

Abstract
Obesity-induced chronic inflammation is associated with metabolic disease. Results from mouse models utilizing a high-fat diet (HFD) have indicated that an increase in activated macrophages, including CD11c+ adipose tissue macrophages (ATMs), contributes to insulin resistance. Obesity primes myeloid cell production from hematopoietic stem cells (HSCs) and Toll-like receptor 4 (TLR4), and the downstream TIR domain–containing adapter protein–inducing interferon-β (TRIF)- and MyD88-mediated pathways regulate production of similar myeloid cells after lipopolysaccharide stimulation. However, the role of these pathways in HFD-induced myelopoiesis is unknown. We hypothesized that saturated fatty acids and HFD alter myelopoiesis by activating TLR4 pathways in HSCs, differentially producing pro-inflammatory CD11c+ myeloid cells that contribute to obesity-induced metabolic disease. Results from reciprocal bone marrow transplants (BMTs) with Tlr4−/− and WT mice indicated that TLR4 is required for HFD-induced myelopoiesis and production of CD11c+ ATMs. Experiments with homozygous knockouts of Irakm (encoding a suppressor of MyD88 inactivation) and Trif in competitive BMTs revealed that MyD88 is required for HFD expansion of granulocyte macrophage progenitors and that Trif is required for pregranulocyte macrophage progenitor expansion. A comparison of WT, Tlr4−/−, Myd88−/−, and Trif−/− mice on HFD demonstrated that TLR4 plays a role in the production of CD11c+ ATMs, and both Myd88−/− and Trif−/− mice produced fewer ATMs than WT mice. Moreover, HFD-induced TLR4 activation inhibited macrophage proliferation, leading to greater accumulation of recruited CD11c+ ATMs. Our results indicate that HFD potentiates TLR4 and both its MyD88- and TRIF-mediated downstream pathways within progenitors and adipose tissue and leads to macrophage polarization.
Keywords: obesity, Toll-like receptor 4 (TLR4), metabolism, myeloid cell, macrophage, TIR domain-containing adapter-inducing interferon-B (TRIF), myeloid differentiation primary response gene 88 (MyD88), metabolic syndrome, insulin resistance, myelopoiesis, glucose intolerance
Introduction
Three decades of research have established the association among obesity, inflammation, and metabolic disease (1). Many of the associated co-morbidities of overnutrition in obese individuals (including insulin resistance, type 2 diabetes, atherosclerosis, cardiovascular disease, and some cancers) have been attributed to chronic low-grade inflammation, also known as meta-inflammation. More specifically, these obesity-induced diseases have been strongly correlated with an increase in myeloid leukocyte populations (macrophages and neutrophils) (2).
Obesity-induced alterations of tissue inflammatory macrophages, both in number and in activation state, have been directly associated with tissue dysfunction. Whereas local macrophage proliferation contributes to this accumulation (3), circulating pro-inflammatory Ly6chi monocyte populations are an important source of tissue macrophages (4). Ly6chi monocytes polarize and traffic into adipose tissue, giving rise to CD11c+ macrophages, specifically in the visceral white adipose tissue (WAT)2 of obese individuals (5). CD11c+ adipose tissue macrophages (ATMs) are distinguished by a CD64+/CD11c+ phenotype in murine models and a CD206+/CD11c+ phenotype in humans and are recruited through chemokines and adipocyte signals that promote chemotaxis of monocytes (6–11). CD11c+ ATMs accumulate disproportionately in visceral WAT and form ring structures known as crown-like structures (CLS) around damaged adipocytes in obese individuals. In contrast, CD11c−, anti-inflammatory resident macrophages predominate in lean individuals (4). The presence of CLS is indicative of a pro-inflammatory state, as these macrophages have been found to secrete cytokines such as tumor necrosis factor-α and interleukin-1β (12) and are thus important contributors to metabolic syndrome, specifically in visceral adipose tissue of males (13).
Given that the associated cytokines produced by ATMs mimic the inflammation from lipopolysaccharide (LPS) stimulation (14, 15), the Toll-like receptor 4 (TLR4) pathway has been implicated in obesity-induced inflammation. TLR complexes recognize pathogen-associated molecular patterns and signal via either an MyD88-dependent or -independent pathway (12). Downstream of TLR4, the MyD88 pathway activates the early phase of NF-κB, whereas the MyD88-independent or TRIF pathway activates interferon-regulatory factor (IRF3) and the late phase of NF-κB activation (16). NF-κB activation plays a key role in the regulation of both innate and adaptive immunity and consequently has been the target for many anti-inflammatory interventions. The role of TLR4 in the context of diet-induced adipose tissue inflammation and metabolic dysfunction is still unclear. Some investigators have reported a metabolic and inflammatory role for TLR4 (17, 18), whereas others have reported that TLR4 knockout does not improve metabolic function (19). The relative contribution of TRIF and MyD88 pathways to TLR4-mediated obesity–induced metabolic impairments and inflammation is unknown.
Whereas it has been shown that in obesity TLR4 deficiency significantly promotes alternative macrophage activation (19) and adipose tissue fibrosis (20), recent work in our laboratory demonstrated an additional role for TLR4 in the expansion of hematopoietic stem cells (HSCs) in obesity (2) and a role for MyD88 in the generation of myeloid progenitors in obesity. TLR4 activation in differentiated ATMs with fatty acids (17) in a high-fat diet environment is one possible mechanism explaining macrophage polarization, but a second mechanism of fatty acid activation within the hematopoietic progenitor cells probably exists as well.
There is a paucity of information on the possible dual role for TLR4, TRIF, and MyD88 pathways in hematopoietic precursors and in mature tissue macrophages. Therefore, we chose to investigate these pathways together in the context of diet-induced obesity. This is critical to understand, as interventions on these inflammatory pathways probably play a role in both the generation of myeloid cells and the activation profile of macrophages in obesity. Based on the literature and prior studies, we sought to evaluate the hypothesis that TLR4, TRIF, and MyD88 are required for hematopoietic stem and progenitor cell responses and tissue macrophage activation with obesity. Studies were carried out using hematopoietic knockout models and in vivo and in vitro assays of progenitor and macrophage activation to determine the contribution of TLR4, TRIF, and MyD88 pathways to diet-induced inflammation. We demonstrate that these pathways contribute significantly to production of CD11c+ ATMs and myelopoiesis during HFD challenge.
Results
Hematopoietic-specific TLR4 knockout animals have reduced meta-inflammation compared with WT animals after HFD challenge
Whereas prior studies have demonstrated that hematopoietic Tlr4−/− mice have improved metabolism (18), it is unclear whether TLR4 deficiency has a direct impact on myeloid cell induction. Because it has been demonstrated that Tlr4−/− animals have fewer activated CD11c+ ATMs with HFD (19), we sought to assess whether the production of CD11c+ ATMs depended upon hematopoietic TLR4 expression by generating bone marrow transplant (BMT) chimeras. Young WT and Tlr4−/− animals were irradiated and given BM of the opposite genotype, generating WT→Tlr4−/− and Tlr4−/−→WT animals. Control animals included WT→WT and Tlr4−/−→Tlr4−/−. 6 weeks after BMT, animals were started on HFD. After 16 weeks of HFD, WT→WT animals were heavier, but there were no differences in weight between the reciprocal groups (WT→Tlr4−/− and Tlr4−/−→WT) (Fig. 1A). The same pattern was seen with gonadal white adipose tissue (GWAT) weight (Fig. 1B), but the percentage of GWAT per whole-body weight was higher in Tlr4−/−→WT animals (Fig. 1C) compared with Tlr4−/− recipient animals. Subcutaneous inguinal white adipose tissue (IWAT) and liver weights were similar in all groups (Fig. S1, A–C), whereas spleen weights were slightly higher in WT recipient animals (WT→WT and Tlr4−/−→WT (Fig. S1D). Although average adipocyte size was not different in the GWAT of both groups, there was a shift toward larger adipocytes in animals with Tlr4−/− marrow (Fig. 1D). These findings suggest that TLR4 deficiency in either BM or non-BM cells impairs HFD response, as demonstrated by differences in weight gain and adiposity.
Figure 1.

Reciprocal BMTs demonstrate that hematopoietic Tlr4−/− animals respond to high-fat diet. WT and Tlr4−/− C57Bl6/J male mice were irradiated at 8 weeks of age and then transplanted with marrow of the opposite genotype: WT marrow into Tlr4−/− (WT→Tlr4−/−) versus Tlr4−/− marrow into WT mice (Tlr4−/−→WT), with appropriate WT→WT and Tlr4−/−→Tlr4−/− controls. 6 weeks after BMT, animals were started on HFD and assessed after 16 weeks for weight (A), GWAT weight (B), percentage GWAT (C), and GWAT adipocyte cross-sectional area distribution at 16 weeks of HFD (D). *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001. n = 6–8 in WT→WT and Tlr4−/−→Tlr4−/− and n = 10–14 in WT→Tlr4−/− and Tlr4−/−→WT groups. Error bars, S.E.
To determine the impact of hematopoietic TLR4 on metabolism, animals were assessed throughout the course of HFD. At 10 weeks of HFD, there were no differences in fasting glucose or fasting insulin levels (Fig. 2, A and B), but fasting free fatty acids (FFAs) were higher in WT→Tlr4−/− compared with Tlr4−/−→WT animals (Fig. 2C). At 12 weeks of HFD, a glucose tolerance test demonstrated that Tlr4−/−→WT animals had overall improved glucose tolerance (Fig. 2E), and after 16 weeks of HFD, even with similar fed glucose levels, insulin levels were lower in Tlr4−/− recipient animals (Fig. 2, D and F). These results suggest that both hematopoietic Tlr4−/− and whole-body Tlr4−/− contribute to the regulation of fatty acid and insulin levels, whereas glucose tolerance is related to hematopoietic TLR4 deficiency.
Figure 2.

Hematopoietic Tlr4−/− animals have improved glucose tolerance. Metabolic assessments were performed on WT→Tlr4−/−, Tlr4−/−→WT animals, and controls. Fasting glucose (A), fasting insulin levels (B), and fasting FFAs (C) were measured after 10 weeks of HFD. D, fed glucose levels at 16 weeks of HFD. E, glucose tolerance testing in WT→Tlr4−/− and Tlr4−/−→WT animals after 12 weeks of HFD, including area under the curve. F, fed insulin values at 16 weeks of HFD. *, p < 0.05; **, p < 0.01; n = 5–8 in WT→WT and TLR4−/− → Tlr4−/− and n = 10–14 in WT→Tlr4−/− and Tlr4−/−→WT groups. (For fed insulin levels, n = 2 for WT→WT and n = 4 for Tlr4−/−→Tlr4−/−.) Error bars, S.E.
Leukocytes were evaluated in blood 2 weeks after BMT, and animals with WT BM had more B220+ cells in circulation, suggesting faster reconstitution (Fig. S2A). Two weeks after HFD exposure, there was a significant increase in circulating B220, Ly6G, and CD115+ myeloid cells in both WT recipient animals, but Ly6chi populations were similar (Fig. 3A). Adipose tissue evaluation at 16 weeks of HFD by flow cytometry (Fig. 3B) demonstrated equal CD45+ leukocytes and ATMs in GWAT (Fig. 3D) but a larger CD11c+CD64+ ATM population in WT→Tlr4−/− compared with Tlr4−/−→WT (Fig. 3E). Further quantification showed a significant increase in CD11c+ ATMs in GWAT of WT donor animals, whereas CD11c− ATMs and dendritic cells (DCs) were equal in both groups (Fig. 3F). Gene expression showed a slight but nonsignificant decrease in Tlr4 expression in Tlr4−/−→WT GWAT (Fig. S2B) due to nonhematopoietic TLR4-expressing cells in adipose tissue.
Figure 3.

Decreased CD11c+ ATMs in hematopoietic Tlr4−/− animals on high-fat diet. A, blood leukocytes were determined by flow cytometry 2 weeks after starting HFD. B, flow cytometry plot of CD45+ cells in stromal vascular fraction. Shown is quantitation of CD45+ cells in SVF (C), ATMs (D), CD11c+ ATMs (E), and CD11c− ATMs, and dendritic cells (DCs) (F). *, p < 0.05; ***, p < 0.005; n = 5–7 in WT→WT and Tlr4−/−→Tlr4−/− and n = 11–14 in WT→Tlr4−/− and Tlr4−/−→WT groups. (For 2-week HFD blood monocytes, n = 2–4 in WT→WT, n = 3–7 in Tlr4−/−→Tlr4−/−, and n = 6–12 in WT→Tlr4−/− and Tlr4−/−→WT groups.) Error bars, S.E.
To confirm appropriate reconstitution of these mice, hematopoietic progenitors and mature leukocytes were evaluated. CD3+ and CD4+ cells were decreased in BM of Tlr4−/−→WT mice compared with WT→Tlr4−/− (Fig. S2C). Hematopoietic stem and progenitor cells were generally similar in both groups of reconstituted mice (Fig. S2D). Myeloid cells, however, were reduced in the spleen (CD11c+ and CD115+) with an increase in CD19+ cells (Fig. S2E). In the blood, CD115+ and Ly6chi monocytes were also reduced, whereas circulating CD4+ cells were increased in mice with TLR4−/− BM (Fig. S2F). Both circulating and splenic monocyte profiles were consistent with a reduction in myeloid response to HFD in the absence of TLR4.
TLR4−/− ATMs proliferate more robustly in response to HFD compared with WT ATMs
Based on our findings of decreased recruited CD11c+ ATMs in animals with Tlr4−/− marrow, we anticipated that resident Tlr4−/− ATMs may expand earlier in HFD challenge, reducing the requirement for recruitment of myeloid cells to the adipose tissue. TLR4 activation in nonhematopoietic cells has been shown to inhibit cell proliferation (21, 22). To understand the initial macrophage response to adipose tissue expansion in TLR4 knockout animals, a short-term diet model was used wherein mice at 10 weeks of age were challenged with HFD until 12 weeks of age. After 2 weeks of HFD, body weights remained consistent with normal diet (ND)-fed animals in both WT and Tlr4−/− groups (Fig. 4A). GWAT pads expanded in both WT and Tlr4−/− animals (Fig. 4B), and GWAT immunofluorescence demonstrated more proliferating (Ki67+) cells in the Tlr4−/− animals (Fig. 4C). Flow cytometry demonstrated that Tlr4−/− mice fed HFD had more ATMs (Fig. S3A and Fig. 4D). CD11c+ ATMs were increased in both WT and Tlr4−/− mice after 2 weeks of HFD, but only Tlr4−/− mice showed expanded CD11c− ATMs (Fig. 4D). DCs were unchanged in either group. When we assessed proliferation, HFD caused proliferation of both ATM types; however, there was enhanced proliferation in CD11c− ATMs of Tlr4−/− animals compared with WT CD11c− ATMs (Fig. 4E). This increase in ATM proliferation was not seen after chronic HFD feeding in GWAT and IWAT, where a large number of both WT and Tlr4−/− leukocytes were proliferating compared with ND-fed animals (Fig. S3, B and C). We further challenged 10-week-old Myd88−/− and Trif−/− mice to 2 weeks of HFD and found that Myd88−/− mice mimic the results seen with the Tlr4−/− animals (Fig. 4E). These data demonstrate that TLR4 and MyD88 inhibit the ability of resident ATMs to proliferate with adipose tissue expansion in short-term HFD exposure.
Figure 4.
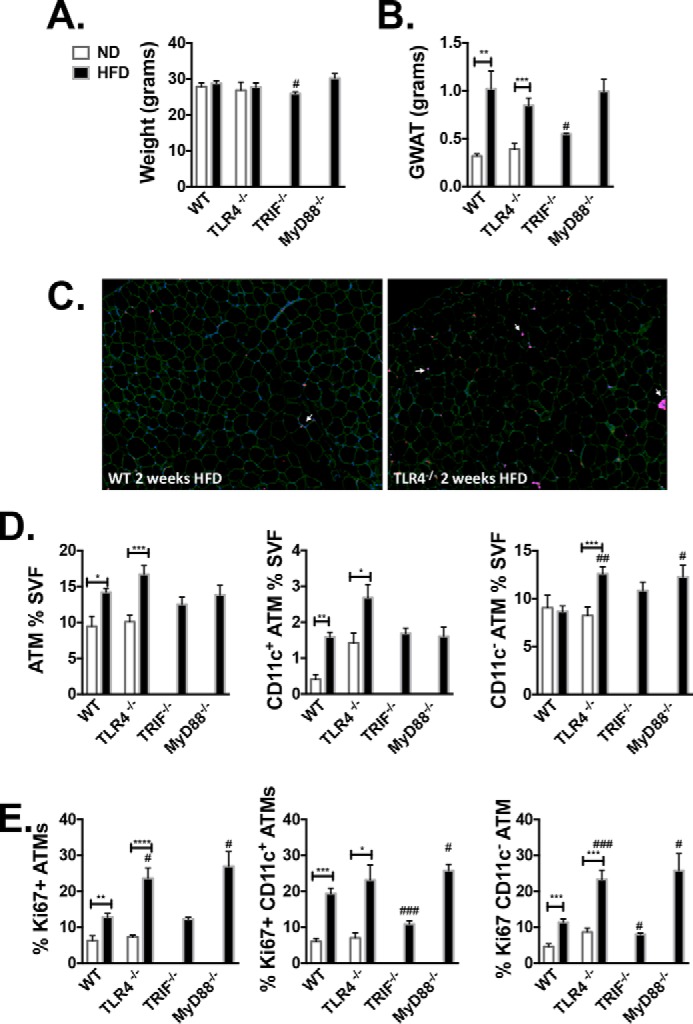
Increased CD11c− ATM proliferation in Tlr4−/− and Myd88−/− animals after short-term high-fat diet. 10-Week-old WT and TLR4−/− animals were challenged to ND or HFD for 2 weeks. At 12 weeks of age, weight (A) and GWAT (B) were assessed. C, immunostaining of GWAT with caveolin (green), Mac 2 (magenta), 4′,6-diamidino-2-phenylindole (blue), and Ki67 (red). D, ATM percentage of SVF, CD11c+ ATM, and CD11c− ATM subsets as determined by flow cytometry. E, Ki67 proliferation as determined by intracellular flow cytometry staining in GWAT ATMs (CD11c+ and CD11c− ATMs). *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001; ##, p < 0.01; ###, p < 0.005 between WT and knockout HFD (n = 4 WT ND, n = 6 Tlr4−/− ND, n = 4 WT HFD, n = 10 Tlr4−/− HFD, n = 4 Trif−/−, and n = 5 Myd88−/− HFD). Error bars, S.E.
Hematopoietic TRIF and MyD88 pathways contribute to myeloid expansion in obesity
We have previously shown that TLR4 and MyD88 pathways both regulate the generation of obesity-induced inflammation. Although TLR4 pathways were mostly necessary for long-term HSC expansion, MyD88 was mostly required for myeloid progenitor expansion, monocyte production, and hence ATM accumulation in obesity (2). Two competitive BMTs were designed to further define the role of MyD88, as well as to determine the role of the noncanonical TRIF (Ticam1) pathway in inflammation. First Trif−/− (CD45.2) and WT (CD45.1) BM was injected in a 1:1 ratio into irradiated CD45.1/CD45.2 heterozygous mice (Fig. 5A). These animals were started on HFD at 6 weeks post-BMT for 16 weeks. TRIF was required to expand pre-GMs, blood monocytes, and ATMs of both types, as seen from lower Trif−/− BM contribution to these cell types. To further delineate the role of MyD88 pathways, IRAKM, a negative regulator of MyD88 activation, was studied. Irakm−/− BM was competed with WT BM (Fig. 5B). Irakm−/− animal BM contributed to a greater number of pre-GMs, blood monocytes, and both types of ATMs, emphasizing a role for MyD88 pathways in generating bone marrow–derived myeloid cells during obesity (Fig. 5B). This same phenomenon for IRAKM pathways has been shown in LPS treatment of animals (23).
Figure 5.

In competitive BMT models, TRIF and MyD88 pathways are necessary for expansion of myeloid progenitors and ATMs in response to HFD. Lethally irradiated CD45.1/CD45.2 heterozygote animals were injected with CD45.1 WT and CD45.2 Trif−/− BM in a 1:1 ratio (A) or CD45.1 WT and CD45.2 Irakm−/− BM in a 1:1 ratio (B). The ratio of quantitation of myeloid progenitors and leukocytes in animals reconstituted after 16 weeks of HFD (long-term hematopoietic stem cells (LT-HSC), granulocyte macrophage progenitor (GMP), pre-granulocyte macrophage (Pre-GM), blood monocytes, and ATMs and CD11c subsets) was calculated. ***, p < 0.005; ****, p < 0.001. n = 9 in TRIF competitive BMT and n = 6 in IRAKM competitive BMT. Error bars, S.E.
TLR4, TRIF, and MyD88 pathway knockouts respond with increased adiposity and glucose intolerance with HFD
WT, Tlr4−/−, Trif−/−, and Myd88−/− mice were placed on 60% HFD for 16 weeks. Animals in all groups gained weight (Fig. 6A), visceral fat (Fig. 6B), and subcutaneous fat (Fig. 6C) when challenged to HFD. Interestingly, liver mass did not increase in Trif−/− and was smaller overall in both Trif−/− and Myd88−/− mice with HFD compared with WT (Fig. 6D). Spleen weights were significantly larger after HFD in WT and Trif−/− mice (Fig. 6E). GWAT demonstrated increased adipocyte hypertrophy in all groups on HFD (Fig. 6F). Hematoxylin and eosin images of GWAT showed similar adipocyte hypertrophy but an increase in CLS and fibrosis in WT HFD-fed mice (Fig. 6G). Purely based on weight and adiposity, all groups were susceptible to obesity with HFD exposure.
Figure 6.

WT, Tlr4−/−, Trif−/−, and MyD88−/− animals respond to HFD challenge. Male animals were started on HFD at 6 weeks of age for 16 weeks. Shown are weight (A), GWAT weight (B), IWAT weight (C), liver weight (D), spleen weight (E), and average cross-sectional adipocyte size (F) of GWAT fat adipocytes. G, representative hematoxylin and eosin images of GWAT. **, p < 0.01; ***, p < 0.005; ****, p < 0.001. n = 23–24 in WT groups, n = 14–21 in Tlr4−/− groups, n = 17–19 in Trif−/−, and n = 5–8 in Myd88−/− (for spleen weight, n = 15 ND and 16 HFD in WT groups, n = 4 ND and 10 HFD in Tlr4−/− groups, n = 17 ND and HFD in Trif−/−, and n = 5 ND and 7 HFD for Myd88−/−; for adipocyte sizing, n = 8–9 in WT groups, n = 3–4 in Tlr4−/− groups, and n = 5 in Trif−/− and Myd88−/−). #, p < 0.05; ##, p < 0.01; ###, p < 0.005; ####, p < 0.001 when knockout groups were compared with WT ND or HFD control. Error bars, S.E.
Fasting glucose levels were not significantly different by genotype after 10 weeks on HFD but were lower in Myd88−/− mice on HFD compared with WT (Fig. 7A). Fasting insulin levels were substantially lower in all genotypes compared with WT (Fig. 7B). Glucose tolerance tests at 12 weeks on HFD were also unaffected by genotype (Fig. 7, C and D). Fed glucose levels were higher in mice on HFD, but insulin levels, although elevated in all groups, were lower in Myd88−/− animals (Fig. S4A). Fed FFAs were increased in WT and Tlr4−/− animals on HFD, whereas HFD-fed Trif−/− mice had lower FFAs in circulation (Fig. S4B) and lower liver TGs and decreased steatosis by histology (Fig. 4, C and D), consistent with the lack of increased liver weight (Fig. 6D).
Figure 7.

WT, Tlr4−/−, Trif−/−, and Myd88−/− animals respond metabolically to HFD challenge. Shown are 10-week fasting glucose (n = 3–8) (A) and insulin (n = 3–7) (B). Shown are 12-week glucose tolerance tests (WT ND, n = 12; WT HFD, n = 21; Tlr4−/− HFD, n = 14; Trif−/− HFD, n = 4; Myd88−/− HFD, n = 6) (C), with calculated area under curve (AUC) (D). *, p < 0.05. Error bars, S.E.
TLR4, TRIF, and MyD88 pathways are required for generation of CD11c+ ATMs
To next evaluate the adipose depot–specific inflammatory responses in the knockout models, ATMs were measured in both GWAT and IWAT by flow cytometry. WT, Tlr4−/−, and Myd88−/− animals expanded total leukocytes and ATMs on HFD (Fig. 8, A and B), but Trif−/− animals preferentially increased DCs in IWAT with no change in ATMs or total leukocytes (Fig. 8E). Whereas WT, Tlr4−/−, and Myd88−/− animals expanded CD11c+ ATMs on HFD, CD11c+ ATMs were significantly higher in WT HFD-fed animals when compared with Tlr4−/− and Myd88−/− HFD-fed animals (p < 0.01) (Fig. 8C). CD11c− ATM populations also expanded in obese Tlr4−/− and Myd88−/− mice similarly to WT mice; however, Trif−/− animals exhibited lower numbers of CD11c− ATMs (Fig. 8D). DCs were not significantly different in HFD-fed animals except for in the IWAT of Tlr4−/− and Trif−/− animals, and levels were increased in GWAT of Myd88−/− mice (Fig. 8E). This pattern of increased CD11c+ ATMs in WT mice was supported by the appearance of more CLS in visceral adipose tissue by immunofluorescence in WT HFD-fed mice (Fig. 8F).
Figure 8.

Tlr4−/−, Trif−/−, and Myd88−/− animals are protected from adipose tissue inflammation. After 16 weeks of HFD challenge, flow cytometry assessments were performed for CD45+ cells (A), ATMs (B), CD11c+ ATMs (C), CD11c− ATMs (D), and DCs (E) (n = 12–21 in WT groups, n = 10–17 in Tlr4−/− groups, n = 7–17 in Trif−/−, and n = 5–8 in Myd88−/−). F, immunofluorescence in GWAT (caveolin (green) and Mac2 (magenta)). G, quantitative real-time PCR (n = 11 WT ND, n = 20 WT HFD, n = 10 Tlr4−/− HFD, n = 5 Trif−/− HFD, and n = 6 MyD88−/− HFD). H, flow cytometry evaluation of hematopoietic progenitors (n = 8 WT ND and HFD, n = 11 Tlr4−/−, n = 5 Trif−/−, and n = 8 Myd88−/−). *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001; #, p < 0.05; ##, p < 0.01; ###, p < 0.005; ####, p < 0.001. Error bars, S.E.
Gene expression analysis was examined in GWAT to confirm lack of Tlr4, Ticam1 (Trif), and Myd88 in the appropriate knockout animals (Fig. 8G). Evaluation of genes associated with adipose function showed increased Tgfb expression in GWAT with HFD, but no significant differences by genotype. Glut4 and Insr expression levels were also reduced equally between genotypes (Fig. 8G), consistent with a decrease in insulin sensitivity. Mcp1 was increased with HFD in WT, Tlr4−/−, Trif−/−, and Myd88−/− animals, although Il1b was highest in WT HFD-fed mice (Fig. 8G). Genes generally activated in CD11c− alternative resident macrophages, such as Arg1 and Tgfb, showed increased expression in all groups. Hematopoietic compartment analysis demonstrated that granulocyte macrophage progenitors were only increased in WT animals on HFD (Fig. 8H), whereas pre-GMs were increased in all groups except the Myd88−/− HFD animals. Overall, these experiments demonstrate that the TLR4, TRIF, and MyD88 pathways are necessary for CD11c+ ATM accumulation and myeloid progenitor expansion with HFD.
TLR4, MyD88, and TRIF are required for hematopoietic responses to saturated fatty acid
We employed in vitro myeloid colony-forming unit (cfu) assays to assess stem cell capacity in the obese knockout mice. Compared with WT HFD-fed controls, TLR4, TRIF, and MyD88 knockouts generated fewer myeloid colonies (Fig. 9A). To understand the mechanism of this effect, we stimulated BM with fatty acids or LPS ex vivo and then performed myeloid CFU assays. For these assays, we used saturated fatty acids (palmitic and stearic acid, 10 μm) and unsaturated fatty acids (linoleic and oleic acid, 10 μm). Whereas WT marrow increased cfu generation in response to saturated fatty acids, Tlr4−/−, Trif−/−, and Myd88−/− marrow did not respond with a similar increase in cfu (Fig. 9B). Similar results were seen with LPS treatment. Overall, oleic acid (an unsaturated fatty acid) decreased the saturated fatty acid response (Fig. 9B).
Figure 9.

TLR4, MyD88, and TRIF are necessary for HFD and saturated fatty acid stimulation of myeloid colonies. After 16 weeks of ND or HFD, bone marrow was collected for myeloid methylcellulose colony-forming assays. A, ratio of HFD to ND myeloid colonies for WT, Tlr4−/−, Trif−/−, and Myd88−/− animals (n = 3–6). B, BM isolated from WT, Tlr4−/−, Trif−/−, and Myd88−/− animals were treated with fatty acid–free BSA or 10 μm fatty acid (PA, stearic acid (SA), linoleic acid (LA), oleic acid (OA), or LPS), and then colonies were counted after 7 days (n = 3 LPS and n = 6–15 for other groups). C, sorted lineage-negative or -positive cfu after BM treated with BSA or PA (n = 9 lineage-positive and n = 18 lineage-negative). D, myeloid colonies from lineage-negative ND and HFD-fed WT and Tlr4−/− animals (n = 6). E, lineage-negative marrow from WT, Trif−/−, and Myd88−/− mice treated with BSA or PA (n = 9–14). F, gene expression from isolated colonies (n = 13). *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001 compared with control. #, p < 0.05; ###, p < 0.005; ####, p < 0.001 when knockout groups were compared with WT. Error bars, S.E.
To determine whether this fatty acid response in myeloid cells was due to stimulation of mature cells or progenitors, we isolated lineage-negative (Lin−) cells (immature noncommitted bone marrow cells), followed by treatment of isolated cells with palmitate. Palmitic acid stimulated WT lineage negative marrow (Fig. 9C), whereas Tlr4−/− lineage-negative BM was resistant to this stimulation and yielded lower colonies compared with WT lineage-negative PA treatment (p = 0.06). Comparison of WT and Tlr4−/− ND and HFD animals showed a brisk response to PA in ND WT mice that was blunted in HFD WT mice. Tlr4−/− mice on ND or HFD failed to induce colony formation in response to PA in Lin− cells (Fig. 9D). Lin− TRIF and MyD88 knockout mice demonstrated that Myd88−, but not Trif, was required for myeloid expansion in response to PA (Fig. 9E); however, both pathways yielded lower myeloid colonies compared with WT mice. This was consistent with our prior research (2) that determined that MyD88 pathways are critical for myeloid inflammation in obesity. Gene expression studies in harvested cells treated with PA (Fig. 9F) demonstrated that palmitate stimulation in WT mice, but not Tlr4−/− animals, induced Mcp1 expression (analysis of variance, p < 0.05).
Discussion
Our findings show that there are distinct roles for TLR4, TRIF, and MyD88 in the inflammatory response to obesity. Prior studies have identified a role for TLR4 and NF-κB (24) in obesity-induced inflammation and insulin resistance (25). Our studies demonstrate that TLR4 reduces local (CD11c−) macrophage proliferation in response to adipose tissue expansion, promotes recruitment of CD11c+ ATM accumulation, and is also necessary for the generation of bone marrow–derived macrophages in response to saturated fatty acid.
Spontaneous mutants lacking TLR4 function are protected from insulin resistance (26), but studies have shown mixed effects of TLR4 knockouts in the metabolic response to HFD. Our reciprocal BMTs suggest that this may be because of opposing TLR4 function in bone marrow–derived and nonhematopoietic compartments, such as adipocytes. Whereas animals reconstituted with Tlr4−/− BM had improved metabolism, glucose tolerance in nonirradiated Tlr4−/− mice on HFD showed similar impairment as WT HFD-fed animals (Fig. 7). Tlr4−/− mice gained significantly more weight when nonirradiated, suggesting that the protection conferred by hematopoietic TLR4 deficiency may be overcome by excess adiposity. TLR4 has a known role in adipocytes (27), especially in short-term HFD, and in recruitment of inflammatory leukocytes (28). Once chronic obesity has led to expansion of adipose tissue, there is probably a secondary role for TLR4 in the generation of fibrosis, promoting further adipose tissue dysfunction (20).
TLR4 and MyD88 have specifically been reported to be critical for ATMs that are “metabolically activated” and play an important role in adipose tissue homeostasis (30). Our results suggest that MyD88 deficiency protects from meta-inflammation because MyD88 is required for accumulation of activated ATMs and enhanced myelopoiesis. Our results are consistent with previous studies (31, 32) that have implicated MyD88 in its contribution to obesity-induced adipose tissue inflammation (31), atherosclerosis (31, 33), and improved glucose sensitivity (34). Studies have previously demonstrated that MyD88 is necessary for TLR2 and TLR4 saturated fatty acid–induced inflammatory activation of mature macrophages (35). Additionally, our results demonstrate that MyD88 pathways are also responsible for the HSC responses to saturated fatty acids in obesity.
Whereas MyD88-independent TRIF signaling has been implicated in hematopoiesis (36) and atherosclerosis (37), there is a paucity of data on the role of TRIF in meta-inflammation. We found that Lin− Trif−/− progenitors produced myeloid colonies in response to palmitate stimulation in vitro. In vivo, however, Trif−/− animals had fewer myeloid progenitors and ATMs with HFD challenge when compared with WT animals. It is interesting to note that although these animals gained weight and adiposity, Trif−/− mice were protected from hepatic steatosis and triglyceride accumulation during HFD. This finding is contrary to what has been observed in models of nonalcoholic steatohepatitis, where Trif−/− animals generally have increased inflammation and fibrosis due to enhanced stellate cell chemokine and cytokine activation of the MyD88 pathway, demonstrating that TRIF is probably important for inhibiting inflammation in the liver (38, 39). However, inflammation is provoked in those studies through amino acid deficiency as opposed to HFD-induced obesity stimulation. This highlights the importance of source and degree of stimulation when evaluating steatosis and hepatic inflammation (40, 41). This also emphasizes that there may be a potential different role for TRIF pathways in the adipose tissue protecting HFD-fed Trif−/− mice from adipose tissue inflammation and dysfunction, leading to protection from fatty acid release and steatosis.
The role of TLR4 pathways in biasing hematopoietic progenitor expansion (42) toward myelopoiesis (43) with LPS stimulation has been demonstrated in acute settings but remains unresolved in the chronic inflammatory state of obesity. The studies here reveal that TLR4 through TRIF and MyD88 signaling cascades are important for hematopoietic contribution to myeloid cells and macrophage polarization in obesity-induced inflammation. The importance of TLR4 in bone marrow–derived myeloid cells for insulin resistance during obesity has only recently been characterized (2, 44, 45), but with a limited understanding of its triggers. Our BM studies indicate that saturated fatty acids have a direct role in enhancing myeloid progenitors via stimulation of TLR4, TRIF, and MyD88 pathways.
Whereas this direct effect of fatty acids on hematopoietic progenitors is novel, it is probably one of many factors altered with HFD, given that increased endotoxins (46) and microbiota changes have also been demonstrated to enhance ATM recruitment via TLR4 (47).
Another limitation of our studies is that the TRIF and MyD88 knockout animal results are possibly confounded by signaling pathways from other TLRs that are activated during an obese state, including TLR2 (48, 49). Also, it is not clear whether this increased TLR4 activation in hematopoietic progenitors through TRIF pathways leads to tolerance or exhaustion in chronic HFD, as has been demonstrated in LPS stimulation (36, 50). Timing and dose of stimulation are critical in LPS studies (51) and hence may not mimic findings in obesity. Further, whereas we were able to characterize the role of TLR4 via MyD88 for inhibition of local CD11c− ATM proliferation and the role of both TRIF and MyD88 for myeloid progenitor expansion, the downstream pathways from this initial signaling are yet to be determined.
Overall, our studies demonstrate that TLR4, TRIF, and MyD88 play critical roles in the profile of ATMs in visceral fat after acute and chronic HFD exposure. In addition, deficiency of any of these pathways reduces myeloid stimulation of bone marrow with saturated fatty acids. These results emphasize that when evaluating obesity-induced inflammation, the tissue-specific macrophage response may only be part of the underlying effect and that there is an additional myeloid progenitor alteration that occurs with obesity.
Experimental procedures
Animal models and treatments
C57Bl/6J (WT), Tlr4−/− (B6.B10ScN-Tlr4lps-del/JthJ; 007227), and Myd88−/− (B6.129P2 (SJL)-Myd88tm.1.1Defr/J; 009088) mice on C57Bl/6J background were purchased from Jackson Laboratories. Trif−/− mice were donated from Dr. Gabriel Nunez's laboratory, originally from the Akira laboratory (52). Irakm−/− mice were donated from a colony bred on a B6 background that was established at the University of Michigan (53). All mice were male and were fed ad libitum either a control ND consisting of 4.5% fat (5001; LabDiet) or an HFD consisting of 60% of calories from fat (Research Diets, Inc., D12492) starting at 6 weeks of age for 16 weeks duration. Glucose tolerance (with 0.7 g/kg) and insulin tolerance (with 1 unit/kg) testing were performed after 6 h of fasting. Animals were housed in a specific pathogen-free facility with a 12-h light/12-h dark cycle and given free access to food and water. Animal protocols were in compliance with the Institute of Laboratory Animal Research Guide for the Care and Use of Laboratory Animals and approved by the University Committee on Use and Care of Animals at the University of Michigan (animal welfare assurance number A3114-01).
Quantitative real-time PCR
RNA extraction was performed with an RNeasy kit (Qiagen) followed by reverse transcription (Applied Biosystems) and real-time PCR analysis using glyceraldehyde-3-phosphate dehydrogenase to normalize (SYBR Green, ABI Prism 7200 Sequence Detection System; Applied Biosystems). Relative expression was assessed by the comparative CT method correcting for amplification efficiency of the primers and performed in duplicate as described previously (2). PCR primers used are reported in Table 1.
Table 1.
Primer sequences
| Forward primer | Reverse primer | |
|---|---|---|
| Gapdh | TGAAGCAGGCATCTGAGGG | CGAAGGTGGAAGAGTGGGAG |
| Il6 | TAGTCCTTCCTACCCCAATTTCC | AAGGAACCCTTAGAGTGCTTACT |
| Mcp1 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| Il1b | AAATACCTGTGGCCTTGGGC | CTTGGGATCCACACTCTCCAG |
| Arg1 | CTCCAAGCCAAAGTCCTTAGAG | AGGAGCTGTCATTAGGGACATC |
| Tgfb | GGACTCTCCACCTGCAAGAC | GACTGGCGAGCCTTAGTTTG |
| Tlr4 | ATGGCATGGCTTACACCACC | GAGGCCAATTTTGTCTCCACA |
| Ticam1 | CCAGCTCAAGACCCCTACAG | CAAGGCACCTAGAATGCCAAA |
| Myd88 | AGGACAAACGCCGGAACTTTT | GCCGATAGTCTGTCTGTTCTAGT |
| Ir | TTTGTCATGGATGGAGGCTA | CCTCATCTTGGGGTTGAACT |
| Glut4 | GTGACTGGAACACTGGTCCTA | CCAGCCACGTTGCATTGTAG |
Adipose tissue stromal vascular fraction (SVF) isolation and flow cytometry
Adipose tissue fractionation and flow cytometry analyses were performed as described previously after Fc blocking of samples (54). SVF cells were stained with CD64 PE, CD45.2 e450, and CD11c-APC-Cy7 or APC (eBioscience) (55) for ATMs.
Flow cytometry assessment of HSC and myeloid progenitors
Bone marrow from one femur was flushed with PBS and made into single-cell suspension using a syringe and then centrifuged. Thereafter, cell pellets were treated with RBC lysis solution for 5 min. After resuspension in PBS, cells were stained with lineage markers on APC (CD4, CD5, CD8, CD11b, B220 (CD45R), Gr1, Ter119), CD117-APC-Cy7, Sca1-PECy7, CD16/32 PerCP5.5 (eBioscience), CD150-PE, Endoglin-Pacific Blue (Biolegend), CD48 FITC, and gating as described by Pronk et al. (56) and Oguro et al. (57).
cfu assays
Bone marrow from a femur was flushed with Iscove's modified Dulbecco's medium. This marrow was then treated with fatty acid–free BSA, with fatty acids (palmitic acid, linoleic acid, oleic acid, stearic acid) (10 μm, purchased from Sigma) in fatty acid–free BSA, or with 10 μg/ml LPS for 1 h at 37 ºC. Cells were then resuspended in MethoCult medium and plated at a density of 10,000 cells/plate/protocol (Stem Cell Technology). After 7 days, colonies were counted. For sorted cfu assays, bone marrow was isolated from animals, and lineage-negative cells were separated using a stem-cell magnet column isolation kit (EasySepTM hematopoietic progenitor cell enrichment kit). After magnetic separation, 1000 cells were used per plate.
Bone marrow transplantation
Bone marrow cells were isolated from donor groups (58) and injected retro-orbitally into lethally irradiated (900 rads) 6-week-old recipient mice (10 million cells/mouse). Animals were treated with antibiotics (polymyxin and neomycin) for 4 weeks after BMT. Following 2 weeks of normal chow diet, they were started on ND or HFD chow. Glucose tolerance testing was performed as described previously (29).
Statistics
Results are presented as mean ± S.E. One-way or two-way analysis of variance was performed with factors of genotype and diet. If there was a main effect for either factor, then t tests were performed for WT versus knockout differences within each diet or for diet groups within each group, respectively.
Author contributions
C. G., L. E., N. L., S. A., M. V., K. M., L. M., J. L., and K. S. data curation; C. G., L. E., N. L., S. A., K. M., J. L., and K. S. formal analysis; C. G., S. A., and K. S. supervision; C. G. and K. S. methodology; C. G., S. A., and K. S. writing-original draft; L. E., N. L., M. V., C. N. L., and K. S. writing-review and editing; S. A. and K. S. investigation; L. M., C. N. L., and K. S. conceptualization; C. N. L. resources; C. N. L. and K. S. funding acquisition; K. S. validation; K. S. visualization; K. S. project administration.
Supplementary Material
Acknowledgments
We thank Nidhi Maley, Dr. Brian Zamarron, Dr. Gabriel Martinez-Santibanez, Dr. Eric Chang, and Devyani Agarwal for assistance during experiments. This work utilized Core Services from the Michigan Nutrition and Obesity Research Center supported by National Institutes of Health Grant DK089503 to the University of Michigan.
This work was supported by the University of Michigan Department of Pediatrics Janette Ferrantino Investigator Award; NIDDK, National Institutes of Health, Grant K08DK101755; and Edith Briskin/SKS Foundation Taubman Emerging Scholar support (to K. S.); National Institutes of Health Grants DK090262 (to C. N. L.); and National Institutes of Health Grant F32DK105676 (to L. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
- WAT
- white adipose tissue
- ATM
- adipose tissue macrophage
- HSC
- hematopoietic stem cells
- TLR
- Toll-like receptor
- TRIF
- TIR domain–containing adapter–inducing interferon-β
- MyD88
- myeloid differentiation primary response 88
- IRAKM
- interleukin-1 receptor–associated kinase-M
- cfu
- colony-forming unit(s)
- HFD
- high-fat diet
- ND
- normal diet
- PA
- palmitic acid
- GWAT
- gonadal white adipose tissue
- IWAT
- inguinal white adipose tissue
- GM
- granulocyte macrophage
- APC
- allophycocyanin
- CLS
- crown-like structures
- LPS
- lipopolysaccharide
- BMT
- bone marrow transplant
- FFA
- free fatty acid
- DC
- dendritic cell
- BM
- bone marrow
- SVF
- stromal vascular fraction.
References
- 1. Saltiel A. R., and Olefsky J. M. (2017) Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Invest. 127, 1–4 10.1172/JCI92035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Singer K., DelProposto J., Morris D. L., Zamarron B., Mergian T., Maley N., Cho K. W., Geletka L., Subbaiah P., Muir L., Martinez-Santibanez G., and Lumeng C. N. (2014) Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol. Metab. 3, 664–675 10.1016/j.molmet.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amano S. U., Cohen J. L., Vangala P., Tencerova M., Nicoloro S. M., Yawe J. C., Shen Y., Czech M. P., and Aouadi M. (2014) Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 19, 162–171 10.1016/j.cmet.2013.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lumeng C. N., Bodzin J. L., and Saltiel A. R. (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117, 175–184 10.1172/JCI29881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crewe C., An Y. A., and Scherer P. E. (2017) The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J. Clin. Invest. 127, 74–82 10.1172/JCI88883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carvalheira J. B., Qiu Y., and Chawla A. (2013) Blood spotlight on leukocytes and obesity. Blood 122, 3263–3267 10.1182/blood-2013-04-459446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wentworth J. M., Naselli G., Brown W. A., Doyle L., Phipson B., Smyth G. K., Wabitsch M., O'Brien P. E., and Harrison L. C. (2010) Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes 59, 1648–1656 10.2337/db09-0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sartipy P., and Loskutoff D. J. (2003) Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 100, 7265–7270 10.1073/pnas.1133870100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cho K. W., Zamarron B. F., Muir L. A., Singer K., Porsche C. E., DelProposto J. B., Geletka L., Meyer K. A., O'Rourke R. W., and Lumeng C. N. (2016) Adipose tissue dendritic cells are independent contributors to obesity-induced inflammation and insulin resistance. J. Immunol. 197, 3650–3661 10.4049/jimmunol.1600820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu X., Grijalva A., Skowronski A., van Eijk M., Serlie M. J., and Ferrante A. W. Jr. (2013) Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 18, 816–830 10.1016/j.cmet.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kratz M., Coats B. R., Hisert K. B., Hagman D., Mutskov V., Peris E., Schoenfelt K. Q., Kuzma J. N., Larson I., Billing P. S., Landerholm R. W., Crouthamel M., Gozal D., Hwang S., Singh P. K., and Becker L. (2014) Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 20, 614–625 10.1016/j.cmet.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li P., Lu M., Nguyen M. T., Bae E. J., Chapman J., Feng D., Hawkins M., Pessin J. E., Sears D. D., Nguyen A. K., Amidi A., Watkins S. M., Nguyen U., and Olefsky J. M. (2010) Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. J. Biol. Chem. 285, 15333–15345 10.1074/jbc.M110.100263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Singer K., Maley N., Mergian T., DelProposto J., Cho K. W., Zamarron B. F., Martinez-Santibanez G., Geletka L., Muir L., Wachowiak P., Demirjian C., and Lumeng C. N. (2015) Differences in hematopoietic stem cells contribute to sexually dimorphic inflammatory responses to high fat diet-induced obesity. J. Biol. Chem. 290, 13250–13262 10.1074/jbc.M114.634568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lawrence T. (2009) The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1, a001651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rossol M., Heine H., Meusch U., Quandt D., Klein C., Sweet M. J., and Hauschildt S. (2011) LPS-induced cytokine production in human monocytes and macrophages. Crit. Rev. Immunol. 31, 379–446 10.1615/CritRevImmunol.v31.i5.20 [DOI] [PubMed] [Google Scholar]
- 16. Akira S., and Takeda K. (2004) Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 10.1038/nri1391 [DOI] [PubMed] [Google Scholar]
- 17. Shi H., Kokoeva M. V., Inouye K., Tzameli I., Yin H., and Flier J. S. (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 116, 3015–3025 10.1172/JCI28898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saberi M., Woods N. B., de Luca C., Schenk S., Lu J. C., Bandyopadhyay G., Verma I. M., and Olefsky J. M. (2009) Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 10, 419–429 10.1016/j.cmet.2009.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Orr J. S., Puglisi M. J., Ellacott K. L., Lumeng C. N., Wasserman D. H., and Hasty A. H. (2012) Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes 61, 2718–2727 10.2337/db11-1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vila I. K., Badin P. M., Marques M. A., Monbrun L., Lefort C., Mir L., Louche K., Bourlier V., Roussel B., Gui P., Grober J., Štich V., Rossmeislová L., Zakaroff-Girard A., Bouloumié A., et al. (2014) Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 7, 1116–1129 10.1016/j.celrep.2014.03.062 [DOI] [PubMed] [Google Scholar]
- 21. Wang Y., Abarbanell A. M., Herrmann J. L., Weil B. R., Manukyan M. C., Poynter J. A., and Meldrum D. R. (2010) TLR4 inhibits mesenchymal stem cell (MSC) STAT3 activation and thereby exerts deleterious effects on MSC-mediated cardioprotection. PLoS One 5, e14206 10.1371/journal.pone.0014206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sodhi C. P., Shi X. H., Richardson W. M., Grant Z. S., Shapiro R. A., Prindle T. Jr., Branca M., Russo A., Gribar S. C., Ma C., and Hackam D. J. (2010) Toll-like receptor-4 inhibits enterocyte proliferation via impaired β-catenin signaling in necrotizing enterocolitis. Gastroenterology 138, 185–196 10.1053/j.gastro.2009.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kobayashi K., Hernandez L. D., Galán J. E., Janeway C. A. Jr., Medzhitov R., and Flavell R. A. (2002) IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110, 191–202 10.1016/S0092-8674(02)00827-9 [DOI] [PubMed] [Google Scholar]
- 24. Gregor M. F., and Hotamisligil G. S. (2011) Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 29, 415–445 10.1146/annurev-immunol-031210-101322 [DOI] [PubMed] [Google Scholar]
- 25. Deng Z. B., Poliakov A., Hardy R. W., Clements R., Liu C., Liu Y., Wang J., Xiang X., Zhang S., Zhuang X., Shah S. V., Sun D., Michalek S., Grizzle W. E., Garvey T., Mobley J., and Zhang H. G. (2009) Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes 58, 2498–2505 10.2337/db09-0216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Poggi M., Bastelica D., Gual P., Iglesias M. A., Gremeaux T., Knauf C., Peiretti F., Verdier M., Juhan-Vague I., Tanti J. F., Burcelin R., and Alessi M. C. (2007) C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 50, 1267–1276 10.1007/s00125-007-0654-8 [DOI] [PubMed] [Google Scholar]
- 27. Norseen J., Hosooka T., Hammarstedt A., Yore M. M., Kant S., Aryal P., Kiernan U. A., Phillips D. A., Maruyama H., Kraus B. J., Usheva A., Davis R. J., Smith U., and Kahn B. B. (2012) Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol-independent mechanism. Mol. Cell. Biol. 32, 2010–2019 10.1128/MCB.06193-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tao C., Holland W. L., Wang Q. A., Shao M., Jia L., Sun K., Lin X., Kuo Y. C., Johnson J. A., Gordillo R., Elmquist J. K., and Scherer P. E. (2017) Short-term versus long-term effects of adipocyte Toll-like receptor 4 activation on insulin resistance in male mice. Endocrinology 158, 1260–1270 10.1210/en.2017-00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Westcott D. J., Delproposto J. B., Geletka L. M., Wang T., Singer K., Saltiel A. R., and Lumeng C. N. (2009) MGL1 promotes adipose tissue inflammation and insulin resistance by regulating 7/4hi monocytes in obesity. J. Exp. Med. 206, 3143–3156 10.1084/jem.20091333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Coats B. R., Schoenfelt K. Q., Barbosa-Lorenzi V. C., Peris E., Cui C., Hoffman A., Zhou G., Fernandez S., Zhai L., Hall B. A., Haka A. S., Shah A. M., Reardon C. A., Brady M. J., Rhodes C. J., et al. (2017) Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet-induced obesity. Cell Rep. 20, 3149–3161 10.1016/j.celrep.2017.08.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu M., Zhou H., Zhao J., Xiao N., Roychowdhury S., Schmitt D., Hu B., Ransohoff R. M., Harding C. V., Hise A. G., Hazen S. L., DeFranco A. L., Fox P. L., Morton R. E., Dicorleto P. E., et al. (2014) MyD88-dependent interplay between myeloid and endothelial cells in the initiation and progression of obesity-associated inflammatory diseases. J. Exp. Med. 211, 887–907 10.1084/jem.20131314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nagareddy P. R., Kraakman M., Masters S. L., Stirzaker R. A., Gorman D. J., Grant R. W., Dragoljevic D., Hong E. S., Abdel-Latif A., Smyth S. S., Choi S. H., Korner J., Bornfeldt K. E., Fisher E. A., Dixit V. D., et al. (2014) Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 19, 821–835 10.1016/j.cmet.2014.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kleinridders A., Schenten D., Könner A. C., Belgardt B. F., Mauer J., Okamura T., Wunderlich F. T., Medzhitov R., and Brüning J. C. (2009) MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 10, 249–259 10.1016/j.cmet.2009.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hosoi T., Yokoyama S., Matsuo S., Akira S., and Ozawa K. (2010) Myeloid differentiation factor 88 (MyD88)-deficiency increases risk of diabetes in mice. PLoS One 5, e12537 10.1371/journal.pone.0012537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang S., Rutkowsky J. M., Snodgrass R. G., Ono-Moore K. D., Schneider D. A., Newman J. W., Adams S. H., and Hwang D. H. (2012) Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 53, 2002–2013 10.1194/jlr.D029546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takizawa H., Fritsch K., Kovtonyuk L. V., Saito Y., Yakkala C., Jacobs K., Ahuja A. K., Lopes M., Hausmann A., Hardt W. D., Gomariz Á., Nombela-Arrieta C., and Manz M. G. (2017) Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell 21, 225–240.e5 10.1016/j.stem.2017.06.013 [DOI] [PubMed] [Google Scholar]
- 37. Richards M. R., Black A. S., Bonnet D. J., Barish G. D., Woo C. W., Tabas I., Curtiss L. K., and Tobias P. S. (2013) The LPS2 mutation in TRIF is atheroprotective in hyperlipidemic low density lipoprotein receptor knockout mice. Innate Immun. 19, 20–29 10.1177/1753425912447130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang L., Miura K., Zhang B., Matsushita H., Yang Y. M., Liang S., Song J., Roh Y. S., and Seki E. (2017) TRIF differentially regulates hepatic steatosis and inflammation/fibrosis in mice. Cell Mol. Gastroenterol. Hepatol. 3, 469–483 10.1016/j.jcmgh.2016.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen J., Li J., Yiu J. H. C., Lam J. K. W., Wong C. M., Dorweiler B., Xu A., and Woo C. W. (2017) TRIF-dependent Toll-like receptor signaling suppresses Scd1 transcription in hepatocytes and prevents diet-induced hepatic steatosis. Sci. Signal. 10, eaal3336 10.1126/scisignal.aal3336 [DOI] [PubMed] [Google Scholar]
- 40. Deng Z. B., Liu Y., Liu C., Xiang X., Wang J., Cheng Z., Shah S. V., Zhang S., Zhang L., Zhuang X., Michalek S., Grizzle W. E., and Zhang H. G. (2009) Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology 50, 1412–1420 10.1002/hep.23148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li L., Chen L., Hu L., Liu Y., Sun H. Y., Tang J., Hou Y. J., Chang Y. X., Tu Q. Q., Feng G. S., Shen F., Wu M. C., and Wang H. Y. (2011) Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology 54, 1620–1630 10.1002/hep.24552 [DOI] [PubMed] [Google Scholar]
- 42. Liu A., Wang Y., Ding Y., Baez I., Payne K. J., and Borghesi L. (2015) Cutting edge: hematopoietic stem cell expansion and common lymphoid progenitor depletion require hematopoietic-derived, cell-autonomous TLR4 in a model of chronic endotoxin. J. Immunol. 195, 2524–2528 10.4049/jimmunol.1501231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boettcher S., Ziegler P., Schmid M. A., Takizawa H., van Rooijen N., Kopf M., Heikenwalder M., and Manz M. G. (2012) Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. J. Immunol. 188, 5824–5828 10.4049/jimmunol.1103253 [DOI] [PubMed] [Google Scholar]
- 44. Razolli D. S., Moraes J. C., Morari J., Moura R. F., Vinolo M. A., and Velloso L. A. (2015) TLR4 expression in bone marrow-derived cells is both necessary and sufficient to produce the insulin resistance phenotype in diet-induced obesity. Endocrinology 156, 103–113 10.1210/en.2014-1552 [DOI] [PubMed] [Google Scholar]
- 45. Liu A., Chen M., Kumar R., Stefanovic-Racic M., O'Doherty R. M., Ding Y., Jahnen-Dechent W., and Borghesi L. (2018) Bone marrow lympho-myeloid malfunction in obesity requires precursor cell-autonomous TLR4. Nat. Commun. 9, 708 10.1038/s41467-018-03145-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim K. A., Gu W., Lee I. A., Joh E. H., and Kim D. H. (2012) High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS One 7, e47713 10.1371/journal.pone.0047713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Caesar R., Tremaroli V., Kovatcheva-Datchary P., Cani P. D., and Bäckhed F. (2015) Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab. 22, 658–668 10.1016/j.cmet.2015.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Davis J. E., Braucher D. R., Walker-Daniels J., and Spurlock M. E. (2011) Absence of Tlr2 protects against high-fat diet-induced inflammation and results in greater insulin-stimulated glucose transport in cultured adipocytes. J. Nutr. Biochem. 22, 136–141 10.1016/j.jnutbio.2009.12.008 [DOI] [PubMed] [Google Scholar]
- 49. Ehses J. A., Meier D. T., Wueest S., Rytka J., Boller S., Wielinga P. Y., Schraenen A., Lemaire K., Debray S., Van Lommel L., Pospisilik J. A., Tschopp O., Schultze S. M., Malipiero U., Esterbauer H., et al. (2010) Toll-like receptor 2-deficient mice are protected from insulin resistance and β cell dysfunction induced by a high-fat diet. Diabetologia 53, 1795–1806 10.1007/s00125-010-1747-3 [DOI] [PubMed] [Google Scholar]
- 50. Schuettpelz L. G., and Link D. C. (2013) Regulation of hematopoietic stem cell activity by inflammation. Front. Immunol. 4, 204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morris M. C., Gilliam E. A., and Li L. (2014) Innate immune programing by endotoxin and its pathological consequences. Front. Immunol. 5, 680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shindou H., Ishii S., Yamamoto M., Takeda K., Akira S., and Shimizu T. (2005) Priming effect of lipopolysaccharide on acetyl-coenzyme A:lyso-platelet-activating factor acetyltransferase is MyD88 and TRIF independent. J. Immunol. 175, 1177–1183 10.4049/jimmunol.175.2.1177 [DOI] [PubMed] [Google Scholar]
- 53. Deng J. C., Cheng G., Newstead M. W., Zeng X., Kobayashi K., Flavell R. A., and Standiford T. J. (2006) Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J. Clin. Invest. 116, 2532–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Morris D. L., Oatmen K. E., Wang T., DelProposto J. L., and Lumeng C. N. (2012) CX3CR1 deficiency does not influence trafficking of adipose tissue macrophages in mice with diet-induced obesity. Obesity 20, 1189–1199 10.1038/oby.2012.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lumeng C. N., DelProposto J. B., Westcott D. J., and Saltiel A. R. (2008) Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 57, 3239–3246 10.2337/db08-0872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pronk C. J., Rossi D. J., Månsson R., Attema J. L., Norddahl G. L., Chan C. K., Sigvardsson M., Weissman I. L., and Bryder D. (2007) Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell 1, 428–442 10.1016/j.stem.2007.07.005 [DOI] [PubMed] [Google Scholar]
- 57. Oguro H., Ding L., and Morrison S. J. (2013) SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 13, 102–116 10.1016/j.stem.2013.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Erickson J. C., Clegg K. E., and Palmiter R. D. (1996) Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature 381, 415–421 10.1038/381415a0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.