

Abstract
C-C motif chemokine receptor 5 (CCR5) is a cell surface–associated, immune-regulatory G protein–coupled receptor (GCPR) with seven transmembrane helices. We previously reported the isolation and initial characterization of short artificial transmembrane protein aptamers, named “traptamers,” that specifically down-regulate CCR5 expression and inhibit infection of human T cells by HIV strains that use CCR5 as a co-receptor. Here, we investigated the mechanism of traptamer-mediated CCR5 down-regulation and show that most of the traptamers (designated class 1 traptamers) form a stable complex with CCR5 and target it for lysosome-mediated degradation. The ability of these traptamers to down-regulate CCR5 depended on Lys197 in the fifth transmembrane helix of CCR5. In the absence of traptamers, substitution of Lys197 to an uncharged amino acid increased CCR5 stability, and introduction of a lysine at the homologous position in CCR2b, a related chemokine receptor, decreased CCR2b levels. The prototypic class 2 traptamer BY6M4 also formed a complex with CCR5, but CCR5 down-regulation caused by class 2 traptamers did not depend on the lysosome or on Lys197. These results demonstrate that traptamers use diverse mechanisms to down-regulate CCR5 and identify a specific amino acid that plays a central role in controlling chemokine receptor stability. Further studies of these traptamers are likely to provide new insights into CCR5 metabolism and biology and may suggest new therapeutic approaches to modulate the levels of CCR5 and other GPCRs.
Keywords: C-C chemokine receptor type 5 (CCR5), G protein-coupled receptor (GPCR), lysosome, chemokine, human immunodeficiency virus (HIV), HIV co-receptor, CCR2b, E5 protein, proximity ligation assay, transmembrane protein, traptamer
Introduction
G protein–coupled receptors (GPCRs)5 are a large family of cell-surface proteins that regulate many intracellular processes. An important GPCR is the C-C chemokine receptor, CCR5, a cell-surface protein with seven hydrophobic transmembrane helices (1). CCR5 binds to several chemokine agonists to coordinate immune cell migration and activation. CCR5 is also the primary co-receptor for HIV during sexual transmission of the virus, although other chemokine receptors, such as CXCR4, can also serve as HIV co-receptors (2). Understanding CCR5 biogenesis and metabolism is likely to inform novel approaches to target this and related receptors to regulate immune function, inhibit HIV infection, and modulate other processes controlled by GPCRs.
During synthesis and trafficking to the cell surface, CCR5 is modified by tyrosine sulfation and O-linked glycosylation in its N-terminal extracellular segment and by palmitoylation of three cysteines in its cytoplasmic C-terminal tail (1, 3, 4). Upon ligand binding, CCR5 is internalized through a process involving phosphorylation of serine residues in the C terminus of CCR5 and an Asp-Arg-Tyr (DRY) motif in the second intracellular loop, followed by binding to β-arrestin and clathrin-mediated endocytosis (5, 6). Internalized CCR5 localizes to recycling endosomes as well as the trans-Golgi network and is rapidly recycled to the cell surface (1, 7–12). Palmitoylation-defective cysteine-to-alanine CCR5 mutants display rapid turnover, reduced cell-surface expression, and impaired internalization in response to ligand (1, 4, 13–15). Lysosome function has not been implicated in CCR5 down-regulation (16), but basal turnover of CCR5 may involve the lysosome, as total CCR5 levels can be modestly increased by lysosomal protease inhibitors, and palmitoylation-deficient CCR5 mutants are subject to lysosomal degradation (15). In contrast, after ligand binding, CXCR4 undergoes mono-ubiquitination in a serine- and lysine-rich C-terminal segment, which directs it to the lysosome, where it is degraded (17, 18). CCR5 lacks this ubiquitination signal.
We previously described the construction and isolation of several 44- or 45-residue artificial transmembrane proteins, named traptamers (for “transmembrane protein aptamers”), that specifically down-regulate expression of CCR5 in murine BaF3 cells and human T cells (Fig. S1) (19). We infected BaF3 cells expressing human CCR5 with retrovirus libraries expressing hundreds of thousands of short, single-pass transmembrane proteins with randomized transmembrane segments and used FACS to isolate traptamers that reduced cell-surface CCR5 expression. These traptamers inhibit infection of T cells by R5-tropic strains of HIV, which use CCR5 as a co-receptor for entry, but do not down-regulate CXCR4 or inhibit X4-tropic HIV. The transmembrane sequences of the traptamers are unrelated to one another, and the mechanism of traptamer-mediated CCR5 down-regulation has not been investigated in detail. The active traptamers do not affect the level of CCR5 mRNA. Most of the traptamers do not cause down-regulation of a chimeric receptor in which the fifth transmembrane helix of CCR5, TM5, is replaced with the corresponding segment from the closely related C-C chemokine receptor CCR2b. In contrast, traptamer BY6 and its derivative BY6M4 down-regulate CCR2b and the TM5 chimeric receptor, suggesting that BY6 and BY6M4 may have a mechanism of action different from that of the other traptamers.
Here, we investigate the mechanism of traptamer-mediated down-regulation of CCR5. We show that most traptamers down-regulate CCR5 by targeting it to the lysosome, where it is degraded, whereas BY6 and BY6M4 instead induce lysosome-independent down-regulation of CCR5. Our studies also identified a specific lysine residue in CCR5 that is required for traptamer-mediated lysosomal degradation of CCR5. Interestingly, the charge at this position also helps set the steady-state level of C-C chemokine receptor expression in the absence of traptamers. These results demonstrate that traptamers utilize diverse mechanisms to down-regulate CCR5 and identify a novel mechanism of traptamer-induced, lysosome-dependent CCR5 down-regulation. In addition, this work provides insight into the normal metabolism of CCR5 and related receptors. Further studies of these unusual artificial proteins are likely to provide additional insights into the biology of CCR5 and other GPCRs.
Results
Traptamer BY1PC2 destabilizes CCR5
We studied the mechanism of traptamer-mediated CCR5 down-regulation primarily in murine BaF3 cells engineered to express human CCR5 (BaF3/CCR5 cells), because down-regulation was more pronounced in these cells than in human T cells (19). We focused on BY1PC2 and BY6M4, which we isolated by introducing random mutations into the original isolates, BY1 and BY6, and screening by FACS to identify optimized traptamer mutants with enhanced ability to down-regulate CCR5. We previously showed that traptamers do not affect the levels of CCR5 mRNA as assessed by quantitative RT-PCR (19). Here, we performed Northern blotting to rule out major structural changes in CCR5 mRNA in cells expressing the traptamers. Total RNA was prepared from parental BaF3 cells and from BaF3/CCR5 cells transduced with an empty retrovirus vector, a vector expressing an inactive traptamer that does not down-regulate CCR5 (US7), or a vector expressing an active traptamer (BY1PC2 or BY6M4). The RNA was electrophoresed and probed for CCR5 sequences. As shown in Fig. S2, similar amounts of full-length CCR5 mRNA were present in control BaF3/CCR5 cells and cells expressing active or inactive traptamers. These results confirm that these traptamers do not affect transcription of the CCR5 gene, destabilize CCR5 mRNA, or cause obvious alterations in CCR5 mRNA processing.
We performed a pulse-chase experiment to determine whether the traptamers caused CCR5 to be rapidly degraded. BaF3/CCR5 cells harboring an empty retrovirus vector or expressing BY1PC2 or BY6M4 were starved of amino acids for 1 h and then metabolically labeled with a 30-min pulse of medium containing [35S]methionine and [35S]cysteine. The cells were harvested immediately after labeling or incubated in the presence of excess unlabeled methionine and cysteine for up to 6 h. Detergent extracts were prepared at the end of the labeling period and at various times during the chase, immunoprecipitated with an anti-CCR5 antibody, subjected to electrophoresis, and examined by autoradiography. As shown in Fig. 1a, immediately following pulse labeling (time 0), two forms of CCR5 were present in control cells. The more rapidly migrating form (labeled p) represents the unmodified primary translation product, whereas the slower migrating form (labeled g) is glycosylated (3, 20). After a 30-min chase, the unmodified form disappeared, but the glycosylated form persisted, suggesting that the unmodified precursor is chased into the glycosylated form during this short time period. With increasing length of the chase period, the glycosylated form displayed a further minor decrease in mobility (labeled f) and then decayed with a half-life of >6 h. In cells expressing BY1PC2, CCR5 detected at the end of the pulse appeared to be the same as in control cells, and the level of the unmodified form of CCR5 was similarly reduced during the 30-min chase. However, during longer chase periods, the glycosylated form of CCR5 showed a markedly shorter half-life (∼2.5 h based on replicate experiments) and was nearly completely absent after a 6-h chase. In addition, the glycosylated form of CCR5 did not display the slight mobility decrease observed in control cells, indicating that CCR5 did not undergo complete modification in cells expressing BY1PC2. These results demonstrated that BY1PC2 accelerated the degradation of CCR5 by engaging CCR5 before it was completely modified.
Figure 1.
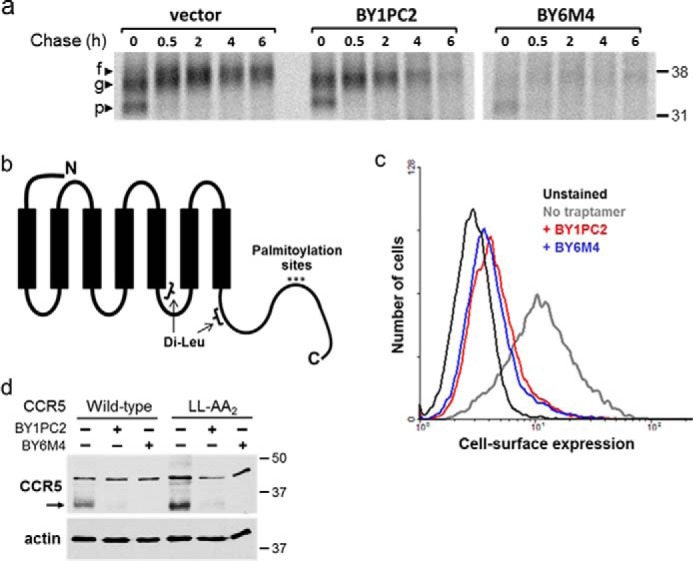
Traptamer BY1PC2 decreases the half-life of CCR5. a, BaF3/CCR5 cells expressing empty vector, BY1PC2 or BY6M4 were incubated for 30 min in medium containing [35S]methionine and [35S]cysteine and then chased with unlabeled medium for the indicated times. Cells were then lysed and immunoprecipitated with R22/7 anti-CCR5 antibody. Immunoprecipitates were subjected to SDS-PAGE followed by autoradiography. Numbers to the right indicate the size in kilodaltons of molecular mass standards. The primary translation product (p), the major glycosylated form (g), and the final fully modified form (f) are indicated. b, schematic of CCR5 showing the position of the dileucine motifs and the three C-terminal cysteines that undergo palmitoylation. The N terminus of CCR5 is extracellular, and the C terminus is intracellular. c, mutant CCR5 in which the three C-terminal cysteines were substituted with alanines was expressed in BaF3 cells in the absence of a traptamer or in the presence of BY1PC2 or BY6M4, as indicated. Cell-surface expression of the mutants was assessed by flow cytometry of nonpermeabilized cells stained with 2D7 anti-CCR5 antibody. d, WT or mutant CCR5 containing leucine-to-alanine substitutions at both of the dileucine repeats (LL-AA2) was expressed in the absence (−) or presence (+) of BY1PC2 or BY6M4 in BaF3 cells, as indicated. Western blotting was performed to determine total CCR5 levels (top). The filters were then stripped and reprobed for actin as a loading control (bottom). In this and subsequent figures, the main band of mature CCR5 is indicated with an arrow. The numbers to the right indicate the size in kilodaltons of molecular mass standards.
BY6M4 had a different effect on CCR5. In cells expressing BY6M4, very little CCR5 was detected at the end of the pulse or at any time thereafter, suggesting that BY6M4 impaired proper translation of CCR5 mRNA or caused the primary translation product to be degraded at an extremely rapid rate. Thus, BY1PC2 and BY6M4 have different effects on CCR5 expression, implying that they are likely to use distinct pathways to mediate CCR5 down-regulation. Additional differences between BY6M4 and the other traptamers are described below. Based on these differences, we designate BY1PC2 and traptamers with similar properties as class 1 traptamers and designate BY6 and its derivative BY6M4 as class 2 traptamers.
Traptamer-mediated CCR5 down-regulation does not require palmitoylation sites or dileucine motifs in CCR5
Palmitoylation of three C-terminal cysteines of CCR5 (Fig. 1b) is involved in CCR5 internalization in response to ligand (4, 13). To determine whether palmitoylation is required for traptamer-mediated CCR5 down-regulation, we mutated these three cysteines to alanines and tested the response of the triple mutant to the traptamers. This CCR5 mutant was expressed on the surface of control BaF3 cells and was markedly down-regulated from the cell surface by both BY1PC2 and BY6M4, as assessed by flow cytometry (Fig. 1c). Thus, the sites of CCR5 palmitoylation are not required for the action of either class 1 or class 2 traptamers.
CCR5 also contains consecutive leucine residues in two of its intracellular domains (positions 221/222 and 308/309; Fig. 1d), which resemble dileucine motifs implicated in internalization and lysosomal degradation of some integral membrane proteins (reviewed in Ref. 21). One of these motifs (positions 308/309) was shown to contribute to CCR5 internalization in response to ligand but did not affect basal CCR5 levels (4). To test the importance of these motifs in traptamer-mediated CCR5 down-regulation, we constructed a CCR5 mutant (LL-AA2) in which both pairs of leucines were mutated to alanines and expressed this mutant in BaF3 cells. Immunoblotting showed that WT CCR5 and the dileucine mutant were significantly down-regulated by BY1PC2 and BY6M4 (Fig. 1d). Thus, these dileucine motifs are not required for traptamer-mediated CCR5 down-regulation.
Lysosomal protease inhibitors restore CCR5 levels in response to class 1 traptamers
Because BY1PC2 decreased the half-life of CCR5, we used immunoblotting to test whether inhibitors of various proteolytic processes affected CCR5 levels in cells expressing the traptamers. BaF3 cells expressing CCR5 alone and cells co-expressing CCR5 plus various traptamers were left untreated or treated with chloroquine or ammonium chloride (NH4Cl), both of which are inhibitors of lysosomal acidification; with 3-methyladenine (3-MA), an inhibitor of autophagy; or with MG132, a proteasome inhibitor. Detergent extracts were prepared from the cells and subjected to SDS-PAGE and Western blotting to determine levels of total CCR5. As shown in Fig. 2a, in the absence of the chemical inhibitors, the traptamers markedly reduced CCR5 levels, as expected (e.g. compare lanes 1, 10, and 13 with lanes 7 and 16). Notably, the inhibitors of lysosomal acidification, chloroquine and ammonium chloride, caused a dramatic increase in CCR5 expression in cells expressing BY1PC2 and BY5 and a modest increase in cells expressing BY3 (e.g. compare lanes 2 and 3 with lane 1). Ammonium chloride also increased expression of CCR5 in cells expressing BY2 and BY4 (Fig. S3a), which, together with BY1PC2, BY3, and BY5, comprise the class 1 traptamers. Additional lysosomal inhibitors, namely bafilomycin A1 and E64d, also increased CCR5 expression in cells expressing BY1PC2 (Fig. 2b). E64d is a membrane-permeable inhibitor of cysteine proteases, not an inhibitor of lysosomal acidification, like the other lysosomolytic agents tested. We also note that five different conformation-specific anti-CCR5 antibodies stained abundant CCR5 in permeabilized, chloroquine-treated cells expressing BY1PC2 or BY5, indicating that these class 1 traptamers did not cause gross misfolding of CCR5 (Fig. S4). In striking contrast, lysosome inhibitors did not increase CCR5 levels in cells expressing BY6 or BY6M4 (Fig. 2a (lanes 14 and 15) and data not shown), demonstrating another difference between BY6M4 and BY1PC2. In contrast to the lysosome inhibitors, inhibitors of proteasomal degradation (MG132) or autophagy (3-MA) did not increase CCR5 expression in cells expressing BY1PC2 or BY6M4 (Fig. 2, c and d), although immunoblotting for the proteasome substrate β-catenin confirmed the efficacy of MG132 in these cells. In fact, we noted that MG132 reproducibly caused a decrease in CCR5 levels in cells with or without traptamers (Fig. 2c). The basis for this effect is not known. Flow cytometry of permeabilized cells confirmed that BY6M4-mediated CCR5 down-regulation was not reversed by chloroquine or MG132 (Fig. S3b). Thus, in BaF3 cells, lysosomal inhibitors reversed the down-regulation of CCR5 by the class 1 but not class 2 traptamers, whereas proteasome and autophagy inhibitors did not reverse CCR5 down-regulation by either class of traptamer. As was the case for human CCR5 in murine BaF3 cells, NH4Cl treatment restored the levels of CCR5 in human CEM.NKR-CCR5 T cells expressing BY1PC2 but not in T cells expressing BY6M4 (Fig. 2e).
Figure 2.

Lysosomal inhibitors increase CCR5 levels in cells expressing class 1 traptamers. a, BaF3/CCR5 cells expressing the empty retroviral vector or the indicated traptamer were untreated (−) or treated (+) with 25 μm chloroquine (CQ) or 10 mm ammonium chloride (NH4Cl) for 24 h, as indicated. Cells were then lysed, and lysates were subjected to SDS-PAGE and Western blotting for CCR5. The filters were then stripped and reprobed for actin as a loading control. b, BaF3/CCR5 cells expressing the empty retroviral vector or BY1PC2 were untreated or treated for 16 h with 25 μm chloroquine, 20 nm bafilomycin A1 (BafA1), or 25 μm E64d and then processed as in a. c and d, BaF3/CCR5 cells expressing vector, BY1PC2, or BY6M4, as indicated, were treated with vehicle or with 4 μm MG132 for 6 h (c) or 2 mm 3-MA for 24 h (d). Cells were then processed as described in a. e, MSCV vector only, BY1PC2, or BY6M4 was expressed in CEM.NKR-CCR5 T cells. Cells were left untreated or treated for 25.5 h with 25 or 30 mm NH4Cl and then processed as described in a. In all panels, the size in kilodaltons of molecular mass standards is shown.
We also used immunofluorescence to determine the effect of chloroquine on CCR5 down-regulation by the class 1 traptamers. BaF3 cells expressing CCR5 and traptamers in various combinations were treated with chloroquine or left untreated. Cells were then spun onto glass slides, permeabilized, and examined by indirect immunofluorescence with an antibody recognizing CCR5. As shown in the top row of Fig. 3a, no CCR5 was detectable in cells lacking the CCR5 gene. In BaF3/CCR5 cells lacking traptamer expression, CCR5 was readily detectable in cells not treated with chloroquine, and there was a modest increase in the level of CCR5 after chloroquine treatment (Fig. 3a, second row of panels). In BaF3/CCR5 cells expressing BY1PC2 or BY5, there was little CCR5 present in the absence of chloroquine, confirming that these traptamers down-regulate CCR5 (Fig. 3a, bottom two rows). Notably, chloroquine caused a marked increase in intracellular CCR5 staining in cells expressing BY1PC2 or BY5, consistent with the flow cytometry and Western blotting results. Taken together, these results indicate that class 1 but not class 2 traptamers target CCR5 for accelerated lysosomal degradation.
Figure 3.

Chloroquine restores intracellular but not cell-surface CCR5 in cells expressing class 1 traptamers. a, parental BaF3 cells expressing the empty retroviral vector MSCV, and BaF3/CCR5-expressing vector, BY1PC2, or BY5 were untreated (left-most column of panels) or treated with 20 μm chloroquine for 24 h, spun onto glass slides, fixed, permeabilized, and stained with the 2D7 anti-CCR5 antibody (green), the C29F4 anti-HA antibody (red, to detect traptamer), and DAPI (blue, to visualize nuclei). Overlapping immune signal in merged panels is colored yellow. The same single confocal z plane is shown in each row of panels in the chloroquine-treated cells. b, BaF3/CCR5 cells expressing vector, BY1PC2, or BY5 were untreated (black bars) or treated (gray bars) with 20 μm chloroquine for 24 h. Nonpermeabilized cells were fixed and stained with a conformation-specific anti-CCR5-FITC conjugate (2D7), and cell-surface CCR5 expression was measured by flow cytometry. The geometric mean fluorescence intensity for each sample was determined and normalized to that of untreated BaF3/CCR5 cells lacking a traptamer. The mean values and S.D. (error bars) from three independent experiments are shown.
We also stained permeabilized cells with an antibody recognizing the HA epitope, which is present on the N terminus of the traptamers. As shown in Fig. 3a, BY1PC2 and BY5 were detected in cells transduced with a traptamer gene. Moreover, the merged panels in Fig. 3a show that there were extensive areas of overlap in the localization of CCR5 and the class 1 traptamers in the chloroquine-treated cells.
Stabilized CCR5 and BY1PC2 are present in the lysosome
We next determined whether chloroquine treatment restored expression of CCR5 at the surface of cells expressing the class 1 traptamers. First, nonpermeabilized cells were stained with anti-CCR5 antibodies and analyzed by flow cytometry for cell-surface CCR5 expression. As shown in Fig. 3b, chloroquine treatment had little effect on the cell-surface expression of CCR5 in cells lacking a traptamer. As reported previously, cell-surface CCR5 levels were markedly reduced when BY1PC2 or BY5 was expressed in the absence of chloroquine (Fig. 3b and Fig. S4) (19). Notably, chloroquine treatment did not restore cell-surface CCR5 expression in cells expressing BY1PC2 or BY5, despite the increased levels of total CCR5 in these cells, indicating that the increased CCR5 in chloroquine-treated cells was not at the cell surface. Thus, chloroquine caused CCR5 to accumulate in an intracellular location in cells expressing class 1 traptamers.
To determine whether CCR5 accumulated in the lysosome in these cells, we used a proximity ligation assay (PLA). The PLA is an antibody-based assay that generates a fluorescent signal only when the two target antigens are within 40 nm of each other (22). We performed PLA with antibodies recognizing CCR5 and LAMP1, a marker of the lysosome. Fig. 4a shows that there was no PLA signal in control cells that did not express CCR5. A low level of lysosomal CCR5 was detected in untreated and chloroquine-treated BaF3/CCR5 cells lacking a traptamer. Lysosomal CCR5 was not detected in BaF3/CCR5 cells expressing BY1PC2 or BY5 in the absence of chloroquine treatment, consistent with efficient down-regulation of CCR5 by these traptamers, whereas chloroquine treatment caused a striking increase in the level of lysosomal CCR5 in cells expressing BY1PC2 or BY5 (Fig. 4a).
Figure 4.
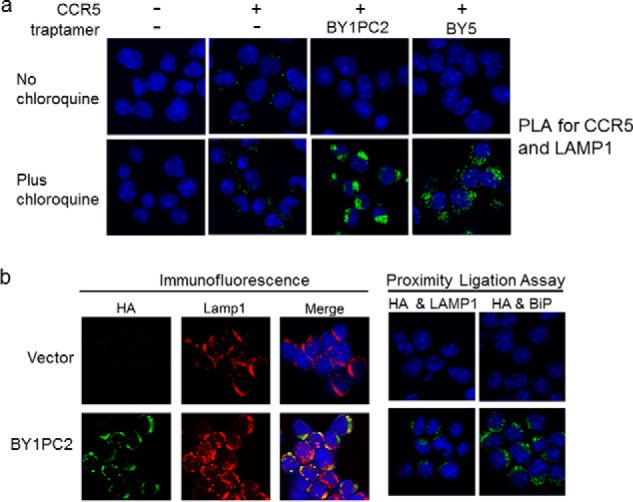
Class 1 traptamers direct CCR5 to the lysosome. a, parental BaF3 cells expressing an empty vector or BaF3/CCR5 cells in the absence of traptamer (−) or the presence of BY1PC2 or BY5 were untreated or treated with 20 μm chloroquine for 24 h. PLA was performed with primary antibodies recognizing CCR5 and the lysosomal marker LAMP1. PLA signal is shown in green. Cells were examined by confocal microscopy. Similar results were obtained in multiple independent experiments. b, left six panels, BaF3/CCR5 cells expressing the retroviral vector MSCV or BY1PC2 were spun onto glass coverslips, permeabilized, and stained with 6E2 anti-HA (green, to stain the traptamer) and anti-LAMP1 (red) antibodies and DAPI to visualize nuclei (blue). Cells were examined by confocal microscopy. Overlapping immune signal in the merged panels is shown in yellow. The same single confocal z plane is shown in each row. Right four panels, BaF3/CCR5 cells expressing empty vector alone or BY1PC2 were spun onto glass coverslips, and PLA was performed with 6E2 anti-HA antibody and either anti-LAMP1 or anti-BiP antibody. PLA signal is shown in green, and DAPI-stained nuclei are shown in blue.
We also used immunofluorescence and PLA to determine whether BY1PC2 itself was localized to the lysosome. As shown in Fig. 4b (left set of panels), there was considerable overlap between staining for BY1PC2 (detected with anti-HA antibody) and LAMP1. Similarly, there was a readily detectable BY1PC2/LAMP1 PLA signal (Fig. 4b, right set of panels), indicating that BY1PC2 localized to lysosomes. We note that a robust PLA signal was also generated with antibodies recognizing BY1PC2 and the ER protein BiP, indicating that BY1PC2 was also present in the ER.
Mapping the transmembrane helices of CCR5 required for traptamer action
Because the traptamers are essentially free-standing transmembrane domains, it seems likely that CCR5 down-regulation is mediated by an interaction of the traptamer with one or more of the transmembrane helices of CCR5. To map the transmembrane segment(s) of CCR5 required for traptamer action, we constructed a series of chimeric receptors in which individual transmembrane helices of CCR5 were replaced by the corresponding segments of CCR2b (Fig. 5a). CCR2b is a C-C chemokine receptor with 95% amino acid identity to CCR5 within the transmembrane helices (23, 24). CCR2b is not down-regulated by the class 1 traptamers but is partially down-regulated by BY6 (19). The chimeric receptors are designated by the transmembrane helix from CCR2b (e.g. “TM5 swap” has transmembrane helix 5 (TM5) of CCR5 replaced by TM5 of CCR2b). In all chimeras, the N-terminal and C-terminal extramembrane segments and the intracellular and extracellular loops were derived from CCR5.
Figure 5.

Class 1 traptamers require lysine 197 of CCR5 for optimal activity. a, schematic diagrams of CCR5/CCR2b chimeric receptors. CCR5 and CCR2b segments are indicated in black and green, respectively. Chimeric receptors are designated by the substituted transmembrane helix from CCR2b. b, the effect of traptamers on cell-surface expression of the chimeric receptors. Individual traptamers were expressed in clonal BaF3 cells expressing WT CCR5 or a chimeric receptor containing a single transmembrane helix from CCR2b. Cell-surface expression of each receptor was analyzed by flow cytometry of nonpermeabilized cells by using R22/7 anti-CCR5 antibody that recognizes an N-terminal epitope present in all of the chimeras. The geometric mean fluorescence intensity for each sample was determined and normalized to 100% in cells expressing the same chimera without a traptamer. Each traptamer/chimera combination was tested at least three times, and the bars show the average results and S.D. (error bars). Each bar is colored according to the chimera expressed, with data for cells expressing WT CCR5 shown in black. For traptamer BY1PC2, BY2, BY3, BY4, and BY5, down-regulation of TM5 swap chimera was significantly impaired compared with WT CCR5 (p value shown), as was down-regulation of TM3 swap by BY6M4. c, class 1 traptamer BY1, BY1PC2, BY3, or BY5 or class 2 traptamer BY6 or BY6M4 was co-expressed with WT CCR5 or the indicated receptor point mutant in BaF3 cells. Cell-surface expression of each receptor was analyzed by flow cytometry. The geometric mean fluorescence intensity for each sample was determined and normalized to 100% in cells expressing the same receptor construct without traptamer. The data for BY1 and BY1PC2 were combined, as were the data for BY6 and BY6M4, and the average mean fluorescence intensity and S.D. are shown. CCR5-K197M was significantly resistant to down-regulation by BY1PC2, BY3, or BY5 (p value shown), but not by BY6. d, vector only or BY1PC2 was co-expressed with WT CCR5, CCR5-K197M, or CCR5-K197R in BaF3 cells. Total CCR5 expression was determined by immunoblotting. Filters were stripped and reprobed for actin as loading control. The first lane in each panel shows parental BaF3 cells without CCR5. The sizes of molecular mass standards are shown in kDa.
Each of the chimeric receptors was stably expressed in BaF3 cells. An active traptamer was subsequently expressed in the cells, and cell-surface expression of the chimera was measured by flow cytometry with an antibody recognizing an N-terminal extracellular CCR5 epitope present in all of the chimeras. Cell-surface expression of each chimera in the presence of a traptamer was normalized to expression of the same chimera in the absence of a traptamer. As shown in Fig. 5b (black bars), the traptamers caused an ∼90% reduction in cell-surface expression of WT CCR5, except for BY5, which caused ∼75% reduction. The class 1 traptamers BY1PC2, BY2, BY3, and BY4 reproducibly caused significant down-regulation of all of the chimeras with the notable exception of the TM5 swap chimera, which contained TM5 of CCR2b and was largely refractory to down-regulation by these traptamers (light green bars). The remaining class 1 traptamer, BY5, displayed a similar pattern, with TM5 swap and TM6 swap being the most resistant to down-regulation, but BY5 down-regulated the TM5 swap more effectively than the other class 1 traptamers and was less active against the other chimeras. To assess whether the TM5 swap chimera was misfolded (thereby accounting for its reduced response to the traptamers), we determined the ability of this chimera to support infection by R5-tropic HIV pseudovirus, which depends on properly expressed and folded CCR5. As shown in Fig. S5a, cells expressing TM5 swap were infected by HIV as efficiently as cells expressing WT CCR5, indicating that in the absence of traptamer, this chimera reached the cell surface and properly displayed the extracellular loops that bind HIV gp120. Thus, the reduced ability of the class 1 traptamers to down-regulate TM5 swap is not due to gross misfolding of this chimera.
CCR5 and CCR2b differ at four amino acid positions in TM5. To determine which of these differences conferred resistance to the class 1 traptamers, we individually replaced each of these amino acids in CCR5 with its counterpart from CCR2b and tested whether these mutations affected the response to the class 1 traptamers. As shown in Fig. 5c, CCR5 mutants containing the I198R, V199N, or V209I substitutions were efficiently down-regulated by BY1PC2, BY3, and BY5. In contrast, the K197M mutant was largely resistant to down-regulation (Fig. 5, c and d). Furthermore, CCR5/K197M supported R5-tropic HIV infection, indicating that it was not grossly misfolded (Fig. S5b). We also tested the ability of BY1PC2 to down-regulate CCR5 in which lysine 197 was replaced with arginine, which, like lysine, has a side-chain positive charge. As shown in Fig. 5d, CCR5/K197R was effectively down-regulated by BY1PC2, suggesting that the positive charge, not the lysine side chain per se, is required for BY1PC2 action.
Compared with the class 1 traptamers, BY6M4 displayed a strikingly different pattern of activity against the chimeric receptors. As shown in Fig. 5b, BY6M4 down-regulated WT CCR5 and TM5 swap to a similar extent, whereas the ability of BY6M4 to down-regulate the TM3 swap chimera (blue bars) was impaired, implying that down-regulation depends on CCR5-specific amino acids in the third transmembrane helix. Consistent with the ability of the class 2 traptamer to down-regulate the TM5 swap, CCR5/K197M and the other TM5 substitution mutants were efficiently down-regulated by BY6M4 and BY6 (Fig. 5c and Fig. S6). Taken together, these results demonstrate that a positive charge at position 197 is required for CCR5 down-regulation by the class 1 traptamers but not by the class 2 traptamers.
Both classes of traptamer are present in a stable protein complex with CCR5
We conducted co-immunoprecipitation experiments to determine whether the active traptamers were present in a stable complex with CCR5 when the two proteins were co-expressed. For these experiments, we used BaF3 cells expressing CCR5 with an N-terminal FLAG epitope tag (FL-CCR5). Control experiments showed that like untagged CCR5, FL-CCR5 was down-regulated by BY1PC2 and BY5 and that chloroquine treatment restored total but not cell-surface FL-CCR5 expression in cells expressing these traptamers (Fig. S7). To analyze complex formation between CCR5 and the class 1 traptamers, we analyzed cells that were treated with chloroquine to stabilize FL-CCR5 so that it could be detected. Cells expressing FL-CCR5 in the presence or absence of BY1PC2 or BY5 were treated with chloroquine and lysed in buffer containing the detergent Triton X-100. The lysates were immunoprecipitated with an antibody that recognized the HA tag on the traptamer, and the immunoprecipitates were subjected to SDS-PAGE and immunoblotted with an anti-CCR5 antibody to visualize FL-CCR5 in the immunoprecipitate. As shown in Fig. 6a, FL-CCR5 was not co-immunoprecipitated by anti-HA antibody from control cells lacking traptamer expression, although FL-CCR5 was abundantly expressed in these cells because of chloroquine treatment (Fig. 6b), demonstrating that the HA antibody did not cross-react with FL-CCR5. Similarly, CCR5 was poorly co-immunoprecipitated from cells co-expressing US7, an unselected traptamer that does not down-regulate CCR5 (Fig. S8). In contrast, CCR5 was co-immunoprecipitated from cells co-expressing BY1PC2 or BY5, suggesting that CCR5 was present in a stable complex with the traptamer (Fig. 6a and Fig. S8). To confirm complex formation, we performed the reciprocal experiment in which FL-CCR5 was immunoprecipitated with anti-FLAG antibody, and traptamer in the immune precipitate was detected by immunoblotting for HA. As shown in Fig. 6b, BY1PC2 and BY5 were co-immunoprecipitated by the anti-FLAG antibody from lysates of cells expressing FL-CCR5 but not from lysates lacking FL-CCR5. Taken together, these experiments demonstrate that when CCR5 and the class 1 traptamers are co-expressed, they exist in a physical complex.
Figure 6.

Class 1 traptamers form a stable complex with CCR5 and the K197M CCR5 mutant. a, BaF3 cells expressing FLAG-tagged CCR5 (FL-CCR5) in the absence (−) or presence (+) of BY1PC2 were treated with 25 μm chloroquine for 24 h and lysed, and extracts were immunoprecipitated (IP) with anti-FLAG or anti-HA antibody, as indicated. Immunoprecipitates were subjected to SDS-PAGE followed by Western blotting using a anti-HA antibody. Similar amounts of FL-CCR5 in both extracts are documented in b, second panel, lanes 1 and 2. b, parental BaF3 cells (lane 4) and BaF3 cells expressing FLAG-tagged CCR5 in the presence or absence of BY1PC2 or BY5, as indicated, were treated with 25 μm chloroquine for 24 h and lysed. Extracts were immunoprecipitated with anti-FLAG antibody and subjected to SDS-PAGE followed by Western blotting with antibody recognizing the traptamer (anti-HA) or CCR5, as indicated. Filters of input extracts were probed with anti-HA or with anti-actin antibody, as indicated. c, detergent extracts from BaF3 cells expressing the CCR5/K197M mutant in the presence or absence of BY1PC2 were immunoprecipitated with an anti-HA antibody, subjected to SDS-PAGE, and Western blotted with an anti-HA antibody or with an anti-CCR5 antibody. Filters of input extracts were probed with an anti-CCR5 antibody or with anti-actin antibody as a loading control. Numbers to the right in all panels indicate the size in kilodaltons of molecular mass standards.
Because the class 1 traptamers were inactive against CCR5/K197M, we tested whether they formed a complex with this mutant. Cells expressing CCR5/K197M in the presence or absence of BY1PC2 were lysed, and anti-HA immunoprecipitation followed by CCR5 Western blotting was performed. As shown in Fig. 6c, CCR5/K197M was readily detected in anti-HA immunoprecipitates from cells expressing BY1PC2 but not from cells expressing the CCR5 mutant alone. Thus, BY1PC2 forms a complex with the K197M mutant despite the inability of this traptamer to down-regulate this mutant.
We also tested whether BY6M4 existed in a stable complex with CCR5. Because CCR5 was not stabilized by lysosome inhibitors in cells expressing BY6M4, we were not able to use chloroquine to elevate CCR5 levels for this analysis. However, the reduced ability of BY6M4 to down-regulate the TM3 swap chimera (Figs. 5b and 7a) allowed us to conduct co-immunoprecipitation experiments with this chimera to assess complex formation. Detergent extracts were prepared from cells co-expressing BY6M4 and FLAG-tagged TM3 swap chimera and subjected to anti-FLAG immunoprecipitation followed by immunoblotting with anti-HA antibody to detect co-immunoprecipitated BY6M4. As shown in Fig. 7b, the anti-FLAG antibody did not co-immunoprecipitate BY6M4 from cells lacking FL-TM3 swap expression, but BY6M4 was readily detected in anti-FLAG immunoprecipitates from cells expressing FL-TM3 swap. BY1PC2 also co-immunoprecipitated with FL-TM3 swap, which is not completely down-regulated by this traptamer (Fig. 7b). Thus, like the class 1 traptamers, the class 2 traptamer BY6M4 is present in a stable complex with CCR5.
Figure 7.

BY6M4 physically interacts with CCR5 and CCR2b. a, parental BaF3 cells (lane 1) or BaF3 cells expressing the FLAG-tagged TM3 swap chimeric receptor in the presence (+) or absence (−) of BY1PC2 (bottom) or BY6M4 (top) were lysed, and lysates were subjected to SDS-PAGE and Western blotting with an anti-CCR5 antibody. b, extracts prepared as in a were subjected to SDS-PAGE directly (input) or after immunoprecipitation with an anti-FLAG antibody. Blots were probed with antibodies recognizing HA or CCR5 or with an antibody recognizing profilin as a loading control. c, extracts were prepared from BaF3 cells expressing untagged CCR2b or FLAG-tagged CCR2b (FL-CCR2b). Anti-FLAG immunoprecipitates (IP) or input extracts were subjected to SDS-PAGE and Western blotting with an anti-HA antibody. For input samples, the filter was stripped and reprobed for profilin as a loading control. Numbers in all panels indicate the size in kilodaltons of molecular mass standards.
Similarly, we determined whether BY1PC2 and BY6M4 interacted with FLAG-tagged CCR2b, which is refractory to down-regulation by the class 1 traptamers (19). As shown in Fig. 7c, BY6M4 co-immunoprecipitated with CCR2b, which is not surprising because BY6M4 causes partial down-regulation of CCR2b (19). This result implies that BY6M4 binds to residues that are shared between CCR5 and CCR2b. In contrast, BY1PC2 did not bind CCR2b, suggesting that the class 1 traptamer binding required residues in CCR5 that are not shared with CCR2b. In addition, as shown in Fig. S9, CXCR4 was not detected in traptamer immunoprecipitates, indicating that neither BY1PC2 nor BY6M4 formed a complex with CXCR4, consistent with our previous observation that CXCR4 is not down-regulated by these traptamers (19). Therefore, BY1PC2 specifically binds CCR5, whereas BY6M4 can also bind to and down-regulate the closely related CCR2b, but neither class of traptamer binds or down-regulates CXCR4.
A side-chain positive charge in transmembrane helix 5 of CCR5 and CCR2b regulates their steady-state levels in the absence of traptamer
In the course of studying the effect of the CCR5 K197M mutation on traptamer-mediated down-regulation of CCR5, we noted that in cells lacking traptamer expression, CCR5/K197M was reproducibly expressed at a higher level than WT CCR5, as assessed by cell-surface flow cytometry (Fig. 8a) and by Western blotting of total, cell-surface, and intracellular CCR5 levels (Fig. 8 (b and c) and Fig. S6). To assess the stability of CCR5/K197M compared with WT CCR5, a cyclohexamide chase experiment was performed. As shown in Fig. 8d, levels of WT CCR5 substantially decreased by 5 h of cyclohexamide treatment, whereas no such decrease in CCR5/K197M was observed after 8.5 h of the treatment, indicating that the K197M mutation increases the stability of CCR5. To explore this phenomenon in more detail, we tested the expression of CCR5 mutants containing arginine, alanine, leucine, or aspartic acid at position 197. As shown in Fig. 8b, the mutant with arginine (K197R), which like WT lysine has a positive charge, was expressed at a similar level as WT CCR5, whereas the leucine, alanine, or aspartic acid mutants were expressed at markedly higher levels, similar to the expression of CCR5/K197M. These experiments indicate that the presence of a positive charge at position 197 in CCR5 regulates the steady-state level of CCR5 by specifying rapid degradation.
Figure 8.

Lysine 197 reduces the stability of CCR5 in the absence of traptamers. a, BaF3 cells expressing empty (MSCVn) vector or BaF3 cells expressing WT or K197M CCR5 were established in parallel, and cell-surface expression of each receptor was analyzed by flow cytometry of nonpermeabilized cells by using 2D7 anti-CCR5 antibody. The geometric mean fluorescence intensity for each sample was determined and normalized to cells expressing WT CCR5. Average and S.D. (error bars) for three experiments are shown. b, BaF3 cells expressing the empty vector, WT CCR5, or the indicated CCR5 point mutant were established in parallel. Detergent lysates were subjected to SDS-PAGE and Western blotting for CCR5 or Hsp90 as a loading control. c, the cells studied in a were subjected to cell-surface biotinylation, and biotinylated cell-surface proteins were recovered from cell extracts with streptavidin beads. The pellet (cell-surface) and supernatant (intracellular) fractions were subjected to SDS-PAGE and immunoblotted using an anti-CCR5 antibody. Samples were also probed for Jag1 or Hsp90 as loading controls. d, BaF3 cells expressing WT CCR5 or CCR5/K197M were treated with cyclohexamide (CHX), and at the indicated times, a portion of the culture was pelleted and lysed. Cell lysates were electrophoresed and immunoblotted for CCR5. The filters were stripped and reprobed for actin as a loading control. Numbers to the right of b, c, and d indicate the size in kilodaltons of molecular mass standards.
CCR2b contains a methionine, not a lysine, at position 205, the position homologous to CCR5 197 (Fig. 9a). To determine whether this position in CCR2b also affected steady-state receptor levels, we replaced methionine 205 with lysine and expressed the M205K mutant in BaF3 cells. As shown in Fig. 9b, CCR2b M205K was expressed at the cell surface at approximately one-third the level of WT CCR2b, as assessed by flow cytometry. To examine CCR2b by Western blotting, we introduced the M205K mutation in a chimeric receptor, designated CCR5/2, in which the N-terminal extracellular domain plus TM1 were derived from CCR5 and the rest of the protein was derived from CCR2b (Fig. 9c). The WT or mutant chimeric receptor was expressed in BaF3 cells, and Western blotting was performed with an antibody that recognizes the CCR5 segment present in these chimeras. As shown in Fig. 9d, the M205K chimera was expressed at a much lower level than the chimera containing the WT methionine at position 205. However, ammonium chloride treatment did not restore high expression levels to the M205K mutant. Similarly, expression of M205K was not elevated by MG132, although MG132 markedly increased the abundance of β-catenin (Fig. S10). Thus, the presence of a positive charge in TM5 at position 197 in CCR5 and position 205 in mutant CCR2b limits the expression of these receptors, most likely by a lysosome-independent and proteasome-independent mechanism.
Figure 9.
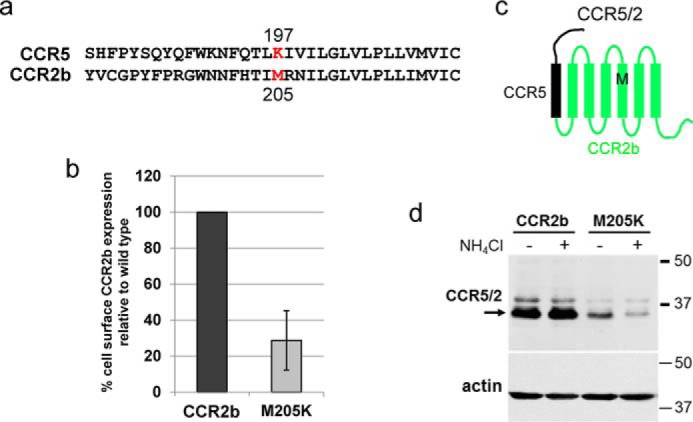
The M205K mutation reduces levels of CCR2b. a, CCR5 amino acids 180–213 aligned with the homologous region in CCR2b. Lysine at position 197 in CCR5 and methionine at position 205 in CCR2b are colored red. b, nonpermeabilized BaF3 cells expressing WT CCR2b or CCR2b/M205K were stained with MAB150 anti-CCR2 antibody, and cell-surface expression of CCR2b was measured by flow cytometry. The geometric mean fluorescence for each sample was determined and normalized to cells expressing WT CCR2b. Averaged results and S.D. (error bar) from three independent experiments are shown. c, schematic diagram of the CCR5/2 chimeric receptor showing the CCR5 segment in black and CCR2b segment in green. The position of CCR2b methionine 205 is indicated by the black M within TM5. d, BaF3 cells expressing the WT chimera (CCR5/2) or the M205K mutant chimera were untreated (−) or treated (+) with 10 mm NH4Cl for 8 h. The cells were then lysed, and extracts were subjected to SDS-PAGE and Western blotting using R22/7 antibody recognizing an N-terminal CCR5 epitope present in the chimera. The blot was stripped and reprobed for actin as a loading control. The numbers on the right indicate the size in kilodaltons of molecular mass standards.
Discussion
We previously isolated traptamers that specifically down-regulate expression of CCR5 without down-regulating CXCR4 (19). Thus, traptamers do not globally modulate GPCR metabolism, but the mechanism of traptamer action was largely unexplored. Here, we show that there are two classes of traptamers that down-regulate CCR5 but differ in their mechanism of action. The class 1 traptamers, such as BY1PC2, target CCR5 to the lysosome, where it undergoes degradation through the action of lysosomal proteases, whereas the class 2 traptamers, namely BY6 and BY6M4, reduce CCR5 expression by a mechanism that does not involve lysosomal degradation. Both classes of traptamers stably interact with CCR5, but binding is not sufficient for CCR5 down-regulation. These results, in conjunction with our previous description of traptamers that specifically induce PDGF β receptor dimerization or an activating conformational change in the erythropoietin receptor (25–27), demonstrate that traptamers can have diverse effects on their targets and highlight the versatility of traptamers despite their simple, related structures.
In cells expressing class 1 traptamers, lysosomal inhibition caused CCR5 to accumulate in the lysosome and not at the cell surface, showing that these traptamers targeted CCR5 to the lysosome for degradation. The class 1 traptamers themselves localize to the lysosome and the ER. The final decrease in the mobility of pulse-labeled CCR5 did not occur in cells expressing BY1PC2 (Fig. 1a), suggesting that the class 1 traptamers prevented the complete modification of CCR5. On the basis of these findings, we hypothesize that the class 1 traptamers intercept CCR5 before it undergoes final modification and redirect it to the lysosome for degradation. The low levels of pulse-labeled CCR5 in cells expressing class 2 traptamers and the inability of the tested inhibitors to restore CCR5 expression in these cells suggest that class 2 traptamers impair synthesis of full-length CCR5. Thus, the class 1 and class 2 traptamers use different mechanisms to down-regulate CCR5.
Several naturally occurring viral membrane proteins have activities similar to the traptamers. Murine cytomegalovirus m06/p48 glycoprotein and human herpesvirus 7 U21 protein target major histocompatibility complex class 1 (MHC1) proteins to the lysosome for degradation (28, 29). These herpesvirus proteins contain large soluble domains, which may enable them to interact with cellular proteins to mediate these effects. The class 1 traptamers may be more analogous to Nef and Vpu, short transmembrane accessory proteins of HIV that facilitate virus infection by down-regulating cellular transmembrane proteins that modulate the immune response, including MHC1 proteins, tetherin, and CD4 (30). Although the mechanism of action of these HIV proteins is not established, some experiments suggest that they direct their target proteins to the lysosome for degradation (31). Similarly, BY6M4 may be analogous to the Rhesus cytomegalovirus Rh178 protein, a small transmembrane protein that interrupts translation of MHC1 proteins before translocation of the ribosome to the membrane of the ER (32). Further study of CCR5 down-regulation by traptamers may shed light on the mechanisms employed by HIV and other viruses to down-regulate cellular proteins or may reveal novel mechanisms used by traptamers.
We used a panel of CCR5/CCR2b chimeric receptors to show that traptamers require specific transmembrane helices of CCR5 for activity. The class 1 traptamers required lysine 197 in the fifth transmembrane helix of CCR5, whereas the activity of class 2 traptamers was not influenced by the fifth transmembrane helix or lysine 197, but required the third transmembrane helix. Because we tested the effects of swapping transmembrane helices of CCR5 one at a time, these experiments have not ruled out the possibility that traptamers may require additional transmembrane helices in various combinations. We also point out that the chimeric receptor approach allowed us to interrogate only those transmembrane elements that differ between CCR2b and CCR5, and thus this approach would not identify important amino acids or structural motifs that lie outside of the transmembrane helices or that are present in both receptors.
Our co-immunoprecipitation results show that both class 1 and class 2 traptamers stably associated with CCR5 in detergent extracts. For WT CCR5, CCR2b, and CXCR4, there is a perfect correlation between traptamer binding and receptor down-regulation: class 1 traptamers bind and down-regulate CCR5 but do not bind or down-regulate CXCR4 and CCR2b; class 2 traptamers bind and down-regulate CCR5 and CCR2b but do not bind or down-regulate CXCR4. This correlation suggests that interaction between the traptamers and the transmembrane helices of CCR5 plays an important role in down-regulation. However, binding of the traptamers to CCR5 was not sufficient for CCR5 down-regulation (e.g. BY1PC2 bound to CCR5/K197M but did not down-regulate it) (Figs. 5 (b and c) and 6c). Further experiments are required to establish the role of binding of the traptamers in CCR5 down-regulation.
The class 1 traptamers may bind to the transmembrane helices of CCR5 and prevent proper folding of the protein or unfold its native packed conformation so that it is recognized as misfolded and targeted by cellular quality-control pathways to the lysosome for degradation. However, the ability of multiple conformation-specific antibodies to recognize chloroquine-stabilized CCR5 in cells expressing class 1 traptamers (Fig. S4) suggests that any such perturbation of CCR5 conformation is relatively subtle. Our mutational studies imply that the traptamers do not induce CCR5 down-regulation by inhibiting CCR5 palmitoylation or the action of dileucine motifs, but it is possible that traptamer binding may affect other modifications of CCR5, which in turn affect its targeting or stability. For example, the class 1 traptamers might induce mono-ubiquitination of CCR5, directing it to the lysosome for degradation. Alternatively, the class 1 traptamers could activate or recruit a cellular protein to CCR5 that targets it to the lysosome or could inhibit or displace a protein that normally binds to CCR5 and limits lysosomal localization. The sequences of the randomized hydrophobic segments of the class 1 traptamers differ from one another (Fig. S1), showing that a variety of different transmembrane interactions can induce lysosomal targeting of CCR5. It seems likely that the different class 1 traptamers use the same mechanism to target CCR5 to the lysosome because they all require TM5 of CCR5, and the three we tested require lysine 197 (Fig. 5,b and c).
Our studies also demonstrated that the charge at position 197 in CCR5 (and at the homologous position 205 in CCR2b) plays an important role in setting the steady-state levels of these receptors in cells lacking traptamers. In these cells, a positive charge at this position appears to target the receptor to a nonlysosomal, nonproteosomal degradation pathway that maintains receptor expression at a relatively low level. The fact that the destabilizing effect of the M205K mutation in CCR2b is lysosome-independent suggests that the class 1 traptamers do not merely accentuate the normal effect of a positive charge at CCR5 position 197. The ability of this positive charge to target chemokine receptors for lysosome-independent degradation in the absence of traptamers and for lysosomal-dependent degradation in the presence of class 1 traptamers demonstrates that this position plays a central role in determining the stability of these receptors.
How do CCR5 lysine 197 and CCR2b methionine 205 determine chemokine receptor stability? The crystal structure of CCR5 bound to the anti-HIV drug maraviroc or an antagonistic ligand showed that lysine 197 is present within the fifth transmembrane helix of CCR5 on the lateral surface of the helical bundle (Fig. S11a) (33, 34). Notably, the overall structures of the WT CCR5 and CCR2b transmembrane helices are very similar despite the differing charge at position 197/205, with a backbone root mean square deviation of <0.8 Å (33–35) (Fig. S11b). This implies that the CCR5 K197M and the CCR2b M205K mutations are not likely to have dramatic effects on the structure of these proteins, a conclusion consistent with the ability of CCR5 K197M to support HIV infection (Fig. 5b). Rather, the charge at position 197/205 exerts a more subtle effect. This lysine does not seem to be a site of ubiquitination required for traptamer-mediated receptor down-regulation or traptamer-independent receptor destabilization because the K197R mutant acts like WT CCR5 in the presence and absence of traptamers (Figs. 5d and 8b), even though arginine cannot be ubiquitinated. This positively charged side chain could be part of a binding or cleavage site for a protease or some other protein that affects CCR5 stability, or it could be the site of a post-translational modification or a structural motif that facilitates CCR5 clearance (33, 34). Published work indicates that lysine 197 is required for optimal co-receptor function with certain dual-tropic HIV strains (36) and that it is within a stretch of nine amino acids (Lys191–Val199) that confers specificity to the CCR5-selective small molecule inhibitor, Schering-C (37). Thus, lysine 197 appears to play an important role in the metabolism and function of CCR5 as well as in its response to class 1 traptamers (33, 34).
Lysine 197 lies within a highly conserved segment of CCR5, and the lysine itself is maintained among a variety of vertebrate species ranging from humans to zebrafish (Fig. S12) (36, 37). The observation that CCR5 evolved to have the relatively destabilizing lysine 197, whereas CCR2b has the stabilizing methionine 205, implies that the levels of these two immunomodulatory receptors have been tightly controlled over evolutionary time. Consistent with this conclusion, no nonsynonymous SNPs have been identified at codon 197 in the CCR5 gene in the more than 30,000 sequenced human genomes compiled in the gnomAD database (38), suggesting the importance of maintaining relatively low levels of CCR5. Our results suggest that this amino acid is an attractive target for the development of small molecules to regulate the abundance of CCR5 and related GPCRs.
Traptamers are a novel class of small artificial proteins that specifically modulate the activity and expression of various cellular transmembrane proteins. Studies of traptamer action may reveal new aspects of translation, intracellular trafficking, metabolism, or activity of transmembrane proteins. Unlike naturally occurring viral proteins, traptamers can be selected that perturb activities that do not necessarily support viral infection. Therefore, traptamers represent a versatile class of proteins that can specifically control cell behavior and serve as tools to study a large number of cellular proteins and processes. The mechanisms traptamers use to modulate their targets may inform the development of new molecules and approaches to regulate cellular transmembrane targets for a variety of research and practical uses.
Experimental procedures
Antibodies
Unconjugated and FITC-conjugated mouse anti-human CCR5 monoclonal antibody 2D7, which recognizes a conformation-specific N-terminal epitope, mouse anti-CXCR4 mAb 12G5, and monoclonal antibodies recognizing profilin, Hsp90 (catalog no. 610418), β-catenin (catalog no. 610153), or Jag1 (catalog no. 612346) were purchased from BD Biosciences. Mouse mAb R22/7 (catalog no. SC-32304), which recognizes an N-terminal epitope of CCR5, was obtained from Santa Cruz Biotechnology (Dallas, TX). Rabbit polyclonal antibodies E164 and 17476-1 recognizing the C-terminal domain of human CCR5 were purchased from Abcam (Cambridge, MA) and Proteintech (Rosemont, IL), respectively. The following conformation-specific anti-human CCR5 monoclonal antibodies were obtained from the National Institutes of Health AIDS Reagent Program: 45531, 45502.111, 45523, and 3A9. The anti-FLAG M2 mAb conjugated to agarose beads as an affinity matrix (catalog no. A2220) was obtained from Sigma-Aldrich. Unconjugated and horseradish peroxidase (HRP)-conjugated anti-HA mouse mAb 6E2 (catalog no. 29995), rabbit anti-HA mAb C29F4 (catalog no. 37245), and anti-actin polyclonal antibodies (catalog no. 49685) were purchased from Cell Signaling Technologies (Danvers, MA). Rabbit anti-LAMP1 (ab24170) and anti-BiP polyclonal (ab21685) antibodies were purchased from Abcam. Rabbit anti-CCR2b mAb MAB150 was obtained from R&D Systems (Minneapolis, MN). Alexa Fluor–conjugated donkey anti-mouse and anti-rabbit secondary antibodies (catalog nos. A21202 and A31512, respectively) used for flow cytometry and/or immunofluorescence were purchased from Invitrogen. HRP-conjugated secondary antibodies used for immunoblotting were purchased from Jackson ImmunoResearch (West Grove, PA).
Plasmid constructs and mutagenesis
The human CCR5 and CCR2b genes were each cloned into the pMSCVneo retroviral plasmid, and traptamers were cloned into the pMSCVpuro retroviral plasmid as described previously (19). The human CXCR4 gene was cloned into the pBABE.fusin plasmid, which was obtained from the National Institutes of Health AIDS Reagent Program (deposited by Nathaniel Landau) and subcloned into pMSCVneo using standard procedures. Site-directed mutagenesis using the QuikChange method (Agilent Technologies, Inc., Santa Clara, CA) was performed to introduce single amino acid substitutions into CCR5 or CCR2b. TM swap CCR5-CCR2b chimeric receptors were generated by one or more rounds of site-directed mutagenesis, in which codons for amino acids in individual transmembrane helices of CCR5 were replaced with the corresponding codons of CCR2b. PCR was used to insert a FLAG epitope tag (DYKDDDDK) after the N-terminal methionine of CCR5, CCR2b, and the TM3 swap chimeric receptor to aid in immunoprecipitation. The CCR5/2b chimera analyzed in Fig. 9 was obtained from Robert Doms (University of Pennsylvania) (39). The Met205 → Lys mutation in CCR2b was introduced by site-directed mutagenesis.
Cells and cell culture
Mouse BaF3 cells were obtained from Alan D'Andrea (Dana Farber Cancer Institute, Boston, MA) and maintained in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 4–7% WEHI-3B cell conditioned medium (as a source of IL-3), 0.05 mm β-mercaptoethanol, 0.5 μg/ml amphotericin B, and antibiotics (RPMI/IL-3). 293T cells were obtained from the American Type Culture Collection (CRL-3216) and maintained in DMEM supplemented with 10% FBS, 20 mm Hepes, 0.5 μg/ml amphotericin B, and antibiotics (DMEM-10). Human CEM.NKR-CCR5 T cells were obtained from the National Institutes of Health AIDS Reagent Program (catalog no. 4376) and maintained in RPM 1640 medium supplemented with 10% FBS (40). Human U87-CD4 astrocytoma cells expressing human CD4 were obtained from the National Institutes of Health AIDS Reagent Program (catalog no. 4031) and maintained in DMEM supplemented with 7.5% fetal bovine serum, 7.5% bovine calf serum, 2 mm l-glutamine, and antibiotics (DMEM-15).
CCR5, CCR2b, CXCR4, and various traptamer constructs were introduced and stably expressed in BaF3 cells by retrovirus-mediated gene transfer as described previously (19). Briefly, a retroviral plasmid encoding the gene of interest was co-transfected with the packaging plasmids pCL-Eco and pVSVG (which encodes the vesicular stomatitis virus G protein (Imgenex, Littleton, CO)) into 293T cells by the calcium phosphate method. The tissue culture supernatant containing retrovirus produced by the cells was collected 48 h later, and 1–2 ml of retrovirus was used to infect 5 × 106 BaF3 cells in 10 ml of RPMI/IL-3 medium containing 4 μg/ml Polybrene. Two days later, cells were split 1:5 in selection medium containing 1 mg/ml G418 or 1 μg/ml puromycin. Alternatively, 5 × 105 BaF3 cells in 0.5 ml of RMP1/IL-3 medium in 12-well plates were infected with 0.5 ml of retrovirus plus Polybrene. Four h later, cells were transfected into T.25 flasks with 5 ml of medium and incubated overnight prior to drug addition. After approximately 1 week of drug selection, cells stably expressing the desired transgene were established. BaF3 cells co-expressing a chemokine receptor and a traptamer were established by sequential infection and selection with the receptor and traptamer-expressing retroviruses. For the experiments shown in Figs. 1a and 2–4 and Fig. S12, we used a clonal BaF3 cell line expressing high levels of cell-surface CCR5, designated BaF3/CCR5, which was isolated by FACS followed by limiting serial dilution as described previously (19). Traptamers and vector were introduced into CEM.NKR.CCR5 cells by retroviral infection and selection with 1 μg/ml puromycin as described above. To assay the response of the TM swap CCR5/CCR2b chimeras and the CCR5 mutants to the traptamers in Fig. 5 (b and c), clonal cell lines expressing high levels of each chimera were isolated as described above. After introduction of the traptamers, cell-surface expression of the chimera was assessed by flow cytometry of puromycin-resistant pooled cell lines. In some experiments, cells were incubated with chloroquine diphosphate (Sigma-Aldrich), ammonium chloride (Sigma-Aldrich), MG132 (Invitrogen and Selleckchem), 3-MA (Sigma-Aldrich), bafilomycin A1 (Sigma-Aldrich), E64d (Sigma-Aldrich), or cycloheximide (Sigma-Aldrich), as indicated.
Northern blotting
Total RNA was extracted from BaF3/CCR5 cells expressing empty MSCVpuro vector, BY1PC2, BY6M4 or the unselected traptamer US7 using TRIzol reagent (Invitrogen) as described by the manufacturer. Briefly, 2.5 × 106 cells were washed in PBS, resuspended in 1 ml of TRIzol reagent, and frozen at −20 °C. After thawing samples at room temperature, RNA was extracted with 200 μl of chloroform, precipitated with 500 μl of isopropyl alcohol, reconstituted in 100 μl of RNase-free H2O, and purified using the RNeasy minikit (Qiagen, Germantown, MD) with on-column DNase treatment. RNA (2 μg/sample) was denatured in 50% formamide, 6.5% formaldehyde, heated at 80 °C, electrophoresed on a 1.2% agarose-formaldehyde denaturing gel, and transferred to nitrocellulose. After cross-linking, the membrane was stained with 0.1% methylene blue and then hybridized to two [32P]dCTP-labeled DNA probes corresponding to nucleotides 1–534 and 532–1059 of the human CCR5 gene, which includes the entire coding region. Radiolabeled probes were generated by standard PCR and purified using Quick Spin columns (Roche Applied Science).
Flow cytometry
For cell-surface staining of CCR5 or CCR2b, 2.5 × 105 to 106 BaF3 cells were washed twice in PBS, blocked in 0.5% BSA in PBS, and incubated with 0.5 mg/ml 2D7 anti-CCR5 antibody, 10 μl of prediluted FITC-conjugated 2D7 antibody, or a 1:100 dilution of anti-CCR2b MAB150 antibody at 4 °C or room temperature for 1 h. For intracellular CCR5 staining, 106 BaF3 cells were first fixed with 1% paraformaldehyde for 20 min, permeabilized in 1% saponin for 1 h, and then incubated with 10 μl of FITC-conjugated 2D7 CCR5 antibody or a 1:100 dilution of the conformation-specific anti-CCR5 monoclonal antibodies obtained from the National Institutes of Health for 1 h at room temperature. Cells were then washed, incubated for 1 h with a 1:100 dilution of Alexa Fluor 488–conjugated donkey anti-mouse secondary antibody, and then washed again. Samples were analyzed on a FACSCalibur or LSRII flow cytometer, and data were analyzed by WinMDI, FlowJo, or DIVA6 software (BD Biosciences).
Immunofluorescence
BaF3 cells were spun onto coated glass slides (Shandon Double Cytoslides, Thermo Scientific, Waltham, MA) using a cytological centrifuge at 400 × g for 5 min. Cells were fixed in 4% paraformaldehyde for 20 min and then permeabilized in 1% saponin for 1 h. Cells were then washed once in PBS; incubated with a 1:100 dilution of primary anti-CCR5, -LMP1, or -HA antibody at room temperature for 1 h; washed again; and incubated with a 1:100 dilution of secondary antibody (donkey anti-mouse Alexa Fluor 488 or donkey anti-rabbit Alexa Fluor 555) at room temperature for 1 h, followed by staining with DAPI at 1 μg/ml. For CCR5 staining, an FITC-conjugated mouse anti-human CCR5 mAb, 2D7, was used without secondary antibody. Images were captured at ×100 magnification using a Leica SP5 confocal microscope and analyzed using the LAS AF (Leica) software.
Proximity ligation assay
The PLA was performed using Duolink reagents (Invitrogen) according to the manufacturer's instructions and as described previously (22, 41). Briefly, BaF3 cells were spun onto glass slides, fixed, permeabilized, and incubated with primary antibody as for immunofluorescence. Cells were incubated with the appropriate pair of PLA probes (at a 1:5 dilution) at 37 °C for 1 h, followed by a 1:40 dilution of ligase at 37 °C for 30 min and the amplification solution (containing the polymerase and fluorescent nucleotides) at 37 °C for 100 min. Nuclei were stained with 5 μg/ml DAPI for 10 min, and images were acquired as described above for immunofluorescence.
Immunoprecipitation and immunoblotting
Cell extracts were prepared by lysing washed cell pellets in cold radioimmune precipitation assay (RIPA) buffer (50 mm Tris·HCl (pH 7.4), 150 mm NaCl, 1% Nonidet P-40, 1 mm EDTA, 1% deoxycholic acid, 0.1% SDS) or Triton lysis buffer (50 mm Tris·HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, and 1% Triton X-100), both containing protease inhibitors. Cell lysates were clarified by centrifugation at 16,000 × g for 30 min, and supernatants were transferred to clean tubes. After measuring the protein concentration of the extracts by a bicinchoninic acid assay (Pierce/Thermo Fisher), a portion of each extract was mixed with 2× or 5× Laemmli sample buffer for electrophoresis and immunoblotting to determine the amount of input receptor. For co-immunoprecipitation analysis to detect traptamer-receptor complexes, extracts were precleared by incubating with protein A-Sepharose and/or protein A/G-agarose beads (Santa Cruz Biotechnology) for 1 h at 4 °C, after which time the beads were pelleted, and the supernatant was transferred to a new tube. To immunoprecipitate traptamer, 1.5–2.5 mg of protein extract was incubated with 12–20 μl of anti-HA antibody C29F4 overnight at 4 °C. Immune complexes were precipitated using protein A-Sepharose, protein A/G-agarose beads, or a mixture of both and then washed five times in NET-N buffer (100 mm NaCl, 20 mm Tris-HCl (pH 8.0), 0.5 mm EDTA, 0.5% Nonidet P-40). To immunoprecipitate FLAG-tagged receptor, ∼1 mg of protein extract was incubated with 100 μl of anti-FLAG affinity matrix for 3–5 h at 4 °C, and then the beads were washed five times in TBS (50 mm Tris·HCl (pH 7.4) and 150 mm NaCl). Both anti-HA and anti-FLAG immune complexes were eluted from beads with 2× Laemmli sample buffer.
To detect a receptor construct or traptamer, eluates from immunoprecipitates or input protein extracts were heated at 60–70 °C for 10 min and electrophoresed on an SDS-10% polyacrylamide gel (for detection of a receptor) or a 17% polyacrylamide gel lacking SDS but containing 0.1% SDS in the running buffer (for detection of traptamer). Gels were then transferred to 0.2 μm polyvinylidene difluoride without SDS for 1–1.5 h. The blots were blocked in blocking buffer (5% nonfat dry milk in TBST (10 mm Tris·HCl (pH 7.4), 167 mm NaCl, and 1% Tween 20)) for 1 h and incubated at 4 °C overnight in primary antibody (anti-CCR5 R22/7, E164 or 17476-1; anti-CXCR4 12G5; or anti-HA-HRP 6E2) diluted 1:1,000 in blocking buffer. Blots were then washed five times in TBST and incubated for 1 h in anti-mouse or anti-rabbit-HRP secondary antibody diluted 1:8000 in blocking buffer and then washed again. The incubation with anti-rabbit-HRP was omitted for samples probed with anti-HA-HRP. Blots were detected by enhanced chemiluminescence. In some cases, blots were stripped by using Restore Western blotting stripping buffer (Pierce/Thermo Fisher), washed, blocked in blocking buffer, and reprobed with anti-actin, anti-profilin, anti-Jag1, or anti-Hsp90 antibody as loading controls and then processed as above.
Pulse-chase and cycloheximide experiments
Approximately 5 × 107 BaF3/CCR5 cells expressing empty MSCVpuro vector, BY1PC2, or BY6M4 were pelleted, washed in PBS, and incubated for 1 h at 37 °C in 10 ml of RPMI 1640 medium lacking cysteine and methionine and supplemented with 2% dialyzed FBS, 25 mm Hepes, 0.06 mm β-mercaptoethanol, l-glutamine, and antibiotics. The cells were then incubated for 30 min at 37 °C in 2.5 ml of the same medium containing 625 μCi of [35S]methionine/[35S]cysteine labeling mix (PerkinElmer Life Sciences). Cells were then pelleted, resuspended in 10 ml of RPMI/IL-3 medium containing unlabeled cysteine and methionine, and divided into five 2-ml aliquots. One aliquot was immediately placed on ice (t = 0), whereas the others were incubated at 37 °C for 0.5, 2, 4, and 6 h and then placed on ice. Immediately after placing on ice, each aliquot was pelleted, washed in PBS, pelleted again, and stored at −80 °C. Cell pellets were lysed by incubating on ice for 10 min in 1 ml of RIPA-MOPS buffer (20 mm MOPS (pH 7.0), 150 mm NaCl, 1% Nonidet P-40, 1 mm EDTA, 1% deoxycholic acid, 0.1% SDS) supplemented with protease inhibitors. After clarifying lysates by centrifugation at 16,000 × g for 15 min at 4 °C, CCR5 was immunoprecipitated by incubating 0.85 ml of each extract with 7.5 μl of R22/7 anti-CCR5 antibody plus 25 μl of protein A/G-agarose beads for 3 h at 4 °C. Beads were washed three times in RIPA-MOPS buffer, and immune complexes were eluted from the beads in 30 μl of 2× Laemmli sample buffer. Samples were heated at 75 °C for 5 min and electrophoresed on a 16-cm SDS-12% polyacrylamide gel. The gel was fixed in acetic acid, dried, and exposed to X-ray film for 3 weeks.
BaF3 cells expressing WT CCR5 or the K197M CCR5 mutant were treated with 100 μg of cycloheximide for various times, after which the cells were washed in PBS, pelleted, and frozen at −80 °C. Cells were then lysed in Triton lysis buffer, and cell lysates were subjected to SDS-PAGE and immunoblotted with an antibody recognizing CCR5 (17476-1 I, Proteintech).
Biotinylation of cell-surface proteins
Surface proteins on intact cells were biotinylated using the EZ-Link-sulfo-NHS-SS-biotin (sulfosuccinimidyl 2-[biotinamido]ethyl-1,3-dithiopropionate) reagent (catalog no. 21331; Thermo Scientific) as described previously with minor modifications (19). Cells were then lysed in RIPA buffer as described (19), and 100 μl of the extract was removed and mixed with 25 μl of 5× protein sample buffer to examine the amount of total cell protein. To precipitate biotinylated proteins, the remainder of the extract was incubated with 100 μl of streptavidin-agarose beads (catalog no. 20349, Pierce/Thermo Fisher) overnight at 4 °C. The streptavidin beads were then pelleted by centrifugation at 14,000 × g for 1 min. The supernatant representing the cytoplasmic fraction was transferred to a new tube, and 100 μl was removed and mixed with 25 μl of 5× protein sample buffer. The pellet containing the biotinylated cell-surface proteins was washed four times in NET-N buffer and then resuspended in 65 μl of 5× sample buffer.
HIV reporter virus assay
U87-CD4 cells expressing CCR5, CCR5/K197M, or the TM5 swap chimeric receptor were established by retrovirus-mediated gene transfer followed by selection with G418. Single-cycle HIV reporter virus expressing yellow fluorescent protein (YFP) and pseudotyped with the R5-trophic ADA Env protein was generated as described previously (19). Briefly, 293T cells were co-transfected with the HIV-eYFP reporter plasmid and a plasmid expressing ADA (gift from Dan Littman, New York University) using the calcium phosphate method. Pseudovirus was harvested from transfected cells and used to infect U87-CD4 cells expressing CCR5, CCR5/K197M, or the TM5 swap chimeric receptor as described (19). YFP expression was then analyzed by flow cytometry, and the percentage of infected cells for each sample was calculated as the fraction of the total number of cells analyzed that were eYFP-positive.
Structure comparisons
Crystal structures of CCR5 (PDB entry 4MBS) and CCR2b (PDB entry 5T1A) were downloaded from the PDB and visualized by using PyMOL. The two structures were superimposed by using the MAPSI software package.
Author contributions
L. M. P., S. A. M., Y. L., E. H. S., A. S., and D. D. conceptualization; L. M. P., S. A. M., Y. L., E. H. S., A. S., and D. D. formal analysis; L. M. P., S. A. M., Y. L., E. H. S., and A. S. investigation; L. M. P. and D. D. writing-original draft; L. M. P., S. A. M., Y. L., E. H. S., A. S., and D. D. writing-review and editing; Y. L. and D. D. funding acquisition; D. D. supervision.
Supplementary Material
Acknowledgments
We thank Jasmine Ayers for assistance with constructing mutations and Robert Doms for reagents. As noted, reagents were obtained from the AIDS Reagents Program, Division of AIDS, NIAID, National Institutes of Health. We thank Jan Zulkeski for assistance in preparing the manuscript.
Note added in proof
The incorrect versions of A and C in Fig. 7 were inadvertently uploaded in the version of this paper that was published as a Paper in Press on April 20, 2018. These errors have now been corrected and do not affect the results or conclusions of this work.
This work was supported by NCI, National Institutes of Health, Grant CA037157 (to D. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S12.
- GPCR
- G protein–coupled receptor
- 3-MA
- 3-methyladenine
- CCR2b
- C-C chemokine receptor 2b
- CCR5
- C-C chemokine receptor 5
- CXCR4
- C-X-C chemokine receptor 4
- ER
- endoplasmic reticulum
- FBS
- fetal bovine serum
- FL-CCR5
- FLAG-tagged CCR5
- HA
- hemagglutinin
- LAMP1
- lysosomal-associated membrane protein 1
- MHC1
- major histocompatibility complex
- PLA
- proximity ligation assay
- RIPA
- radioimmune precipitation assay
- YFP
- yellow fluorescent protein
- eYFP
- enhanced YFP
- TM
- transmembrane domain
- HRP
- horseradish peroxidase
- IL
- interleukin
- PDB
- Protein Data Bank
- DAPI
- 4′,6-diamidino-2-phenylindole
- DMEM
- Dulbecco's modified Eagle's medium.
References
- 1. Oppermann M. (2004) Chemokine receptor CCR5: insights into structure, function, and regulation. Cell. Signal. 16, 1201–1210 10.1016/j.cellsig.2004.04.007 [DOI] [PubMed] [Google Scholar]
- 2. Berger E. A., Murphy P. M., and Farber J. M. (1999) Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 17, 657–700 10.1146/annurev.immunol.17.1.657 [DOI] [PubMed] [Google Scholar]
- 3. Bannert N., Craig S., Farzan M., Sogah D., Santo N. V., Choe H., and Sodroski J. (2001) Sialylated O-glycans and sulfated tyrosines in the NH2-terminal domain of CC chemokine receptor 5 contribute to high affinity binding of chemokines. J. Exp. Med. 194, 1661–1673 10.1084/jem.194.11.1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kraft K., Olbrich H., Majoul I., Mack M., Proudfoot A., and Oppermann M. (2001) Characterization of sequence determinants within the carboxyl-terminal domain of chemokine receptor CCR5 that regulate signaling and receptor internalization. J. Biol. Chem. 276, 34408–34418 10.1074/jbc.M102782200 [DOI] [PubMed] [Google Scholar]
- 5. Huttenrauch F., Nitzki A., Lin F. T., Höning S., and Oppermann M. (2002) β-Arrestin binding to CC chemokine receptor 5 requires multiple C-terminal receptor phosphorylation sites and involves a conserved Asp-Arg-Tyr sequence motif. J. Biol. Chem. 277, 30769–30777 10.1074/jbc.M204033200 [DOI] [PubMed] [Google Scholar]
- 6. Signoret N., Hewlett L., Wavre S., Pelchen-Matthews A., Oppermann M., and Marsh M. (2005) Agonist-induced endocytosis of CC chemokine receptor 5 is clathrin dependent. Mol. Biol. Cell 16, 902–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Escola J. M., Kuenzi G., Gaertner H., Foti M., and Hartley O. (2010) CC chemokine receptor 5 (CCR5) desensitization: cycling receptors accumulate in the trans-Golgi network. J. Biol. Chem. 285, 41772–41780 10.1074/jbc.M110.153460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borroni E. M., Mantovani A., Locati M., and Bonecchi R. (2010) Chemokine receptors intracellular trafficking. Pharmacol. Ther. 127, 1–8 10.1016/j.pharmthera.2010.04.006 [DOI] [PubMed] [Google Scholar]
- 9. Delhaye M., Gravot A., Ayinde D., Niedergang F., Alizon M., and Brelot A. (2007) Identification of a postendocytic sorting sequence in CCR5. Mol. Pharmacol. 72, 1497–1507 10.1124/mol.107.038422 [DOI] [PubMed] [Google Scholar]
- 10. Hüttenrauch F., Pollok-Kopp B., and Oppermann M. (2005) G protein-coupled receptor kinases promote phosphorylation and β-arrestin-mediated internalization of CCR5 homo- and hetero-oligomers. J. Biol. Chem. 280, 37503–37515 [DOI] [PubMed] [Google Scholar]
- 11. Oppermann M., Mack M., Proudfoot A. E., and Olbrich H. (1999) Differential effects of CC chemokines on CC chemokine receptor 5 (CCR5) phosphorylation and identification of phosphorylation sites on the CCR5 carboxyl terminus. J. Biol. Chem. 274, 8875–8885 10.1074/jbc.274.13.8875 [DOI] [PubMed] [Google Scholar]
- 12. Mueller A., Kelly E., and Strange P. G. (2002) Pathways for internalization and recycling of the chemokine receptor CCR5. Blood 99, 785–791 10.1182/blood.V99.3.785 [DOI] [PubMed] [Google Scholar]
- 13. Blanpain C., Wittamer V., Vanderwinden J. M., Boom A., Renneboog B., Lee B., Le Poul E., El Asmar L., Govaerts C., Vassart G., Doms R. W., and Parmentier M. (2001) Palmitoylation of CCR5 is critical for receptor trafficking and efficient activation of intracellular signaling pathways. J. Biol. Chem. 276, 23795–23804 10.1074/jbc.M100583200 [DOI] [PubMed] [Google Scholar]
- 14. Venkatesan S., Petrovic A., Locati M., Kim Y. O., Weissman D., and Murphy P. M. (2001) A membrane-proximal basic domain and cysteine cluster in the C-terminal tail of CCR5 constitute a bipartite motif critical for cell surface expression. J. Biol. Chem. 276, 40133–40145 10.1074/jbc.M105722200 [DOI] [PubMed] [Google Scholar]
- 15. Percherancier Y., Planchenault T., Valenzuela-Fernandez A., Virelizier J. L., Arenzana-Seisdedos F., and Bachelerie F. (2001) Palmitoylation-dependent control of degradation, life span, and membrane expression of the CCR5 receptor. J. Biol. Chem. 276, 31936–31944 10.1074/jbc.M104013200 [DOI] [PubMed] [Google Scholar]
- 16. Venkatesan S., Rose J. J., Lodge R., Murphy P. M., and Foley J. F. (2003) Distinct mechanisms of agonist-induced endocytosis for human chemokine receptors CCR5 and CXCR4. Mol. Biol. Cell 14, 3305–3324 10.1091/mbc.E02-11-0714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marchese A., and Benovic J. L. (2001) Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem. 276, 45509–45512 10.1074/jbc.C100527200 [DOI] [PubMed] [Google Scholar]
- 18. Marchese A., Raiborg C., Santini F., Keen J. H., Stenmark H., and Benovic J. L. (2003) The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell 5, 709–722 10.1016/S1534-5807(03)00321-6 [DOI] [PubMed] [Google Scholar]
- 19. Scheideman E. H., Marlatt S. A., Xie Y., Hu Y., Sutton R. E., and DiMaio D. (2012) Transmembrane protein aptamers that inhibit CCR5 expression and HIV coreceptor function. J. Virol. 86, 10281–10292 10.1128/JVI.00910-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Farzan M., Mirzabekov T., Kolchinsky P., Wyatt R., Cayabyab M., Gerard N. P., Gerard C., Sodroski J., and Choe H. (1999) Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 96, 667–676 10.1016/S0092-8674(00)80577-2 [DOI] [PubMed] [Google Scholar]
- 21. Bonifacino J. S., and Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 10.1146/annurev.biochem.72.121801.161800 [DOI] [PubMed] [Google Scholar]
- 22. Lipovsky A., Zhang W., Iwasaki A., and DiMaio D. (2015) Application of the proximity-dependent assay and fluorescence imaging approaches to study viral entry pathways. Methods Mol. Biol. 1270, 437–451 10.1007/978-1-4939-2309-0_30 [DOI] [PubMed] [Google Scholar]
- 23. Raport C. J., Gosling J., Schweickart V. L., Gray P. W., and Charo I. F. (1996) Molecular cloning and functional characterization of a novel human CC chemokine receptor (CCR5) for RANTES, MIP1β, and MIP1α. J. Biol. Chem. 271, 17161–17166 10.1074/jbc.271.29.17161 [DOI] [PubMed] [Google Scholar]
- 24. Charo I. F., Myers S. J., Herman A., Franci C., Connolly A. J., and Coughlin S. R. (1994) Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc. Natl. Acad. Sci. U.S.A. 91, 2752–2756 10.1073/pnas.91.7.2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cammett T. J., Jun S. J., Cohen E. B., Barrera F. N., Engelman D. M., and Dimaio D. (2010) Construction and genetic selection of small transmembrane proteins that activate the human erythropoietin receptor. Proc. Natl. Acad. Sci. U.S.A. 107, 3447–3452 10.1073/pnas.0915057107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He L., Steinocher H., Shelar A., Cohen E. B., Heim E. N., Kragelund B. B., Grigoryan G., and DiMaio D. (2017) Single methyl groups can act as toggle switches to specify transmembrane protein-protein interactions. eLife 6, e27701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Freeman-Cook L. L., Dixon A. M., Frank J. B., Xia Y., Ely L., Gerstein M., Engelman D. M., and DiMaio D. (2004) Selection and characterization of small random transmembrane proteins that bind and activate the platelet-derived growth factor beta receptor. J. Mol. Biol. 338, 907–920 10.1016/j.jmb.2004.03.044 [DOI] [PubMed] [Google Scholar]
- 28. Glosson N. L., Gonyo P., May N. A., Schneider C. L., Ristow L. C., Wang Q., and Hudson A. W. (2010) Insight into the mechanism of human herpesvirus 7 U21-mediated diversion of class I MHC molecules to lysosomes. J. Biol. Chem. 285, 37016–37029 10.1074/jbc.M110.125849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reusch U., Muranyi W., Lucin P., Burgert H. G., Hengel H., and Koszinowski U. H. (1999) A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 18, 1081–1091 10.1093/emboj/18.4.1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dubé M., Bego M. G., Paquay C., and Cohen É. A. (2010) Modulation of HIV-1-host interaction: role of the Vpu accessory protein. Retrovirology 7, 114 10.1186/1742-4690-7-114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arias J. F., Iwabu Y., and Tokunaga K. (2012) Sites of action of HIV-1 Vpu in BST-2/tetherin downregulation. Curr. HIV Res. 10, 283–291 10.2174/157016212800792423 [DOI] [PubMed] [Google Scholar]
- 32. Richards R., Scholz I., Powers C., Skach W. R., and Früh K. (2011) The cytoplasmic domain of rhesus cytomegalovirus Rh178 interrupts translation of major histocompatibility class I leader peptide-containing proteins prior to translocation. J. Virol. 85, 8766–8776 10.1128/JVI.05021-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tan Q., Zhu Y., Li J., Chen Z., Han G. W., Kufareva I., Li T., Ma L., Fenalti G., Li J., Zhang W., Xie X., Yang H., Jiang H., Cherezov V., Liu H., Stevens R. C., Zhao Q., and Wu B. (2013) Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 341, 1387–1390 10.1126/science.1241475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng Y., Han G. W., Abagyan R., Wu B., Stevens R. C., Cherezov V., Kufareva I., and Handel T. M. (2017) Structure of CC chemokine receptor 5 with a potent chemokine antagonist reveals mechanisms of chemokine recognition and molecular mimicry by HIV. Immunity 46, 1005–1017.e5 10.1016/j.immuni.2017.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zheng Y., Qin L., Zacarias N. V., de Vries H., Han G. W., Gustavsson M., Dabros M., Zhao C., Cherney R. J., Carter P., Stamos D., Abagyan R., Cherezov V., Stevens R. C., IJzerman A. P., et al. (2016) Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 540, 458–461 10.1038/nature20605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Doranz B. J., Lu Z. H., Rucker J., Zhang T. Y., Sharron M., Cen Y. H., Wang Z. X., Guo H. H., Du J. G., Accavitti M. A., Doms R. W., and Peiper S. C. (1997) Two distinct CCR5 domains can mediate coreceptor usage by human immunodeficiency virus type 1. J. Virol. 71, 6305–6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lau G., Labrecque J., Metz M., Vaz R., and Fricker S. P. (2015) Specificity for a CCR5 inhibitor is conferred by a single amino acid residue: role of Ile198. J. Biol. Chem. 290, 11041–11051 10.1074/jbc.M115.640169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., O'Donnell-Luria A. H., Ware J. S., Hill A. J., Cummings B. B., Tukiainen T., Birnbaum D. P., Kosmicki J. A., Duncan L. E., Estrada K., et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rucker J., Samson M., Doranz B. J., Libert F., Berson J. F., Yi Y., Smyth R. J., Collman R. G., Broder C. C., Vassart G., Doms R. W., and Parmentier M. (1996) Regions in β-chemokine receptors CCR5 and CCR2b that determine HIV-1 cofactor specificity. Cell 87, 437–446 10.1016/S0092-8674(00)81364-1 [DOI] [PubMed] [Google Scholar]
- 40. Trkola A., Matthews J., Gordon C., Ketas T., and Moore J. P. (1999) A cell line-based neutralization assay for primary human immunodeficiency type 1 isolates that use either the CCR5 or the CXCR4 coreceptor. J. Virol. 73, 8966–8974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Popa A., Zhang W., Harrison M. S., Goodner K., Kazakov T., Goodwin E. C., Lipovsky A., Burd C. G., and DiMaio D. (2015) Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog. 11, e1004699 10.1371/journal.ppat.1004699 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.