

Abstract
The binding of photosystem I (PS I) from Thermosynechococcus elongatus to the native cytochrome (cyt) c6 and cyt c from horse heart (cyt cHH) was analyzed by oxygen consumption measurements, isothermal titration calorimetry (ITC), and rigid body docking combined with electrostatic computations of binding energies. Although PS I has a higher affinity for cyt cHH than for cyt c6, the influence of ionic strength and pH on binding is different in the two cases. ITC and theoretical computations revealed the existence of unspecific binding sites for cyt cHH besides one specific binding site close to P700. Binding to PS I was found to be the same for reduced and oxidized cyt cHH. Based on this information, suitable conditions for cocrystallization of cyt cHH with PS I were found, resulting in crystals with a PS I:cyt cHH ratio of 1:1. A crystal structure at 3.4-Å resolution was obtained, but cyt cHH cannot be identified in the electron density map because of unspecific binding sites and/or high flexibility at the specific binding site. Modeling the binding of cyt c6 to PS I revealed a specific binding site where the distance and orientation of cyt c6 relative to P700 are comparable with cyt c2 from purple bacteria relative to P870. This work provides new insights into the binding modes of different cytochromes to PS I, thus facilitating steps toward solving the PS I–cyt c costructure and a more detailed understanding of natural electron transport processes.
Keywords: photosynthesis, cytochrome c, complex, docking, crystallography, photosystem I
Introduction
Photosystem I (PS I)4 from the thermophilic cyanobacterium Thermosynechococcus elongatus is a membrane-bound, trimeric, 1-MDa multipigment protein supercomplex. It converts light to electrochemical energy with a quantum efficiency of almost 100%. Because of its high stability, it is a suitable system for biotechnological applications. Thus, the protein complex has been used in photobioelectrodes for the generation of photocurrents and production of biofuels (1–4). The structure of PS I from T. elongatus was solved at 2.5-Å resolution in 2001 (5), and that from plants was solved very recently at 2.6-Å resolution (6). The cyanobacterial PS I consists of nine transmembrane and three cytoplasmic subunits harboring 127 cofactors per monomer. The two core subunits, PsaA and PsaB, bind the majority of cofactors, including reaction center (RC) and antenna pigments. In the RC, light-induced charge separation starts at the primary electron donor P700, a dimer of strongly interacting chlorophylls (Chls). The electron transport chain consists of two branches with one of the branches being more active than the other (7). From either branch, the electrons are transferred to the iron–sulfur cluster FX. The electrons are finally accepted by the terminal iron–sulfur clusters FA and FB bound by the extrinsic subunit PsaC.
In cyanobacteria and green algae, the soluble redox mediators cytochrome c6 (cyt c6) and plastocyanin (PC) donate electrons to oxidized P700 at the luminal side of the thylakoid membrane. The alternative expression of these homologous proteins is regulated by the availability of copper (8). In plants, however, solely PC occurs, whereas the cyanobacterium T. elongatus contains only cyt c6 (8, 9). Mutagenesis studies indicated that optimal binding of both electron mediators for electron transfer to P700+ occurs at a hydrophobic binding site, which is formed by two parallel tryptophan residues, Trp-655 from PsaA (Trp-A655) and Trp-632 from PsaB (Trp-B632) (10). Besides this hydrophobic site, a second binding site exists in plant and algal PS I that is based on electrostatic interactions due to positively charged side chains of PsaF. After binding to the charged site, PC reorients itself to bind to the hydrophobic area and form the active complex (11, 12). This binding model is based on kinetic data. Because of the strong, charged binding site, plant and algal PSs I form a stable complex with PC (13). In contrast, PsaF does not contribute to the binding of PC or cyt c6 in most cyanobacteria.
For cyanobacteria, kinetic data and NMR perturbation experiments (14) allowed elucidating the binding patch on cyt c6 for binding to PS I. However, no detailed structural information about the binding of cyt c6 to PS I is available. Such information is not only of fundamental scientific interest, but it could also be helpful to improve biotechnological applications. PS I from different organisms has been used in this context for creating photobioelectrodes or light-switchable biosensors. In some of these systems, cytochromes have been utilized to achieve electron transport to PS I (3, 15). Recently, it was found that mitochondrial cyt c from horse heart (cyt cHH) can be used to couple PS I to electrodes in an efficient way (1, 2, 16). On account of the demonstrated functionality of these non-native hybrid systems, the questions arise of how cyt cHH interacts with PS I and whether this interaction is different from native cyt c6 under physiological conditions.
Structural information is a prerequisite for answering these questions. In particular, X-ray crystallography requires cyt c–PS I cocrystals in which cyt c is located in the specific binding site for electron transfer. For cocrystallization, conditions must be found under which a stable complex is formed. To this end, in this study we focused on an investigation of the binding properties of PS I from T. elongatus with cyt cHH and cyt c6 under a variety of buffer conditions for elucidating the binding site.
In particular, we utilized analysis of oxygen reduction measurements and isothermal titration calorimetry (ITC). Based on these binding studies, cyt cHH was cocrystallized with PS I, and the crystal structure, in which, however, cyt cHH was not visible, was analyzed. Hence, as an alternative, binding of cyt cHH and cyt c6 was theoretically modeled using rigid body docking combined with electrostatic calculations of binding energies. Docking complexes were found for both cytochromes that are likely to resemble the actual cyt c–binding site of cyanobacterial PS I.
Results
Purity of isolated proteins
Dynamic light scattering (DLS) reveals that the hydrodynamic radius (RH) of the purified PS I ranges from 9 to 10 nm, with a polydispersity of less than 5%, as expected for monodisperse, trimeric PS I (17, 18). The absence of PS I monomers and dimers was further verified by blue native PAGE (Fig. S1). The subunit composition of each PS I preparation was analyzed by MS (Table S1). 10 subunits of the PS I protein complex could be detected. Most of them were post-translationally modified (for more details, see Ref. 19). However, subunits A and B could only be detected by SDS-PAGE because of their high mass (data not shown).
The cloning of the ORF tll1383 encoding cyt c6 resulted in a recombinant protein that carried a His6 tag at the C terminus. The protein was extracted from the periplasm of Escherichia coli and purified using a nickel-nitrilotriacetic acid column and subsequently anionic exchange chromatography. 1 liter of E. coli cells yielded 5 mg of protein. The purified protein was analyzed by SDS-PAGE (Fig. S2) and MS. The mass of the purified cyt c6 determined by MALDI-TOF shows the presence of a single peak at 11,063 m/z, which is in good agreement with the calculated mass of 11,061 g/mol (cyt c6 with a His6 tag and a heme group).
Interaction of PS I with cyt cHH and cyt c6: dependence on pH and ionic strength
To evaluate the interaction of cyt cHH and cyt c6 with PS I, we analyzed their capability to act as an electron donor for the photocatalytic complex. Here, oxygen reduction was used as a detection tool. We investigated a pH range from pH 6 corresponding to physiological conditions (luminal pH) (20) to pH 8 for potential crystallization setups. This range also includes conditions under which photobioelectrodes are often used (1, 2).
By analyzing the concentration-dependent behavior of both proteins, it was found that the Michaelis–Menten model is well-suited to describe the kinetics. Here, the enzyme is PS I, the substrate is cytochrome, the pre-equilibrium is between PS I and cytochrome, and the catalytic reaction involves all electron transfer reactions. The turnover number (kcat) is represented by the rate of oxygen reduction. For cyt cHH, kcat and Km are highly dependent on pH (Table 1). In phosphate buffer at pH 6–8, kcat increases from 7 to 35 s−1, and Km increases from 12 to 31 μm. Besides pH, the type of buffer also affects the binding affinity. In Tricine buffer, pH 8, Km was decreased by a factor of 6 compared with phosphate buffer. The turnover number was identical in both buffer types at pH 7.5 and 8.
Table 1.
Oxygen consumption measurements of PS I with cyt cHH and cyt c6 as electron donor in either 5 mm phosphate buffer or 25 mm Tricine buffer at specified pH and addition of ions
Kinetic constants were determined by applying the Michaelis–Menten model. Standard deviations were determined from three independent measurements.
| Buffer | pH | Km | kcat |
|---|---|---|---|
| μm | s−1 | ||
| Cyt cHH | |||
| Phosphate buffer | 6.0 | 11.5 ± 2.8 | 7.1 ± 1.3 |
| Phosphate buffer | 6.5 | 14.2 ± 2.7 | 8.5 ± 0.7 |
| Phosphate buffer | 7.0a | 22.8 ± 2.8 | 20.3 ± 1.0 |
| Phosphate buffer | 7.5 | 23.5 ± 2.3 | 28.9 ± 1.1 |
| Tricine | 7.5 | 4.9 ± 0.5 | 27.7 ± 1.5 |
| Phosphate buffer | 8.0 | 30.5 ± 3.0 | 34.9 ± 3.1 |
| Tricine | 8.0 | 5.0 ± 0.7 | 34.3 ± 1.9 |
| Tricine + 25 mm NaCl | 8.0 | 10.8 ± 1.7 | 34.8 ± 1.4 |
| Tricine + 10 mm MgSO4 | 8.0 | 44.7 ± 3.6 | 31.2 ± 0.9 |
| Cyt c6 | |||
| Tricine | 8.0 | 290 ± 45 | 55 ± 9 |
| Tricine + 10 mm MgSO4 | 8.0 | 65 ± 9 | 143 ± 11 |
| Tricine + 200 mm MgSO4 | 8.0 | 33.3 ± 1.3 | 159 ± 2 |
a Value taken from Ref. 2.
To assess which buffer type is suitable to achieve high kcat and/or low Km, the oxygen reduction rate of PS I with 16 μm cyt cHH was analyzed in different buffer types at pH 8 (Fig. S3). Because kcat remains constant, the change in the reduction rate results from the change in the Km value. For each buffer used, the reduction rate decreased linearly with increasing buffer concentration in the range from 5 to 100 mm. The rate was highest in Tricine and Tris buffer followed by HEPES, MOPS, and lastly phosphate buffer. This order seems to correlate with the ionic strength of the buffer solutions. Ions of different charge affect the binding properties between proteins differently. Therefore, the reduction rate of PS I with cyt cHH was analyzed in the presence of NaCl, KCl, NH4Cl, Na2SO4, CaCl2, MgCl2, and MgSO4. None of these ions induced a specific effect but rather resulted in a decreased reduction rate. This appears to originate from the increasing ionic strength (Fig. 1). Consequently, divalent ions led to a stronger decrease than monovalent ions at identical molar concentration.
Figure 1.
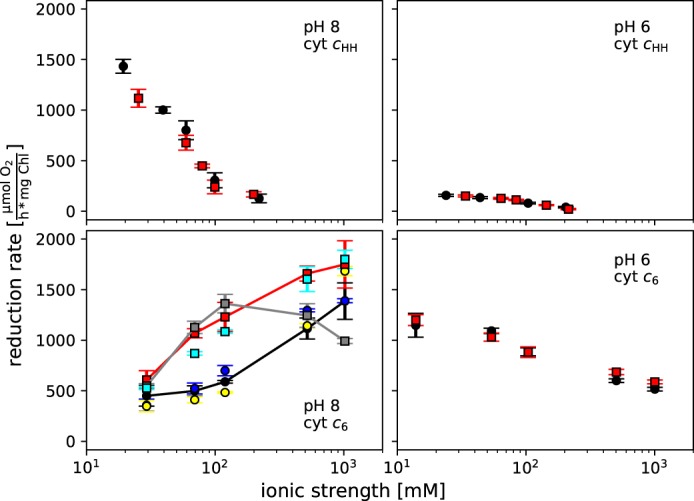
Oxygen reduction rates of PS I with 16 μm cyt cHH (top) and cyt c6 (bottom) at pH 8 (left) and pH 6 (right) as a function of ionic strength. Monovalent (NaCl; black) and divalent (MgCl2; red) cations are depicted as circles and squares, respectively. For cyt c6, pH 8 (bottom, left) differences between the applied salts become prominent. Therefore, a further differentiation of the salts is shown: NaCl (black), Na2SO4 (yellow), NH4Cl (blue), MgCl2 (red), CaCl2 (gray), and MgSO4 (cyan). NaCl, MgCl2, and CaCl2 are connected by a line in their corresponding color. All measurements were performed in either 25 mm Tricine-NaOH, pH 8, or 5 mm MES-NaOH, pH 6, with 2 mm ascorbic acid and 300 μm methyl viologen at 20 °C. The concentration of buffer ions and counterions, which contribute to the ionic strength, was calculated using the Henderson–Hasselbalch equation with a pKa of 8.2 and 6.2 for Tricine and MES buffers, respectively. Standard deviations (error bars) were determined from three to nine independent measurements.
All these experiments demonstrate that increasing salt concentrations decrease the reduction rate by strongly altering Km, whereas kcat still remains constant. This clearly points to an electrostatic nature of the interaction between PS I and cyt cHH.
An opposing trend was found for the interaction of PS I with its native electron donor, cyt c6. In this case, the oxygen reduction rates of PS I in the presence of cyt c6 without additional salt ions can be increased by decreasing the pH (Fig. 1). An increase of the ionic strength at pH 6 led to a decrease of the reduction rate, whereas at pH 8 an increase of the reduction rate was measured with a larger magnitude for divalent cations at 100 mm ionic strength than for monovalent ions or divalent anions. The addition of CaCl2 led to a decreased reduction rate above 100 mm. Therefore, the highest reduction rate can be obtained at pH 8 at high ionic strength except for CaCl2. The increase in reduction rate originated from an increasing kcat as well as a decreasing Km (Table 1).
Cocrystal structure of PS I with cyt cHH
PS I possesses a high affinity for cyt cHH at low ionic strength, and it can be crystallized by “salting in” at low pH (21, 22). We combined this knowledge and crystallized PS I in the presence of cyt cHH with MES-NaOH, pH 6.0, and low MgSO4 concentrations. Green crystals grew within a week. Each crystal contained both PS I and cyt cHH as analyzed by MALDI-TOF (Fig. S4). The cyt cHH content of the crystals was analyzed for crystal batches grown at different cyt cHH:PS I ratios (Fig. S5). Crystals containing a 1:1 ratio of both proteins were achieved by growing at a 5-fold excess of cyt cHH. Crystals did not grow at higher cyt cHH concentration. At a 10-fold excess of cyt cHH, no nucleation occurred even at 0 mm MgSO4.
The crystals diffracted to 3.4-Å resolution with 97% completeness. Unit cell parameters are identical to those from PS I crystals grown without cyt cHH (Table S2). We cannot yet assign an electron density for cyt cHH at 3.4-Å resolution (supporting Fig. S6). Nevertheless, we were able to detect the subunit cyt cHH after X-ray measurements of PS I–cyt cHH crystals by subsequent MALDI-TOF analysis of these crystals (Fig. S7). In contrast to cyt cHH, no PS I–cyt c6 cocrystals with high cyt c6 saturation were achieved.
Different binding modes of cyt cHH and cyt c6
We used ITC to analyze the binding behavior of cyt c to PS I. The proteins need to be soluble throughout the measurement and in high concentration. 25 mm NaCl at pH 8.0 was found suitable for ITC measurements (Table S3).
To test the influence of the redox state of cyt cHH on the binding, the measurements were performed either in the presence (reduced cyt cHH and PS I) or absence (oxidized cyt cHH and PS I) of sodium ascorbate. Due to the low binding affinity and protein concentration, the number of binding sites (n) cannot be derived with certainty. For both redox conditions, a fit to the binding curve with n = 1 or n = 2 binding sites resulted in a large error (Fig. 2). This means that at least a second type of binding site is necessary to describe the experimental data (Table 2). The heterogeneity of the binding can also be visualized by depicting the data in a logarithmic binding curve (Fig. S8). Assuming the dissociation constant (KD) of the specific binding site, where the electron transfer occurs, to be equal to the Km value from the Michaelis–Menten kinetic analysis, a reasonable set of parameters for a model of two types of binding sites can be obtained (Table 2). These data suggest that the majority of the produced heat originates from the specific binding site. For the second type of binding sites, n2 > 1 was obtained, suggesting a rather complex binding behavior. The cyt cHH binding seems to be independent of the redox state with equal numbers of binding sites and dissociation constants of 19 and 25 μm for the oxidized and reduced proteins, respectively.
Figure 2.
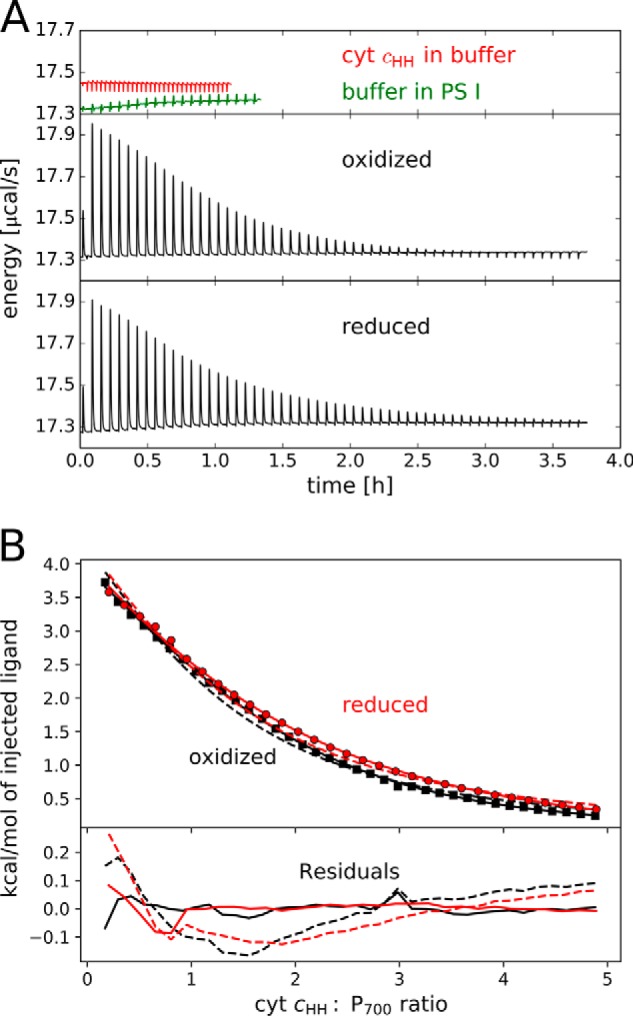
Isothermal titration calorimetry of PS I with cyt cHH. A, thermogram for exemplary background measurements (top) and oxidized (middle) and reduced (bottom) proteins. B, integrated heats of titrations after background subtraction in the presence (red) or absence (black) of 5 mm ascorbate. High cyt cHH:P700 ratios are omitted for a better overview. Fits (top) and residuals (bottom) are shown for one set of binding sites with n = 1.0 (dashed line) and for one set of binding sites with n = 1.5 (solid line). Parameters obtained from the models are shown in Table 2. Measurements were performed at 20 °C in 25 mm Tricine buffer, pH 8.0, with 25 mm NaCl and 0.02% DDM. Each titration step consisted of a 5-μl injected volume from 1 mm cyt cHH.
Table 2.
Binding parameters derived from ITC measurements
Data sets for oxidized and reduced proteins were analyzed with either one or two sets of binding sites. Standard deviations were determined from three independent measurements (n, number of binding sites; KD, dissociation constant; ΔH, binding enthalpy).
| Cyt cHH oxidized |
Cyt cHH reduced |
Cyt c6 reduced |
|||
|---|---|---|---|---|---|
| 1a | 2a | 1a | 2a | 1a | |
| n1 | 1.5 ± 0.1 | 1b | 1.5 ± 0.2 | 1b | 1b |
| KD1 (μm) | 18.8 ± 0.2 | 11b | 24.8 ± 1.2 | 11b | 21 ± 3 |
| ΔH1 (cal/mol) | 5320 ± 70 | 8500 ± 280 | 4530 ± 250 | 7290 ± 360 | −1370 ± 40 |
| n2 | 2.0 ± 0.2 | 6.4 ± 1.2 | |||
| KD2 (μm) | 28.4 ± 1.0 | 39.4 ± 5.4 | |||
| ΔH2 (cal/mol) | −480 ± 120 | −220 ± 100 | |||
a Number of types of binding sites presumed in the fit.
b n1 and KD1 were set to 1 and 11 μm, respectively, as derived from Michaelis–Menten kinetics.
In contrast to cyt cHH, the heat of cyt c6 binding is exothermic, indicating a different binding mechanism. Also, the binding properties of cyt c6 to PS I are dependent on the oxidation state: although binding is found for reduced cyt c6, the thermogram of oxidized cyt c6 equals the heat of dilution (Fig. 3). The integrated heat signals of reduced proteins saturate at a lower cyt c6:PS I ratio compared with cyt cHH, indicating a higher affinity. The values calculated from a fit assuming one binding site are found in Table 2, but due to the low heat of binding compared with the high heat of dilution, absolute values should be taken with care. An increased PS I concentration at elevated ionic strength (200 mm MgSO4) did not improve the signal (Fig. S9).
Figure 3.
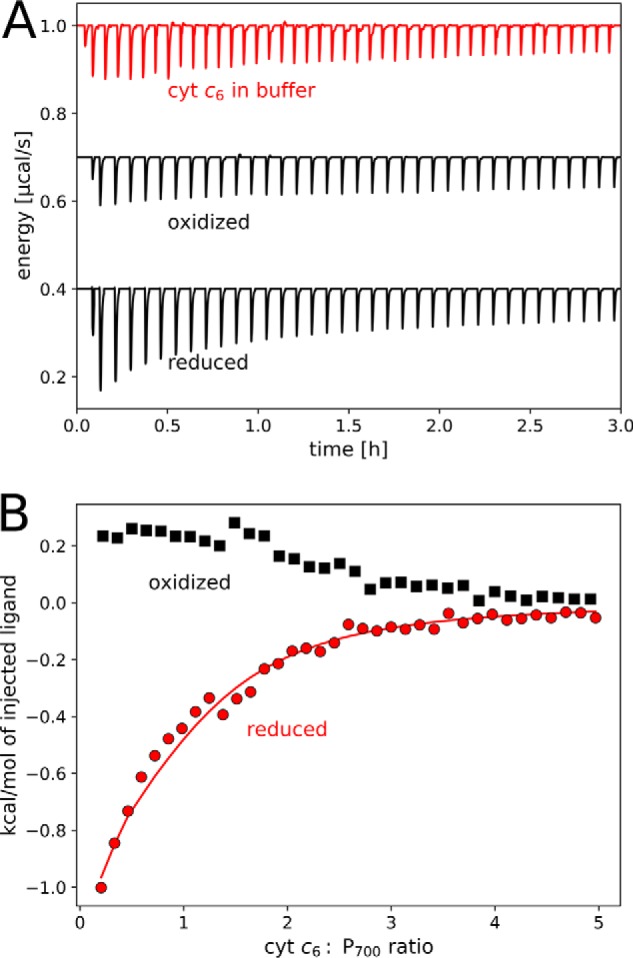
Isothermal titration calorimetry of PS I with cyt c6. A, thermogram for exemplary background measurements (top, red) and oxidized (middle) and reduced (bottom) proteins. B, integrated heats of titrations after background subtraction in the presence (red; reduced) or absence (black; oxidized) of 5 mm ascorbate. The fit of the data for reduced cyt c6 is shown for one set of binding sites with n = 1.0. Parameters obtained from the model are shown in Table 2. After substraction of the heat of dilution, the data for oxidized cyt c6 converge to negative values at high cyt c6:PS I ratio (−0.1 kcal/mol; not shown) and are thus not analyzed by a model. Measurements were performed at 20 °C in 25 mm Tricine-NaOH, pH 8.0, with 25 mm NaCl and 0.02% DDM. Each titration step consisted of a 5-μl injected volume from 1 mm cyt c6.
Analysis of unspecific binding sites of cyt cHH and cyt c6
To investigate this complex binding behavior, potential binding sites (further referred to as docking sites) were calculated by FTDock and pyDock3 (23, 24). Fig. 4 and Fig. S10 give an overview of the positions of docking sites with negative binding energy. The binding energy ranged from −14.4 to +123.4 kcal/mol and from −28.3 to +55.8 kcal/mol for docking sites of cyt cHH located at the cytoplasmic and luminal side, respectively. This result suggests that binding of cyt cHH to PS I occurs preferentially at the luminal side. Accordingly, the binding sites identified by ITC, including both specific and unspecific binding sites, can be expected to be located at the luminal side. Although cyt cHH is a non-native electron donor to PS I, an accumulation of docking sites (henceforth denoted as a cluster) at the luminal side of PS I close to P700 was found (Fig. 4, left).
Figure 4.

Molecular docking simulation of monomeric PS I with cyt cHH (left) and cyt c6 (right). Each sphere represents the position of a docked cyt c. The binding energy, calculated by pyDock, is highlighted by a color code. Docking states with less than −20 kcal/mol are highlighted by an increased sphere size.
Similarly, docking sites of cyt c6 were found on both the luminal side (−31.6 to +51.0 kcal/mol) and cytoplasmic side (−20.7 to +65.9 kcal/mol). The docking sites of cyt c6 at the luminal side with strongly negative binding energies are less dispersed compared with those for cyt cHH with the majority of these sites organized in a cluster close to P700 (Fig. 4, right). As expected for cyanobacterial PS I, none of the docking sites are in close vicinity to PsaF.
Elucidating the specific cyt c–binding site of PS I
The most interesting binding site is that where the electron transfer from cyt c to P700 occurs (specific binding site). At this site, the heme group of cyt c and P700 have to be in close proximity. In the case of cyt c6, the 100 docking sites with the strongest interaction display binding energies in the range of −31 to −15 kcal/mol. For 25 of these 100 sites, the smallest distance between carbon atoms of the heme group of cyt c6 and tryptophan residues Trp-A655 and Trp-B631 of PS I is below 10 Å. An NMR analysis of cyt c6–PS I interaction in Nostoc sp. PCC 7119 revealed certain amino acid residues of cyt c6 that are likely part of the binding interface (14). Nostoc sp. cyt c6 shares a high sequence identity with cyt c6 of T. elongatus. 13 of the 25 docking sites identified above are in agreement with the NMR results with heme–tryptophan distances of 2.5–8.9 Å.
Binding of cyt c6 and PS I depends on ionic strength and pH as shown above. Therefore, the electrostatic binding energy for these 13 docking sites was calculated for three different values of ionic strengths at pH 6 and 8 using the Poisson–Boltzmann equation (Fig. S11). Recalculating the electrostatic binding energy revealed that the binding energy of most of the docking sites is decreased to less positive values by increasing the ionic strength at pH 8 but not at pH 6 (Fig. S11). The closest of these docking sites has a heme–tryptophan distance of 2.5 Å and a binding energy of −15.5 kcal/mol (Fig. 5, bottom). The distance between the iron from the heme group and the magnesium of the two P700 chlorophylls is 21.4 and 21.3 Å, respectively. In this specific docking site, cyt c6 is in close proximity to a luminal loop of PsaA. The negatively charged amino acid residue Asp-628 from this loop is at a 7.4-Å distance from the negatively charged residue Glu-34 from cyt c6, leading to a repulsive interaction at low ionic strength (Fig. 5). The amino acid residues that form the interface between T. elongatus cyt c6 and PS I are shown in Table S4. It must be mentioned that the absolute distances shown in Table S4 have to be taken with caution because the expected perturbation of amino acid residues upon binding is not described by rigid body docking. Of the 19 amino acid residues shown in Table S4, only three are not perturbed in cyt c6 from Nostoc sp. upon binding to PS I (14).
Figure 5.
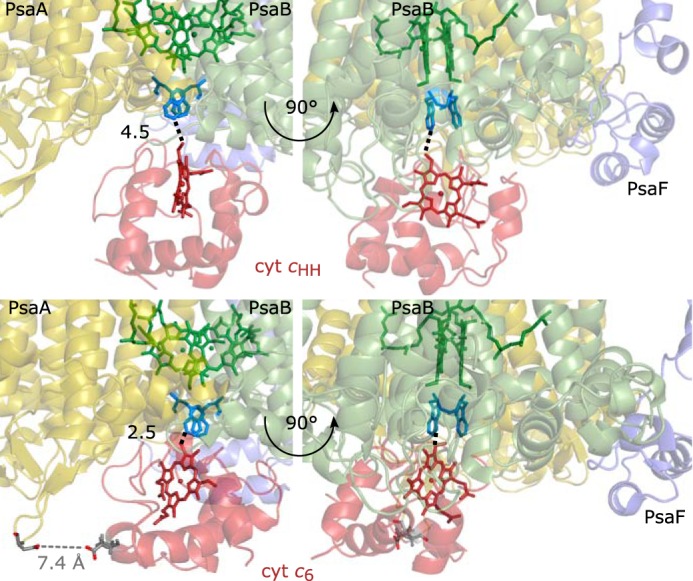
Potential cyt cHH (top)– and cyt c6 (bottom)–binding site of PS I. Shown are the docking sites that most likely resemble the specific cyt c–binding site of PS I. The heme group (red) of cyt cHH and cyt c6 points toward the luminal tryptophan residues Trp-A655 and Trp-B631 (blue) and P700 (green) of PS I. The distances between the heme groups and the closest tryptophan are highlighted by a black dotted line. Cyt c6 does not interact with PsaF (purple) but is close to the luminal loop of PsaA (yellow). The carboxyl group of Glu-34 from cyt c6 is at a distance of 7.4 Å from the carboxyl group of Asp-628 from PsaA (gray dotted line).
Because cyt cHH is a non-native binding partner, it does not necessarily have to bind in the native binding site. The 300 cyt cHH docking sites with strongest binding have binding energies in the range of −28 to −15 kcal/mol. 36 of these 300 docking sites have heme–tryptophan distances of less than 10 Å between carbon atoms. After recalculating the electrostatic binding energy using the Poisson–Boltzmann equation, seven docking sites remain; these docking sites show pH and ionic strength dependence in good agreement with the analysis of kinetic parameters (see above). The electrostatic binding energy was strongly negative in the absence of salt ions and increased to about 0 kcal/mol at an ionic strength corresponding to 100 mm MgSO4 (Fig. S11). Of these seven cyt cHH docking sites, the one with the most negative binding energy (−25.8 kcal/mol) is that in closest proximity to P700. Here, the distance between the heme group and the parallel tryptophan residues Trp-A655/Trp-B631 is 4.5 Å (Fig. 5, top). The distances of the iron from the heme group and the magnesium ions from P700 is 24.3 and 24.9 Å, respectively. The distances between the closest side chains of PS I and cyt cHH are shown in Table S5. There is no salt bridge between residues, suggesting that the electrostatic interactions are mainly nonspecific.
Discussion
Activity and affinity of PS I for cyt c
The PS I oxygen reduction rate with both cytochromes is highly dependent on the pH and ionic strength. These effects are in agreement with the P700+ re-reduction rates from time-resolved spectroscopy with cyt c6 (25, 26). The binding affinity of PS I for cyt c6 is increased by increasing ionic strength at pH 8 but not at pH 6, which is close to the physiological pH (20). The isoelectric point (pI) of His6-tagged cyt c6 can be estimated to 6.5 based on the amino acid sequence and assuming a reduced heme group using the compute pI tool from ExPASy (27). Without the His tag, the pI is estimated to be 5.5. Thus, in both cases, cyt c6 is close to zero net charge at pH 6, whereas it is negatively charged at pH 8. If we assume that the luminal side of PS I is negatively charged at both pH values (given that it is negatively charged at pH 7 (16)), it follows that there is a repulsive interaction between PS I and cyt c6 at pH 8 that is almost absent at pH 6. This can explain the ionic strength dependence found for the two different pH values.
At both pH 8 and 6, increasing the ionic strength decreases the binding affinity of cyt cHH to PS I. As the pI of cyt cHH is 10.5 (28), the protein is positively charged at the investigated pH values. Therefore, decreasing the ionic strength favors binding of cyt cHH. In this study, Km values of T. elongatus PS I of up to 33 (pH 8, high ionic strength) and 5 μm (pH 8, low ionic strength) could be achieved for the native and non-native cytochrome, respectively (Table 1). Both values are comparable with the affinity of plastocyanins and cytochromes in plants, algae, and other cyanobacteria (7–125 μm (14, 29–32)).
The binding affinity of the homologous cyt c2 to photosynthetic bacterial reaction centers (bRCs) is 1 μm (33). In this case, cocrystallization of cyt c2 with bRC was successful (34). These results indicate how strong the affinity must be for successful cocrystallization. The present data confirm that cyt cHH can form a stable complex with PS I at low ionic strength (2). This motivated us to attempt a cocrystallization of cyt cHH with PS I. The low ionic strength necessary for complex formation matches the known crystallization conditions of PS I (5).
Binding affinity of oxidized and reduced cyt c to PS I
To elucidate why cyt cHH is not identified in the crystal structure, we analyzed the binding behavior of cyt cHH by ITC. Cyt cHH binds to PS I at more than one site. The positive enthalpy (Table 2) reveals that the binding of cyt cHH to PS I is endothermic. Positive enthalpies for the electrostatic binding of cyt cHH were reported and are likely to originate from the displacement of bound water molecules (35). Another observation by ITC is that the binding is independent of its redox state in contrast to cyt c6. This behavior renders cyt cHH a suitable redox mediator in biotechnological applications.
Cocrystal structure of PS I with cyt cHH
A cocrystal structure of cyt cHH with PS I was solved, but no electron density was found for cyt cHH. This may be explained by the following reasons.
In the crystal, cyt cHH is highly disordered or flexible. In Fig. S12, a part of the PS I crystal lattice is shown. Here, the PS I trimers form layers with the membrane planes oriented parallel to each other. The crystal contacts are formed by the cytoplasmic subunit PsaE and luminal helices of the subunit PsaF. A volume is present between the trimers in which no electron density is visible. Part of this volume is occupied with detergent belts (17). The remaining volume contains an aqueous phase, including an area close to the luminal surface of PS I, highlighted in blue in Fig. S12. The cyt cHH is expected to be located in this volume. As illustrated by the randomly chosen docking state shown in Fig. S12, cyt cHH cannot form protein contacts with other PS I trimers. In such flexible environments, a high-resolution crystal structure is usually necessary to visualize the cocrystallized protein (36). If there is more than one binding site for cyt cHH, the occupancy of the specific binding site at P700 will not be 100% even in a 1:1 cocrystal. In this respect, variation of the protein ratio could have an influence as shown for cyt cHH–peroxidase cocrystals (37, 38). By using isothermal titration calorimetry and rigid body docking, we revealed that there is more than one cyt cHH–binding site at PS I under low ionic strength. These binding sites are likely to spread over the whole luminal side of PS I (Fig. 4) and mostly would not interfere with the crystal contacts. Increasing the cyt cHH:PS I ratio might be necessary to achieve full occupancy for the binding site at P700. However, cyt cHH disturbs the crystal formation. Therefore, saturating the binding site at P700 is not possible under the crystallization conditions used in this study.
Even if cyt cHH is bound to the site close to P700 at 100% occupancy, cyt cHH could occur in different conformations or orientations, rendering it invisible in the crystal structure. This possibility is supported by the theoretical binding studies (Fig. S10).
The specific cyt c–binding site at PS I
The specific binding sites of cyt c6 and cyt cHH are those with closest proximity of the heme group to Trp-A655 as analyzed by rigid body docking. Calculating the binding energy of the closest docking sites at different pH values and ionic strengths resulted in changes that are in good agreement with the measured oxygen reduction rates for both cytochromes.
Both cytochromes bound more strongly to Trp-A655 than to Trp-B631 (Fig. 5) as was also shown for cyt c6 from Chlamydomonas reinhardtii (39). It was found that the PS I–cyt cHH complex has a more negative binding energy than the PS I–cyt c6 complex, which is in good agreement with the higher affinity of PS I for cyt cHH. The distance between the heme group and P700 is smaller for the PS I–cyt c6 complex than for the PS I–cyt cHH complex. As the positioning of the heme group is slightly different for both complexes, different turnover numbers can be expected. Indeed, PS I has a higher turnover number using cyt c6 as electron donor (Table 1).
At low ionic strength and pH 8, the binding energy of PS I and cyt c6 is repulsive. This repulsive interaction partially arises from negatively charged side chains on the luminal loop of PsaA and cyt c6 as was shown for the interaction of PS I with PC (40) and as revealed by rigid body docking (Fig. 5). The screening effect is stronger for divalent cations than for monovalent ions of the same ionic strength (Fig. 1), suggesting that divalent cations can form a bridge between these side chains.
Previously, a costructure of PS I with cyt c6 was achieved by rigid body docking for the diatom Phaeodactylum tricornutum (31); here, the docking sites with the most negative energy result from interaction of cyt c6 with PsaF. However, the closest docking site is different and has less negative binding energy. In contrast to this diatom, cyt c6 from T. elongatus does not show the complex kinetics that can be explained with an additional docking site at PsaF (25, 26, 41). Indeed, the docking sites described in the present work that show short heme–P700 distances are found within the top 100 ranks with the most negative binding energies, and no binding site close to PsaF with a high binding energy can be identified. This is in agreement with the binding properties in most cyanobacteria (12, 42).
In contrast to cyanobacterial and algal PSs I, the cocrystal structure of the bRC with cyt c2 from Rhodobacter sphaeroides is known (34). bRCs are structurally homologous to cyanobacterial photosystems (43). Fig. 6 shows a comparison between the modeled PS I–cyt c6 complex and the cocrystal structure of the bRC–cyt c2 complex. Both complexes differ only in a small rotation of the cytochrome but have identical heme–P700/P870 distances. This suggests that the specific binding site, where the electron transfer occurs, diverged only slightly during evolution. The positioning of the heme group relative to the active center remains conserved, whereas the sequence identity of the amino acid residues on the protein surface is low.
Figure 6.
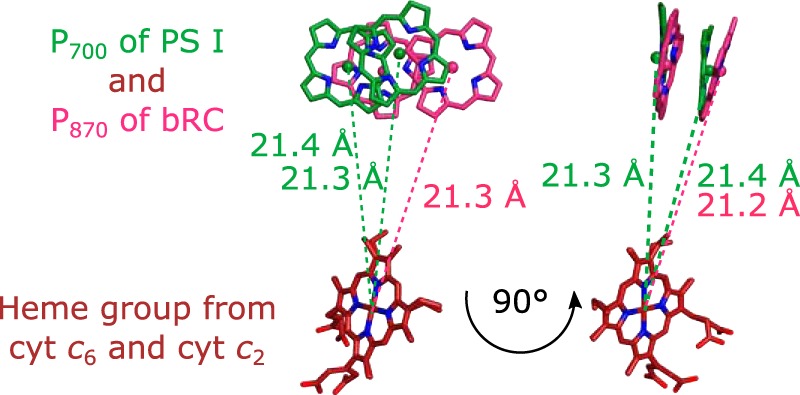
Superposition of the potential cyt c6–binding site to the known cyt c2–binding site of the bRC from R. sphaeroides (Protein Data Bank code 1l9b (34)). The superposition was achieved by aligning the heme groups. The right view is rotated by 90° with respect to the left view. The distances of the heme iron from cyt c6 to the Mg2+ ions of P700 are 21.4 and 21.3 Å, respectively. These distances are identical to the distances between the heme iron of cyt c2 and the Mg2+ ions of P870 (pink) from bRC (21.3 and 21.2 Å, respectively).
Conclusions and outlook
We analyzed the binding behavior of a native and a non-native cytochrome to PS I from the cyanobacterium T. elongatus. Although the highest turnover number was found for the cyt c6–PS I complex, the highest affinity was detected for cyt cHH. Both proteins show a very different dependence of the interaction with PS I on the ionic strength. For cyt cHH, this points to a mainly electrostatically determined mode of binding to the photoactive protein complex.
This information is not only of fundamental interest but can also be used to improve biotechnological applications. Because self-assembled photobioelectrodes often need low ionic strength, cyt cHH is well-suited as a mediator for the assembly of PS I. Other arguments for the use of cyt cHH are the high turnover number and the similar binding behavior of oxidized and reduced protein.
Theoretical modeling of cyt c–PS I interactions revealed docking sites for cyt c6 that highly resembles the native binding site of cyt c2 with bRC. In addition, the modeling provides a rationale for the inability to detect cyt cHH in cocrystals as it suggests a variety of binding sites. To improve the modeling with regard to pH dependence and accuracy of computed binding energies, future work will also consider the protonation states of titratable groups in the proteins that may be different from those assumed in the present work. Improved cocrystal structures will ultimately serve to understand the electron transfer reaction. The present data suggest that PS I should be cocrystallized with cyt cHH at higher cyt cHH concentration with low ionic strength at pH 6, which might be achieved by using an alternative precipitation agent such as PEG. First crystals diffracting at medium resolution have been obtained. Although cyt c6 binds to PS I at a conserved binding site and no unspecific binding occurs, the binding affinity of cyt c6 to PS I is weaker, and further investigations are needed to find suitable conditions for the cocrystallization. The present results serve as a guideline in this respect.
Experimental procedures
Chemicals and enzymes
All chemicals were of analytical grade or higher and purchased from Sigma-Aldrich. Cytochrome c from horse heart was purchased from Sigma-Aldrich with 95% purity for the majority of experiments and 99% purity for crystal structure analysis. The detergent n-dodecyl β-d-maltoside (DDM) was purchased from Glycon (Germany). The plasmid pEC86, harboring the genes for heme maturation, was kindly provided by L. Thöny-Meyer (44).
Isolation of proteins
Cultivation of T. elongatus and membrane protein extraction were performed as reported previously (45). For the purification of PS I, the protein extract was applied to two steps of anion exchange chromatography. In the first step, PS I was separated from PS II by a column packed with Toyo Pearl DEAE 650 S (GE Healthcare) equilibrated with buffer A (20 mm MES-NaOH, pH 6.0, 5% (v/v) glycerol, 20 mm CaCl2, 0.02% (w/v) DDM). After washing with buffer A containing 5 mm MgSO4, proteins were eluted with a linear gradient from 5 to 100 mm MgSO4 in buffer A. PS II was eluted at 30 mm MgSO4, whereas PS I was eluted at 55 mm MgSO4. The PS I fractions were pooled and diluted with buffer B (5 mm MES-NaOH, pH 6.0, 0.02% DDM) to a conductivity of 6 mS/cm and applied to a Q-SepharoseTM (GE Healthcare) column pre-equilibrated with buffer B containing 60 mm MgSO4. The column was washed with 2 column volumes of buffer B containing 60 mm MgSO4, and the proteins were eluted with a linear gradient of MgSO4 in buffer B. Trimeric PS I eluted at 150 mm MgSO4. The PS I trimer fractions were concentrated in an Amicon stirred filtration cell using a Biomax 100 membrane (Millipore, Germany) and crystallized by slowly diluting against buffer B (protocol modified from Ref. 21). The crystals were washed in buffer B, redissolved by addition of 30 mm MgSO4, and again crystallized as before. The concentration of the RC (equivalent to the concentration of P700) was determined by ϵ680 = 5.5 μm−1 cm−1, and the concentration of PS I–bound Chl a was determined by ϵ680 = 57.1 mm−1 cm−1 in 25 mm Tris-HCl, pH 8.0, 100 mm NaCl, 0.02% DDM (46).
Cloning and expression of cytochrome c6 in E. coli
The coding gene for cytochrome c6 from T. elongatus (tll1283) was amplified by PCR using the primers 5′-CTCGAGGCCTGCCCAACCCTT-3′ and 5′-CATATGGCTGACCTAGCCCATGGT-3′ containing restriction sites for NdeI and XhoI (underlined), respectively. Chromosomal DNA isolated from T. elongatus served as a template. The resulting PCR product was subcloned in pJET1.2 vector (Thermo Fisher, Germany) and verified by DNA sequencing (Services in Molecular Biology, Germany). Subsequently, cloning was performed in pET22b expression vector (Novagen, Germany) and transformed into E. coli BL21-Star strain. For maturation of cytochrome c6 in E. coli, the pEC86 vector was also introduced. For heterologous expression, cells were grown in 1 liter of LB medium containing 100 μg ml−1 ampicillin and 10 μg ml−1 chloramphenicol at 37 °C for 16 h. The addition of isopropyl 1-thio-β-d-galactopyranoside was not necessary. Harvested cells were incubated in 20% (w/v) sucrose, 1 mm EDTA, 25 mm Tris-HCl, pH 8.0, for 30 min on ice. Subsequently, the cells were centrifuged at 12,000 × g for 10 min at 4 °C. The cell pellet was resolubilized in cold 10 mm Tris-HCl, pH 8.0, containing 5 mm MgSO4 to isolate the periplasmatic proteins. After centrifugation (10,000 × g, 10 min, 4 °C), the supernatant was adjusted to buffer C (500 mm NaCl, 20 mm imidazole, 20 mm phosphate buffer, pH 7.5) and applied to a nickel-nitrilotriacetic acid column (Rotigarose-His/Ni, Carl Roth, Germany). The column was washed with 10 volumes of buffer C, and the protein was eluted with a linear gradient at 140 mm imidazole. The cyt c6–containing fractions were pooled and dialyzed against 1 mm sodium ascorbate in 25 mm Tricine-NaOH, pH 7.2. For further purification, cyt c6 was applied to Toyo Pearl DEAE 650 S (GE Healthcare) and washed with 5 column volumes of 25 mm Tricine-NaOH, pH 7.2, 10 mm NaCl. Cyt c6 was eluted with a linear gradient of 10–30 mm NaCl in 25 mm Tricine-NaOH, pH 7.2. Cyt c concentration was spectrophotometrically determined in the presence of 5 mm sodium ascorbate (ϵ550 = 29.5 mm−1 cm−1 for cyt cHH and ϵ553 = 25 mm−1 cm−1 for cyt c6 (42, 47)).
Polyacrylamide gel electrophoresis
Purity of the isolated cyt c6 was verified by SDS-PAGE with 15% polyacrylamide using 0.5–10 μg of protein according to Laemmli (48). PS I was analyzed by blue native PAGE with a polyacrylamide gradient from 3 to 9% according to Wittig et al. (49). PS I crystals corresponding to 5 μg of Chl were dissolved in solubilization buffer containing 0.2% DDM and 100 mm NaCl. The gel was destained by 10% acetic acid.
DLS
Homogeneity of purified trimeric PS I samples was verified by DLS. PS I crystals were dissolved in 25 mm Tricine-NaOH, pH 8.0, 100 mm NaCl, 0.02% DDM to a protein concentration between 5 and 10 μm P700 and filtered through a 0.45-μm membrane. Measurements were performed on a DynaPro NanoStar (Wyatt) with a 787 nm laser at 20 °C in a disposable 4-μl cuvette.
MS analysis
The subunit composition of PS I samples was analyzed by MALDI-TOF. 0.5 μl of 2 μm purified PS I was mixed with 0.5 μl of sinapinic acid in 40% (w/v) acetonitrile, 0.1% (v/v) TFA on the target. MALDI-TOF mass spectra were recorded on a Microflex spectrometer (Bruker, Germany) in linear, positive-ion mode.
PS I activity measurements
The oxygen reduction rate of PS I was measured with a Clark-type electrode (Oxygraph+, Hansatech, Germany). Except where stated, the standard reaction mixture contained 25 mm Tricine-NaOH, pH 8.0, 0.02% DDM, 2 mm sodium ascorbate, 300 μm methyl viologen with either 16 μm cyt cHH or 16 μm cyt c6 at 20 °C. The reaction mixture was stirred under illumination of >500 μmol of photons m−2 s−1 for 30 s. The reaction was started by addition of PS I (5 μg ml−1 chlorophyll), and the initial velocity of the reaction was determined. For the determination of Km and kcat, the data were analyzed in terms of Michaelis–Menten kinetics using cyt cHH or cyt c6 as substrate. All measurements were done with three different PS I preparations.
ITC
For the ITC measurements, PS I crystals were washed twice with H2O containing 0.02% DDM and subsequently dissolved to buffer D (25 mm Tricine-NaOH, pH 8.0, 0.02% DDM, 25 mm NaCl). To remove remaining PS I crystals in the solution, the sample was centrifuged for 5 min and filtered through a 0.45-μm membrane. Cyt cHH powder was dissolved in buffer D. Cyt c6 was dialyzed against buffer D in a centrifugal concentrator (5000 molecular weight cutoff) except for the 0.02% DDM, which was added after the final concentrating step. Experiments under reducing conditions were carried out by addition of 5 mm sodium ascorbate (final concentration) in the dark. Experiments were performed at 20 °C with a VP-ITC (MicroCal, Northampton, MA) in a 1.45-ml cell. Baseline subtraction was done by NITPIC 1.1.5 (50, 51), and data analysis was performed with Origin 7 software.
Cocrystallization of the PS I–cyt cHH supercomplex and X-ray diffraction analysis
For cocrystallization, 7.5 μm P700 was mixed with 37.5 μm cyt cHH in buffer B containing 40 mm MgSO4. Samples were dialyzed successively against buffer B containing 5, 3, and 2 mm MgSO4 in dialysis buttons (Hampton Research) with a 2000 molecular weight cutoff membrane (Carl Roth) at 20 °C. 200–500-μm-long, but thin and often hollow, needle-shaped crystals grew within 3–5 days and were used for microseeding. The crystals were crushed in a seed tool kit (Hampton Research) by vortexing for 5 min. The seed stock was centrifuged for 5 min at 16,000 × g, and the supernatant was diluted a 100-fold with buffer B. 1 μl of the diluted supernatant was added to 40 μl of 7.5 μm P700 with 37.5 μm cyt cHH in buffer B containing 3 mm MgSO4. Crystals grown overnight were needle-shaped with dimensions from 200 × 30 to 800 × 100 μm and diffracted up to 3.4-Å resolution. For cryoprotection, the buffer was exchanged against buffer B containing 0.25–1.75 m sucrose in 0.25 m steps with 5–10-min incubation time for each step and 2-h incubation time for the last step. Crystals were frozen in liquid nitrogen.
Diffraction data were collected from single crystals at beamline 14.1 at BESSY II electron storage ring operated by Helmholtz-Zentrum Berlin (Berlin-Adlershof, Germany) and at beamline 8.2.1 at the Advanced Light Source (Berkeley, CA) (52). Data were integrated by XDS and XDSAPP (53, 54). The model was based on the 2.5-Å resolution structure of PS I (Protein Data Bank code 1jb0) (5) and refined by iterative cycles of REFMAC5 (55) followed by hand building in Coot (56).
Analysis of cyt cHH content in PS I–cyt cHH cocrystals
The presence of cyt cHH in the cocrystals was qualitatively analyzed by MALDI-MS. Single PS I–cyt cHH cocrystals with sizes from 200 × 30 to 800 × 100 μm from different batches were selected and transferred to buffer B. Each crystal was washed six times by exchanging the supernatant against buffer B (dilution factor >10 for each step) and subsequently dissolved in buffer B containing 20 mm MgSO4. The supernatant from these crystallization batches was treated in the same manner as control samples. MS analysis was done as described above. Additionally, for one crystal, an X-ray diffraction data set at 3.5-Å resolution was measured, and the sucrose concentration in the crystal was successively reduced afterward by washing with 1.5, 1.0, 0.5, 0, and 0 m sucrose in buffer B for analysis by MALDI-TOF. The supernatant of the single crystals was exchanged for 10 μl of buffer B containing 10 mm MgSO4 for solubilization. 0.5 μl of each sample was measured as described under “MS analysis.”
Quantitative analysis of the cyt cHH content was carried out by washing a batch of crystals six times in buffer B, subsequently dissolving the batch in buffer B containing 100 mm MgSO4, and separating the cyt cHH from PS I using a centrifugal concentrator with a 100,000 molecular weight cutoff membrane (Sartorius Stedim, Germany). The PS I and cyt cHH concentrations were analyzed by their chlorophyll and heme absorption bands at 680 and 550 nm, respectively.
Protein–protein docking simulation
The cyt c–binding site of PS I was analyzed by rigid body docking using FTDock and rescoring by pyDock3 (23, 24). The crystal structure of cyt cHH (Protein Data Bank code 1hrc), the solution structure of cyt c6 (Protein Data Bank code 1c6s), and the crystal structure of PS I (Protein Data Bank code 1jb0) were prepared as topology files using tLEaP in AmberTools16 with the ff99SB force field, including force field information for the heme group. After FTDock sampling, a palmitoyloleolyphosphatidylglycerol membrane was simulated around PS I using CHARMM membrane builder (57). Cytochromes that have a center to membrane distance of less than their radius of gyration are considered to clash with the membrane and were therefore neglected.
The electrostatic binding energies of selected docking sites were recalculated using the Adaptive Poisson-Boltzmann Solver for different MgSO4 concentrations and pH values (APBS 1.4 (58)). The temperature was set to 293.15 K, the dielectric constant of the particles was set to 10, and that of water was set to 80.1. The Protein Data Bank files were prepared by pdb2pqr 2.1 (59). The electrostatic binding energy was calculated as follows.
| (Eq. 1) |
Author contributions
A. K. and A. Z. conceptualization; A. K., M. H., J. F. K., F. M., and A. Z. data curation; A. K., J. F. K., and A. Z. formal analysis; A. K., S. C. F., J. F. K., and F. M. validation; A. K., M. H., K. R. S., and S. C. F. investigation; A. K. and M. H. methodology; A. K., M. H., and A. Z. writing-original draft; F. M., F. L., and H. L. writing-review and editing; F. L., H. L., and A. Z. funding acquisition; F. L., H. L., and A. Z. project administration; A. Z. supervision.
Supplementary Material
Acknowledgments
We thank Prof. Holger Dobbek (Humboldt-Universität zu Berlin, Germany) for kindly providing access to ITC and Dr. Joerg Fettke (University of Potsdam, Germany) for kind access and support to the Bruker Microflex spectrometer. We thank Dr. Brian Jimenez-Garcia and Dr. Juan Fernandez-Recio for support with the docking calculations. We thank the beamline staff of the BESSY II electron storage ring (Berlin-Adlershof, Germany) and Martin Bommer (Humboldt-Universität zu Berlin, Germany) for support. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract DE-AC02-05CH11231.
This work was supported in part by the Bundesministerium für Bildung und Forschung (BMBF), Germany (Biotechnologie 2020+, Project 031A154B) and the Deutsche Forschungsgemeinschaft through the cluster of excellence “Unifying Concepts in Catalysis” coordinated by the Technische Universität Berlin (Project E2/E3). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S12, Tables S1–S5, and supporting theoretical models.
- PS I
- photosystem I
- bRC
- bacterial reaction center
- Chl
- chlorophyll
- cyt cHH
- cytochrome c from horse heart
- cyt c
- cytochrome c
- DDM
- dodecyl β-d-maltoside
- DLS
- dynamic light scattering
- ITC
- isothermal titration calorimetry
- RC
- reaction center
- Trp-A655 and Trp-B632
- tryptophan 655 and 632 from PsaA and PsaB, respectively
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- PC
- plastocyanin
- mS
- millisiemens.
References
- 1. Stieger K. R., Feifel S. C., Lokstein H., Hejazi M., Zouni A., and Lisdat F. (2016) Biohybrid architectures for efficient light-to-current conversion based on photosystem I within scalable 3D mesoporous electrodes. J. Mater. Chem. A 4, 17009–17017 10.1039/C6TA07141D [DOI] [Google Scholar]
- 2. Stieger K. R., Ciornii D., Kölsch A., Hejazi M., Lokstein H., Feifel S. C., Zouni A., and Lisdat F. (2016) Engineering of supramolecular photoactive protein architectures: the defined co-assembly of photosystem I and cytochrome c using a nanoscaled DNA-matrix. Nanoscale 8, 10695–10705 10.1039/C6NR00097E [DOI] [PubMed] [Google Scholar]
- 3. Lubner C. E., Applegate A. M., Knörzer P., Ganago A., Bryant D. A., Happe T., and Golbeck J. H. (2011) Solar hydrogen-producing bionanodevice outperforms natural photosynthesis. Proc. Natl. Acad. Sci. 108, 20988–20991 10.1073/pnas.1114660108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nowaczyk M. M., and Plumeré N. (2016) Short circuit at the chlorophyll. Nat. Chem. Biol. 12, 990–991 10.1038/nchembio.2240 [DOI] [PubMed] [Google Scholar]
- 5. Jordan P., Fromme P., Witt H. T., Klukas O., Saenger W., and Krauss N. (2001) Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature 411, 909–917 10.1038/35082000 [DOI] [PubMed] [Google Scholar]
- 6. Mazor Y., Borovikova A., Caspy I., and Nelson N. (2017) Structure of the plant photosystem I supercomplex at 2.6 Å resolution. Nat. Plants 3, 17014 10.1038/nplants.2017.14 [DOI] [PubMed] [Google Scholar]
- 7. Guergova-Kuras M., Boudreaux B., Joliot A., Joliot P., and Redding K. (2001) Evidence for two active branches for electron transfer in photosystem I. Proc. Natl. Acad. Sci. U.S.A. 98, 4437–4442 10.1073/pnas.081078898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wood P. M. (1978) Interchangeable copper and iron proteins in algal photosynthesis. Eur. J. Biochem. 87, 9–19 10.1111/j.1432-1033.1978.tb12346.x [DOI] [PubMed] [Google Scholar]
- 9. Nakamura Y., Kaneko T., Sato S., Ikeuchi M., Katoh H., Sasamoto S., Watanabe A., Iriguchi M., Kawashima K., Kimura T., Kishida Y., Kiyokawa C., Kohara M., Matsumoto M., Matsuno A., et al. (2002) Complete genome structure of the thermophilic cyanobacterium Thermosynechococcus elongatus BP-1. DNA Res. 9, 123–130 10.1093/dnares/9.4.123 [DOI] [PubMed] [Google Scholar]
- 10. Sommer F., Drepper F., and Hippler M. (2002) The luminal helix l of PsaB is essential for recognition of plastocyanin or cytochrome c6 and fast electron transfer to photosystem I in Chlamydomonas reinhardtii. J. Biol. Chem. 277, 6573–6581 10.1074/jbc.M110633200 [DOI] [PubMed] [Google Scholar]
- 11. Hervás M., De la Rosa M., and Tollin G. (1992) A comparative laser-flash absorption spectroscopy study of algal plastocyanin and cytochrome c552 photooxidation by photosystem I particles from spinach. Eur. J. Biochem. 203, 115–120 10.1111/j.1432-1033.1992.tb19835.x [DOI] [PubMed] [Google Scholar]
- 12. Hervás M., Navarro J. A., Díaz A., Bottin H., and De la Rosa M. A. (1995) Laser-flash kinetic analysis of the fast electron transfer from plastocyanin and cytochrome c6 to photosystem I. Experimental evidence on the evolution of the reaction mechanism. Biochemistry 34, 11321–11326 10.1021/bi00036a004 [DOI] [PubMed] [Google Scholar]
- 13. Haehnel W., Ratajczak R., and Robenek H. (1989) Lateral distribution and diffusion of plastocyanin in chloroplast thylakoids. J. Cell Biol. 108, 1397–1405 10.1083/jcb.108.4.1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Díaz-Moreno I., Díaz-Quintana A., Molina-Heredia F. P., Nieto P. M., Hansson O., De la Rosa M.A., and Karlsson B. G. (2005) NMR analysis of the transient complex between membrane photosystem I and soluble cytochrome c6. J. Biol. Chem. 280, 7925–7931 10.1074/jbc.M412422200 [DOI] [PubMed] [Google Scholar]
- 15. Utschig L. M., Silver S. C., Mulfort K. L., and Tiede D. M. (2011) Nature-driven photochemistry for catalytic solar hydrogen production: a photosystem I–transition metal catalyst hybrid. J. Am. Chem. Soc. 133, 16334–16337 10.1021/ja206012r [DOI] [PubMed] [Google Scholar]
- 16. Stieger K. R., Feifel S. C., Lokstein H., and Lisdat F. (2014) Advanced unidirectional photocurrent generation via cytochrome c as reaction partner for directed assembly of photosystem I. Phys. Chem. Chem. Phys. 16, 15667–15674 10.1039/C4CP00935E [DOI] [PubMed] [Google Scholar]
- 17. Golub M., Hejazi M., Kölsch A., Lokstein H., Wieland D. C. F., Zouni A., and Pieper J. (2017) Solution structure of monomeric and trimeric photosystem I of Thermosynechococcus elongatus investigated by small-angle X-ray scattering. Photosynth. Res. 133, 163–173 10.1007/s11120-017-0342-6 [DOI] [PubMed] [Google Scholar]
- 18. Mukherjee D., May M., and Khomami B. (2011) Detergent–protein interactions in aqueous buffer suspensions of Photosystem I (PS I). J. Colloid Interface Sci. 358, 477–484 10.1016/j.jcis.2011.03.070 [DOI] [PubMed] [Google Scholar]
- 19. El-Mohsnawy E., Kopczak M. J., Schlodder E., Nowaczyk M., Meyer H. E., Warscheid B., Karapetyan N. V., and Rögner M. (2010) Structure and function of intact photosystem 1 monomers from the cyanobacterium Thermosynechococcus elongatus. Biochemistry 49, 4740–4751 10.1021/bi901807p [DOI] [PubMed] [Google Scholar]
- 20. Kramer D. M., Sacksteder C. A., and Cruz J. A. (1999) How acidic is the lumen? Photosynth. Res. 60, 151–163 10.1023/A:1006212014787 [DOI] [Google Scholar]
- 21. Krauβ N., Schubert W.-D., Klukas O., Fromme P., Witt H. T., and Saenger W. (1996) Photosystem I at 4 Å resolution represents the first structural model of a joint photosynthetic reaction centre and core antenna system. Nat. Struct. Mol. Biol. 3, 965–973 10.1038/nsb1196-965 [DOI] [PubMed] [Google Scholar]
- 22. Fromme P., and Witt H. T. (1998) Improved isolation and crystallization of photosystem I for structural analysis. Biochim. Biophys. Acta 1365, 175–184 10.1016/S0005-2728(98)00059-0 [DOI] [Google Scholar]
- 23. Gabb H. A., Jackson R. M., and Sternberg M. J. (1997) Modelling protein docking using shape complementarity, electrostatics and biochemical information1. J. Mol. Biol. 272, 106–120 10.1006/jmbi.1997.1203 [DOI] [PubMed] [Google Scholar]
- 24. Cheng T. M., Blundell T. L., and Fernandez-Recio J. (2007) pyDock: electrostatics and desolvation for effective scoring of rigid-body protein–protein docking. Proteins 68, 503–515 10.1002/prot.21419 [DOI] [PubMed] [Google Scholar]
- 25. Hatanaka H., Sonoike K., Hirano M., and Katoh S. (1993) Small subunits of photosystem I reaction center complexes from Synechococcus elongatus. I. Is the psaF gene product required for oxidation of cytochrome c-553? Biochim. Biophys. Acta 1141, 45–51 10.1016/0005-2728(93)90187-K [DOI] [PubMed] [Google Scholar]
- 26. Nguyen K., Vaughn M., Frymier P., and Bruce B. D. (2017) In vitro kinetics of P700+ reduction of Thermosynechococcus elongatus trimeric photosystem I complexes by recombinant cytochrome c6 using a Joliot-type LED spectrophotometer. Photosynth. Res. 131, 79–91 10.1007/s11120-016-0300-8 [DOI] [PubMed] [Google Scholar]
- 27. Bjellqvist B., Hughes G. J., Pasquali C., Paquet N., Ravier F., Sanchez J.-C., Frutiger S., and Hochstrasser D. (1993) The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis 14, 1023–1031 10.1002/elps.11501401163 [DOI] [PubMed] [Google Scholar]
- 28. Malmgren L., Olsson Y., Olsson T., and Kristensson K. (1978) Uptake and retrograde axonal transport of various exogenous macromolecules in normal and crushed hypoglossal nerves. Brain Res. 153, 477–493 10.1016/0006-8993(78)90333-5 [DOI] [PubMed] [Google Scholar]
- 29. Drepper F., Hippler M., Nitschke W., and Haehnel W. (1996) Binding dynamics and electron transfer between plastocyanin and photosystem I. Biochemistry 35, 1282–1295 10.1021/bi951471e [DOI] [PubMed] [Google Scholar]
- 30. Zygadlo A., Jensen P. E., Leister D., and Scheller H. V. (2005) Photosystem I lacking the PSI-G subunit has a higher affinity for plastocyanin and is sensitive to photodamage. Biochim. Biophys. Acta 1708, 154–163 10.1016/j.bbabio.2005.02.003 [DOI] [PubMed] [Google Scholar]
- 31. Bernal-Bayard P., Pallara C., Carmen Castell M., Molina-Heredia F. P., Fernández-Recio J., Hervás M., and Navarro J. A. (2015) Interaction of photosystem I from Phaeodactylum tricornutum with plastocyanins as compared with its native cytochrome c6: reunion with a lost donor. Biochim. Biophys. Acta 1847, 1549–1559 10.1016/j.bbabio.2015.09.006 [DOI] [PubMed] [Google Scholar]
- 32. van Thor J. J., Geerlings T. H., Matthijs H. C., and Hellingwerf K. J. (1999) Kinetic evidence for the PsaE-dependent transient ternary complex photosystem I/ferredoxin/ferredoxin:NADP+ reductase in a cyanobacterium. Biochemistry 38, 12735–12746 10.1021/bi9903502 [DOI] [PubMed] [Google Scholar]
- 33. Moser C. C., and Dutton P. L. (1988) Cytochrome c and c2 binding dynamics and electron transfer with photosynthetic reaction center protein and other integral membrane redox proteins. Biochemistry 27, 2450–2461 10.1021/bi00407a031 [DOI] [PubMed] [Google Scholar]
- 34. Axelrod H. L., Abresch E. C., Okamura M. Y., Yeh A. P., Rees D. C., and Feher G. (2002) X-ray structure determination of the cytochrome c2: reaction center electron transfer complex from Rhodobacter sphaeroides. J. Mol. Biol. 319, 501–515 10.1016/S0022-2836(02)00168-7 [DOI] [PubMed] [Google Scholar]
- 35. Pettigrew G. W., Goodhew C. F., Cooper A., Nutley M., Jumel K., and Harding S. E. (2003) The electron transfer complexes of cytochrome c peroxidase from Paracoccus denitrificans. Biochemistry 42, 2046–2055 10.1021/bi027125w [DOI] [PubMed] [Google Scholar]
- 36. Shimada S., Shinzawa-Itoh K., Baba J., Aoe S., Shimada A., Yamashita E., Kang J., Tateno M., Yoshikawa S., and Tsukihara T. (2016) Complex structure of cytochrome c–cytochrome c oxidase reveals a novel protein–protein interaction mode. EMBO J. 10.15252/embj.201695021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Poulos T. L., Sheriff S., and Howard A. J. (1987) Cocrystals of yeast cytochrome c peroxidase and horse heart cytochrome c. J. Biol. Chem. 262, 13881–13884 [PubMed] [Google Scholar]
- 38. Pelletier H., and Kraut J. (1992) Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science 258, 1748–1755 10.1126/science.1334573 [DOI] [PubMed] [Google Scholar]
- 39. Sommer F., Drepper F., Haehnel W., and Hippler M. (2004) The hydrophobic recognition site formed by residues PsaA-Trp651 and PsaB-Trp627 of photosystem I in Chlamydomonas reinhardtii confers distinct selectivity for binding of plastocyanin and cytochrome c6. J. Biol. Chem. 279, 20009–20017 10.1074/jbc.M313986200 [DOI] [PubMed] [Google Scholar]
- 40. Navarro J. A., Hervás M., Sun J., De la Cerda B., Chitnis P. R., and De la Rosa M. A. (2000) Negatively charged residues in the H loop of PsaB subunit in photosystem I from Synechocystis sp. PCC 6803 appear to be responsible for electrostatic repulsions with plastocyanin. Photosynth. Res. 65, 63–68 10.1023/A:1006404621724 [DOI] [PubMed] [Google Scholar]
- 41. Proux-Delrouyre V., Demaille C., Leibl W., Sétif P., Bottin H., and Bourdillon C. (2003) Electrocatalytic investigation of light-induced electron transfer between cytochrome c6 and photosystem I. J. Am. Chem. Soc. 125, 13686–13692 10.1021/ja0363819 [DOI] [PubMed] [Google Scholar]
- 42. Hervás M., Ortega J. M., Navarro J. A., De la Rosa M. A., and Bottin H. (1994) Laser flash kinetic analysis of Synechocystis PCC 6803 cytochrome c6 and plastocyanin oxidation by photosystem I. Biochim. Biophys. Acta 1184, 235–241 10.1016/0005-2728(94)90228-3 [DOI] [Google Scholar]
- 43. Fischer W. W., Hemp J., and Johnson J. E. (2016) Evolution of oxygenic photosynthesis. Annu. Rev. Earth Planet. Sci. 44, 647–683 10.1146/annurev-earth-060313-054810 [DOI] [Google Scholar]
- 44. Arslan E., Schulz H., Zufferey R., Künzler P., and Thöny-Meyer L. (1998) Overproduction of the Bradyrhizobium japonicum c-type cytochrome subunits of the cbb3 oxidase in Escherichia coli. Biochem. Biophys. Res. Commun. 251, 744–747 10.1006/bbrc.1998.9549 [DOI] [PubMed] [Google Scholar]
- 45. Kern J., Loll B., Lüneberg C., DiFiore D., Biesiadka J., Irrgang K.-D., and Zouni A. (2005) Purification, characterisation and crystallisation of photosystem II from Thermosynechococcus elongatus cultivated in a new type of photobioreactor. Biochim. Biophys. Acta 1706, 147–157 10.1016/j.bbabio.2004.10.007 [DOI] [PubMed] [Google Scholar]
- 46. Müh F., and Zouni A. (2005) Extinction coefficients and critical solubilisation concentrations of photosystems I and II from Thermosynechococcus elongatus. Biochim. Biophys. Acta 1708, 219–228 10.1016/j.bbabio.2005.03.005 [DOI] [PubMed] [Google Scholar]
- 47. van Gelder B. F., and Slater E. C. (1962) The extinction coefficient of cytochrome c. Biochim. Biophys. Acta 58, 593–595 10.1016/0006-3002(62)90073-2 [DOI] [PubMed] [Google Scholar]
- 48. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 49. Wittig I., Braun H.-P., and Schägger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 10.1038/nprot.2006.62 [DOI] [PubMed] [Google Scholar]
- 50. Keller S., Vargas C., Zhao H., Piszczek G., Brautigam C. A., and Schuck P. (2012) High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 84, 5066–5073 10.1021/ac3007522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Scheuermann T. H., and Brautigam C. A. (2015) High-precision, automated integration of multiple isothermal titration calorimetric thermograms: new features of NITPIC. Methods 76, 87–98 10.1016/j.ymeth.2014.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mueller U., Darowski N., Fuchs M. R., Förster R., Hellmig M., Paithankar K. S., Pühringer S., Steffien M., Zocher G., and Weiss M. S. (2012) Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 19, 442–449 10.1107/S0909049512006395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krug M., Weiss M. S., Heinemann U., and Mueller U. (2012) XDSAPP: a graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Crystallogr. 45, 568–572 10.1107/S0021889812011715 [DOI] [Google Scholar]
- 55. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jo S., Kim T., Iyer V. G., and Im W. (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 10.1002/jcc.20945 [DOI] [PubMed] [Google Scholar]
- 58. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 10.1093/nar/gkh381 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.