within this time frame (Figure 1E). As predicated (Wang et al., 2005), intra-femoral injection provided an engraftment advantage to retain HPCs in the injected femur, while a subpopulation of somatic HPCs could still home to the contralateral femur (Figure 1F) but not to extramedullary sites such as the lung (Figures S1D–S1F) or spleen (Figures S1G–S1I). In contrast, hPSC-HPCs were not able to persist even at 24 hr post transplant in any location, yet CB HPCs were capable of robust and exponential expansion in the BM at both cellular doses (Figures 1G and 1H; comparison of fits test, Prism software). Due to the rarity of these cells at early time points post transplantation, evaluation of HSC function by secondary transplantation was not feasible for both somatic and hPSC-derived HPCs. However, we were able to extract BM and compare early engraftment kinetics at the progenitor level using the CFU assay in vitro. Functional CB HPCs were retained in injected femur BM and continued progenitor output over the first week, while putative hPSC-HPCs failed to be retained as measured by erythroid-myeloid CFU assays (Figures 1J–1L). Strikingly, BM retention of CB HPCs was strongly correlated between phenotypic and functional measures, whereas no such relationship existed for hPSC-HPCs (Figure 1M). These results suggest a reduction of somatic HPCs within the first 24 hr post injection, followed by a rapid increase from 24 hr to 5 days in vivo. This behavior is in sharp contrast with hPSC-HPCs, which fail to recover and proliferate in vivo. These experimental observations reveal an unappreciated deficiency contributing to hPSC-HPC engraftment failure that occurs upon initial transplantation.
Somatic and hPSC-Derived HPCs Are Functionally Similar In Vitro
Based on the inability of hPSC-HPCs to survive and proliferate in vivo, we examined their behavior in vitro across a broad range of hPSC differentiation methodologies, and compared them with adult/somatic control sources of HPCs. Somatic HPCs can be harvested from human BM, adult mobilized peripheral blood (MPB), and neonatal CB sources, and are enriched in the CD34+CD45+ subpopulation (Figure 2A). Similarly, the past decade has provided several methodologies to derive hPSC-HPCs, and so we investigated three very different protocols and approaches: (1) cytokines and BMP4 treatment of EBs (Chadwick et al., 2003), (2) OP9 co-culture (Vodyanik et al., 2006), and (3) endothelial-hematopoietic transition (EHT) (Lee et al., 2017) (Figure 2B). We routinely observed hPSC-derived hematopoietic CFU morphology similar to somatic HPCs when cultured for 14 days in vitro (Figures 2C and 2D). To control for variations in CD34+CD45+ frequency, we determined the CFU output and lineage distribution per 1,000 CD34+CD45+ cells, which was similar across adult BM, adult MPB, and neonatal CB donor samples (Figure 2E). Similarly, while the three differentiation methods produced varying frequencies of hPSC-HPCs (Figure 2B), CFU per 1,000 HPCs was also similar (Figure 2F). hPSC-HPCs readily survive and proliferate in vitro (Figure 2) and yet failed to survive very short time frames in vivo (Figure 1). Even the same hPSC-HPCs tested in parallel in these two environments (in vivo versus in vitro) led to sharply contrasting effects in progenitor capacity (Figures 1B and 1K). These comparative results indicate hPSC-HPCs may be insufficiently responding to BM environmental cues in vivo that prevent the survival that can be readily demonstrated in vitro.
Figure 2.

Shared Progenitor Capacity of Somatic and hPSC-Derived HPCs
(A) Phenotypic (CD34+CD45+) somatic HPCs, by flow cytometry. Negative stain in black.
(B) Phenotypic (CD34+ with CD45+ and/or CD43+) hPSC-HPCs, by flow cytometry, with: cytokines and BMP4 (Chadwick et al., 2003); OP9 co-culture (Vodyanik et al., 2006); and EHT in normoxia or hypoxia (Lee et al., 2017) differentiation methods. Negative stain in black.
(C and D) Somatic (C), and hPSC-derived (D) hematopoietic colonies (CFU); picked CFU were spun onto slides for Wright-Giemsa staining (Cytospin). White scale bar represents 500 μm; black scale bar represents 30 μm.
(E and F) Total CFU relative to 1.0 × 103 input phenotypic HPCs from (E) somatic HPCs; data points represent n = 4 (BM, CB) and n = 8 (MPB) independently assayed wells, pooled from independently performed healthy donors (BM n = 1, CB n = 2, MPB n = 3); and (F) hPSC-HPCs; data points represent n = 10 (cytokines and BMP4, OP9 co-culture) and n = 3 (EHT, hypoxia [hyp.], and normoxia [nor.]) independently assayed wells, pooled from independently performed experiments. One-way ANOVA, no statistical differences. Data are represented as means ± SEM.
BFU-E, burst-forming unit-erythroid; E, erythrocyte; G, granulocyte; GEM, granulocyte, erythrocyte, and monocyte; GM, granulocyte and monocyte; and M, monocyte.
Deficient Chemokine Receptor Expression Is a Consistent Feature of hPSC-HPCs
To understand potential underlying interactions that differ between hPSC-HPCs and BM versus adult/somatic HPCs and BM, we applied global transcriptome analysis of primitive hematopoietic populations enriched for HSC/HPCs (CD45+CD34+CD38–), from 15 healthy donors, including BM, MPB, CB, and fetal blood (FB) sources (Figure 3A, see also Supplemental Experimental Procedures) and collectively compared these with hPSC-HPCs-derived samples using the cytokines and BMP4 (Chadwick et al., 2003), and EHT method of differentiation (Lee et al., 2017), as well as using gene expression data for TF-expressing hPSC-HPCs (Doulatov et al., 2013). Gene set enrichment analysis (GSEA) of global transcriptional profiles identified several gene sets relevant to environmental cues in vivo, including chemokine receptors, extracellular matrix interactions, integrins, and cell surface interactions at the vascular wall (Figure 3B, Table S1), which were enriched in hPSC-HPCs. Closer observation identified two consistent differentially expressed chemokine receptor genes, CXCR4 and CX3CR1 (Figure 3C), which are associated with BM retention and egress, respectively (Nakano et al., 2017). We next performed global transcriptome analysis comparing the four different sources of hPSC-HPCs individually with somatic HPCs (MPB, BM, CB, and FB), which identified 666 consistently differentially expressed genes (Figure 3D, Table S2). Unsupervised hierarchical clustering segregated 120 as exclusively expressed by somatic HPCs (Figure 3E). Within these 120 genes, we observed HOXA5, HOXA10, FLT3, and PROM1, and epigenetic regulators such as HDAC7, KAT6A, and MLLT3, as well as CXCR4, which are down-regulated in hPSC-HPCs compared with somatic HPCs (Table S2). STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) protein-protein interaction database (Szklarczyk et al., 2015) analysis identified potential interactions of the 120 HSC-related genes, where CXCR4 is prominently linked to key HSC/HPC-related genes (Figure 3F).
Figure 3.

Identification of Aberrant Chemokine Receptor Expression by hPSC-HPCs
(A) Principal component analysis (PCA) of global transcriptome from fluorescence-activated cell sorting (FACS)-purified CD45+CD34+CD38± HPCs, including samples from GEO: GSE49938 (Doulatov et al., 2013), and from our lab (GEO: GSE3823 and GSE106721), diamond and circle symbols, respectively. Further details in Supplemental Experimental Procedures.
(B) Gene sets identified by GSEA as enriched in hPSC- versus somatic HPCs, using meaned groups described in Figure 1A. GSEA reports in Table S1.
(C) Blue-Pink O′ Gram heat maps from GSEA report using meaned groups described in Figure 1A.
(D) ANOVA comparing the four sources of hPSC-HPCs individually with somatic HPCs, depicted as a Venn diagram.
(E) Unsupervised hierarchical clustering of hPSC- and somatic HPCs using 666 genes identified in Figure 3D.
(F) STRING analysis of 120 somatic HPC up-regulated genes, where 43 are shown; 77 disconnected nodes were removed.
(G and H) Flow cytometry of CXCR4 and CX3CR1 of CD34+CD45+ somatic HPCs (G), and hPSC-HPCs (H); HPC phenotypes in Figure 2B. Negative stain in black.
(I and J) Summary of CXCR4+CX3CR1– (I), and CX3CR1+CXCR4– (J), expression of phenotypic HPCs, as assessed by flow cytometry. Data points represent n independent healthy donors (somatic HPCs) or independent biological replicates (hPSC-HPCs) (precise n values indicated in the figure), pooled from independently performed experiments. Triangle symbols indicate biological replicates cultured in hypoxia. Data are represented as means ± SEM.
These consistent observations suggested a potentially broad deficiency that we further investigated with flow cytometric analysis, which revealed that CXCR4 and CX3CR1 proteins are mutually exclusively expressed on the cell surface (Figures S2A–S3C). Furthermore, CXCR4 is enriched on CB HPCs (Figure S2A), while CX3CR1 is enriched on hPSC-HPCs (Figures S2B and S2C). We expanded upon these observations with three diverse sources each of somatic and hPSC-derived HPCs (Figures 3G–3J). We observed a highly reproducible CXCR4+CX3CR1– phenotype expressed by un-cultured somatic HPCs and absence of this subset from hPSC-HPCs, which instead were CX3CR1+CXCR4–. Importantly, somatic HPCs engrafted in mice at 5 weeks continued to highly express CXCR4 (Figure S2D). Low expression of cutaneous lymphocyte antigen (CLA, HECA452 clone) was recently suggested to have a role in hPSC-HPC transplantation deficiency (Lee et al., 2017). While we could replicate low CLA expression relative to a somatic HPC source, we again did not observe CXCR4 expression by transcriptional or protein analyses of EHT-derived hPSC-HPCs (Figures S2E and S2F), suggesting both may be important factors. CXCR4– and CX3CR1+ phenotypes of hPSC-HPCs have been previously noted (Ng et al., 2016, Salvagiotto et al., 2008, Tian et al., 2006) and reinforce our observations that low CXCR4 expression is a consistent deficiency of hPSC-HPCs across a broad range of methodologies and hPSC cell lines.
To move beyond phenotypic observations, we physically isolated CB and hPSC-derived hematopoietic cells based exclusively on chemokine receptor expression, and determined biological hematopoietic progenitor function. As predicted, hematopoietic progenitors measured by CFU potential were enriched within CXCR4+ CB cells, and not observed from CX3CR1+ cells (Figures 4A–4C), as CX3CR1 expression is restricted to mature hematopoietic cells (Nakano et al., 2017). In stark contrast, CFU potential was observed from CX3CR1+ hPSC-derived hematopoietic cells, while CXCR4+ hPSC-derived cell CFU potential was significantly reduced (Figures 4D–4F). CXCR4 and CX3CR1 are chemokine receptors that bind only one cytokine: CXCL12 and CX3CL1, respectively (Balkwill, 2004). CXCL12 is most highly expressed in BM tissue, whereas CX3CL1 is more highly expressed in non-hematopoietic and extramedullary sites such as the lungs, kidneys, and spleen (Uhlén et al., 2015). We therefore sought to determine whether the CXCR4 or CX3CR1 receptors expressed by hPSC-hematopoietic cells (Figure 3E) were functionally capable of initiating chemotaxis toward their cognate ligands (Figure 4G), to investigate whether hPSC-HPCs may be receiving BM cues for retention or egress. Serving as a positive control, bulk somatic hematopoietic cells were able to migrate toward both CXCL12 and CX3CL1 (Figure 4H), as both receptors were observed on bulk CB cells (Figure 3D). MPB was an exception, which exhibited a blunted response toward CXCL12 (Figure 4H), which is likely correlated to CXCR4-antagonist treatment (AMD3100) in donors to mobilize healthy HSCs and HPCs out of the BM and into circulation (Broxmeyer et al., 2005). Despite multiple experiments, hPSC-HPCs did not migrate toward CXCL12 or CX3CL1 (Figure 4H). In vivo BM engraftment requires signals mediating BM retention and limiting egress. Due to the lack of chemotactic response exhibited by hPSC-HPCs (Figure 4H), we suggest the lack of CXCL12-CXCR4 function is preventing BM retention, while CX3CL1-CX3CR1 binding is not actively participating in BM egress (Nakano et al., 2017). Together, these data suggest that promoting CXCR4 expression may be beneficial to improve hPSC-HPC function.
Figure 4.

Differential Segregation of Progenitors by CXCR4 and CX3CR1
(A) Sort purities from CB cells FACS-purified by CXCR4 and CX3CR1 expression. Quadrant (Q) 2 was not detected (ND).
(B) Composite well images of Q1- and Q4-sorted CB. 5 × 102 Q1 and 1 × 103 Q4 sorted cells were seeded per well. Arrowheads indicate individual CFU. Scale bar represents 5 mm.
(C) CFU frequency of FACS-purified CB based on chemokine receptor expression. Data points represent n independently assayed wells (precise n values indicated in the figure). Ø is zero. Q2 was ND. Data are represented as means ± SEM.
(D) Sort purities from hPSC-derived hematopoietic cells FACS-purified by CXCR4 and CX3CR1 expression. Q2 was ND.
(E) Composite well images of Q1- and Q4-sorted hPSC-derived hematopoietic cells; 2 × 104 sorted cells were seeded per well. Arrowheads indicate individual CFU. Scale bar represents 5 mm.
(F) CFU frequency of FACS-purified hPSC-derived hematopoietic cells based on chemokine receptor expression. Data points represent n independently assayed wells (precise n values indicated in the figure). Q2 was ND. Data are represented as means ± SEM.
(G) Transwell assay was conducted with 200 ng/mL CXCL12, or 200 ng/mL CX3CL1, or control (0.001% BSA).
(H) Transwell migration was quantified by flow cytometry. Data points represent n independently assayed wells (precise n values indicated in the figure), pooled from four independently performed experiments. CB used as positive control in every hPSC-HPC experiment. Two-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. Data are represented as means ± SEM.
Inducing CXCL12-Dependent CXCR4 Signaling
On the basis of a beneficial functional relationship between CXCR4 and somatic HPCs (Brenner et al., 2004, Kahn et al., 2004), we next sought to pharmacologically induce CXCR4 expression by hPSC-HPCs. We tested several pharmacological agents reported to upregulate CXCR4, including cyclic AMP (cAMP) agonists, forskolin and PGE2 (Goessling et al., 2011, Saxena et al., 2016), and hormones (Flonase and estrogen; Guo et al., 2017, Rodriguez-Lara et al., 2017). While these compounds effectively increased the number of CXCR4+ CB HPCs, this was not observed from hPSC-HPCs (Figures 5A, 5B, S3A, and S3B). GSEA pathway analysis demonstrated that networks targeted by these compounds are not equally active (Figure 5C), supporting our observation of an inability to upregulate CXCR4 by hPSC-HPCs.
Figure 5.

Overcoming Resistance to CXCR4 Expression Enables CXCL12-Dependent Signaling
(A) Flow cytometric of CXCR4 within CD34+CD45+ CB or hPSC-HPCs treated with indicated compounds; see also Supplemental Experimental Procedures.
(B) A summary of total CXCR4+CD34+CD45+ cells from CB and hPSC-HPCs, relative to control (0.01% DMSO or BSA). Data points represent n independently assayed wells (precise n values indicated in Supplemental Experimental Procedures), pooled from three independently performed experiments. Data are represented as means ± SEM. Two-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(C) GSEA analysis of pathways that regulate CXCR4. Samples: CB and H9-HPC; CD34+CD45+CD38–, shown in Figure 3A. GO, Gene Ontology; NES, normalized enrichment score.
(D) Human CXCR4 was cloned into pHIV-EGFP, which has an internal ribosome entry site (IRES) translation link to GFP (CXCR4+). Site-directed mutagenesis of CXCR4 (N123K) was termed CXCR4(off)+. Vector control, expressing GFP, was used in parallel for all experiments.
(E) Flow cytometry at 48 hr post transduction on EB day 16, showing comparable hPSC-HPC frequency (CD34+CD45+), and robust CXCR4+GFP+ co-expression.
(F) Transwell assay was conducted with 200 ng/mL CXCL12 or control (0.001% BSA), and quantified by flow cytometry at 48 hr post transduction on EB day 16. Data points represent n independently assayed wells (precise n values indicated in the figure), pooled from three independently performed experiments. Two-way ANOVA, ∗∗∗∗p < 0.0001. Data are represented as means ± SEM.
(G) Calcium flux transients were monitored in response to CXCL12 (200 ng/mL) in the absence (top row) or presence (bottom row) of CXCR4 inhibitor (AMD3100, 10 μM) at 48 hr post transduction on EB day 16. Ionomycin (10 μM) treatment was used as a positive control to identify live cells.
(H and I) The frequency of cells that responded to CXCL12 treatment (transient >150% of baseline) was quantified in the absence (H) or presence (I) of CXCR4 inhibitor, AMD3100. Data points represent n = 4 independently assayed wells per sample, pooled from two independently performed experiments. Ø is zero. Data are represented as means ± SEM.
(J) Total CFU counts from transduced hPSC-HPCs seeded into MethoCult ±150 ng/mL CXCL12 at 48 hr post transduction on EB day 16. Data points represent n independently assayed wells (precise n values indicated in the figure), pooled from three independently performed experiments. Two-way ANOVA, ∗∗∗∗p < 0.0001. Data are represented as means ± SEM.
Therefore, lentiviral ectopic CXCR4 expression was developed and functionally validated (Figure 5D). We developed an additional vector, which expressed CXCL12-unresponsive CXCR4 (N123K mutation; Zhang et al., 2002; termed CXCR4(off)+), in order to identify CXCL12-dependent biological effects. A third vector expressed only GFP (vector control). Transduction with our CXCR4 vectors resulted in robust expression of CXCR4, which could also be indirectly monitored using GFP expression (Figure 5E). Upon transduction, CXCR4+ hPSC-derived hematopoietic cells could robustly transmigrate toward CXCL12 in Transwells in vitro (Figure 5F), a classic feature of adult/somatic HPCs (Wright et al., 2002). CXCR4+ and CXCR4(off)+ hPSC-HPCs expressed similar cell surface CX3CR1 compared with control (Figure S3C), and transduced hPSC-HPCs remained unresponsive to CX3CL1 in chemotaxis assays (Figure S3D). Intracellular calcium signaling was induced by CXCL12 in CXCR4+ hPSC-derived hematopoietic cells (Figure 5G), achieving similar frequencies to that observed in CB (Figure 5H). The response to CXCL12 was not observed in CXCR4(off) transduced cells and could be inhibited by pre-treatment with the CXCR4 antagonist AMD3100 (Figures 5G and 5I). CXCR4+ hPSC-HPC calcium flux responses were achieved at similar frequencies to CB (Figure 5H) and were inhibited with the CXCR4 antagonist (Figures 5G and 5I) (Broxmeyer et al., 2005), further demonstrating the functional integration of CXCR4 and the ability to pharmacologically regulate this network within hPSC-HPCs. Additionally, CXCL12 treatment resulted in a CXCR4 dependent 2-fold increase in progenitor capacity (Figure 5J). The enhancement in CFU potential is in line with a previous report where CXCL12 supplementation in MethoCult increased CB progenitor propagation (Broxmeyer et al., 2007). Overall, these data demonstrate that both CXCL12 and CXCR4 are critically involved in hPSC-HPCs, and that lentiviral expression of CXCR4 enables its functional integration into networks supporting biological processes for which hPSC-HPCs were deficient compared with somatic HPC sources.
Enhanced BM Progenitor Retention of CXCR4+ hPSC-HPCs
Ectopic CXCR4 has previously been shown to enhance CB engraftment (Kahn et al., 2004). We therefore transplanted GFP-tagged CXCR4+ hPSC-HPCs in vivo and assessed survival and proliferation. Equivalent numbers of hPSC-HPCs transduced with CXCR4 or vector control were injected into large groups of mice (n ≥ 20), in parallel with somatic HPCs (Figures 6A and 6B). Encouragingly, CXCR4+ hPSC-HPCs exhibited enhanced BM retention at the fifth day post transplant (Figure 6C), and led to a significant increase in phenotypic hPSC-HPC retention overall (Figures 6D and S4A) and also enabled CXCR4+ hPSC-derived HPCs to migrate to the contralateral femur BM similarly to somatic HPCs (Figure S4B). This finding was specific to the BM, as we did not observe HPCs in the spleen (Figures S4C–S4E). Highest frequencies of GFP were observed from the BM of femurs injected with CXCR4+ hPSC-HPCs (Figure S4F). The retention of phenotypic hPSC-HPCs was also paralleled with retention of functional progenitors. Impressively, CXCR4 alone was sufficient to rescue progenitor function following transplantation (Figures 6E, 6F, S4G, and S4H). CFU were manually picked and validated for human origin and lentivirus-based CXCR4 and GFP sequences by PCR (Figures 6G and 6H). We ascertained a loss of erythroid progenitors from CXCR4+ hPSC-HPCs in vivo (Figure S4G), consistent with previous reports of HPCs derived from non-human primate M. nemestrina-induced PSCs (Gori et al., 2015). CFU retention, measured as a fraction of CFU injected, was equivalent between somatic and CXCR4+ hPSC-derived HPCs, and significantly higher than control hPSC-HPCs (Figure 6I). Unfortunately, CXCR4 was not sufficient to confer sustained engraftment tested at 4 weeks (Figures 6J–6L). This lack of prolonged hematopoietic reconstitution is possibly due to the silencing of CXCR4 vector (loss of GFP) over time (Figure S4I). Our results suggest the importance of identifying hPSC-HPCs that exhibit dynamic auto-regulation of the CXCR4 pathway in response to extracellular stimuli similar to adult/somatic HPCs.
Figure 6.

CXCR4 Rescues Transplantation Deficiency of hPSC-HPCs
(A) Transplant groups: 5x104 CD34+CD45+ cells were transplanted from CBCD34 and both hPSC-HPC groups (48 hr post transduction, on EB day 16). BM from saline-injected mice were collected at every harvest. n numbers represent transplanted mice, pooled from two independently performed experiments with four harvest analyses.
(B) Injection fraction phenotyping.
(C) Flow cytometry of BM harvested on day 5.
(D) Total mCD45–hCD45+CD34+ cells per IF. CXCR4+ and vector in purple and black, respectively. CB in inset (red). Data points represent n transplanted mice. Two-tailed t test, ∗∗p < 0.01.
(E) CFU analysis from harvested BM. Arrowheads indicate individual CFU. Scale bar represents 100 μm.
(F) Total human CFU per IF. CXCR4+ and vector in purple and black, respectively. CB in inset (red). Each data point represents one mouse. Two-tailed t test, ∗∗p < 0.01.
(G) 1–5 colonies (2 ± 2) per transplanted BM samples were picked by micropipette, and the non-colony remainder of the well was kept separately (rest). If no CFU were observed, the entire well was collected (all).
(H) CFU were analyzed for human and vector sequences by PCR. Samples from BM harvested on day 5. Each box is one NSG mouse. PCR was completed with one to five colonies (2 ± 2) per sample. Two representative CXCR4+ mice are shown, with two (left) and one (right) colony shown. CB PCR was completed with one colony. gen, genomic.; vir., viral; sal., saline.
(I) CFU retention per IF as a percentage of input CFU. To assess BM retention separately from cellular proliferation and expansion, only 24 hr retention data for CB shown. Ø is zero. Data are represented as means ± SEM.
(J) Flow cytometry of CB and hPSC-HPC transplanted IF, harvested 4 weeks post transplant.
(K and L) Human chimerism (hCD45+mCD45–) and (L) GFP frequency at 4 weeks. Data points represent n transplanted mice. Data are represented as means ± SEM.
CXCR4 Networks Are a Feature of Somatic HSC/HPCs
To investigate CXCR4 auto-regulation, sustained activation, and network responsiveness, global transcriptome analysis of CXCR4+ and CXCR4– hPSC-HPCs (CD34+CD45+GFP±) were compared with somatic and seven TF-hPSC-derived HSC/HPCs with long-term engraftment capacity, three TF- and five TF-expressing hPSC-HPCs with little to no engraftment capacity, and related BM/niche cell types (Figure 7A). CXCR4+ hPSC-HPCs clustered as an intermediate between CXCR4– hPSC-HPCs and somatic HPCs. Furthermore, GSEA identified four independent HSC signatures that were significantly enriched in CXCR4+ versus CXCR4– hPSC-HPCs, in addition to other cellular adhesion molecules (Figures 7B and 7C). Global transcriptome analysis of CXCR4+ and CXCR4– hPSC-HPCs revealed genes that may shift hPSC-HPCs closer toward somatic HPC profiles as well as genes that continue to be aberrantly expressed (Figure 7D, Table S3). We observe classical HSC genes, PROM1 and CD34, and adhesion genes, SELL and ICAM2, remaining highly expressed by somatic HPCs compared with CXCR4+ hPSC-HPCs. Interestingly, CXCR4+ hPSC-HPCs express equivalent levels of the epigenetic regulator MLLT3 compared with somatic HPCs, whereas CXCR4– continue to lack equivalent MLLT3 expression (Figure 3F and Table S3). Three-hundred and twenty-five genes remain aberrantly expressed, which include mobilization-inducing MMP9 and epigenetic regulator EZH2, in both CXCR4+ and CXCR4– hPSC-HPCs, and prevent complete transcriptional overlap with somatic HPCs. Deeper molecular analysis of the transcriptome was performed using Pearson correlation and scoring pathway expression using normalized enrichment scores (NES) from GSEA analyses where BM HPCs were used as reference (Figure 7E). CXCR4+ hPSC-HPCs were most similar to BM HPCs compared with other hPSC-derived cells (Pearson correlation). However, negative NES of CXCR4 regulatory pathways suggests ectopic CXCR4 did not induce auto-regulation. In contrast, while seven TF hPSC-HSC/HPCs are molecularly distinct from somatic HSCs (Pearson correlation of 0.77, similar to original report of >0.7; Sugimura et al., 2017), they appear to overexpress CXCR4-related pathways, using the NES as an indicator. The observation of active CXCR4-related pathways in seven TF hPSC-HSC/HPCs (Sugimura et al., 2017) and BM/niche cell types suggests that, during in vivo-programming, BM may select for hPSC-HSCs and HPCs capable of receiving extracellular cues for survival. This reinforces the hypothesis of CXCR4 signaling as necessary, but not sufficient, to produce hPSC-HSCs and HPCs. To functionally investigate auto-regulation, we treated CXCR4+ hPSC-HPCs with several CXCR4-inducing agents (Figure 7F). Indeed, pathways upstream of CXCR4, such as PGE2 and glucocorticoids (flonase) (Goessling et al., 2011, Guo et al., 2017), were unable to synergize with ectopic CXCR4 (Figures 7F–7H). However, forskolin, a cAMP agonist, increased total CXCR4+ HPCs while total cellular yield was equivalent, suggesting incomplete CXCR4 auto-regulation.
Figure 7.

CXCR4 Auto-regulation as a Target for hPSC-HSC Development
(A) Data points in PCA correlation biplot represent the global transcriptome of n biological replicates pooled from the following sources of FACS-purified HPCs (CD45+CD34+CD38±); GEO: GSE83719, triangles; GEO: GSE49938, diamonds; GEO: GSE3823 and GSE92778, circles; and GEO: GSE106721, hexagons. Ellipses are 2 SDs.
(B and C) Enrichment plots (B) and NES (C) of gene sets identified by GSEA as enriched in CD34+CD45+CXCR4+ hPSC-HPCs versus CD34+CD45+CXCR4– hPSC-HPCs. Of 3,685 C2 (MSigDB) gene sets, 195 were significantly enriched in CD34+CD45+CXCR4+ hPSC-HPCs at nominal p value <0.05.
(D) ANOVA comparing CXCR4+ and CXCR4– hPSC-HPCs with somatic HPCs depicted with Venn diagram.
(E) Sample groups described in (A) were assessed at the global level (all genes) using Pearson’s correlation coefficient, and within gene sets; see also Supplemental Experimental Procedures. NES from GSEA of indicated groups (biological replicates, not averaged) compared with BM HPCs. GPCR, G protein-coupled receptor.
(F–H) CXCR4+ hPSC-HPCs were exposed to CXCR4-inducing compounds for 48 hr; see also Supplemental Experimental Procedures. CXCR4 staining of CD34+CD45+ cells (F), and quantification of total CXCR4+CD34+CD45+ (G), and bulk cells (H). Data points represent three independently assayed biological replicates. One-way ANOVA, ∗∗∗p<0.001. Data are represented as means ± SEM.
(I) A model of CXCR4 function in hPSC-HPCs.
Discussion
Our study demonstrates that CXCR4 is necessary for early transplantation survival but not sufficient to confer long-term engraftment. We suggest that the loss of CXCR4 leads hPSC-HPCs to revert to CXCR4– characteristics instead of auto-regulating CXCR4 in situ (Figure 7H). Altogether, CXCL12-mediated CXCR4 signaling by hPSC-HPCs promotes progenitor proliferation, survival, migration in vitro, and increased BM retention in vivo. Progress toward hPSC-HSC long-term engraftment has largely been pursued using ectopic TF expression. In contrast, somatic HSCs receive non-cell-autonomous signals in the BM directing cell fate (Boyd and Bhatia, 2014, Boyd et al., 2017), therefore we investigated these cues in the context of hPSC-HPCs. Our findings identified hPSC-HPC transplantation deficiencies, which complements previous reports of limited engraftment at 4 weeks (Gori et al., 2015, Lee et al., 2017, Ng et al., 2016, Risueño et al., 2012, Wang et al., 2005), while at a much earlier time frame within the first week post transplantation.
Cell-autonomous approaches to produce hPSC-HSC/HPCs have made significant progress using ectopic HSC-specific TFs (Doulatov et al., 2013, Doulatov et al., 2017, Ran et al., 2013, Sugimura et al., 2017). However, all four of these reports required TFs with leukemogenic potential, most prominently RUNX1 (AML1) and ERG (Crans and Sakamoto, 2001). Furthermore, the additional effects of in vivo BM programming during multiplexed TF expression appear to support higher chimerism frequencies of hPSC-derived hematopoietic cells and multilineage differentiation potential (Sugimura et al., 2017). CXCR4 function is critical for BM retention and HSC function (Lapidot and Kollet, 2002, Nagasawa et al., 1996, Sugiyama et al., 2006); however, similar to TF expression, it must be tightly regulated. Gain-of-function mutations in CXCR4 are associated with a rare primary immunodeficiency called WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis) syndrome (Heusinkveld et al., 2017). Clinical features include abnormal retention of mature hematopoietic cells in the BM of patients, which suggest constitutive knockin or long-term induced expression systems may be deleterious for promoting balanced multilineage engraftment from hPSC-HPCs. This points to the importance of producing hPSC-HPCs that can auto-regulate CXCR4 activity, without prolonged ectopic expression.
Our study establishes a proof of principle that supporting CXCR4 expression is a viable target not only to enhance progenitor survival and proliferation when exposed to CXCL12 in vitro or BM in vivo but also to promote a more normal hematopoietic transcriptional shift toward somatic HSCs and HPCs. Independent of ectopic chemokine receptor modulation, several other lines of evidence provide support for required functional connectivity between BM niche and hPSC-HPCs for sustained engraftment. The injection of undifferentiated human PSCs into mice has led to the rare production of hPSC-HSCs isolated from the BM (Suzuki et al., 2013), when the original cells were injected as heterotopic teratomas. Conversely, once isolated from their niche, somatic HSCs are difficult to propagate in vitro, and prolonged culture leads to diminished engraftment potential (Brenner et al., 2004, Hofmeister et al., 2007). Strikingly, the first clinical report of a pharmacological agent able to expand HSCs in vitro involved Notch signaling, which operates in a non-cell-autonomous role in the BM (Delaney et al., 2010). Together, these relationships underscore the importance of BM cues for hPSC-HPCs as an unappreciated area of biological interaction that requires further investigation for future clinical applications of human HPCs derived from hPSC sources.
Experimental Procedures
Somatic Blood Samples
Informed consent was obtained from full-term umbilical CB and MPB donors with protocols approved by the Research Ethics Board at McMaster University. Human BM was purchased from Lonza (Cedarlane, #1M-105). Mononuclear cells (MNCs) were recovered by density gradient centrifugation (Ficoll-Paque, GE Healthcare), and red blood cells were lysed using ammonium chloride (StemCell Technologies). Lineage depletion was performed by magnetic cell separation using a lineage antibody kit (StemCell Technologies, #19309C). Cells were cryopreserved in 10% DMSO in fetal bovine serum (FBS) until use.
Xenotransplantation
Immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were bred in a barrier facility, and all experimental protocols were approved by the Animal Research Ethics Board of McMaster University. Mouse sex and age were controlled within each experiment, and mice were randomly assigned to experimental groups, which included male and female mice. No statistical method was used to predetermine sample size. Mice were sublethally irradiated (single dose of 315 cGy, 137Cs) 24 hr before transplant. Cells were transplanted by intra-femoral injection as previously described (Wang et al., 2005), at doses described in the figure legends. At harvest, mice were killed and BM from the injected and contralateral femurs were collected separately. Spleens were separately harvested. Lungs were harvested in a subset of experiments. Cells were recovered by mechanical dissociation in IMDM supplemented with 3% FBS (HyClone, Canada), and 1 mM EDTA (Invitrogen). Red blood cells were lysed using ammonium chloride. BM samples were counted and plated for CFU frequency. BM cells from NSG mice injected with saline were used as negative controls for CFU. Phenotyping was analyzed using fluorescence minus one gating. Data points were combined from all independent experiments and outliers were not excluded.
Statistics
Data are represented as means ± SEM. Prism (6.0c, GraphPad) software was used for all statistical analyses, and the criterion for statistical significance was p < 0.05. Statistics are described in figure legends.
For details of all other procedures, see Supplemental Experimental Procedures.
Author Contributions
J.C.R., B.T., D.G., A.L.B., and D.P.P. performed experiments. T.J.C. provided technical assistance for the calcium flux assay. J.C.R. and M.B. designed experiments, interpreted data, and wrote the manuscript. M.B. directed the study. B.T. and D.G. contributed equally.
Acknowledgments
J.C.R. was supported by the Canadian Institutes of Health Research (CIHR) Doctoral Award, the Michael G. DeGroote Doctoral Scholarship, and an Ontario Graduate Fellowship. D.G. and D.P.P. were supported by the Jans Graduate Scholarship in Stem Cell Research. This work was supported by a research grant to M.B. from CIHR (MOP-142223) and support to MB from the Canada Research Chair Program (Tier 1) in Human Stem Cell Biology and Michael G. DeGroote Chair in Stem Cell Biology. We acknowledge M. Graham for human sample processing; M.P. Mathieu and A. Fiebig-Comyn for animal husbandry and in vivo experimental assistance; Z. Shapovalova, M. Subapanditha, and N. McFarlane for their assistance with cell sorting experiments; and Dr. S. Raha and M. Wong for procedures related to hypoxia. We also thank members of the Labor and Delivery Ward at the McMaster Children's Hospital for supplying human cord blood samples.
Published: May 8, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.04.003.
Accession Numbers
Microarray data from this study have been deposited in the GEO (NCBI) under the accession number GEO: GSE106721.
Supplemental Information
References
- Balkwill F. Cancer and the chemokine network. Nat. Rev. Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Boyd A.L., Bhatia M. Bone marrow localization and functional properties of human hematopoietic stem cells. Curr. Opin. Hematol. 2014;21:249–255. doi: 10.1097/MOH.0000000000000055. [DOI] [PubMed] [Google Scholar]
- Boyd A.L., Reid J.C., Salci K.R., Aslostovar L., Benoit Y.D., Shapovalova Z., Nakanishi M., Porras D.P., Almakadi M., Campbell C.J.V. Acute myeloid leukaemia disrupts endogenous myelo-erythropoiesis by compromising the adipocyte bone marrow niche. Nat. Cell Biol. 2017;19:1336–1347. doi: 10.1038/ncb3625. [DOI] [PubMed] [Google Scholar]
- Brenner S., Whiting-Theobald N., Kawai T., Linton G.F., Rudikoff A.G., Choi U., Ryser M.F., Murphy P.M., Sechler J.M., Malech H.L. CXCR4-transgene expression significantly improves marrow engraftment of cultured hematopoietic stem cells. Stem Cells. 2004;22:1128–1133. doi: 10.1634/stemcells.2003-0196. [DOI] [PubMed] [Google Scholar]
- Broxmeyer H.E., Mejia J.A., Hangoc G., Barese C., Dinauer M., Cooper S. SDF-1/CXCL12 enhances in vitro replating capacity of murine and human multipotential and macrophage progenitor cells. Stem Cells Dev. 2007;16:589–596. doi: 10.1089/scd.2007.0044. [DOI] [PubMed] [Google Scholar]
- Broxmeyer H.E., Orschell C.M., Clapp D.W., Hangoc G., Cooper S., Plett P.A., Liles W.C., Li X., Graham-Evans B., Campbell T.B. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J. Exp. Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick K., Wang L., Li L., Menendez P., Murdoch B., Rouleau A., Bhatia M. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood. 2003;102:906–915. doi: 10.1182/blood-2003-03-0832. [DOI] [PubMed] [Google Scholar]
- Copeland E. Hematopoietic stem-cell transplantation. N. Engl. J. Med. 2006;354:1813–1826. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- Crans H., Sakamoto K. Transcription factors and translocations in lymphoid and myeloid leukemia. Leukemia. 2001;15:313–331. doi: 10.1038/sj.leu.2402033. [DOI] [PubMed] [Google Scholar]
- Cutler C., Multani P., Robbins D., Kim H.T., Le T., Hoggatt J., Pelus L.M., Desponts C., Chen Y.B., Rezner B. Prostaglandin-modulated umbilical cord blood hematopoietic stem cell transplantation. Blood. 2013;122:3074–3081. doi: 10.1182/blood-2013-05-503177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney C., Heimfeld S., Brashem-Stein C., Voorhies H., Manger R.L., Bernstein I.D. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat. Med. 2010;16:232–236. doi: 10.1038/nm.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S., Vo L.T., Macari E.R., Wahlster L., Kinney M.A., Taylor A.M., Barragan J., Gupta M., McGrath K., Lee H.Y. Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aah5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S., Vo L., Chou S., Kim P., Arora N., Li H., Hadland B., Bernstein I., Collins J., Zon L. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell. 2013;13:459–470. doi: 10.1016/j.stem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster K., Lassailly F., Anjos-Afonso F., Currie E., Rouault-Pierre K., Bonnet D. Different motile behaviors of human hematopoietic stem versus progenitor cells at the osteoblastic niche. Stem Cell Reports. 2015;5:690–701. doi: 10.1016/j.stemcr.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman E. History of cord blood transplantation. Bone Marrow Transplant. 2009;44:621–626. doi: 10.1038/bmt.2009.280. [DOI] [PubMed] [Google Scholar]
- Goessling W., Allen R.S., Guan X., Jin P., Uchida N., Dovey M., Harris J.M., Metzger M.E., Bonifacino A.C., Stroncek D. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell. 2011;8:445–458. doi: 10.1016/j.stem.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gori J.L., Butler J.M., Chan Y.-Y., Chandrasekaran D., Poulos M.G., Ginsberg M., Nolan D.J., Elemento O., Wood B.L., Adair J.E. Vascular niche promotes hematopoietic multipotent progenitor formation from pluripotent stem cells. J. Clin. Invest. 2015;125:1243–1254. doi: 10.1172/JCI79328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratwohl A., Pasquini M., Aljurf M., Atsuta Y., Baldomero H., Foeken L., Gratwohl M., Bouzas L., Confer D., Frauendorfer K. One million haemopoietic stem-cell transplants: a retrospective observational study. Lancet Hematol. 2015;2:e91–e100. doi: 10.1016/S2352-3026(15)00028-9. [DOI] [PubMed] [Google Scholar]
- Guo B., Huang X., Cooper S., Broxmeyer H.E. Glucocorticoid hormone-induced chromatin remodeling enhances human hematopoietic stem cell homing and engraftment. Nat. Med. 2017;23:424–428. doi: 10.1038/nm.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusinkveld L.E., Yim E., Yang A., Azani A.B., Liu Q., Gao J.L., McDermott D.H., Murphy P.M. Pathogenesis, diagnosis and therapeutic strategies in WHIM syndrome immunodeficiency. Expert Opin. Orphan Drugs. 2017;5:813–825. doi: 10.1080/21678707.2017.1375403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmeister C.C., Zhang J., Knight K.L., Le P., Stiff P.J. Ex vivo expansion of umbilical cord blood stem cells for transplantation: growing knowledge from the hematopoietic niche. Bone Marrow Transplant. 2007;39:11–23. doi: 10.1038/sj.bmt.1705538. [DOI] [PubMed] [Google Scholar]
- Jetmore A., Plett P.A., Tong X., Wolber F.M., Breese R., Abonour R., Orschell-Traycoff C.M., Srour E.F. Homing efficiency, cell cycle kinetics, and survival of quiescent and cycling human CD34(+) cells transplanted into conditioned NOD/SCID recipients. Blood. 2002;99:1585–1593. doi: 10.1182/blood.v99.5.1585. [DOI] [PubMed] [Google Scholar]
- Kahn J., Byk T., Jansson-Sjostrand L., Petit I., Shivtiel S., Nagler A., Hardan I., Deutsch V., Gazit Z., Gazit D. Overexpression of CXCR4 on human CD34+ progenitors increases their proliferation, migration, and NOD/SCID repopulation. Blood. 2004;103:2942–2949. doi: 10.1182/blood-2003-07-2607. [DOI] [PubMed] [Google Scholar]
- Lapidot T., Kollet O. The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homing and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2m(null) mice. Leukemia. 2002;16:1992–2003. doi: 10.1038/sj.leu.2402684. [DOI] [PubMed] [Google Scholar]
- Ledran M.H., Krassowska A., Armstrong L., Dimmick I., Renström J., Lang R., Yung S., Santibanez-Coref M., Dzierzak E., Stojkovic M. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell. 2008;3:85–98. doi: 10.1016/j.stem.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Lee J., Dykstra B., Spencer J.A., Kenney L.L., Greiner D.L., Shultz L.D., Brehm M.A., Lin C.P., Sackstein R., Rossi D.J. mRNA-mediated glycoengineering ameliorates deficient homing of human stem cell-derived hematopoietic progenitors. J. Clin. Invest. 2017;127:2433–2437. doi: 10.1172/JCI92030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa T., Hirota S., Tachibana K., Takakura N., Nishikawa S., Kitamura Y., Yoshida N., Kikutani H., Kishimoto T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635–638. doi: 10.1038/382635a0. [DOI] [PubMed] [Google Scholar]
- Nakano H., Lyons-Cohen M.R., Whitehead G.S., Nakano K., Cook D.N. Distinct functions of CXCR4, CCR2, and CX3CR1 direct dendritic cell precursors from the bone marrow to the lung. J. Leukoc. Biol. 2017;101:1143–1153. doi: 10.1189/jlb.1A0616-285R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng E.S., Azzola L., Bruveris F.F., Calvanese V., Phipson B., Vlahos K., Hirst C., Jokubaitis V.J., Yu Q.C., Maksimovic J. Differentiation of human embryonic stem cells to HOXA+ hemogenic vasculature that resembles the aorta-gonad-mesonephros. Nat. Biotechnol. 2016;34:1168–1179. doi: 10.1038/nbt.3702. [DOI] [PubMed] [Google Scholar]
- Ramos-Mejia V., Navarro-Montero O., Ayllon V., Bueno C., Romero T., Real P.J., Menendez P. HOXA9 promotes hematopoietic commitment of human embryonic stem cells. Blood. 2014;124:3065–3075. doi: 10.1182/blood-2014-03-558825. [DOI] [PubMed] [Google Scholar]
- Ran D., Shia W.J., Lo M.C., Fan J.B., Knorr D.A., Ferrell P.I., Ye Z., Yan M., Cheng L., Kaufman D.S. RUNX1a enhances hematopoietic lineage commitment from human embryonic stem cells and inducible pluripotent stem cells. Blood. 2013;121:2882–2890. doi: 10.1182/blood-2012-08-451641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risueño R.M., Sachlos E., Lee J.-H., Lee J.B., Hong S.-H., Szabo E., Bhatia M. Inability of human induced pluripotent stem cell-hematopoietic derivatives to downregulate microRNAs in vivo reveals a block in xenograft hematopoietic regeneration. Stem Cells. 2012;30:131–139. doi: 10.1002/stem.1684. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lara V., Ignacio G.S., Cerbon Cervantes M.A. Estrogen induces CXCR4 overexpression and CXCR4/CXL12 pathway activation in lung adenocarcinoma cells in vitro. Endocr. Res. 2017;42:219–231. doi: 10.1080/07435800.2017.1292526. [DOI] [PubMed] [Google Scholar]
- Salvagiotto G., Zhao Y., Vodyanik M., Ruotti V., Stewart R., Marra M., Thomson J., Eaves C., Slukvin I. Molecular profiling reveals similarities and differences between primitive subsets of hematopoietic cells generated in vitro from human embryonic stem cells and in vivo during embryogenesis. Exp. Hematol. 2008;36:1377–1389. doi: 10.1016/j.exphem.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S., Ronn R.E., Guibentif C., Moraghebi R., Woods N.B. Cyclic AMP signaling through Epac Axis modulates human hemogenic endothelium and enhances hematopoietic cell generation. Stem Cell Reports. 2016;6:692–703. doi: 10.1016/j.stemcr.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheurer S.B., Rybak J.N., Rosli C., Neri D., Elia G. Modulation of gene expression by hypoxia in human umbilical cord vein endothelial cells: a transcriptomic and proteomic study. Proteomics. 2004;4:1737–1760. doi: 10.1002/pmic.200300689. [DOI] [PubMed] [Google Scholar]
- Sugimura R., Jha D.K., Han A., Soria-Valles C., da Rocha E.L., Lu Y.-F., Goettel J.A., Serrao E., Rowe R.G., Malleshaiah M. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature. 2017;545:432–438. doi: 10.1038/nature22370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T., Kohara H., Noda M., Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Suzuki N., Yamazaki S., Yamaguchi T., Okabe M., Masaki H., Takaki S., Otsu M., Nakauchi H. Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol. Ther. 2013;21:1424–1431. doi: 10.1038/mt.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X., Woll P.S., Morris J.K., Linehan J.L., Kaufman D.S. Hematopoietic engraftment of human embryonic stem cell-derived cells is regulated by recipient innate immunity. Stem cells. 2006;24:1370–1380. doi: 10.1634/stemcells.2005-0340. [DOI] [PubMed] [Google Scholar]
- Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Vodyanik M., Thomson J.A., Slukvin I. Leukosialin (CD43) defines hematopoietic progenitors in human embryonic stem cell differentiation cultures. Blood. 2006;108:2095–2105. doi: 10.1182/blood-2006-02-003327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.M., Menendez P., Shojaei F., Li L., Mazurier F., Dick J.E., Cerdan C., Levac K., Bhatia M. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J. Exp. Med. 2005;201:1603–1614. doi: 10.1084/jem.20041888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Yu S., Myers J., Wang Y., Xin W.W., Alkabri M., Xin A.W., Li M., Huang A.Y., Xin W. Notch2 blockade enhances hematopoietic stem cell mobilization and homing. Haematologica. 2017;102:1785–1795. doi: 10.3324/haematol.2017.168674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisel K.C., Gao Y., Shieh J.-H., Moore M.A.S. Stromal cell lines from the aorta-gonado-mesonephros region are potent supporters of murine and human hematopoiesis. Exp. Hematol. 2006;34:1505–1516. doi: 10.1016/j.exphem.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Wright D.E., Bowman E.P., Wagers A.J., Butcher E.C., Weissman I.L. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J. Exp. Med. 2002;195:1145–1154. doi: 10.1084/jem.20011284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.B., Navenot J.M., Haribabu B., Tamamura H., Hiramatu K., Omagari A., Pei G., Manfredi J.P., Fujii N., Broach J.R. A point mutation that confers constitutive activity to CXCR4 reveals that T140 is an inverse agonist and that AMD3100 and ALX40-4C are weak partial agonists. J. Biol. Chem. 2002;277:24515–24521. doi: 10.1074/jbc.M200889200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.