

Abstract
Antibody (Ab) fragments have great clinical potential as cancer therapeutics and diagnostics. Their small size allows for fast clearance from blood, low immunoreactivity, better tumor penetration, and simpler engineering and production. The smallest fragment derived from a full-length IgG that retains binding to its antigen, the single-chain variable fragment (scFV), is engineered by fusing the variable light and variable heavy domains with a peptide linker. Along with switching the domain orientation, altering the length and amino acid sequence of the linker can significantly affect scFV binding, stability, quaternary structure, and other biophysical properties. Comprehensive studies of these attributes in a single scaffold have not been reported, making design and optimization of Ab fragments challenging. Here, we constructed libraries of 3E8, an Ab specific to tumor-associated glycoprotein 72 (TAG-72), a mucinous glycoprotein overexpressed in 80% of adenocarcinomas. We cloned, expressed, and characterized scFVs, diabodies, and higher-order multimer constructs with varying linker compositions, linker lengths, and domain orientations. These constructs dramatically differed in their oligomeric states and stabilities, not only because of linker and orientation but also related to the purification method. For example, protein L–purified constructs tended to have broader distributions and higher oligomeric states than has been reported previously. From this library, we selected an optimal construct, 3E8.G4S, for biodistribution and pharmacokinetic studies and in vivo xenograft mouse PET imaging. These studies revealed significant tumor targeting of 3E8.G4S with a tumor-to-background ratio of 29:1. These analyses validated 3E8.G4S as a fast, accurate, and specific tumor-imaging agent.
Keywords: antibody engineering, in vivo imaging, immunohistochemistry, oligomerization, protein stability, colorectal cancer, tumor marker, mucin, drug development
Introduction
Despite decades of research, cancer is the second leading cause of death worldwide. Last year alone, over 1.5 million new cancer cases and over 500,000 cancer deaths were projected to occur in the United States (1). The inherent diversity and complexity of the disease makes the design of a single drug or cure impossible. The most widely used therapies for cancer include radiation and chemotherapy, increasingly augmented with some form of immunotherapy, as well as surgery. For solid tumors, the most successful treatments depend on the complete surgical resection of tumors and any metastases (2). Adenocarcinomas, which arise from glandular epithelial tissues, are the most common solid tumor, frequently found in colon, breast, pancreas, prostate, and lung.
There are a handful of methods used in surgical oncology to ensure that a solid tumor has been completely removed. Knowledge of tumor progression pathways, visual identification, and physical palpation are used with imaging methods such computerized tomography, positron emission tomography (PET),2 and single-photon emission computed tomography to identify locations and margins of tumors and metastases. PET and single-photon emission computed tomography are two forms of nuclear imaging that involve the injection of a radiolabeled tracer agent into a patient. The agent is then detected by an imager, and the three-dimensionally rendered image is often overlaid with a three-dimensional image from co-registered computerized tomography. The effectiveness of these techniques is limited by the specificity of the radiolabeled agent. The most widely used PET agent, [18F]fluorodeoxyglucose, is taken up by rapidly metabolizing cells, which is a characteristic of some tumors (3). However, [18F]fluorodeoxyglucose has limited use in that it targets only highly metabolic tumors and also targets other noncancerous tissues such as the brain, liver, and damaged or healing tissue. These shortcomings are in part what has inspired the use of molecular targeting agents, such as antibodies and antibody fragments, for better tissue specificity.
Antibodies function as targeting agents by binding specifically to cancer markers called tumor-associated antigens (TAA) (4). Some TAAs have direct roles in cancer growth or metastasis and may have potential as therapeutic targets (5). Other antigens have no known physiological function. One such TAA, tumor-associated glycoprotein (TAG)-72, is a high-molecular-weight mucin expressed in 80% of adenocarcinomas (6). The antibody 3E8 is an engineered, humanized derivative of CC49, which is believed to bind a disaccharide, sialyl-Tn, that is found on TAG-72 (7). We have developed an scFV antibody fragment of 3E8 (3E8.scFV),3 which we use here as a model protein.
Radiolabeled antibody fragments have the potential to produce improved cancer-specific images. Unlike a full-length IgGs, antibody fragments are small, and many do not contain constant domains, allowing for increased tumor penetration and less severe host immune response (8, 9). Most importantly, their small size allows for faster clearance from the blood, producing little circulating background and higher signal-to-noise ratios and therefore higher quality images. The serum half-lives of these antibody fragments can be specifically tuned to optimize tumor penetration and clearance from blood by controlling their size through chemical modification or other means. Clearance time can be matched to radionuclide half-life to select the desired imaging time for patients (10, 11). Fragment size can be controlled by modification with polyethyleneglycol chains or, as in our study, through designed manipulation of oligomeric state.
Despite these advantages, antibody fragments often suffer from instability, aggregation, reduced binding affinity, and substantially decreased serum half-life (12). In the case of the variable fragment (FV), the loss in stability is due to the removal of the constant domains that have interdomain disulfide bonds. This leaves only the weak noncovalent interaction between the VL and VH domains. Engineering disulfide bridges (dsFV) or, more commonly, the genetic fusion of a flexible peptide linker between domains (scFV) have been shown to improve overall stability (12, 13). These linkers tend to be simple, rich in glycine and serine repeats, biologically inspired (e.g. the interdomain linker region of the fungal cellulase cellobiohydrolase I (CBHI)), or discovered via high throughput methods such as phage display (14, 15). The reduction in binding affinity observed for antibody fragments is often related to stability but also can be related to antibody fragment monovalency (16).
ScFV antibodies can form dimeric, trimeric, and even higher order oligomeric species based on the size of their engineered linkers. The tunability of quaternary structure is due to the added constraints on intramolecular domain interactions, which can then only be satisfied intermolecularly. The rule of thumb is that monomeric species form with 12 or more amino acids in the linker, whereas 5–12 amino acids will produce a dimeric species or diabody (17). Shorter linkers or the direct fusion of variable domains will produce triabodies in some studies but tetramers in others (18). In general, the studies leading to these rules include fairly small data sets, limited biophysical characterization, and often compare disparate scaffold molecules.
In this study, through an expansive series of constructs, we elucidate the importance of antibody fragment linkers and domain orientation. Specifically, we have cloned, expressed, and characterized scFVs, diabodies, and higher order multimeric constructs with varying linker compositions, sizes, and domain orientations. These constructs have been characterized by surface plasmon resonance (SPR) to test for antigen binding, by differential scanning fluorimetry (DSF) to test for thermal stability, and by gel chromatography to test for quaternary structure and homogeneity. We observed deviations in oligomeric forms from previously reported trends in linker design. We believe purification by protein L binding gives a more accurate representation of oligomeric distributions than commonly used immobilized metal affinity chromatography (IMAC) purification.
In addition, we have selected a single construct (VH–G4S–VL) from the 3E8 linker library with ideal biophysical characteristics and pharmacokinetic (PK) properties to be used as a possible lead candidate for cancer imaging. After further characterization, production optimization, and in vitro tissue staining, we have successfully produced xenograft mouse images with a remarkably high 29:1 tumor-to-background contrast.
Results
In designing our library, we primarily used glycine and serine repeat linkers, because their small side chains allow flexibility, and serine increases solubility and decreases immunotoxicity (19). In addition, we included a non-Gly-Ser repeat linker, 205C, which was designed to be helical and possibly advantageous over nonstructured linkers if less susceptible to proteolysis (20, 21).
Construction of His6-tagged 3E8 library
The 17-member 3E8 antibody fragment library was cloned using overlap PCR and two different templates (Fig. 1A). For all VL–VH domain–oriented constructs, our original 3E8 construct (VL–205C–VH, manuscript in preparation) was used as the template (22). For the cloning of VH–VL orientation constructs, a gene containing a G4S linker (custom synthesis by Genewiz) was used as the template. For all cloning and purification, our lab's pHLIC plasmid vector was used and contained the features shown in Fig. 1D (23). Two separate PCR products were created for each construct that contained altered but overlapping linker regions between domains and joined together to create a full-length gene (Fig. 1, B and C). At first, our constructs were purified from cell lysate by IMAC purification. The concentration and purity of these constructs were determined by SDS–PAGE (Fig. 1E), typically in the range of 95% purity.
Figure 1.

Library design and construction. A, list of library members with varying linker lengths from 25 amino acids (205C) to a single glycine. B, detailed schematic of 3E8 gene coding region along with location of amplification, I and IV, and mutagenic primers, II and III. C, agarose gel displaying DNA fragments created during initial mutagenic PCR and full-length 3E8 gene when those products are amplified together. D, vector map of T7 expression vector, pHLIC, containing ColE1 replication origin, ampicillin resistance gene, Lac repressor, and 3E8 cloning region between NdeI (red) and BamHI (green) restriction sites. E, representative SDS–PAGE gel showing a subset of purified library members purified with IMAC purification.
Gel filtration of His6-tagged 3E8 library
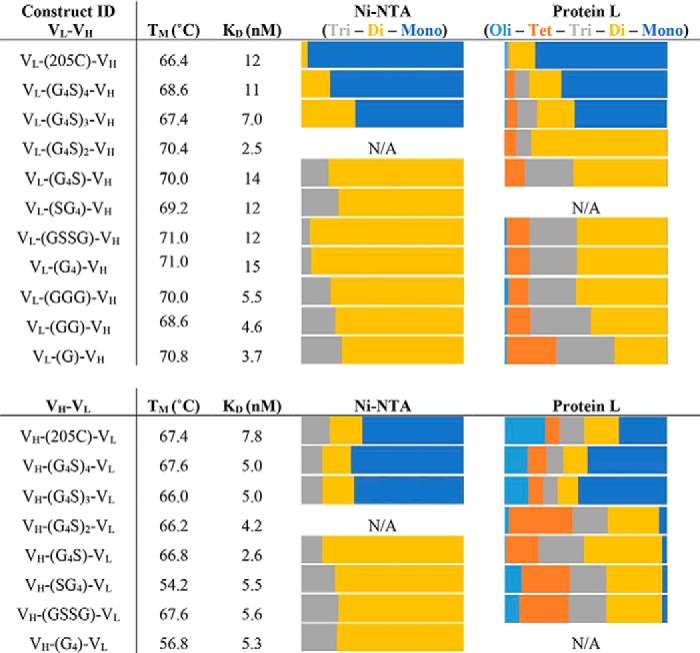
Gel filtration analysis of the IMAC-purified constructs showed a distribution of oligomeric states of monomer, dimer, trimer, tetramer, to higher order oligomer (Fig. 2, A and B). The primary oligomeric state of our constructs was either a monomer or a dimer and depended on the linker length. Constructs with 15–25 amino acids produced monomers and constructs with 1–5 produced dimers (Table 1). The overall pattern of quaternary structure was also found to be independent of domain orientation.
Figure 2.

Biophysical characterization of library constructs by domain orientation. A and B, gel filtration chromatography of library members for determination of oligomeric state using analytical grade Superdex 75 column. Antibody fragment library members elute from the analytical Superdex 75 column according to size in solution. Soluble aggregates elute from the column followed by tetramer, trimer, dimer, and monomer. Antibody library members are grouped based on domain orientation with VL–VH orientation on the top and VH–VL domain orientation on the bottom. C and D, normalized DSF of 3E8 library members. The hydrophobic dye, SYPRO Orange, exhibits increased fluorescence when binding denatured 3E8. Relative stability can be assessed by melting temperature, which is determined from fluorescence inflection points as 3E8 is thermally denatured. The constructs are grouped based on orientation where VH–VL orientation (bottom) appears to be generally less stable than VL–VH (top).
Table 1.
Antibody library biophysical characterization summary
The list in the table shows each antibody construct and its measured melting temperature (°C), apparent binding dissociation constant (nm), and oligomeric state (relative area calculated under each gel filtration peak). Oligomeric states are divided into monomers (Mono), dimers (Di), trimers (Tri), tetramers (Tet), and higher-order oligomers (Oli).
Stability characterization
The thermostability of the seventeen antibody fragment constructs was analyzed by DSF (Fig. 2, C and D). The melting point for each fragment was calculated by finding the maxima of the first derivative of each curve (Table 1). The fragments exhibited a range in stability of 17 °C. We found that constructs in the VH–VL orientation displayed a lower stability of ∼5 °C compared with their VL–VH counterparts. We also noted a non–two-state melting pattern that was more prevalent in the VH–VL orientation.
Binding affinity characterization
All of these antibody fragments were found to bind to immobilized bovine submaxillary mucin (a source of sialyl-Tn) with low nanomolar affinity (Table 1). Specifically, these constructs ranged in apparent affinity from 3 to 15 nm, when fitting to a simple 1:1 binding model. Higher order oligomers may appear to bind more tightly because of avidity, but we chose this simple model to compare the binding regardless of oligomeric state. Antibody fragments in the VH–VL orientation were tighter binders (Fig. S1).
Summary of His6-tagged library
The data from the IMAC-purified constructs demonstrate the versatility of scFvs with respect to linker length, amino acid composition, and domain orientation. All 17 antibody fragments were well folded, stable, and functional binders. We did see a tendency for greater instability arising from the VH–VL orientation. This decrease in stability is also correlated to constructs with higher oligomeric states, but as these molecules increase in valency, increasing the binding avidity, we can detect slower off rates and slightly lower apparent KD (Table 1). The patterns we see in the oligomeric states of these molecules correlate with the linker length observations Wörn and Plückthun (24).
Design of tagless 3E8 library
After completion of the antibody linker library, a construct was selected for further in vivo characterization. The 3E8 construct VH–G4S–VL (termed 3E8.G4S) showed enhanced apparent binding and stability. In addition, the dimeric nature of this construct created a complex greater than 50 kDa and was predicted to have a longer serum half-life than its monomeric counterparts.
For future commercial production of 3E8.G4S, alterations in the expression plasmid and method of purification were made. The resistance gene from pHLIC was changed from ampicillin to kanamycin to create the new expression vector pHLICK. Additionally, the His6 tag was removed from the sequence, and an alternate method of purification by protein L was adapted. This new expression format and purification process increased our protein yields substantially. The protein eluted from a single protein L column was equal, if not greater, in purity than our previously described two column Ni–NTA purification method (Fig. S2).
A sample of 3E8.G4S purified using the new methodology was recharacterized and was shown to have comparable binding and stability. Surprisingly, gel filtration showed an increased presence of higher order oligomeric states (i.e. trimers and tetramers). After further investigation, we determined that the reason for these differences was not an artifact of protein L purification or the absence of the His6 tag but rather is a more accurate representation of the oligomeric state. Additionally, we found that having the His6 tag on multimeric constructs prevented some of them from binding to protein L. For this reason, a library of tagless 3E8 linker and domain constructs was created for protein L purification and oligomeric state analysis.
Construction of tagless 3E8 library
The tagless 3E8 constructs were created using several different cloning methods (Fig. S4). Two new linker constructs were created using overlap PCR that contained a 10-amino acid linker (G4S)2 while removing the His6 tag and TEV site from the N-terminal portion of the gene. VH–VL domain oriented sequences were created using a double digest scheme of existing restriction sites. The VL–VH orientation sequences did not contain these sites, and therefore alternate PCR methods were used for His6 tag and TEV site removal.
Gel filtration of tagless 3E8 library
Genetic removal of the His6 tag and purification using protein L affinity columns allowed for higher protein yield and purity. This produced a substantially different composition of oligomeric states (Fig. 3, A and B). Those constructs, which were previously monomeric, contained more dimeric and trimeric species. Likewise, constructs that were previously dimeric contained more trimeric and tetrameric species. It also became clear that the VH–VL orientation contained larger amounts of soluble aggregate.
Figure 3.
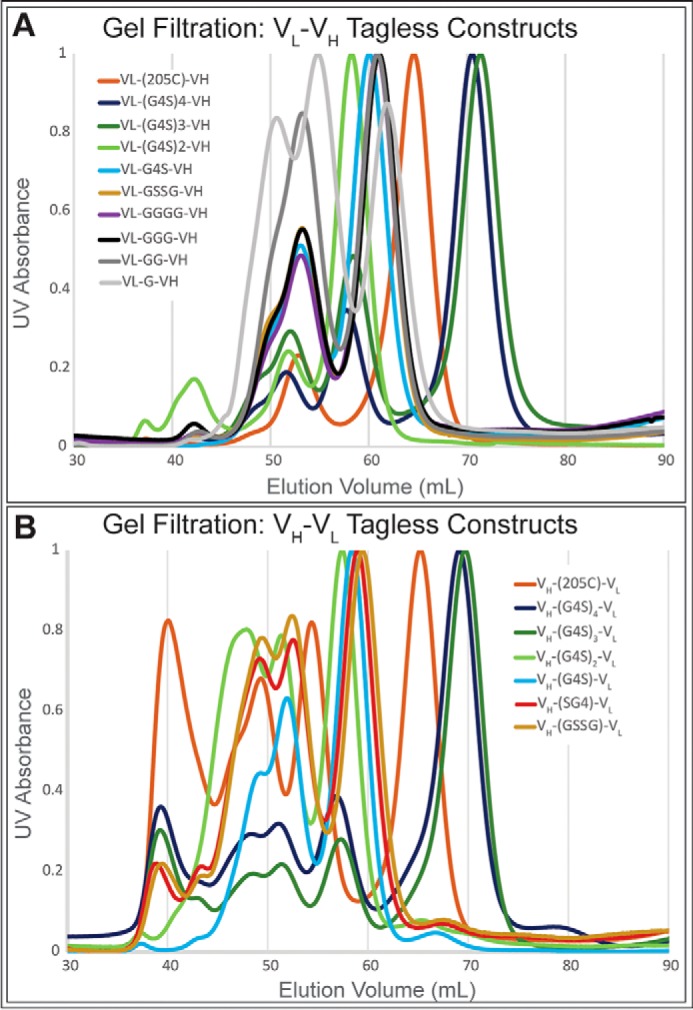
Gel filtration of tagless constructs using a prep grade Superdex 75 column. Antibody fragment library members elute from the column according to size in solution. Soluble aggregates elute from the column followed by tetramer, trimer, dimer, and monomer. Antibody library members are grouped based on domain orientation with VL–VH orientation (A) and VH–VL domain orientation (B). Substantial differences in higher order oligomers were seen between previous gel filtration data of library members.
Analytical ultracentrifugation
A subset of the tagless antibody constructs were analyzed using AUC. The results correlated to the gel filtration analysis in both predicted species molecular weights and relative distribution of oligomeric species (Fig. S5).
Further explorations in Ni–NTA IMAC versus protein L purification
To understand the contrasting gel filtration results between Ni–NTA IMAC and protein L purification, several additional experiments were performed. The first experiment was conducted to determine the effects of the His6 tag on the oligomeric states of the constructs. Gel filtration on protein L–purified construct VL-(G4S)4-VL before and after TEV cleavage of the His6 tag revealed a small shift caused by the decrease in size but no change in the relative distribution of oligomeric states (Fig. S7B). Next, the concentration dependence of the oligomeric states was tested by concentrating three different antibody constructs and comparing their gel filtration chromatographs before and after concentration. No difference in the relative oligomeric distribution was seen (Fig. S7A). The reproducibility of the oligomeric state distribution was determined by 12 parallel 1-liter purifications of 3E8.G4S. Gel-filtration analysis of these small batch purifications showed less than 10% deviation of dimer to higher oligomeric state ratio (Fig. S3).
Analysis of 3E8.G4S
After the initial analysis of both His6-tagged and tagless library members, the construct 3E8.G4S showed promising biophysical characteristics including a low apparent KD of 3.6 nm, high stability, and a favorable distribution of oligomeric states (Fig. 4, A, B, D, and E). Sed-fit (25) analysis of the AUC data determined this construct to be 47% dimer, 42% trimer, and 11% tetramer (Fig. 4C). We selected this variant for further in vitro and in vivo testing.
Figure 4.

Biophysical characterization summary of tagless 3E8.G4S. A, SDS–PAGE gel of 3E8.G4S. B and C, oligomeric states of tagless 3E8.G4S are determined by gel filtration (B) and analytical ultracentrifugation (C). D, raw SPR binding curves (colored) and Biacore fitted curves (black) show tight binding of 3.6 nm to antigen. E, comparable stabilities were seen between tagged (light blue) and tagless (dark blue) 3E8.G4S constructs.
Separation of 3E8.G4S, enriched diabody, and enriched oligomers
Using a prep-grade gel-filtration column, a sample of 3E8.G4S was separated by size into two fractions. These fractions included an enriched diabody sample and an enriched oligomer (trimer and tetramer) sample. These samples were analyzed by gel filtration 24 h after their original purification from 3E8.G4S. No change in oligomeric state was seen, suggesting that domain swapping equilibration does not occur freely in solution (Fig. S6). These enriched fractions were also analyzed by DSF and SPR. Both diabody- and oligomer-enriched fractions were stable and active binders.
Immunohistochemistry
The application of targeting adenocarcinomas was tested by immunohistochemistry. A C-terminal FLAG tag was cloned into 3E8.G4S to create 3E8.G4S.FLAG. A single-chain monomer version (VL–205C–VH) of the FLAG tagged 3E8 was also created, 3E8.scFV.FLAG, to see the effects of multivalency on detection sensitivity. The resulting staining showed enhanced detection ability in both antibody fragments over the commercially available anti-sialyl-Tn IgG, B-72.3 (Fig. 5A) without an increase in background staining of healthy tissue. Additionally, the polyvalency of antibody fragments allowed for greater dilution without loss in signal, because we saw comparable staining with 500 nm of monomeric 3E8.scFV.FLAG to 15 nm of multimeric 3E8.G4S.FLAG.
Figure 5.

Tissue and in vivo studies of 3E8 construct VH–G4S–VL. A, immunohistochemistry of human adenocarcinomas by two FLAG-tagged versions of select library members. The far-left panel shows a monomeric FLAG-tagged 3E8 at 1:150 dilution from a 2 mg/ml stock. The middle-left panel shows 3E8.G4S.FLAG (dimeric/trimeric/tetrameric mixture) at 1:2400 dilution of a 1.7 mg/ml stock. The middle-right panel shows commercially available B-72.3 IgG at 1:100 dilution of stock. The far-right panel shows a negative control in which the same staining protocol was used with removal of primary antibody. B, 125I activity change in blood postinjection of 125I-labeled 3E8 dimer, oligomers, and unfractionated 3E8.G4S. The blood was collected at 0.5, 1, 5, 24, 48, and 72 h after injection for the unfractionated sample and 0.5, 1.5, 3, 6, 24, 48, and 72 h for dimer and oligomer fractions. The data were converted to % ID of whole blood pool and presented as averages ± S.D. (n = 5). C, 125I activity in mouse organs after 72 h postinjection. Unfractionated 3E8.G4S (red) abundance falls between the smaller molecular weight diabody fraction (gray) and the higher order oligomeric trimer and tetramer fraction (black). D, LS174T xenograft mouse Micro-PET image of 124I-3E8.G4S. Images at 48 h show iodinated 3E8.G4S antibodies accumulating at the tumor site.
Pharmacokinetics
To see the effect of oligomeric state on the PK properties of 3E8.G4S, the trimeric and tetrameric species were again separated from the dimeric species using prep scale gel filtration. Unfractionated 3E8.G4S, 3E8.G4S purified dimer, and 3E8.G4S purified oligomers were separately 125I-labeled and injected into mice via tail vein (n = 5). The half-lives were determined to be 0.67, 2.0, and 3.3 h for dimeric, unaltered, and oligomeric 3E8.G4S species, respectively (Fig. 5B). As expected, the smaller molecules (dimer fraction) cleared the blood faster than the higher order oligomers (trimer and tetramer). Furthermore, the half-life of unfractionated 3E8.G4S fell in between its isolated components.
Biodistribution
The distributions of the three distinct 125I-labeled 3E8.G4S oligomeric samples in mice were analyzed 72 h postinjection. The higher-order oligomeric states of 3E8.G4S, in general, were found in higher amounts throughout the mouse compared with the dimeric fraction of 3E8.G4S. The unfractionated 3E8.G4S was found to fall between the values of the oligomeric and dimeric fractions of 3E8.G4S (Fig. 5C). The biodistribution results correlated well to the PK analysis, and we saw no greater than 0.6% ID/g (percentage of injected dose per gram) accumulation of higher order oligomers in any organ including the kidneys or liver.
In vivo imaging of xenograft mouse model
For final proof of concept, 3E8.G4S purified by protein L was labeled using 124I and injected into xenograft mice bearing the TAG-72–positive human colorectal adenocarcinoma cell line, LS174T. Micro-PET imaging of 3E8.G4S 48 h after intravenous administration showed a low background of 0.21% ID/g and significant targeting of all tumors, averaging 6.61% ID/g in tumor and yielding a final tumor to background ratio of 29:1 based on biodistribution (Fig. 5D).
Discussion
Antibody fragments have great potential for clinical use as enhanced cancer imaging tools, but the widespread use of engineered fragments is hindered by their decreased binding affinity and stability and by the resulting difficulty of large-scale production. The genetic fusion of the VH and VL domains by peptide linker not only allows for the expression of a single polypeptide but also reduces the entropic penalty of folding, causing increased domain interaction. Throughout the literature, single chain FVs have been constructed with wide variability in their domain orientation, and linker length and composition. Linker length has generally been shown to dictate oligomeric state, where linkers greater than 15 residues produce monomers, 6–15 residues produce dimers, and fewer than five residues produce higher multimers (17, 18, 26). This can be explained by the inability, caused by short linker size, of the variable light and variable heavy domains to properly orient themselves for interaction within a monomer. Instead, one antibody fragment's VL and VH chains interact with another molecule's complementary domains in what is known as domain swapping (27). Additionally, the order in which the heavy and light domains are fused varies throughout the literature. In some cases, VL–linker–VH rather than VH–linker–VL shows favorable biophysical characteristics, and in other cases the reverse is true (28, 29). The complexity of these results motivated our comprehensive study in 3E8.
Previous reports have shown success in the adaptation of other anti–TAG-72 IgGs into the single-chain variable fragments. These studies include B72.3 scFV production using Gly4-Ser linker repeats and a CC49 scFV using the 25-amino acid 205C linker (20, 29). Interestingly, both studies revealed the tendency for antibody fragment multimerization despite linker lengths that would predict monomer formation. The B72.3 scFV was purified by affinity chromatography to mucin (bovine submaxillary mucin (BSM)) with acid elution, and CC49 scFV was purified by anion exchange chromatography (MonoQ) (30). Pharmacokinetic and target binding studies of CC49 have shown that dimeric and even higher order oligomeric forms are more favorable than the scFV monomers (20, 31). These multivalent species have increased avidity yet maintained their ability for rapid in vivo tumor localization and blood clearance (32). Initial studies in our lab, however, found the CC49 scFV to be highly destabilized and aggregation prone (data not shown). This led to use of an alternate platform, 3E8, in the engineering of a humanized, stable, anti-TAG-72 scFV.
Our linker library study is consistent with previously observed linker length rules for our Ni–NTA (IMAC) purified His6 tag constructs. Larger linkers from 15–25 amino acids in length formed mostly monomers (with low amounts of dimer), whereas smaller linkers from 1–5 amino acids formed mostly dimers (with low amounts of trimer). However, we saw drastic changes in the quaternary structure that did not align with the established linker length rules when we analyzed protein L–purified fragments. More heterogeneous compositions were observed, with increased presence of higher order oligomers. Additionally, we observed shifts in oligomeric distributions as the linker is decreased in size rather than a sharp transition between discrete oligomeric states. For example, the maximum fraction of monomer is seen with 20–25 amino acids in the linker, but at 20 amino acids nearly 25% of the material is dimer, trimer, and trace amounts of tetramer. At 5 amino acids, two-thirds of the material is dimer, but significant amounts of trimer and tetramer are observed, and the maximum amount of dimer is seen at 10 amino acids. It is important to realize that different antibodies may not have precisely the same distributions at these linker lengths because of differences in the sequences and structures of those antibodies. The generality of these relationships bears further investigation.
The main reason for the contrast between purification methods, we believe, is the multivalency of the His6 tagged higher order oligomers in interaction with the Ni–NTA resin. The Ni–NTA column purification method, which is used in most scFV linker studies, enriches monomeric and dimeric forms of 3E8 because higher multimers bind more tightly to the Ni–NTA resin in competitive elution with imidazole. Protein L purification, on the other hand, elutes nearly all protein when the shift to low pH results in a nonbinding condition. This gives a more unbiased view of the oligomeric states of the protein as it was expressed. Therefore, we must reconsider linker length rules in light of how the molecules have been purified.
The biophysical properties of 3E8 are heavily dependent on the domain orientation. The most notable difference between the two sets of constructs is their oligomeric states. The VH–VL domain orientation produces large amounts of higher order oligomers. This is correlated to the decrease in overall stability, and the oligomers' increased polyvalency is the reason we see lower apparent KD values in many of these constructs. The long possibly helical linker 205C is of interest because it produced one of the most ideal constructs in the VL–VH orientation but formed soluble aggregates in the VH–VL orientation. The two domains are not identical, and there is no a priori reason that two possible linker connection topologies should be optimal with the same linker length. This optimal orientation for a particular peptide length may simply be idiosyncratic to the construct.
Immunohistochemistry with two FLAG-tagged library members was consistent with SPR binding data. Qualitative analysis reveals darker staining from 3E8.G4S.FLAG compared with 3E8.scFV.FLAG despite higher dilution, suggesting tighter binding of multivalent over monomeric constructs in the tissue. These studies also validate the applicability of 3E8 antibody fragments for future immunohistochemistry studies and the potential for linker engineered antibody fragments as improved immunohistochemistry agents.
Our most optimal molecule, 3E8.G4S, is a multivalent anti-TAG 72 antibody fragment with excellent biophysical characteristics for imaging. Its thermal stability is greater than most other reported scFVs and is comparable with that of the full IgG. PK and biodistribution data reveal the mixture of dimer, trimer, and tetramer produces serum half-life compatible with same-day or next-day imaging, and the molecule does not bind nonspecific targets. The 48-h PET image produced in a TAG-72–positive xenograft mouse model shows the potential of this cancer diagnostic and a substantial improvement over current technologies available in the clinic.
Experimental procedures
Design of linker library
The library of 3E8.scFV constructs was designed to include a variety of linkers. The sequences of the VH and VL domains of 3E8 are as reported by Yoon et al. (7). The linkers include a long, 25-amino acid structured linker, 205C, and a variety of glycine and serine rich linkers. The Gly-Ser repeat and nonrepeat linkers composed of (G4S)4, (G4S)3, (G4S)2, G4S, SG4, G4, and GSSG were included and were predicted to form monomers and dimers, respectively. The order of the variable domains was also varied with these seven linkers. Lastly, smaller linkers G, GG, and GGG in the VL–linker–VH orientation were chosen to test for higher order multimer formation.
Construction of linker library
3E8.scFV constructs were cloned into a pHLIC plasmid with lacI cassette, ColE1 origin, and ampicillin resistance (Fig. 1D). The original 3E8.scFV construct was designed to contain a PelB leader sequence for periplasmic expression, a PAMA cleavage site, a His6 tag, and a TEV protease cleavage site. This is followed by the variable light domain, 205C linker, and the variable heavy domain. The 3E8 constructs VL–205C–VH and VH–G4S–VL were used as templates in overlap PCR for VL–VH and VH–VL constructs, respectively (Table S1). Mutagenic primers were designed to include portions of the VH or VL domain, as well as the desired linker (primers II and III in Fig. 1B). Mutagenic primers were paired with amplification primers that flanked the 3E8 gene on each side. The two PCR products were then combined with amplification primers I and IV to produce the full gene, coding for the new linker (Fig. 1, B and C). The gene was then ligated into a pHLIC vector between NdeI and BamHI sites and transformed into DH10B Escherichia coli, and the sequence was confirmed.
Cloning of VH–VL tagless variants
The constructs with VH–VL domain orientation were cloned using a series of PCR and restriction digest steps. Initially, a tagless version of VH–G4S–VL was made by PCR using the tagged VH–G4S–VL library member. The forward primers contained the pelB leader sequence and ended with an 18-bp overlap of the VH domain, omitting the His6 tag and TEV protease site. This construct was then used to create the remaining tagless variants by restriction digest of the tagged and tagless plasmids with EagI and SpeI (Fig. S4A). This digest separated all features upstream of the VH domain from the rest of the gene, and all previously tagged plasmids could be ligated with a DNA sequence removed from an identical double digest of the tagless VH–G4S–VL. The tagless construct VH–(G4S)2–VL was added to the library and cloned using the same overlap PCR methods as described in the construction of the tagged variants.
Cloning of VL–VH tagless variants
The constructs with VL–VH domain orientation did not contain a SpeI restriction site and therefore were cloned using PCR with primers containing flanking BsaI restriction sites. Because BsaI restriction enzymes cut outside of their recognition sites, after primer digestion the restriction site is removed from the final product. The primers were designed to remove the His6 tag and TEV protease site. The tagless construct VL–(G4S)2–VH was added to the library and cloned using the same overlap PCR methods as described in the tagged variants.
Expression of linker library
The sequence confirmed constructs were transformed into the C43(DE3) Walker E. coli expression strain (33). Seed cultures were grown and inoculated into 1.5-liter cultures at 37 °C. Induction of cultures with 0.05 mm isopropyl β-d-thiogalactopyranoside was performed when cell density measured 0.6 A600. The cultures were then cold shocked at 4 °C and expressed at 16 °C overnight. The cells were centrifuged at 9,000 × g for 5 min, and pellets were either stored at −80 °C or purified immediately.
IMAC purification of His-tagged antibodies
The cell pellets were resuspended in 25 ml of a lysis buffer (50 mm Tris-HCl, pH 8, 300 mm NaCl, 15 mm imidazole). Additives to the cell suspension were then: 75 μl of 2 m MgCl2, 150 μl of 150 mm CaCl2, 5 μl of 10 mg/ml RNase, 50 units of DNase (Roche Diagnostics), 30 mg of lysozyme, and 300 μl of 10% Triton X-100. The suspension was then lysed using an Avestin EmulsiFlex-C3 homogenizer and centrifuged at 40,000 × g for 45 min. The supernatant was collected and incubated with 1 ml of Ni–NTA–agarose resin for 2 h at 4 °C. The cell lysate and resin were then poured onto a fritted filter and washed with 5 ml of 50 mm Tris-HCl, 300 mm NaCl, and 30 mm imidazole buffering solution, pH 8. Protein was eluted from the column with 5 ml of 300 mm imidazole Tris-HCl buffering solution. Eluted protein was then incubated overnight at room temperature with 200 μl of 0.4 mg/ml TEV protease. Proteins were then dialyzed out of imidazole into lysis buffer, and incubated again with Ni–NTA resin for 2 h to remove the cleaved His6 tag and His6-tagged TEV protease. The flow-through of the second Ni–NTA column was evaluated by SDS–PAGE for relative quantification and purity.
Protein L purification of tagless antibodies
The cell pellets were resuspended in 25 ml of protein L binding buffer (150 mm sodium phosphate buffer, 150 mm NaCl, pH 7). The same additives as with Ni–NTA purification were mixed into the resuspension. The suspension was then lysed using Emulsiflex and centrifuged twice at 40,000 × g for 45 min (decanting supernatant into new centrifuges tubes between runs). The lysate was then hand-pumped into prepacked 1 ml of GE Healthcare Lifesciences HiTrap protein L columns at ∼1 ml/min. Five column volumes of binding buffer were pumped through the column as a washing step. Protein was eluted from the column using a 100 mm glycine buffer, pH 3.
Differential scanning fluorimetry
Purified antibodies were heated with a hydrophobic dye, SYPRO Orange, and the resulting fluorescence was measured. A working stock of SYPRO orange was made by diluting in PBS from 5000× to 350×. 1 μl of working stock dye was added to 19 μl of antibody at 0.2 and 0.04 mg/ml concentrations. Both dilutions of each antibody were measured in triplicate. The samples were heated, and fluorescence was measured every 0.2 °C from 25 to 95 °C in a Bio Rad C1000 Thermocyler using the FRET channel.
Surface plasmon resonance
SPR experiments were performed on a Biacore T100. BSM was conjugated to a CM5 censor chip by amine coupling. Antibody fragments were diluted in HEPES-buffered saline to concentrations of 5, 10, 25, 50, 75, 100, 150, and 200 mm. A buffer control and a replicate at 75 mm were also measured for binding. Chip regeneration was performed between each injection using a 6 m guanidine hydrochloride, 200 mm acetate solution. A 1:1 binding model was globally fit to the data. This model produces an apparent KD, because higher oligomeric state species may exhibit binding avidity to multiple sialyl-Tn epitopes in BSM. Such avidity could result in lower apparent KD values with the same binding sites. The small spread of the KD values and the goodness of fit statistics for the 1:1 binding curves suggest that this is a relatively small effect for these molecules with BSM.
Analytical Superdex 75 column
IMAC–purified antibody fragments were analyzed by SuperdexTM 75 10/300 GL gel filtration column. 500 μl of 0.2 mg/ml sample was injected onto the column and run at 0.4 ml/min. The gel filtration running buffer contained 50 mm Tris-HCl, pH 8, and 100 mm NaCl. Protein standards used to determine molecular weights of antibody constructs are noted in Fig. S8A.
Prep grade Superdex 75 column
Protein L–purified antibody fragments were separated by HiLoadTM 16/60 SuperdexTM 75 prep grade gel filtration column. 500 μl of 0.2 mg/ml sample was injected onto the column and run at 0.5 ml/min. The gel filtration running buffer contained 50 mm Tris, pH 8, and 100 mm NaCl. Protein standards used to determine molecular weights of antibody constructs are noted in Fig. S8B.
Analytical ultracentrifugation
A subset of the tagless antibody fragments were measured by AUC (Beckman Coulter ProteomeLabTM XL-I at the Ohio State University Biophysical Interaction and Characterization Facility). Concentration of antibody fragments was normalized to 0.2 mg/ml. The oligomeric states and relative concentrations were analyzed by Sedfit software.
Gel filtration of His6 tag cleaved antibody
To determine the effects of the His6 tag on quaternary states, the antibody construct VL–(G4S)4–VL, containing a TEV cleavable His6 tag, was purified by protein L. Purified protein was then separated into two aliquots. One sample was fully digested by TEV protease, as determined by an apparent SDS–PAGE molecular weight shift. Both samples were analyzed on the prep grade Superdex 75 column for changes in oligomeric states.
Concentration dependence of oligomeric states
The concentration dependence of oligomeric state distribution was determined by gel filtration analysis of three antibody constructs, VH–205C–VL, VH–(G4S)4–VL, and VH–(G4S)3–VL. The constructs were purified via protein L to a final concentration of 0.2 mg/ml (8 μm). A portion of each purified antibody was then further concentrated to 0.8 mg/ml (31 μm). All six samples were then injected onto the prep grade Superdex 75 column. Chromatographs were normalized to a maximum value of 1 to better illustrate quaternary structure distribution independent of variations in concentration.
Reproducibility of protein L purification
The reproducibility of the oligomeric distribution of 3E8.G4S was examined by purification of twelve 1-liter cultures. The samples were purified by protein L as previously described. Final concentration of the two samples in PBS was determined by SDS–PAGE gel to be 0.2 mg/ml (8 μm). Replicates were then injected onto the analytical Superdex 75 column. Chromatographs were normalized to a maximum value of 1 to better illustrate quaternary structure distribution independent of variations in concentration.
Separation of oligomeric states in 3E8.G4S
Using the analytical Superdex 75 column, protein L–purified 3E8.G4S was separated by oligomeric state. The resulting chromatograph showed two distinct size distributions correlating to dimer and higher order oligomers (later determined as trimer and tetramer). Eluted fractions containing only dimer were collected separately from fractions containing higher oligomers. Final concentrations of these fractions were determined by SDS–PAGE gel to be 0.7 mg/ml (27 μm) for enriched diabody and 1 mg/ml (38 μm) for enriched higher oligomers. After 24 h, these samples were reanalyzed by gel filtration. A 1 mg/ml (38 μm) sample of 3E8.G4S that was not size-fractioned was reserved for later comparative biophysical analysis.
Radioiodination of proteins
Enriched diabody, 3E8.G4S, and higher oligomers were labeled with 124I for PET imaging and biodistribution and with 125I for PK experiments by a standard iodogen method (34). For a typical reaction, 0.5–1 mg of protein was transferred into an iodogen tube (Pierce) containing 100 μl of phosphate buffer (0.1 m), pH 7.4, followed by the addition of known amounts of 125INa (PerkinElmer Life Sciences) or 124INa (IBA Molecular, Dulles, VA) in NaOH base. An additional 50 μl of phosphate buffer (0.1 m), pH 7.4, was then added to the mixture and covered with parafilm, and the mixture was incubated at room temperature for 30–45 min with occasional swirling. The 125I- or 124I-labeled protein was loaded onto a Sephadex G-25 (PD-10) size-exclusion column and eluted with PBS for separation of labeled protein from the free iodide. Several fractions of ∼10 drops were collected, and the fractions containing the highest radioactivity were combined in a preweighed plastic vial. The amount of radioactivity was determined using a dose calibrator. The percent yield of radiolabeling was calculated by dividing the total radioactivity of the combined sample by the amount of radioactivity added to the iodogen tube.
Pharmacokinetics
Normal 6–8-week old female BALB/c mice were used for blood clearance studies. 5 μCi of 125I-labeled enriched diabody or 3E8.G4S or higher oligomer were injected in 100 μl of PBS in the tail veins. Blood samples (5 μl) were drawn from the saphenous vein by puncture, using a 30-gauge syringe needle, at 0.5, 1, 5, 24, 48, and 72 h postinjection for 3E8.G4S and 0.5, 1.5, 3, 6, 24, 48, and 72 h for enriched diabody and higher oligomers, and blood samples were collected into a capillary tube. The radioactivity of the blood samples was counted using an automated well counter (PerkinElmer Life Sciences Wizard II, model 2480). A blood factor of 78 ml/kg was used to calculate % ID (percent injected dose) in blood for each mouse based on the individual weight of the mouse. Mean % ID was determined for each dose group at each time point, and area under the mean % ID versus time curve was calculated between 1 and 72 h (area under the curve, for 1–72 h) using the trapezoid rule and expressed as % ID × h. Blood clearance was calculated as dose/area under the curve and expressed as ml/h/kg.
Biodistribution
For tissue distribution, the mice were sacrificed at 72 h after administration. The organs and tissues were dissected, including heart, lungs, spleen, liver, kidneys, pancreas, gastrointestinal tract, muscle, skin, blood, tail, and carcass. Organs and tissues were then weighed, and radioactivity was counted using well counter. The percentage of injected dose per gram (% ID/g) for each tissue was calculated.
PET imaging
PET imaging studies were conducted on athymic nu/nu mice bearing TAG-72 expressing LS174T human colon carcinoma xenograft tumors. The animals were administered nonradioactive potassium iodide ad libitum 24 h prior to tracer injection. 236 ± 2 μCi of 124I-labeled 3E8.G4S fragment was injected via tail vein into tumor bearing mice (n = 5). The animals were anesthetized using isoflurane inhalation with 5% of dial vaporizer for induction and 1.5–2% for maintenance. Micro-PET (Inveon, Siemens Preclinical, Knoxville, TN) imaging of mice was carried out at 24 and 48 h after intravenous administration. The Micro-PET images were reconstructed with OSEM2D algorithm to reduce possible artifacts from bladder contents and low localization counts.
Animal studies
Animal studies described were performed under a protocol approved by the Institutional Animal Care and Use Committee of Ohio State University (Columbus, OH).
Author contributions
N. E. L., B. J. S., E. W. M., M. F. T., and T. J. M. conceptualization; N. E. L. and B. J. S. data curation; N. E. L., B. J. S., H. D., C. L. H., and T. J. M. formal analysis; N. E. L. validation; N. E. L., B. J. S., S. D., M. A. R., K. K., and T. J. M. investigation; N. E. L. visualization; N. E. L., B. J. S., H.D., S.D., M. A. R., K. K., and T. J. M. methodology; N. E. L. writing-original draft; N. E. L., E. W. M., M. F. T., and T. J. M. project administration; N. E. L., B. J. S., S.D., M. A. R., K. K., M. F. T., and T. J. M. writing-review and editing; B. J. S. and T. J. M. supervision; E. W. M. and T. J. M. funding acquisition; M. F. T. and T. J. M. resources.
Supplementary Material
Acknowledgments
We thank Michelle Williams and Karen Woolum for assistance with xenograft mouse Micro-PET imaging.
This work was supported by a research contract from Enlyton, Ltd. (to T. J. M. and M. F. T.). Protein stability studies were supported by National Institutes of Health Grant R01 GM083114 (to T. J. M.). N. E. L., B. J. S., C. L. H., E. W. M., and T. J. M. are inventors on patents related to this work that have been licensed by Enlyton, Ltd. E. W. M. and T. J. M. are officers of Enlyton, Ltd., and C. L. H., E. W. M., and T. J. M. are shareholders of Enlyton, Ltd. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1–S8.
B. J. Sullivan, E. D. Alten, N. E. Long, C. Wachnowsky, C. L. Hitchcock, E. W. Martin, Jr., and T. J. Magliery, manuscript in preparation.
- PET
- positron emission tomography
- scFv
- single-chain variable fragment
- TAG-72
- tumor-associated glycoprotein 72
- TAA
- tumor-associated antigen
- SPR
- surface plasmon resonance
- DSF
- differential scanning fluorimetry
- IMAC
- immobilized metal affinity chromatography
- PK
- pharmacokinetics
- Ni–NTA
- nickel-nitrilotriacetic acid
- ID
- injected dose
- TEV
- tobacco etch virus
- AUC
- analytical ultracentrifugation
- BSM
- bovine submaxillary mucin.
References
- 1. Siegel R. L., Miller K. D., and Jemal A. (2016) Cancer statistics, 2016. CA Cancer J. Clin. 66, 7–30 10.3322/caac.21332 [DOI] [PubMed] [Google Scholar]
- 2. Mammen J. M., James L. E., Molloy M., Williams A., Wray C. J., and Sussman J. J. (2007) The relationship of lymph node dissection and colon cancer survival in the Veterans Affairs Central Cancer Registry. Am. J. Surg. 194, 349–354 10.1016/j.amjsurg.2006.08.092 [DOI] [PubMed] [Google Scholar]
- 3. Shankar L. K., Hoffman J. M., Bacharach S., Graham M. M., Karp J., Lammertsma A. A., Larson S., Mankoff D. A., Siegel B. A., Van den Abbeele A., Yap J., Sullivan D., and National Cancer Institute (2006) Consensus recommendations for the use of F-18-FDG PET as an indicator of therapeutic response in patients in national cancer institute trials. J. Nucl. Med. 47, 1059–1066 [PubMed] [Google Scholar]
- 4. Brannon-Peppas L., and Blanchette J. O. (2004) Nanoparticle and targeted systems for cancer therapy. Adv. Drug Deliv. Rev. 56, 1649–1659 10.1016/j.addr.2004.02.014 [DOI] [PubMed] [Google Scholar]
- 5. Coulie P. G., Van den Eynde B. J., van der Bruggen P., and Boon T. (2014) Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat. Rev. Cancer 14, 135–146 10.1038/nrc3670 [DOI] [PubMed] [Google Scholar]
- 6. Julien S., Videira P. A., and Delannoy P. (2012) Sialyl-Tn in cancer: (how) did we miss the target? Biomolecules 2, 435–466 10.3390/biom2040435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoon S. O., Lee T. S., Kim S. J., Jang M. H., Kang Y. J., Park J. H., Kim K. S., Lee H. S., Ryu C. J., Gonzales N. R., Kashmiri S. V., Lim S. M., Choi C. W., and Hong H. J. (2006) Construction, affinity maturation, and biological characterization of an anti-tumor-associated glycoprotein-72 humanized antibody. J. Biol. Chem. 281, 6985–6992 10.1074/jbc.M511165200 [DOI] [PubMed] [Google Scholar]
- 8. Todorovska A., Roovers R. C., Dolezal O., Kortt A. A., Hoogenboom H. R., and Hudson P. J. (2001) Design and application of diabodies, triabodies and tetrabodies for cancer targeting. J. Immunol. Methods 248, 47–66 10.1016/S0022-1759(00)00342-2 [DOI] [PubMed] [Google Scholar]
- 9. Moran N. (2011) Boehringer splashes out on bispecific antibody platforms. Nat. Biotechnol. 29, 5–6 10.1038/nbt0111-5 [DOI] [PubMed] [Google Scholar]
- 10. Chapman A. P., Antoniw P., Spitali M., West S., Stephens S., and King D. J. (1999) Therapeutic antibody fragments with prolonged in vivo half-lives. Nat. Biotechnol. 17, 780–783 10.1038/11717 [DOI] [PubMed] [Google Scholar]
- 11. Hitchcock C. L., Magliery T. J., Mojzisik C., Johnson M., Arnold M. W., and Martin E. W. (2017) Evolution of a system to increase precision in the surgical management of colorectal carcinoma. Clin. Surg. 2, 1491 [Google Scholar]
- 12. Plückthun A. (1991) Antibody engineering: advances from the use of Escherichia coli expression systems. Biotechnology 9, 545–551 [DOI] [PubMed] [Google Scholar]
- 13. Pantoliano M. W., Bird R. E., Johnson S., Asel E. D., Dodd S. W., Wood J. F., and Hardman K. D. (1991) Conformational stability, folding, and ligand-binding affinity of single-chain-Fv immunoglobulin fragments expressed in Escherichia coli. Biochemistry 30, 10117–10125 10.1021/bi00106a007 [DOI] [PubMed] [Google Scholar]
- 14. Alfthan K., Takkinen K., Sizmann D., Söderlund H., and Teeri T. T. (1995) Properties of a single-chain antibody containing different linker peptides. Protein Eng. 8, 725–731 10.1093/protein/8.7.725 [DOI] [PubMed] [Google Scholar]
- 15. Tang Y., Jiang N., Parakh C., and Hilvert D. (1996) Selection of linkers for a catalytic single-chain antibody using phage display technology. J. Biol. Chem. 271, 15682–15686 10.1074/jbc.271.26.15682 [DOI] [PubMed] [Google Scholar]
- 16. Cuesta A. M., Sánchez-Martin D., Blanco-Toribio A., Villate M., Enciso-Álvarez K., Alvarez-Cienfuegos A., Sainz-Pastor N., Sanz L., Blanco F. J., and Alvarez-Vallina L. (2012) Improved stability of multivalent antibodies containing the human collagen XV trimerization domain. mAbs 4, 226–232 10.4161/mabs.4.2.19140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Atwell J. L., Breheney K. A., Lawrence L. J., McCoy A. J., Kortt A. A., and Hudson P. J. (1999) scFv multimers of the anti-neuraminidase antibody NC10: length of the linker between VH and VL domains dictates precisely the transition between diabodies and triabodies. Protein Eng. 12, 597–604 10.1093/protein/12.7.597 [DOI] [PubMed] [Google Scholar]
- 18. Dolezal O., Pearce L. A., Lawrence L. J., McCoy A. J., Hudson P. J., and Kortt A. A. (2000) ScFv multimers of the anti-neuraminidase antibody NC10: shortening of the linker in single-chain Fv fragment assembled in V-L to V-H orientation drives the formation of dimers, trimers, tetramers and higher molecular mass multimers. Protein Eng. 13, 565–574 10.1093/protein/13.8.565 [DOI] [PubMed] [Google Scholar]
- 19. Huston J. S., Mudgetthunter M., Tai M. S., Mccartney J., Warren F., Haber E., and Oppermann H. (1991) Protein engineering of single-chain Fv analogs and fusion proteins. Method Enzymol. 203, 46–88 10.1016/0076-6879(91)03005-2 [DOI] [PubMed] [Google Scholar]
- 20. Pavlinkova G., Beresford G. W., Booth B. J., Batra S. K., and Colcher D. (1999) Pharmacokinetics and biodistribution of engineered single-chain antibody constructs of MAb CC49 in colon carcinoma xenografts. J. Nucl. Med. 40, 1536–1546 [PubMed] [Google Scholar]
- 21. Whitlow M., Bell B. A., Feng S. L., Filpula D., Hardman K. D., Hubert S. L., Rollence M. L., Wood J. F., Schott M. E., Milenic D. E., Yokota T., and Schlom J. (1993) An improved linker for single-Chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng. 6, 989–995 10.1093/protein/6.8.989 [DOI] [PubMed] [Google Scholar]
- 22. Gong L., Ding H., Long N. E., Sullivan B. J., Martin E. W. Jr., Magliery T. J., and Tweedle M. F. (2018) A 3E8.scFv.Cys-IR800 conjugate targeting TAG-72 in an orthotopic colorectal cancer model. Mol. Imaging Biol. 20, 47–54 10.1007/s11307-017-1096-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Durani V., Sullivan B. J., and Magliery T. J. (2012) Simplifying protein expression with ligation-free, traceless and tag-switching plasmids. Protein Expr. Purif. 85, 9–17 10.1016/j.pep.2012.06.007 [DOI] [PubMed] [Google Scholar]
- 24. Wörn A., and Plückthun A. (2001) Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 305, 989–1010 10.1006/jmbi.2000.4265 [DOI] [PubMed] [Google Scholar]
- 25. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kortt A. A., Lah M., Oddie G. W., Gruen C. L., Burns J. E., Pearce L. A., Atwell J. L., McCoy A. J., Howlett G. J., Metzger D. W., Webster R. G., and Hudson P. J. (1997) Single-chain Fv fragments of anti-neuraminidase antibody NC10 containing five- and ten-residue linkers form dimers and with zero-residue linker a trimer. Protein Eng. 10, 423–433 10.1093/protein/10.4.423 [DOI] [PubMed] [Google Scholar]
- 27. Holliger P., Prospero T., and Winter G. (1993) Diabodies: small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. U.S.A. 90, 6444–6448 10.1073/pnas.90.14.6444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dorai H., McCartney J. E., Hudziak R. M., Tai M. S., Laminet A. A., Houston L. L., Huston J. S., and Oppermann H. (1994) Mammalian-cell expression of single-chain Fv (Sfv) antibody proteins and their C-terminal fusions with interleukin-2 and other effector domains. Biotechnology 12, 890–897 [DOI] [PubMed] [Google Scholar]
- 29. Desplancq D., King D. J., Lawson A. D., and Mountain A. (1994) Multimerization behaviour of single chain Fv variants for the tumour-binding antibody B72.3. Protein Eng. 7, 1027–1033 10.1093/protein/7.8.1027 [DOI] [PubMed] [Google Scholar]
- 30. King D. J., Byron O. D., Mountain A., Weir N., Harvey A., Lawson A. D., Proudfoot K. A., Baldock D., Harding S. E., and Yarranton G. T. (1993) Expression, purification and characterization of B72.3 Fv fragments. Biochem. J. 290, 723–729 10.1042/bj2900723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goel A., Colcher D., Baranowska-Kortylewicz J., Augustine S., Booth B. J., Pavlinkova G., and Batra S. K. (2000) Genetically engineered tetravalent single-chain Fv of the pancarcinoma monoclonal antibody CC49: improved biodistribution and potential for therapeutic application. Cancer Res. 60, 6964–6971 [PubMed] [Google Scholar]
- 32. Goel A., Baranowska-Kortylewicz J., Hinrichs S. H., Wisecarver J., Pavlinkova G., Augustine S., Colcher D., Booth B. J., and Batra S. K. (2001) 99mTc-labeled divalent and tetravalent CC49 single-chain Fv's: novel imaging agents for rapid in vivo localization of human colon carcinoma. J. Nucl. Med. 42, 1519–1527 [PubMed] [Google Scholar]
- 33. Miroux B., and Walker J. E. (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–298 10.1006/jmbi.1996.0399 [DOI] [PubMed] [Google Scholar]
- 34. Bailey G. S. (1996) The Iodogen Method for Radiolabeling Protein, Humana Press, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.