

Abstract
The vacuolar H+-ATPase (V-ATPase) is an ATP-driven proton pump involved in many cellular processes. An important mechanism by which V-ATPase activity is controlled is the reversible assembly of its two domains, namely the peripheral V1 domain and the integral V0 domain. Although reversible assembly is conserved across all eukaryotic organisms, the signaling pathways controlling it have not been fully characterized. Here, we identify glucose starvation as a novel regulator of V-ATPase assembly in mammalian cells. During acute glucose starvation, the V-ATPase undergoes a rapid and reversible increase in assembly and activity as measured by lysosomal acidification. Because the V-ATPase has recently been implicated in the activation of AMP kinase (AMPK), a critical cellular energy sensor that is also activated upon glucose starvation, we compared the time course of AMPK activation and V-ATPase assembly upon glucose starvation. We observe that AMPK activation precedes increased V-ATPase activity. Moreover, the starvation-induced increase in V-ATPase activity and assembly are prevented by the AMPK inhibitor dorsomorphin. These results suggest that increased assembly and activity of the V-ATPase upon glucose starvation are dependent upon AMPK. We also find that the PI3K/Akt pathway, which has previously been implicated in controlling V-ATPase assembly in mammalian cells, also plays a role in the starvation-induced increase in V-ATPase assembly and activity. These studies thus identify a novel stimulus of V-ATPase assembly and a novel signaling pathway involved in regulating this process. The possible function of starvation-induced increase in lysosomal V-ATPase activity is discussed.
Keywords: vacuolar ATPase, proton transport, AMP-activated kinase (AMPK), phosphatidylinositide 3-kinase (PI 3-kinase), Akt PKB, glucose starvation, regulated assembly
Introduction
Vacuolar H+-ATPases (V-ATPases)2 are a family of multisubunit, ATP-dependent proton pumps present in many intracellular membranes and the plasma membrane of specialized cells (1–5). The V-ATPase is composed of two domains: the peripheral V1 domain (subunits A–H), which carries out ATP hydrolysis, and the integral V0 domain (subunits a, d, e, c, c′, and c″), which is responsible for proton transport (2).
One of the major ways that cells modulate V-ATPase activity is through the rapid and reversible dissociation of V1 from the V0 domain, also known as regulated assembly. Regulated assembly has been most thoroughly studied in yeast, where reversible dissociation occurs rapidly in response to glucose depletion, likely as a means to conserve cellular ATP stores (3). Assembly in yeast requires the glycolytic enzyme aldolase (6) and the heterotrimeric RAVE complex (7, 8), whereas disassembly requires a catalytically active enzyme and intact microtubules but not new protein synthesis (9–12). Our lab has previously shown that the Ras/cAMP/protein kinase A (PKA) pathway is involved in glucose-regulated assembly of the V-ATPase in yeast (13).
A number of stimuli have been reported to alter V-ATPase assembly in higher eukaryotes. V-ATPase assembly increases upon dendritic cell maturation (14, 15), during influenza infection (16, 17), in response to increased glucose concentrations (16–19), during amino acid starvation (20), and upon EGF stimulation (21). In dendritic cells, the V-ATPase provides the acidic lysosomal environment required for antigen processing, such that increased assembly upon maturation facilitates enhanced rates of antigen processing (14, 15). Our lab has shown that this increased assembly and activity of the V-ATPase in dendritic cells is PI3K- and mechanistic target of rapamycin complex I (mTORC1)-dependent (15). During influenza infection, endosomal acidification is increased as a way to increase the fusion of the viral coat with the endosomal membrane, thus facilitating entry of the viral nucleic acid into the host cell (16, 17, 22, 23). In this case, increased assembly of the V-ATPase depends upon increased activity of PI3K and extracellular signal-regulated kinase (ERK) (17). The function of the EGF-induced increase in V-ATPase assembly and activity is thought to be in providing adequate levels of amino acids derived from lysosomal protein degradation to allow maximal activation of mTORC1 signaling (21). Elevated glucose concentrations have also been shown to stimulate V-ATPase assembly and acidification of intracellular compartments in a PI3K-dependent manner (18). This increase in V-ATPase assembly and activity at high glucose concentrations may provide cells with a mechanism to reduce the increased acid load during periods of enhanced glycolysis. Finally, during amino acid starvation, we have shown that the V-ATPase also undergoes increased assembly on lysosomal membranes (20). We hypothesize that this represents a primitive cellular response to starvation that increases lysosomal protein degradation as a way to restore cellular stores of amino acids. Interestingly, unlike the cases described above, amino acid-dependent assembly of the V-ATPase is independent of both PI3K and mTORC1 (20).
Recently, V-ATPases have been shown to play a critical role in regulating the activity of the cellular energy sensor 5′-AMP-activated protein kinase (AMPK) (24). AMPK is activated in response to glucose starvation. Because V-ATPase assembly in yeast changes in response to glucose starvation, we hypothesized that regulated assembly of the V-ATPase may play a role in AMPK activation during glucose starvation in mammalian cells. Here, we demonstrate that glucose starvation does indeed increase both V-ATPase assembly and activity in mammalian cells, and we examine the relationship between AMPK activation and assembly changes. Furthermore, we sought to identify the signaling pathways that are involved in glucose-mediated changes in V-ATPase assembly and activity in mammalian cells.
Results
Glucose starvation increases V-ATPase assembly in mammalian cells
To determine whether glucose starvation affects V-ATPase assembly, HEK293T cells conditioned to serum-free DMEM containing 5 mm glucose were either subjected to acute glucose starvation by replacement of the media with serum-free DMEM containing 0 mm glucose for 10 min or were maintained in fresh serum-free DMEM containing 5 mm glucose. Because exposure to elevated glucose has previously been reported to increase V-ATPase assembly after 1 h (17), cells were also treated with serum-free DMEM containing 25 mm glucose for 1 h as a positive control. After treatment, cells were harvested, homogenized, and separated into their membrane and cytosolic fractions by sedimentation. Samples were then separated by SDS-PAGE, and Western blotting was performed using an antibody directed against subunit A of the V1 domain. The amount of V1 present in the membrane fraction is a reflection of the level of V-ATPase assembly. As shown in Fig. 1A, glucose starvation led to an increase in V-ATPase assembly as reflected by the increase in subunit A staining in the membrane fraction following incubation of cells at 0 mm glucose relative to cells maintained at 5 mm glucose. Consistent with previous results, we also observed an increase in assembly upon incubation of cells at elevated glucose. It should be noted that a relatively large fraction of subunit A remains in the cytosol under all conditions. Although it is possible that this may be due to a relatively large pool of cytosolic V1 domains, it is also possible that some V1 dissociates from V0 during cell homogenization and fractionation. Thus, the absolute degree of in vivo V-ATPase assembly may be underestimated in our experiments. Quantitation of Western blots was performed by densitometric analysis using ImageJ software. To correct for any variations in protein loading, the intensity of staining of subunit A relative to a membrane marker (subunit d) was determined. Values were then expressed relative to those obtained at 5 mm glucose (defined as 1.0). As shown in Fig. 1B, glucose starvation increased assembly by 1.9 ± 0.5-fold (p < 0.02) whereas incubation at elevated glucose concentrations increased assembly by 1.4 ± 0.1-fold (p < 0.02). Glucose re-addition for 10 min returned V-ATPase assembly to baseline levels (0.9 ± 0.1), suggesting V-ATPase assembly changes rapidly and reversibly in response to glucose levels.
Figure 1.
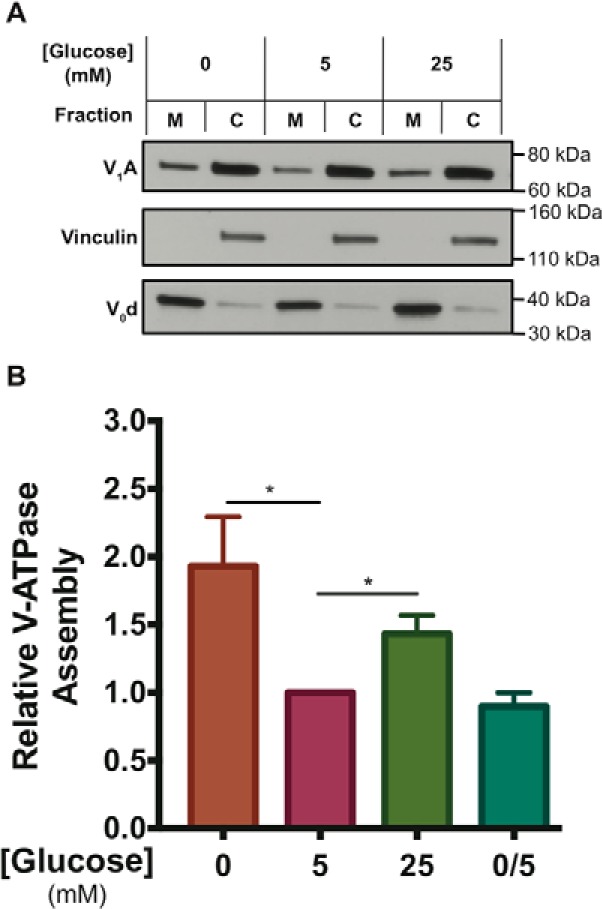
Incubation of HEK293T cells at low or high glucose increases V-ATPase assembly relative to physiological concentrations. A, HEK293T cells were maintained in serum-free DMEM containing 5 mm (physiological) glucose for ∼6 h and then treated with serum-free DMEM containing 0 mm glucose (10 min), 5 mm glucose (1 h), 25 mm glucose (1 h), or 0 mm glucose (10 min) followed by 5 mm glucose (10 min). Incubation at high glucose was performed for longer times to allow comparison with previously published results (16–18). Following treatment, cells were fractioned into membrane (M) and cytosolic (C) fractions as described under “Experimental procedures.” Samples were subjected to SDS-PAGE and Western blotting using an antibody against subunit A of the V1 domain as a measure of assembly. Antibody staining of subunit d of the V0 domain was used as a loading control for the membrane fraction, and staining for vinculin was used as a loading control for the cytosolic fraction. The quantity of subunit A present in the membrane fraction is an indication of the level of V-ATPase assembly. Shown is a representative Western blotting. B, Western blots performed as described in A were quantified using ImageJ software to determine the amount of V-ATPase assembly. Band intensities of subunit A in the membrane fraction were normalized to band intensities of the membrane loading control (subunit d). Results were then normalized to the baseline assembly levels observed at 5 mm glucose, which was defined as 1.0 for each individual trial. We find that the actual fraction of membrane bound versus cytosolic V1 is 0.33 ± 0.16 (n = 6) in HEK293T cells under normal physiological glucose concentrations (5 mm). Relative to the degree of assembly at 5 mm glucose (defined as 1.0 for this comparison), the average level of assembly for cells treated with 0 mm glucose was 1.9 ± 0.5 (p < 0.02, n = 9), whereas the average level of assembly for cells treated with 25 mm glucose was 1.4 ± 0.1 (p < 0.02, n = 5). Glucose re-addition for 10 min after starvation (labeled 0/5) returned assembly levels to 0.9 ± 0.1 (n = 3). The error bars represent standard error. The asterisks indicate statistically significant differences at the indicated p values.
To confirm that these results were not specific to HEK293T cells, the porcine epithelial cell line LLC-PK1 was treated as described above (using Medium 199 in place of DMEM). We found that glucose starvation increased assembly by 2.6 ± 1.0-fold (n = 2), whereas elevated glucose increased assembly 1.9 ± 0.2-fold (n = 2) in these cells (Fig. S1, A and B). Taken together, these results indicate that acute glucose starvation is a novel positive regulator of V-ATPase assembly in mammalian cells.
Lysosomal V-ATPase activity increases both in vitro and in vivo upon glucose starvation
To determine whether the increase in V-ATPase assembly observed upon glucose starvation results in a corresponding increase in V-ATPase activity, we used FITC-dextran, a pH-dependent probe, to quantify the rate of proton pumping into lysosomes. FITC-dextran was loaded into HEK293T cells through fluid-phase endocytosis and was chased to lysosomes by incubation in dye-free media. Cells were then incubated under the conditions described above followed by cell homogenization and sedimentation to obtain the membrane fraction. Although lysosomes were not isolated, this was not necessary for these experiments because the FITC fluorescence signal was localized exclusively to lysosomes present in the membrane fraction. Because the FITC signal is quenched at low pH, the rate of V-ATPase-dependent proton pumping into lysosomes was measured as the rate of ATP-dependent, concanamycin A-sensitive fluorescence quenching (concanamycin A is a specific V-ATPase inhibitor (25)). After normalizing the data to the initial fluorescence reading to control for the number of lysosomes added to the cuvette, the initial rate of fluorescence quenching was determined using linear regression analysis, and activity values are expressed relative to those observed for lysosomes obtained from cells incubated at 5 mm glucose (defined as 1.0). As shown in Fig. 2A, glucose starvation led to a 17 ± 2% increase in lysosomal V-ATPase-dependent proton transport (p < 0.01, n = 20), whereas elevated glucose increased activity by 13 ± 2% (p < 0.01, n = 20). The effect of glucose starvation was again reversed upon re-addition of 5 mm glucose to cells. To confirm that these results were not specific to HEK293T cells, LLC-PK1 cells were treated as described above. We found that glucose starvation increased lysosomal V-ATPase activity by 35% (n = 2; Fig. S1C). We conclude that both glucose starvation and elevated glucose cause an increase in lysosomal V-ATPase activity through increased assembly.
Figure 2.
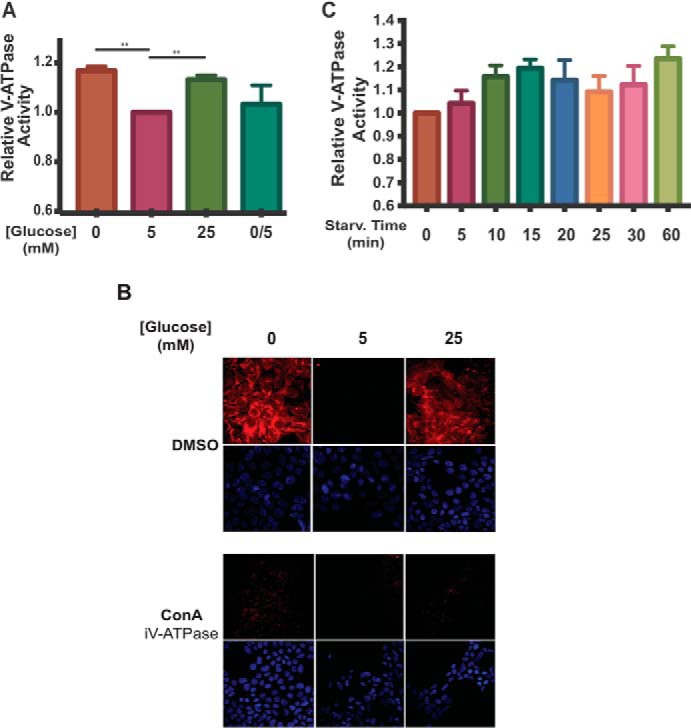
Incubation of HEK293T cells at low or high glucose increases V-ATPase-dependent acidification of lysosomes. A, HEK293T cells were incubated with FITC-dextran to load lysosomes by fluid-phase endocytosis as described under “Experimental procedures.” Cells were then maintained in serum-free DMEM containing 5 mm glucose for ∼6 h and then treated with serum-free DMEM containing 0 mm glucose (10 min.), 5 mm glucose (1 h), 25 mm glucose (1 h), or 0 mm glucose (10 min.) followed by 5 mm glucose (10 min.). Following treatment, lysosomes were isolated from cells as described under “Experimental procedures.” Fluorescence intensity at 520 nm (excitation at 490 nm) was measured as a function of time following addition of Mg-ATP. The rate V-ATPase-dependent proton transport activity for each condition was determined by performing a linear regression analysis on the initial rate of concanamycin A-sensitive fluorescence quenching (where present, concanamycin A was added at 1 μm). Rates of fluorescence quenching from independent trials were normalized to the baseline value observed for lysosomes isolated from cells maintained at 5 mm glucose (defined as 1.0). The average lysosomal V-ATPase activity for lysosomes from cells treated with 0 mm glucose was 1.17 ± 0.02 (p < 0.01, n = 20), and for cells treated with 25 mm glucose the average activity was 1.13 ± 0.02 (p < 0.01, n = 20). There was no significant difference in activity between lysosomes from cells treated with 5 mm glucose compared with lysosomes from cells treated with 0 mm glucose followed by 5 mm glucose (0/5). The error bars represent the mean ± S.E. The asterisks indicate statistically significant differences at the indicated p values. B, HEK293T cells were treated with glucose as described in A. Concanamycin A (ConA) at 5 μm or DMSO was added to the cells 1 h before collection. 10 min prior to cell collection, cells were incubated with LysoTracker to stain for acidic cellular compartments and then fixed as described under “Experimental procedures.” Staining was detected by confocal fluorescence microscopy (red, LysoTracker; blue, DAPI), and representative images are shown, n = 2. C, FITC-dextran loaded HEK293T cells were maintained in serum-free DMEM containing 5 mm glucose for ∼6 h and then treated with 0 mm glucose for the indicated amounts of time. Rates of fluorescence quenching were measured as in A. n = 3.
Although the results described above clearly indicate an increase in V-ATPase-dependent proton transport into lysosomes isolated from glucose-starved cells, we wished to determine whether this resulted in a detectable change in lysosomal pH in intact cells. To accomplish this, we stained cells with LysoTracker Red DND-99, which is a cell-permeable red fluorescent dye linked to a weak base that accumulates in acidic compartments in vivo. As intracellular compartments become more acidic, accumulation of the dye generates a more intense fluorescence signal. HEK293T cells were treated as described above, and LysoTracker was added to cells 10 min before collection. As shown in Fig. 2B, incubation of cells at both 0 and 25 mm glucose increased the number and intensity of fluorescent puncta relative to that observed at 5 mm glucose. To confirm that the observed increase in intracellular acidification was V-ATPase-dependent, the above experiments were repeated following incubation of cells with concanamycin A (5 μm) for 1 h. As shown in Fig. 2B, concanamycin completely abolished LysoTracker staining of cells, indicating that the observed fluorescence signal is due to the activity of the V-ATPase. These results indicate that glucose starvation results in a measurable increase in acidification of intracellular compartments in HEK293T cells. To better characterize the observed changes in V-ATPase activity, the time course of this change was monitored following glucose starvation. As shown in Fig. 2C, lysosomal V-ATPase activity reaches a maximum within 10–15 min of glucose deprivation, with a small but not statistically significant reduction at later times. These results suggest that the observed changes are too rapid to involve new protein synthesis, consistent with glucose-dependent changes in assembly in yeast.
ERK and AMPK signaling are activated upon glucose starvation
Previous work in our lab has shown that, in yeast, activation of the Ras/cAMP/PKA pathway, which plays a crucial role in metabolic control, prevents V-ATPase disassembly upon glucose starvation (13). In mammalian cells, both PI3K and ERK, major regulators of cell homeostasis and growth, have been implicated in V-ATPase assembly in response to a variety of stimuli (15, 17, 18). Because of their known involvement in the V-ATPase assembly process, the activity of these signaling molecules was determined upon glucose starvation. Additionally, we tested the activity of AMPK, a key regulator of energy homeostasis, because this pathway is known to be activated upon glucose starvation. HEK293T cells were conditioned to serum-free DMEM containing 5 mm glucose and then subjected to either acute glucose starvation or continued incubation in fresh serum-free DMEM at 5 mm glucose. Following treatment, cell homogenates were prepared and subjected to SDS-PAGE and Western blotting to detect levels of P-Akt (as a measure of PI3K/Akt signaling), P-ERK (as a measure of mitogen-activated protein kinase (MAPK) signaling), P-vasodilator-stimulated phosphoprotein (P-VASP, as a measure of PKA signaling), or P-acetyl-CoA carboxylase (P-ACC, as a measure of AMPK signaling). We found that glucose starvation led to a dramatic increase in AMPK activity and a detectable increase in ERK activation as measured by P-ACC and P-ERK, respectively (Fig. 3). By contrast, PI3K and PKA signaling as measured by P-Akt and P-VASP, respectively, did not change under conditions of glucose starvation. We have also tested activity of the p38 MAPK and c-Jun N-terminal kinase (JNK), both members of the MAPK family of signaling proteins involved in cell growth homeostasis, and mTORC1 (a major regulator of cell growth and metabolism that is downstream of the PI3K/Akt signaling pathway), and we see no activation of these pathways upon glucose starvation (Fig. S2B). We see a similar pattern of AMPK and ERK activation upon acute glucose starvation in LLC-PK1 cells (Fig. S2A). Collectively, these data demonstrate that AMPK and ERK are activated in response to glucose starvation and suggest that these kinases may be involved in the observed changes in V-ATPase assembly and activity.
Figure 3.
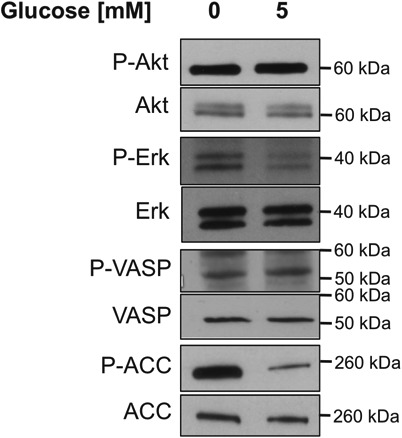
Effect of glucose starvation on various signaling pathways in HEK293T cells. HEK293T cells were maintained in serum-free DMEM containing 5 mm glucose for ∼6 h and then treated with serum-free DMEM containing 0 or 5 mm glucose for 10 min. Following treatment, whole-cell lysates were prepared as described under “Experimental procedures.” Samples were subjected to SDS-PAGE, and Western blotting was performed using antibodies directed against the phosphorylated substrates of various signaling pathways, including PI3K (P-Akt), ERK (P-ERK), PKA (P-VASP), and AMPK (P-ACC). Total levels of the substrates were also detected using the corresponding antibodies. Representative images are shown. n = 3.
Increased V-ATPase assembly upon glucose starvation is not required for AMPK activation
Because the V-ATPase was recently shown to be required for activation of AMPK (24), we wanted to determine whether increased V-ATPase assembly upon glucose starvation was necessary for AMPK activation. V-ATPase-dependent lysosomal acidification and AMPK activity were measured as described above at various times after removal of glucose from the media. As shown in Fig. 4, the starvation-induced increase in lysosomal V-ATPase activity is not observed until 3–4 min after removal of glucose, while an increase in AMPK activity is observed within 1 min of glucose starvation. These results suggest that the starvation-induced increase in V-ATPase activity is not required for activation of AMPK. By contrast, ERK activation occurs over a time course similar to increased V-ATPase activity, leaving open the question of the relationship between these two processes.
Figure 4.
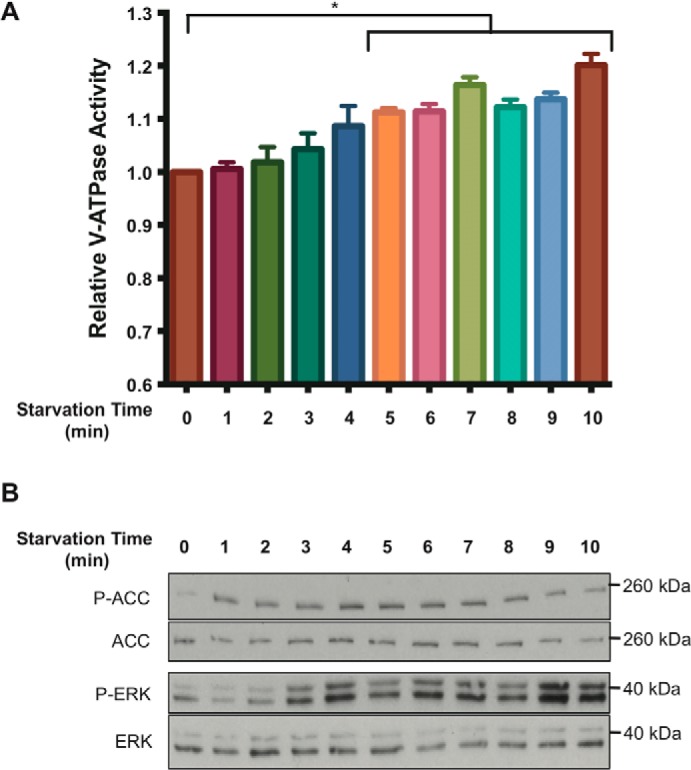
Time dependence of V-ATPase–dependent lysosomal acidification, AMPK activity, and ERK activity following glucose starvation of HEK293T cells. A, HEK293T cells were loaded with FITC-dextran and maintained in serum-free DMEM containing 5 mm glucose for ∼6 h and then treated with DMEM containing 0 mm glucose for the indicated times. Lysosomes were isolated and analyzed for V-ATPase-dependent lysosomal acidification as described under “Experimental procedures.” The error bars represent the standard error (n = 3). The asterisk represents a p value less than 0.05 comparing activity at t = 0 with each of the time points indicated under the horizontal bar. B, HEK293T cells were maintained in serum-free DMEM containing 5 mm glucose for ∼6 h then treated with DMEM containing 0 mm glucose for the indicated times. Whole-cell lysates were then prepared as described under “Experimental procedures,” and samples were subjected to SDS-PAGE and Western blotting using antibodies against the indicated proteins. Representative images are shown.
PI3K, Akt, and AMPK are involved in increased V-ATPase activity upon glucose starvation
To identify the signaling pathways responsible for increased V-ATPase activity upon glucose starvation, we measured V-ATPase-dependent lysosomal acidification as described above in the absence and presence of various signaling pathway inhibitors. HEK293T cells were treated with inhibitors of AMPK (dorsomorphin), PI3K (LY294002), PKA (H89), or mitogen-activated protein kinase kinase 1/2 (MEK1/2, upstream of ERK; AZD6244), during incubation of cells in DMEM containing 0, 5, or 25 mm glucose. As shown in Fig. 5A, treatment with either dorsomorphin or LY294002 led to elimination of the increase in V-ATPase-dependent lysosomal acidification upon glucose starvation, suggesting the involvement of AMPK and PI3K in this process. Additionally, inhibition of either AMPK or PI3K also led to reduced V-ATPase activity at all glucose concentrations tested (p < 0.01), suggesting that these kinases are also important in supporting basal V-ATPase-dependent acidification of lysosomes. By contrast, neither inhibitor blocked the increase in activity observed at high glucose. Similarly, there was no effect of H89 or AZD6244 on the increase in activity at either high or low glucose, suggesting that the PKA and ERK pathways are not involved in these processes.
Figure 5.
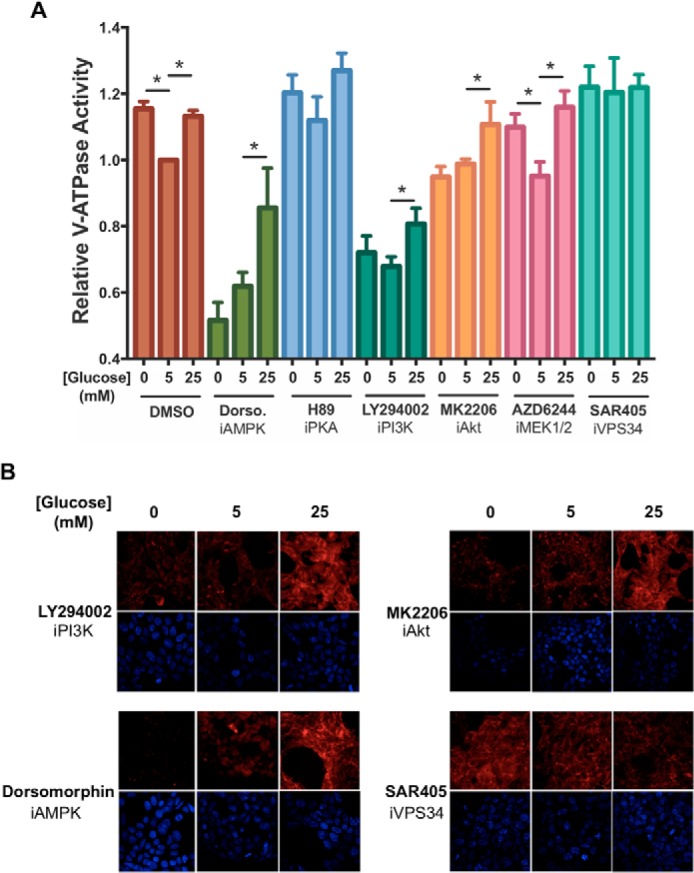
Effect of inhibitors of AMPK, PKA, PI3K, Akt, ERK, or Vps34 on starvation-dependent increases in V-ATPase-dependent lysosomal acidification in HEK293T cells. A, HEK293T cells were loaded with FITC-dextran and maintained in serum-free DMEM containing 5 mm glucose for ∼6 h and then treated with serum-free DMEM containing 0 mm glucose (10 min), 5 mm glucose (1 h), or 25 mm glucose (1 h). Where present, the cells were incubated with indicated inhibitors for 1 h prior to cell homogenization. Inhibitors included dorsomorphin (5 μm, an AMPK inhibitor), H89 (50 μm, a PKA inhibitor), LY294002 (50 μm, a PI3K inhibitor), MK2206 (1 μm, an Akt inhibitor), AZD6244 (1 μm, a MEK1/2 inhibitor), and SAR405 (10 μm, a Vps34 inhibitor). After treatment, cells were homogenized, and V-ATPase-dependent lysosomal acidification was tested as described in Fig. 2A. The error bars represent standard error. The asterisks represent statistically significant differences with a p value less than 0.05 (n ≥ 3). B, HEK293T cells were maintained in serum-free DMEM containing 5 mm glucose for ∼6 h and then treated with serum-free DMEM containing 0 mm glucose (10 min.), 5 mm glucose (1 h), or 25 mm glucose (1 h) in the presence or absence of various signaling pathway inhibitors. Inhibitors were added at the concentrations indicated in A and, where present, were incubated with cells for a total of 1 h. 10 min prior to cell collection, cells were incubated with LysoTracker to stain for acidic intracellular compartments and then fixed as described under “Experimental procedures.” Staining was detected by confocal fluorescence microscopy (red, LysoTracker; blue, DAPI). Drug-free and concanamycin A-treated controls for the experiment are shown in Fig. 2B. Representative images are shown, n = 2.
To determine what class of PI3K is involved in starvation-induced changes in V-ATPase activity, we tested the effects of an inhibitor of Akt (downstream of class I PI3K; MK2206) on this process. As shown in Fig. 5A, MK2206 prevented the increased activity observed upon glucose starvation but not that observed at high glucose, supporting the involvement of class I PI3K in the starvation-induced response. Interestingly, MK2206 did not decrease the baseline activity observed at 5 mm glucose the way that LY294002 did, suggesting that PI3K plays some additional role in controlling V-ATPase activity that is independent of Akt. To test the involvement of the class III PI3K Vps34 (a major regulator of autophagy that is downstream of AMPK signaling) in glucose-regulated changes in V-ATPase activity, we tested the effect of the class III inhibitor SAR405. As shown in Fig. 5A, SAR405 prevented the increase in activity at either low or high glucose by increasing the baseline activity at 5 mm glucose. We speculate that this increase in activity might be due to activation of AMPK upon Vps34 inhibition. In fact, we observe an increase in P-ACC in the presence of SAR405, consistent with this explanation (Fig. S2C). This result suggests the decrease in baseline V-ATPase activity observed upon PI3K inhibition with LY294002 is not due to inhibition of the class III PI3K. To confirm that the results observed by measuring V-ATPase-dependent acidification of lysosomes in vitro is reflective of the in vivo response, lysosomal acidification was measured in intact cells using LysoTracker staining, as described above. Consistent with the results shown in Fig. 5A, the increase in lysosomal acidification upon glucose starvation was blocked by treatment of cells with inhibitors of AMPK, PI3K, and Akt, whereas the increase observed at high glucose was unaffected (Figs. 5B and 2B). Similarly, treatment of cells with the Vps34 inhibitor prevented any change in staining at either high or low glucose. Although it may appear that LysoTracker staining using the Vps34 inhibitor was lower than DMSO-treated cells, staining was not reproducibly different between these conditions. These results suggest that the observed changes in activity measured in lysosomes from broken cells are reflective of similar changes in intact cells.
Finally, we wanted to determine whether the effects of inhibitors on glucose-dependent changes in V-ATPase activity were due to changes in V-ATPase assembly. As shown in Fig. 6, inhibition of AMPK, PI3K, or Akt largely prevented the increased assembly upon glucose starvation, consistent with the observed activity changes being due to changes in assembly. Taken together, these results demonstrate that changes in V-ATPase assembly and activity upon glucose starvation are under the control of both the AMPK and PI3K/Akt signaling pathways.
Figure 6.
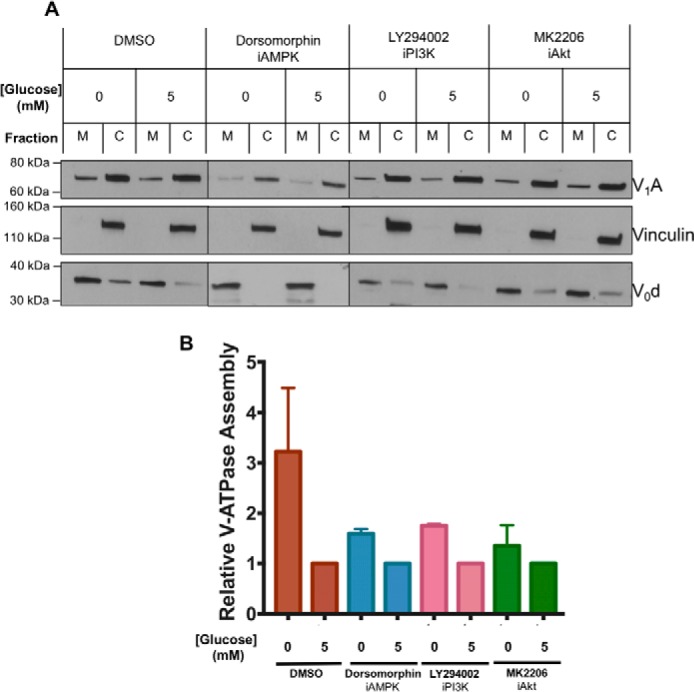
Effect of inhibitors of AMPK, PI3K, and Akt on starvation-dependent increase in V-ATPase assembly in HEK293T cells. A, HEK293T cells were maintained in serum-free DMEM containing 5 mm glucose for ∼6 h and then treated with serum-free DMEM containing 0 mm glucose (10 min) or 5 mm glucose (10 min) in the presence or absence of the indicated signaling pathway inhibitors. Inhibitors were present at the concentrations indicated in Fig. 5 and, where present, were incubated with cells for 1 h prior to analysis of assembly. Following treatment, assembly was assayed as described in Fig. 1A. Representative Western blots are shown. M indicates membrane and C indicates cytosolic fractions. B, Western blotting quantitation was performed as described in Fig. 1B, with values expressed relative to assembly measured at 5 mm glucose under each set of conditions. Error bars represent standard error, n = 2.
Discussion
Sensing and responding to changes in the availability of nutrients is essential to the viability of all cells. The V-ATPase has recently been identified as an essential part of the activation machinery for two critical nutrient sensors, namely mTORC1 and AMPK (24, 26, 27). mTORC1 integrates signals from growth factors and amino acid availability to control cell growth. AMPK senses energy levels in the cell to control the balance between energy utilizing and energy-generating processes (28). In the presence of adequate levels of amino acids, mTORC1 is recruited to a complex containing the V-ATPase and Ragulator present on the lysosomal membrane where it is activated by Rheb (26, 27, 29–31). In addition, V-ATPase activity is required for proper amino acid-dependent activation of mTORC1. Similarly, AMPK, together with the scaffolding protein Axin, is recruited to the same lysosomal V-ATPase–Ragulator complex under conditions of glucose starvation, where it is activated by phosphorylation of liver kinase B1 (LKB1) (24, 32). Given the role of the V-ATPase in controlling these key nutrient sensors, and the observation that both glucose and amino acid levels have been shown to control V-ATPase assembly in mammalian cells, we wished to test whether glucose-dependent changes in V-ATPase assembly are important in controlling AMPK activity.
We first sought to determine how V-ATPase assembly varies in response to changes in glucose concentration in mammalian cells. In agreement with previous reports (16–18), elevating glucose concentrations to high levels (25 mm) relative to normal physiological concentrations (5 mm) resulted in increased assembly of the V-ATPase in HEK293T and LLC-PK1 cells. Surprisingly, glucose starvation of cells similarly increased assembly of the V-ATPase relative to cells maintained at physiological glucose concentrations. This is in contrast to yeast, where glucose starvation causes dissociation of the V-ATPase, presumably as a means to conserve cellular stores of ATP under nutrient-limiting conditions (12). We further observed that glucose-dependent changes in V-ATPase assembly resulted in corresponding changes in V-ATPase activity, both in living cells and in isolated lysosomes (the site where both AMPK and mTORC1 are activated). Although the magnitude of this increase in V-ATPase-dependent acidification of lysosomes upon glucose starvation is quantitatively smaller than the increase observed in assembly, suggesting that not all of the assembled V-ATPases may be active, the exact relationship between the degree of assembly and the amount of proton transport is not known, making this latter conclusion uncertain. Nevertheless, glucose starvation of cells clearly leads to both increased assembly of the V-ATPase and increased activity of the V-ATPase in lysosomes.
To determine whether the starvation-induced increase in lysosomal V-ATPase activity is involved in the activation of AMPK upon glucose withdrawal, the time course for these two events was compared. We found that activation of AMPK actually precedes the starvation-induced increase in V-ATPase activity, ruling out the possibility that changes in AMPK depend upon the glucose-induced changes in the V-ATPase. The observation that AMPK activation precedes the increase in V-ATPase assembly suggests that the basal level of assembled and functional lysosomal V-ATPase present at normal physiological levels of glucose is sufficient to participate in the previously reported starvation-dependent recruitment to lysosomes and activation of AMPK by the V-ATPase–Ragulator complex (24). If not involved in activation of AMPK, what might be the function of the observed increase in lysosomal acidification upon glucose starvation? Among the processes activated in response to glucose starvation is autophagy (33). The lipid and protein breakdown that occurs during autophagy contributes to energy generation in the cell by providing breakdown products (fatty acids and amino acids) that can be further metabolized through fatty acid oxidation and the citric acid cycle to generate ATP via respiration. Importantly, the lysosomal lipases and proteases that are responsible for this breakdown are acid-activated. The rapid decrease in lysosomal pH may represent a primitive cellular response to glucose starvation that helps shift cellular energy production toward nonglycolytic sources.
We next wished to determine what signaling pathways participate in the starvation-induced increase in V-ATPase assembly and activity. We focused on those pathways that have previously been shown to be activated by glucose starvation and/or involved in changes in V-ATPase assembly. As expected, glucose starvation led to a strong activation of AMPK as well as a modest increase in ERK activity. No change in PKA or PI3K activity upon glucose starvation was observed. This latter result could be due to the use of HEK293T cells, which have a constitutively active PI3K. As noted above, the change in AMPK activity following glucose starvation occurs rapidly enough to be involved in the observed change in V-ATPase activity. By contrast, the change in ERK activity occurs with a similar time course, making its role uncertain.
To further examine the role these signaling pathways play in the observed changes in V-ATPase activity, the effect of pharmacological inhibitors was tested. We find that the starvation-induced increase in V-ATPase activity is completely blocked by the AMPK inhibitor dorsomorphin, whereas a MEK1/2 inhibitor, which blocks ERK signaling, as well as a PKA inhibitor, had no effect. Surprisingly, the increase in V-ATPase activity was also blocked by inhibitors of both PI3K and Akt. Similar results were obtained for assembly of the V-ATPase upon inhibition of the various signaling pathways. Thus, inhibition of PI3K, Akt, and AMPK all prevented the increase in assembly upon glucose starvation. Although no change in PI3K or Akt activity is observed in HEK293T cells upon glucose starvation, it is likely that there is a glucose-dependent change in a substrate of these pathways (possibly associated with the pump itself), which is upstream of activity and assembly. It is interesting to note that inhibition of both AMPK and PI3K but not Akt significantly reduces the baseline level of V-ATPase activity at physiological glucose concentrations. In the case of PI3K signaling, this suggests that the increase in V-ATPase activity observed upon glucose starvation depends upon Akt signaling downstream of PI3K, but that there is additional non-Akt-dependent PI3K signaling that supports V-ATPase activity under basal conditions. Interestingly, the role of AMPK in V-ATPase activation appears to be independent of autophagy (which is activated by AMPK), because inhibition of Vps34 (which plays a crucial role in activation of autophagy) does not inhibit and instead increases pump activity. This activation of lysosomal acidification upon Vps34 inhibition may be due to activation of AMPK. These results clearly show that both AMPK and PI3K play a role in controlling V-ATPase activity in response to glucose starvation.
These findings are in contrast to the results in yeast, where glucose-mediated changes in pump assembly and activity are dependent upon PKA signaling (13). Although we know that V-ATPase activity is necessary for proper autophagic flux (34, 35), it is possible that glucose starvation in yeast reduces V-ATPase assembly and activity to a level that still produces a pH sufficient for autophagy to take place. By contrast, mammalian cells may require a lower pH for proper autophagic flux to occur, thus requiring increased pump activity during glucose starvation and necessitating the involvement of a different signaling pathway in controlling V-ATPase activity in response to changes in glucose levels. PKA has also been previously implicated in regulation of V-ATPase assembly in insect cells (36–38), where phosphorylation of subunit C has been proposed to play a key role (39). Although PKA-dependent phosphorylation of subunit A has been demonstrated in renal cells, this is reported to participate in control of V-ATPase trafficking rather than assembly (40). Similarly, AMPK-dependent phosphorylation of subunit A appears to regulate trafficking of the pump in renal cells (41). Thus, ours is the first report of AMPK involvement in control of V-ATPase assembly.
Previous studies have demonstrated a role for PI3K in increased V-ATPase activity at elevated glucose concentrations and upon maturation of dendritic cells (15, 18). By contrast, no effect of inhibition of either PI3K or Akt is observed on increased V-ATPase activity at elevated glucose concentrations in our studies. One possible explanation for this is that the PI3K pathway is already constitutively activated in HEK293T cells as a result of expression of the polyomavirus large T antigen. Thus, it is possible that any dependence of V-ATPase assembly and activity on PI3K is masked by the artificially high activity of the PI3K pathway in these cells even at physiological glucose concentrations, such that no further increase in assembly and activity can occur at elevated glucose concentrations. In addition, these previous studies reported that incubation of HK-2 or LLC-PK1 cells overnight at 0.1 mm glucose reduced acidification relative to cells maintained at 5 mm glucose, and this effect was reversed upon incubation at elevated (10–20 mm) glucose (18). We are not certain of the basis for this apparent discrepancy, but several factors may account for the observed differences. First, in their experiments, cells were subjected to glucose starvation for 18 h, and no information was presented concerning the changes that occur acutely upon glucose withdrawal. It is possible that any increase in lysosomal acidification that occurs rapidly after starvation may be reversed at longer times in the interest of preserving cellular stores of ATP. In addition, no measurements were performed in their study on cells restored to normal (5 mm) glucose following overnight starvation, so it is not possible to directly compare their treatment of cells with those employed in this study. Similarly, in the studies of Marjuki et al. (17), lysosomal pH was measured in cells incubated at 0 and 25 mm glucose without reference to the pH observed at normal physiological glucose concentrations. Furthermore, in many cases, cells were subjected to changes in both glucose and serum during the experiments (18), despite the likely effects of serum factors on lysosomal acidification (21). By contrast, we chose to perform all experiments in cells maintained in the absence of serum throughout, to avoid this complication.
In summary, our studies have demonstrated an increase in V-ATPase-dependent lysosomal acidification upon glucose starvation that involves both the AMPK and PI3K/Akt signaling pathways. The ability to sense and respond to levels of nutrients is crucial for maintaining proper cellular homeostasis. Understanding the role of the V-ATPase as an energy sensor and the function of regulated assembly in energy homeostasis may provide new therapeutic tools for the treatment of diseases, such as diabetes, neurodegenerative disorders, and cancer, in which cellular energy sensing is aberrant. Future work in this area will be focused on understanding the link between AMPK signaling and the V-ATPase during glucose starvation.
Experimental procedures
Materials and equipment
FalconTM T-75 flasks (353136), Corning steriflip cups (430758), Beckman Coulter® ultra-clear centrifuge tubes (344057), and FalconTM polystyrene 10-cm plates (353003) were purchased from Thermo Fisher Scientific. HEK293T (CRL-3216) and LLC-PK1 (CL-101) cells were obtained from the ATCC. FBS (12306C), anti-vinculin antibodies (V9131; 1:500,000), DMSO (D2650), PMSF (P7626), and FITC-dextran (FD40S) were purchased from Sigma. Pre-cast 4–15% Mini-PROTEAN® TGDTM gels (456-1084) and peroxidase-conjugated anti-mouse antibodies (170-6516; 1:5,000) were purchased from Bio-Rad. Penicillin/streptomycin (15140-122), MEM (11095-080), DMEM (11885-076), Medium 199 (11150-067), PBS (20012-027), dextrose (215510), leupeptin (78435), and Amicon®Ultra 0.5-ml centrifugal filters (UFC501096) were purchased from Thermo Fisher Scientific. Glucose-free DMEM (D9800-02) and glucose-free Medium 199 powder (M2851-01) were purchased from US Biologicals, and aprotinin (10981532001) and pepstatin (11524488001) were purchased from Roche Applied Science. Poly-d-lysine (A-003-E) was purchased from Millipore. RT Prolong Gold Fixative with DAPI (P36935) and LysoTracker® Red DND-99 (L7528) were purchased from Life Technologies, Inc. Drugs LY294002 (S1105), dorsomorphin (S7306), H89 (S1582), MK2206 (S1078), and AZD6244 (S1008) were purchased from Selleckchem. Concanamycin A (BVT-0237) was purchased from BioViotica, and SAR405 (A8883) was purchased from APExBIO. Chemiluminescent substrate (RPN2106) was purchased from GE Healthcare, and autoradiography film (E3012) was purchased from Denville Scientific Inc. Antibodies against the following were purchased from Cell Signaling Technologies: P-Akt (4060; 1:2,000); Akt (4691; 1:1,000); P-ERK (4370; 1:500); ERK (4695; 1:1,000); P-VASP (3111; 1:1,000); VASP (3112; 1:1,000); P-ACC (3661; 1:1,000); ACC (3662; 1:1,000); P-AMPK (2535; 1:500); AMPK (2532; 1:1,000); P-S6K (9234; 1:2,000); S6K (2708; 1:2,000); P-VASP (3111; 1:1,000); VASP (3112; 1:1,000); PKA substrate (9624; 1:1,000); P-p38 (9211; 1:500); p38 (9212; 1:1,000); P-JNK (9255; 1:500); and JNK (9252; 1:1,000). Mammalian V0d (ab56441; 1:1,000) and peroxidase-conjugated anti-rabbit (ab97051; 1:5,000) antibodies were purchased from Abcam, and the mammalian V1A antibody (H00000523-A01; 1:1,000) was purchased from Abnova.
Cell culture
HEK293T cells were maintained in FalconTM T-75 flasks containing MEM supplemented with 1% penicillin/streptomycin and 10% FBS. LLC-PK1 cells were and grown in FalconTM T-75 flasks with Medium 199 supplemented with 1.5 g/liter sodium bicarbonate, 1% penicillin/streptomycin, and 3% FBS. Cells were grown at 37 °C with 5% CO2 in a humidified environment.
Glucose-containing media for treating HEK293T cells
Powdered, glucose-free DMEM was purchased from US Biologicals and dissolved in water according to the manufacturer's instructions. Final pH was adjusted to 7.3, and dextrose was added to make solutions with final glucose concentrations of 0, 5, or 25 mm. Media were filtered through a 0.22-micron membrane filter and stored at 4 °C.
Glucose media for treating LLC-PK1 cells
Powdered, glucose-free Medium 199 was purchased from US Biologicals and dissolved in water according to the manufacturer's instructions. Final pH was adjusted to 7.3, and dextrose was added to make solutions with final glucose concentrations of 0, 5, or 25 mm. Media were filtered through a 0.22-micron membrane filter and stored at 4 °C.
LysoTracker staining
2 × 104 cells were plated into 24-well plates containing glass coverslips that had been coated by treatment with poly-d-lysine and allowed to attach overnight. The following day, cells were rinsed twice with warm PBS and then treated with serum-free DMEM containing 5 mm glucose for at least 6 h. Next, cells were again washed twice with warm PBS and then treated with serum-free DMEM containing 0, 5, or 25 mm glucose for 10 min, 1, and 1 h, respectively, at 37 °C in the presence of DMSO, concanamycin A (5 μm), dorsomorphin (5 μm), MK2206 (1 μm), SAR405 (10 μm), or LY294002 (50 μm) for 1 h prior to collection. LysoTracker was added to the wells at a 1:1,000 dilution 10 min before sample collection. After the appropriate treatment time, cells were rinsed with PBS and then fixed at room temperature for 15 min using 4% paraformaldehyde. Coverslips were then fixed onto slides using RT ProLong Gold Fixative with DAPI. Slides were left to cure overnight and then stored at −20 °C. All images were obtained using a Leica TCS SPE microscope and LAS AF Software.
FITC-dextran loading of lysosomes and concanamycin A-dependent proton pumping
1 × 106 cells were plated into 10-cm poly-d-lysine-coated plates. The next day, the medium was replaced with media containing 2 mg/ml FITC-dextran, and cells were incubated overnight to allow for endocytosis of the fluorescent dye. The following day, the cells were washed twice with warm PBS then treated with serum-free DMEM (Medium 199 for LLC-PK1 cells) containing 5 mm glucose for at least 6 h. After the 6-h treatment, cells were again rinsed twice with warm PBS and then treated with serum-free DMEM (Medium 199 for LLC-PK1 cells) containing 5, 25, or 0 mm glucose for 1 h, 1 h, or 10 min, respectively, at 37 °C in the presence of DMSO, AZD6244 (5 μm), dorsomorphin (5 μm), H89 (50 μm), MK2206 (1 μm), SAR405 (10 μm), or LY294002 (50 μm) for 1 h prior to collection. To test for the reversibility of the change in activity during glucose starvation, cells were serum-starved as stated above, then treated with DMEM containing 0 mm glucose for 10 min, washed twice with warm PBS, and treated again with DMEM containing 5 mm glucose for 10 min.
Treatments for glucose starvation time course experiments were performed as follows. After the 6-h incubation in serum-free DMEM containing 5 mm glucose as described above, cells were rinsed twice with warm PBS and then treated with serum-free DMEM containing 0 mm glucose over the course of 10 min, where one plate of cells was removed and placed on ice every minute. This provided a time course for the first 10 min. Alternatively, cells were treated with serum-free DMEM containing 0 mm glucose over the course of 60 min, where incubations were started so that all plates finished at the same time. Treatment times for this time course were 0, 5, 10, 15, 20, 25, 30, and 60 min. Cells at the 0-min time point were maintained in fresh serum-free DMEM containing 5 mm glucose for the entirety of the experiment.
After glucose treatment, cells were placed on ice and rinsed with ice-cold PBS. Cells were scraped into 500 μl of fractionation buffer (125 mm KCl, 50 mm sucrose, 1 mm EDTA, 20 mm HEPES, 1 mm PMSF, 2 g/ml aprotinin, 5 g/ml leupeptin, and 1 g/ml pepstatin) and centrifuged at 1,200 × g for 5 min. Next, cells were resuspended in 750 μl of fractionation buffer and lysed by passing the cell suspension through a 27-gauge needle eight times. To clear lysates of intact cells and nuclei, samples were centrifuged at 2,000 × g for 10 min. The resulting supernatant was centrifuged for an additional 15 min at 16,100 × g to pellet membranes, including FITC-dextran–containing lysosomes, and pellets were resuspended in 100 μl of fractionation buffer. To measure concanamycin A-sensitive proton pumping, ∼30 μl of sample was added to 500 μl of pre-warmed (37 °C) fractionation buffer. Samples were excited at a wavelength of 490 nm, whereas emission fluorescence at 520 nm was continuously recorded in a PerkinElmer Life Sciences luminescence spectrophotometer model LS50B using Flwinlab software. After recording an initial fluorescence reading, V-ATPase proton pumping was activated by adding 1 mm ATP and 2 mm magnesium. As protons were pumped into the lysosomal lumen, the FITC-dextran signal was quenched. To verify that this quenching was specific to V-ATPase activity, the assay was performed in the presence of 1 μm concanamycin A, which completely inhibited fluorescence quenching after Mg-ATP addition. To verify that the signaling pathway inhibitors were not directly affecting lysosomal V-ATPase activity, the quenching assay was performed in the presence of AZD6244 (1 mm), dorsomorphin (1 mm), H89 (10 mm), MK2206 (200 μm), SAR405 (2 mm), or LY294002 (10 mm). Despite their presence at much higher concentrations than were added to intact cells, none of the inhibitors had any effect on the rate of V-ATPase-dependent proton transport activity when added to the assay. To control for the number of lysosomes added to the cuvette, the data collected for each sample was normalized to its respective initial fluorescence reading taken before the addition of Mg-ATP. Because lysosomes were loaded with FITC-dextran by endocytosis, using the initial fluorescence signal before ATP addition gives an accurate estimate of intact lysosomes in the preparation. The rate of proton pumping was determined by fitting the data to a linear regression using GraphPad Prism 7.0a software.
Cell fractionation
3 × 106 cells were plated into 10-cm poly-d-lysine-coated plates. The following day, cells were washed twice with warm PBS and then treated with serum-free DMEM (Medium 199 for LLC-PK1 cells) containing 5 mm glucose for at least 6 h. After the 6-h treatment, cells were washed twice with warm PBS and then treated with serum-free DMEM (Medium 199 for LLC-PK1 cells) containing 0, 5, or 25 mm glucose for 10 min, 1 h, and 1 h, respectively. Cells treated with inhibitors were treated with dorsomorphin (5 μm), MK2206 (1 μm), or LY294002 (50 μm) for 1 h prior to collection. To test for the reversibility of assembly during glucose starvation, cells were serum-starved as stated above, treated with DMEM containing 0 mm glucose for 10 min, washed twice with warm PBS, and treated again with DMEM containing 5 mm glucose for 10 min.
After treatment, cells were placed on ice and washed twice with ice-cold PBS. Cells were scraped into 650 μl of homogenization buffer (250 mm sucrose, 1 mm EDTA, 10 mm HEPES, 1 mm PMSF, 2 μg/ml aprotinin, 5 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mm NaF, and 1 mm glycerophosphate) and lysed by passing the cell suspension through a 27-gauge needle eight times. Next, cell lysates were cleared of nuclei and intact cells by centrifuging the samples at 500 × g for 10 min. The supernatant was ultracentrifuged at 100,000 × g for 30 min to pellet the membrane fraction. Using Amicon® Ultra 10K centrifugal filters, the supernatant was concentrated to give the cytosolic fraction. The membrane pellet was rinsed once with homogenization buffer and resuspended in homogenization buffer supplemented with 1% SDS.
Whole-cell lysis
3 × 106 cells were plated into 10-cm poly-d-lysine-coated plates. The following day, cells were washed twice with warm PBS and then treated with serum-free DMEM containing 5 mm glucose for at least 6 h. After the 6-h treatment, cells were again rinsed twice with warm PBS and then treated with serum-free DMEM containing 0 or 5 mm glucose for 10 min at 37 °C.
Treatments for the glucose starvation time course were performed as follows. After the 6-h incubation in serum-free DMEM containing 5 mm glucose as described above, cells were rinsed twice with warm PBS and then treated with serum-free DMEM containing 0 mm glucose over the course of 10 min, where one plate of cells was removed and placed on ice every minute. This resulted in a time course for the first 10 min following glucose starvation. Alternatively, for the 0–60-min time course, cells were treated with serum-free 0 mm glucose DMEM containing 0 mm glucose over the course of 60 min, where incubations were timed to all finish at the same time. One plate of cells was treated at a time and placed in the incubator resulting in all plates removed at the same time. Treatment times for this time course were 0, 5, 10, 15, 20, 25, 30, and 60 min. Cells at the 0-min time point were maintained in serum-free DMEM containing 5 mm glucose for the entirety of the experiment.
After treatment, cells were placed on ice and rinsed twice with ice-cold PBS. Cells were scraped into 300 μl of ice-cold lysis buffer (150 mm NaCl, 1% Triton X-100, 50 mm Tris-HCl (pH 7.5), 1 mm PMSF, 2 μg/ml aprotinin, 5 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mm NaF, and 1 mm glycerophosphate). Samples were continuously agitated for 30 min to lyse the cells and then centrifuged at 500 × g for 10 min to clear lysates of unbroken cells and nuclei.
Western blotting
Following fractionation or whole-cell lysis, protein concentrations were determined using the Lowry method (42). Samples were diluted in SDS-containing sample buffer, and proteins were separated by SDS-PAGE on 4–15% precast gels. Following separation, proteins were transferred to a nitrocellulose membrane. After blocking with 1.5% milk in TBS-T, the appropriate antibodies followed by secondary antibodies were added for protein detection using chemiluminescent substrate and X-ray film. Protein levels for fractionation experiments were quantified using ImageJ Version 10.2 software and the intensity of subunit A of the V1 domain present in the membrane fraction was normalized for the amount of membrane protein (subunit d of the V0 domain). Ratios were normalized for each individual experiment to those observed for cells maintained at 5 mm glucose (which was set to 1). Because it was not possible to run all inhibitor experiments on the same gel, each individual fractionation experiment using signaling pathway inhibitors was normalized to the signal observed at 5 mm glucose in the absence of the inhibitor.
Author contributions
C. M. M. and M. F. conceptualization; C. M. M. data curation; C. M. M. formal analysis; C. M. M. and M. F. funding acquisition; C. M. M. investigation; C. M. M. methodology; C. M. M. writing-original draft; C. M. M. and M. F. writing-review and editing; M. F. supervision; M. F. project administration.
Supplementary Material
Acknowledgments
We thank Michael Collins, Laura Stransky, and Kristina Cotter-Cross-for many helpful discussions.
* This work was supported by the National Institutes of Health Grant GM34478 (to M. F.) and a Sackler Families Collaborative Cancer Biology Award from Tufts University (to C. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, Mitzutani Foundation, National Science Foundation, and Ministerio de Economia de Spain.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1 and S2.
- V-ATPase
- vacuolar proton-translocating adenosine triphosphatase
- mTORC1
- mechanistic target of rapamycin complex I
- AMPK
- 5′-AMP-activated protein kinase
- VASP
- vasodilator-stimulated phosphoprotein
- ACC
- acetyl-CoA carboxylase
- LKB1
- liver kinase B1
- DMEM
- Dulbecco's modified Eagle's medium
- PMSF
- phenylmethylsulfonyl fluoride
- DAPI
- 4′,6-diamidino-2-phenylindole
- MEM
- minimum essential medium
- FBS
- fetal bovine serum.
References
- 1. Forgac M. (2007) Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917–929 10.1038/nrm2272 [DOI] [PubMed] [Google Scholar]
- 2. Cotter K., Stransky L., McGuire C., and Forgac M. (2015) Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem. Sci. 40, 611–622 10.1016/j.tibs.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kane P. M. (2012) Targeting reversible disassembly as a mechanism of controlling V-ATPase activity. Curr. Protein Pept. Sci. 13, 117–123 10.2174/138920312800493142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Breton S., and Brown D. (2013) Regulation of luminal acidification by the V-ATPase. Physiology 28, 318–329 10.1152/physiol.00007.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun-Wada G.-H., and Wada Y. (2013) Vacuolar-type proton pump ATPases: acidification and pathological relationships. Histol. Histopathol. 28, 805–815 [DOI] [PubMed] [Google Scholar]
- 6. Lu M., Ammar D., Ives H., Albrecht F., and Gluck S. L. (2007) Physical interaction between aldolase and vacuolar H+-ATPase is essential for the assembly and activity of the proton pump. J. Biol. Chem. 282, 24495–24503 10.1074/jbc.M702598200 [DOI] [PubMed] [Google Scholar]
- 7. Smardon A. M., Tarsio M., and Kane P. M. (2002) The RAVE complex is essential for stable assembly of the yeast V-ATPase. J. Biol. Chem. 277, 13831–13839 10.1074/jbc.M200682200 [DOI] [PubMed] [Google Scholar]
- 8. Seol J. H., Shevchenko A., Shevchenko A., and Deshaies R. J. (2001) Skp1 forms multiple protein complexes, including RAVE, a regulator of V-ATPase assembly. Nat. Cell Biol. 3, 384–391 10.1038/35070067 [DOI] [PubMed] [Google Scholar]
- 9. MacLeod K. J., Vasilyeva E., Merdek K., Vogel P. D., and Forgac M. (1999) Photoaffinity labeling of wild-type and mutant forms of the yeast V-ATPase A subunit by 2-azido-[32P]ADP. J. Biol. Chem. 274, 32869–32874 10.1074/jbc.274.46.32869 [DOI] [PubMed] [Google Scholar]
- 10. Xu T., and Forgac M. (2001) Microtubules are involved in glucose-dependent dissociation of the yeast vacuolar [H+]ATPase in vivo. J. Biol. Chem. 276, 24855–24861 10.1074/jbc.M100637200 [DOI] [PubMed] [Google Scholar]
- 11. Parra K. J., and Kane P. M. (1998) Reversible association between the V1 and V0 domains of yeast vacuolar H+-ATPase is an unconventional glucose-induced effect. Mol. Cell. Biol. 18, 7064–7074 10.1128/MCB.18.12.7064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kane P. M. (1995) Disassembly and reassembly of the yeast vacuolar H+-ATPase in vivo. J. Biol. Chem. 270, 17025–17032 [PubMed] [Google Scholar]
- 13. Bond S., and Forgac M. (2008) The Ras/cAMP/protein kinase a pathway regulates glucose-dependent assembly of the vacuolar H+-ATPase in yeast. J. Biol. Chem. 283, 36513–36521 10.1074/jbc.M805232200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Trombetta E. S., Ebersold M., Garrett W., Pypaert M., and Mellman I. (2003) Activation of lysosomal function during dendritic cell maturation. Science 299, 1400–1403 10.1126/science.1080106 [DOI] [PubMed] [Google Scholar]
- 15. Liberman R., Bond S., Shainheit M. G., Stadecker M. J., and Forgac M. (2014) Regulated assembly of vacuolar ATPase is increased during cluster disruption-induced maturation of dendritic cells through a phosphatidylinositol 3-kinase/mTOR-dependent pathway. J. Biol. Chem. 289, 1355–1363 10.1074/jbc.M113.524561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kohio H. P., and Adamson A. L. (2013) Glycolytic control of vacuolar-type ATPase activity: a mechanism to regulate influenza viral infection. Virology 444, 301–309 10.1016/j.virol.2013.06.026 [DOI] [PubMed] [Google Scholar]
- 17. Marjuki H., Gornitzky A., Marathe B. M., Ilyushina N. A., Aldridge J. R., Desai G., Webby R. J., and Webster R. G. (2011) Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion: ERK and PI3K regulate V-ATPase activity. Cell. Microbiol. 13, 587–601 10.1111/j.1462-5822.2010.01556.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sautin Y. Y., Lu M., Gaugler A., Zhang L., and Gluck S. L. (2005) Phosphatidylinositol 3-kinase-mediated effects of glucose on vacuolar H+-ATPase assembly, translocation, and acidification of intracellular compartments in renal epithelial cells. Mol. Cell. Biol. 25, 575–589 10.1128/MCB.25.2.575-589.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dechant R., Binda M., Lee S. S., Pelet S., Winderickx J., and Peter M. (2010) Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V-ATPase. EMBO J. 29, 2515–2526 10.1038/emboj.2010.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stransky L. A., and Forgac M. (2015) Amino acid availability modulates vacuolar H+-ATPase assembly. J. Biol. Chem. 290, 27360–27369 10.1074/jbc.M115.659128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu Y., Parmar A., Roux E., Balbis A., Dumas V., Chevalier S., and Posner B. I. (2012) Epidermal growth factor-induced vacuolar H+-ATPase assembly: a role in signaling via mTORC1 activation. J. Biol. Chem. 287, 26409–26422 10.1074/jbc.M112.352229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ochiai H., Sakai S., Hirabayashi T., Shimizu Y., and Terasawa K. (1995) Inhibitory effect of bafilomycin A1, a specific inhibitor of vacuolar-type proton pump, on the growth of influenza A and B viruses in MDCK cells. Antiviral Res. 27, 425–430 10.1016/0166-3542(95)00040-S [DOI] [PubMed] [Google Scholar]
- 23. Guinea R., and Carrasco L. (1995) Requirement for vacuolar proton-ATPase activity during entry of influenza virus into cells. J. Virol. 69, 2306–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang C.-S., Jiang B., Li M., Zhu M., Peng Y., Zhang Y.-L., Wu Y.-Q., Li T. Y., Liang Y., Lu Z., Lian G., Liu Q., Guo H., Yin Z., Ye Z., et al. (2014) The lysosomal v-ATPase-ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 20, 526–540 10.1016/j.cmet.2014.06.014 [DOI] [PubMed] [Google Scholar]
- 25. Dröse S., Bindseil K. U., Bowman E. J., Siebers A., Zeeck A., and Altendorf K. (1993) Inhibitory effect of modified bafilomycins and concanamycins on P- and V-type adenosinetriphosphatases. Biochemistry 32, 3902–3906 10.1021/bi00066a008 [DOI] [PubMed] [Google Scholar]
- 26. Efeyan A., Zoncu R., Chang S., Gumper I., Snitkin H., Wolfson R. L., Kirak O., Sabatini D. D., and Sabatini D. M. (2013) Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493, 679–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zoncu R., Bar-Peled L., Efeyan A., Wang S., Sancak Y., and Sabatini D. M. (2011) mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+ -ATPase. Science 334, 678–683 10.1126/science.1207056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Inoki K., Kim J., and Guan K.-L. (2012) AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 52, 381–400 10.1146/annurev-pharmtox-010611-134537 [DOI] [PubMed] [Google Scholar]
- 29. Sancak Y., Peterson T. R., Shaul Y. D., Lindquist R. A., Thoreen C. C., Bar-Peled L., and Sabatini D. M. (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 10.1126/science.1157535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim E., Goraksha-Hicks P., Li L., Neufeld T. P., and Guan K.-L. (2008) Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 10, 935–945 10.1038/ncb1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Efeyan A., and Sabatini D. M. (2013) Nutrients and growth factors in mTORC1 activation. Biochem. Soc. Trans. 41, 902–905 10.1042/BST20130063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y.-L., Guo H., Zhang C.-S., Lin S.-Y., Yin Z., Peng Y., Luo H., Shi Y., Lian G., Zhang C., Li M., Ye Z., Ye J., Han J., Li P., et al. (2013) AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 18, 546–555 10.1016/j.cmet.2013.09.005 [DOI] [PubMed] [Google Scholar]
- 33. Feng Y., He D., Yao Z., and Klionsky D. J. (2014) The machinery of macroautophagy. Cell Res. 24, 24–41 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mauvezin C., and Neufeld T. P. (2015) Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 11, 1437–1438 10.1080/15548627.2015.1066957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoshimori T., Yamamoto A., Moriyama Y., Futai M., and Tashiro Y. (1991) Bafilomycin A1, a specific inhibitor of vacuolar-type H+ -ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266, 17707–17712 [PubMed] [Google Scholar]
- 36. Rein J., Voss M., Blenau W., Walz B., and Baumann O. (2008) Hormone-induced assembly and activation of V-ATPase in blowfly salivary glands is mediated by protein kinase A. Am. J. Physiol. Cell Physiol. 294, C56–C65 10.1152/ajpcell.00041.2007 [DOI] [PubMed] [Google Scholar]
- 37. Sumner J. P., Dow J. A., Earley F. G., Klein U., Jäger D., and Wieczorek H. (1995) Regulation of plasma membrane V-ATPase activity by dissociation of peripheral subunits. J. Biol. Chem. 270, 5649–5653 10.1074/jbc.270.10.5649 [DOI] [PubMed] [Google Scholar]
- 38. Zimmermann B., Dames P., Walz B., and Baumann O. (2003) Distribution and serotonin-induced activation of vacuolar-type H+-ATPase in the salivary glands of the blowfly Calliphora vicina. J. Exp. Biol. 206, 1867–1876 10.1242/jeb.00376 [DOI] [PubMed] [Google Scholar]
- 39. Voss M., Vitavska O., Walz B., Wieczorek H., and Baumann O. (2007) Stimulus-induced phosphorylation of vacuolar H+-ATPase by protein kinase A. J. Biol. Chem. 282, 33735–33742 10.1074/jbc.M703368200 [DOI] [PubMed] [Google Scholar]
- 40. Alzamora R., Thali R. F., Gong F., Smolak C., Li H., Baty C. J., Bertrand C. A., Auchli Y., Brunisholz R. A., Neumann D., Hallows K. R., and Pastor-Soler N. M. (2010) PKA regulates vacuolar H+-ATPase localization and activity via direct phosphorylation of the A subunit in kidney cells. J. Biol. Chem. 285, 24676–24685 10.1074/jbc.M110.106278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alzamora R., Al-Bataineh M. M., Liu W., Gong F., Li H., Thali R. F., Joho-Auchli Y., Brunisholz R. A., Satlin L. M., Neumann D., Hallows K. R., and Pastor-Soler N. M. (2013) AMP-activated protein kinase regulates the vacuolar H+-ATPase via direct phosphorylation of the A subunit (ATP6V1A) in the kidney. Am. J. Physiol. Renal Physiol. 305, F943–F956 10.1152/ajprenal.00303.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lowry O. H., Rosebrough N. J., Farr A. L., and Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.