

Abstract
Many Gram-negative bacterial pathogens use a type III secretion system to infect eukaryotic cells. The injection of bacterial toxins or protein effectors via this system is accomplished through a plasma membrane channel formed by two bacterial proteins, termed translocators, whose assembly and membrane-insertion mechanisms are currently unclear. Here, using purified proteins we demonstrate that the translocators PopB and PopD in Pseudomonas aeruginosa assemble heterodimers in membranes, leading to stably inserted hetero-complexes. Using site-directed fluorescence labeling with an environment-sensitive probe, we found that hydrophobic segments in PopD anchor the translocator to the membrane, but without adopting a typical transmembrane orientation. A fluorescence dual-quenching assay revealed that the presence of PopB changes the conformation adopted by PopD segments in membranes. Furthermore, analysis of PopD's interaction with human cell membranes revealed that PopD adopts a distinctive conformation when PopB is present. An N-terminal region of PopD is only exposed to the host cytosol when PopB is present. We conclude that PopB assists with the proper insertion of PopD in cell membranes, required for the formation of a functional translocon and host infection.
Keywords: type III secretion system (T3SS), protein translocation, protein assembly, membrane protein, membrane transport, Pseudomonas aeruginosa (P. aeruginosa), infection, fluorescence, translocation, Perfringolysin O, PopB, PopD, quenching, translocon
Introduction
Pseudomonas aeruginosa is an opportunistic pathogen that poses severe threats to immunocompromised individuals and hospitalized patients because of its ability to develop resistance to antibiotics. Like many bacterial pathogens, P. aeruginosa has been shown to exploit the type III secretion (T3S)3 system to establish infection (1, 2). T3S is activated upon direct cell contact (3), and functions as a conduit through which effector proteins are secreted (4). P. aeruginosa uses the T3S system to transport up to four different effector proteins (ExoT, ExoY, ExoS, or ExoU) (5) into the target cells to trigger apoptosis, disrupt the actin cytoskeleton, and cause cell death. Insertion of the translocon and pore formation have been recently shown to have downstream effects resulting ultimately in modifications to the host epigenome (6). Because of such a critical role in pathogen infection, the T3S system constitutes an excellent target for the development of novel therapeutic agents (7).
The T3S system consists of a multimeric protein complex that can be divided into four major structural elements: (i) a cytosolic platform that delivers and sorts proteins to be secreted, (ii) a basal body that spans the two bacterial membranes and the periplasmic space, (iii) a hollow needle that extends more than 50 nm from the surface of the outer membrane, and (iv) a translocon complex that is required for protein translocation across the target cell plasma membrane. Structural information is available for the T3S basal body and needle (the injectisome) (8), which was visualized together with the cytosolic platform in situ for different pathogens including Yersinia enterocolitica (9), Shigella flexneri (10), and Salmonella typhimurium (11). However, structural information on the translocon complex, which is essential for protein translocation into the host cytosol, has remained elusive.
In P. aeruginosa, it has been hypothesized that the translocon is formed by two T3S-secreted proteins PopB and PopD (12). Bacteria lacking PopB or PopD lose the ability to translocate effector proteins into the target cell, despite the fact that proteins are still being secreted through the needle. PopB and PopD have been detected on mammalian cell membranes after incubations with P. aeruginosa (12) and both proteins can bind and oligomerize into homo- or hetero-complexes with discrete stoichiometry on liposomal membranes (13). However, how PopB and PopD interact with membranes and the mechanism behind the insertion remains largely unknown.
In this work, we provide specific insights about the interaction of the PopD translocator with membranes and the mechanism of PopD insertion into cell membranes. PopB and PopD are inserted into membranes as integral membrane proteins. Analysis of the primary sequence of PopD reveals one hydrophobic segment long enough to cross the lipid bilayer. Here we identified a second conserved segment in PopD that could become a transmembrane helix upon protonation of its acidic residues. Combining site-specific fluorescent labeling and a series of biophysical techniques, we found that both segments interact with the membrane but do not adopt a typical transmembrane orientation in the PopB and PopD hetero-complexes reconstituted in liposomes. Interaction with PopB not only redirected the oligomerization of PopD from homo-oligomers to hetero-oligomers, but also changed the conformation adopted by these membrane-interacting segments. To specifically study the assembly of P. aeruginosa translocators in mammalian cells, we established a procedure to isolate translocators inserted into cell membranes. Using this method we demonstrated that PopD associated with cell membranes also underwent a structural rearrangement when PopB was present. Together, our findings provide an explanation, in the perspective of protein insertion, for the requirement of both PopB and PopD to form a functional translocon.
Results
Protonation of acidic residues in PopD showed a second potential transmembrane segment
We have shown that the association of purified PopB and PopD with liposomal membranes was facilitated by incubations at acidic pH. We have also shown that the presence of PopB promotes PopD binding to membranes at higher pH, but the reasons for this low pH requirement are unknown (13). Analysis of the primary sequence of PopD showed only one hydrophobic segment long enough to cross-the lipid bilayer (Leu119–Val137 or H1, Fig. 1A). Interestingly, a second putative transmembrane helix segment (Met63–Phe81 or H2) appeared when the hydropathy analysis was performed with protonated Asp and Glu residues (simulating the acidic conditions required for membrane binding, Fig. 1B). The hydropathy of H2 increased 13.77 kcal/mol after protonation, as calculated using the Wimley-White octanol scale, or 7.39 kcal/mol when using the interfacial scale (14). The observed increase of hydropathy in H2 would indicate a more favorable partition into the lipid bilayer, and this could explain the increased membrane association observed at acidic pH. Inspection of other T3S PopD homologues showed a similar pH-sensitive hydrophobicity for this segment, suggesting that this characteristic has been conserved among T3S systems (Fig. S1). We reasoned that elimination of the negative charges present in this segment will promote the binding and insertion of PopD into membranes at higher pH. The simultaneous modification of the E69A, D71A, and E75C in PopD shifted the pore formation activity to higher pH, indicating that protonation of these acidic residues could facilitate protein-membrane interaction (Fig. 1C). The increase in the pH range at which PopDE69A,D71A,E75C perforated model membranes was modest, and we could not discard that other factors may be required to efficiently assemble a translocon complex into the membrane at neutral pH, for example, the association of PopD with PopB.
Figure 1.

Identification of a second potential transmembrane segment after protonation of acidic residues in PopD. A, schematic of the primary structure of P. aeruginosa T3S translocator PopD. Predicted hydrophobic segments 1 (H1) and 2 (H2) are shown. The segment H2, whose hydrophobicity increased upon protonation of acidic residues, is underlined. B, hydropathy plot of PopD before (black) and after neutralization of acidic residues (red). The graph was generated using membrane protein explorer (MPEx) with a 19-amino acid sliding window. ΔG was defined as the free energy to transfer amino acids from lipid bilayer to water. The position of segment H2 is indicated with a line. C, pore formation activity of WT PopD and PopDE69A/D71A/E75C at the indicated pH. Pore formation was determined as the fraction of encapsulated Tb(DPA)33− quenched by EDTA as described previously (37). PopD was incubated with liposomes at 20–23 °C for 20 min. The protein:lipid ratio was 1:1000. The mean from two independent experiments are reported with error bars corresponding to the range. D, glutaraldehyde cross-linking of PopD homo-oligomers and PopB and PopD hetero-oligomers formed on liposomes. PopD alone or premixed with an excess of PopB were incubated with liposomes in buffer B for 20 min with a protein:lipid ratio of ∼1:5000. Proteoliposomes were pelleted and subjected to immunoblotting using an anti-PopD antibody. Expected molecular masses for PopD n-mers (left) and PopD:PopB n-mers (right) were estimated using the molecular mass for PopD (31.3 kDa) and PopB (40.1 kDa) monomers.
Formation of PopB and PopD heterodimers leads to stable membrane-inserted hetero-complexes
PopB alters the pH-dependent binding of PopD to membranes, and as mentioned above, the interaction of PopD with PopB modifies the stoichiometry of the formed oligomers (13). This suggested that the formation of a PopB–PopD heterodimer could be responsible for the change in the stoichiometry of the complexes, however, the existence of such a heterodimer has remained elusive. To detect the presence of a PopB–PopD heterodimer, PopD was incubated with liposomes with or without PopB, and the resulting oligomers were reacted with the nonspecific cross-linker glutaraldehyde. The presence of cross-linked proteins was detected by immunoblotting using anti-PopD antibodies. When PopD was allowed to form homo-oligomers in liposomes, cross-linking captured several complexes with apparent molecular masses that corresponds to PopD dimers (62.6 kDa), trimers (93.9 kDa), and pentamers (156.5 kDa). Other complexes with higher molecular mass were also observed but it is difficult to estimate apparent molecular masses in this region. In the presence of an excess of PopB, a band that corresponds to a heterodimer (71.4 kDa) was observed. Additionally, oligomers with molecular masses similar to a dimer of heterodimers (142.7 kDa) and a trimer of heterodimers (214.1 kDa) were also observed (Fig. 1D). Despite the fact that the composition of the complexes observed in the presence of PopB cannot be precisely assessed from this SDS-PAGE analysis, it is clear that addition of PopB redirected the formation of PopD homo-complexes toward formation of PopD–PopB heterodimers.
Because the cross-linking reaction of membrane-associated complexes required neutral pH, we tested whether neutralizing the pH released the translocators from the liposomal membranes (Fig. 2A). We also tested conditions commonly used to distinguish peripherally associated proteins from membrane-inserted proteins (15, 16). Proteoliposomes containing inserted PopB and PopD were pelleted and resuspended in the indicated buffers (Fig. 2, B and C). In the end, liposome-bound proteins were separated from free proteins by sucrose gradient centrifugation. Proteoliposomes float to the top of the gradient due to their low density, whereas unbound free proteins sediment to the bottom. As expected, binding of PopB and PopD to membranes at pH 7.5 was not favorable, but both PopB and PopD remained bound if the pH was raised to neutral (Fig. 2A). When treated with a chaotropic agent, alkaline pH, or high salt buffers, about 54 ± 20% of PopD was dissociated from the membrane when treated with 6 m urea in the absence of PopB (Fig. 2B). In contrast, PopB and PopD remained mostly membrane-bound when forming hetero-complexes. In the presence of PopB, only 16 ± 7% of PopD was dissociated by urea (Fig. 2C). These observations indicated that after stable insertion at acidic pH, PopB and PopD remained stably inserted in membranes at neutral pH.
Figure 2.

Assessment of the stability of membrane-associated PopB and PopD. A, PopB and PopD remained bound to membranes after pH neutralization. Purified PopD and PopB (1:7 molar ratio) were added to liposomes suspended in 20 mm sodium acetate, pH 4, or buffer C (HBS). Sample neutralization was achieved by addition of 2 m Trizma base. Membrane-associated proteins were separated from unbound proteins using a membrane flotation assay. The top fractions containing membrane-bound proteins were collected, precipitated with TCA, and analyzed for PopB and PopD by immunoblotting. Band intensities were quantified using ImageJ. Membrane-bound proteins in lanes labeled 7.5, 4–7, and 4 (no liposome) were quantified by normalizing the detected PopB and PopD to that in lane 4 (plus liposomes). Data are reported as the mean from two independent experiments and their range. Representative blots are shown. B, some dissociation of PopD from homo-oligomers was observed when proteoliposomes were subjected to the indicated treatments. Proteoliposomes containing PopD alone were treated with buffer C, 6 m urea, pH 8.0, 0.1 m sodium carbonate, pH 11.5, or 1 m NaCl in 10 mm Hepes, pH 7.5, for 30 min on ice. Membrane-bound proteins (top fractions: T) were separated from dissociated proteins (middle: M, and bottom: B fractions) using a floatation assay. All fractions (T, M, and B) were collected, precipitated with TCA, and PopD was detected by immunoblotting. The amount of dissociated proteins was quantified and shown as the percentage of total protein ((M + B)/(M + B + T)) in each condition. Data are reported as the mean from two independent experiments and the range. Representative blots are shown. C, PopB and PopD hetero-complexes were stably inserted in the membranes. Extraction of PopB and PopD on liposomes were performed as described in B. PopB and PopD were detected by immunoblotting using the indicated primary antibodies and peroxidase-conjugated secondary antibodies. The amount of dissociated protein was analyzed as described in B.
The hydrophobic segments of PopD did not adopt typical transmembrane orientation in PopB/PopD hetero-complexes
Stable association of PopB and PopD with membranes suggests that the translocators may insert one or more hydrophobic segments across the membrane, as observed for integral membrane proteins. Therefore, we targeted H1 and H2 segments (Fig. 1A) for site-directed fluorescence labeling and used multiple biophysical techniques to determine the interaction and location of the segments at the membrane (17). We labeled Cys residues (introduced one at a time on different locations in these segments) with the environment-sensitive fluorescent probe (7-nitrobenz-2-Oxa-1,3-Diazol-4-yl) (NBD). NBD has been successfully used to probe the location of amino acids in membrane proteins because of its small size, its ability to locate in polar and nonpolar environment, and the distinct fluorescent properties displayed in those environments (18, 19). The NBD emission intensity, maximum emission wavelength (λmax), and fluorescence lifetime (τ) are excellent reporters for the microenvironment around the targeted amino acid (20). In an aqueous environment, NBD exhibits red-shifted λmax and lower τ; compared with a hydrophobic environment. For example, the emission spectrum of NBD in water has λmax at 551 nm, whereas NBD-labeled cholesterol in membranes showed a λmax of 519 nm (Fig. 3). Single NBD-labeled PopD derivatives were mixed with an excess of PopB to ensure the incorporation of labeled PopD into hetero-complexes. Excess PopB will form some PopB homo-oligomers (13), but PopB is not labeled with NBD and therefore will not interfere with the analysis of PopD membrane interaction.
Figure 3.

The hydrophobic segments of PopD did not adopt a typical transmembrane orientation in hetero-complexes. Multiple residues in segments H1 and H2 were replaced by Cys and labeled with the environment-sensitive probe NBD. Each single NBD-labeled PopD was premixed with a 10 times molar excess of PopB WT before incubation with liposomes. Reported data correspond to the mean of two independent measurements and error bars indicate the range. A, λmax of NBD attached to the indicated residue in segment H2. B, λmax of NBD attached to the indicated residue in segment H1. C, average τ; for NBD-labeled residues in segment H2. D, average τ for NBD-labeled residues in segment H1. The λmax and τ for NBD in water (red lines) and NBD-cholesterol in liposomes (blue lines) serve as references for expected values of λmax and τ for NBD located in a polar or hydrophobic environment, respectively.
After inserting into membranes, the λmax of NBD-labeled PopD derivatives in the hetero-complexes showed a value ranging from 527 to 532 nm for both segments (Fig. 3, A and B). The λmax of PopD-NBD derivatives implies that both hydrophobic segments lie in a relatively nonpolar environment. Intensity weighted average τ of NBD within segment H2 spanned from 4.5 to 6.5 ns, whereas segment H1 from 6 to 8 ns was similar to the measured τ of cholesterol-NBD (Fig. 3, C and D). Consistent with λmax results, τ of PopD-NBD also suggests a nonpolar location for both segments. Overall, NBD in segment H1 exhibited a slightly more hydrophobic environment than H2. Furthermore, flanking residues A53C, H104C, and T154C displayed a more red-shifted λmax and low τ, suggesting a polar surrounding.
The data on λmax and τ describe the polarity of the environment around each labeled residue, but it is not a direct indication of the exposure and location of the residue in the membrane bilayer. Alternatively, NBD probes could be located in a hydrophobic pocket provided by a nonpolar protein cavity or at a protein–protein interface. Fluorescence quenching studies are useful to confirm the location of NBD moieties (17, 20). For example, exposure to the aqueous solvent could be assessed using water-soluble iodide ions as quenchers, and membrane exposure using nitroxide moieties covalently attached to the acyl chain of a phospholipid (e.g. 1-palmitoyl-2-stearoyl-(12-doxyl)-sn-glycero-3-phosphocholine (12-doxyl-PC)) (18, 21). The quenching ratio calculated from iodide and 12-doxyl-PC quenching was used to report on the relative exposure of each NBD-labeled residue to the membrane. A high quenching ratio (defined as exposure to water/exposure to membrane core) indicates a location close to the surface of lipid bilayer, and low quenching ratio indicates proximity to the nonpolar membrane core. PopDA53C-NBD showed the highest quenching ratio, with a value of 4.6 ± 0.2, suggesting that Ala53 is very solvent-exposed, in agreement with the polar environment indicated by fluorescence values reported in Fig. 3. The quenching ratio of residues in segment H2 ranged from 1.42 ± 0.005 to 2.21 ± 0.02, residues in segment H1 ranged narrowly from 0.66 ± 0.01 to 1.18 ± 0.01 (Fig. 4, A and B). Low values in both segments implies that indeed segment H1 and H2 interact with the membrane, and H1 is inserted deeper into the membrane than H2. What's more, neither of these segments showed a quenching pattern of a typical hydrophobic transmembrane segment, in which residues in the middle position display lower quenching ratios compared with residues at the ends of the segment.
Figure 4.

Dual quenching of the emission of NBD-labeled PopD in homo-oligomers or hetero-oligomers with PopB. A and B, quenching ratio of NBD-labeled PopD residues in segments H2 (A) and H1 (B) when PopD was reconstituted into hetero-complexes. Quenching ratio was calculated using kq/(1 − Fdoxyl/F0), where kq is the bimolecular quenching constant obtained from iodide quenching (a measure of NBD exposure to the aqueous solvent) and the amount of quenching observed when 12-doxyl-PC was incorporated into the liposomes (a measure of NBD exposure to the membrane core). C and D, quenching ratio of NBD-labeled PopD residues in segments H2 (C) and H1 (D) in PopD homo-oligomers. In this case, exposure to the membrane core was calculated using 10-DN and quenching ratio was determined using kq/(1 − F10-DN/F0). Data shown are the mean of two measurements and error bars correspond to the range.
The hydrophobic segments of PopD adopt different conformations in the presence of PopB
Given that PopB redirects the formation of PopD homo-oligomers to hetero-oligomers, we compared the quenching ratio for PopD-NBD derivatives in the absence and presence of excess PopB. Initial quenching ratio determinations for PopD-NBD in homo-oligomers were calculated using iodide and 10-doxylnonadecane (10-DN) as quenchers. 10-DN is a quencher located close to the center of the membrane bilayer (22). The quenching ratios for PopD in hetero-oligomers were calculated using 12-doxyl-PC, a quencher that also locates close to the bilayer center, but with a wider quenching radius compared with 10-DN. Although both quenchers provide essentially the same information on NBD accessibility, it is not feasible to directly compare the absolute values between sets of quenching ratios obtained with different membrane-restricted quenchers. However, in these experiments we focused on conformational changes observed in the presence or absence of PopB. When PopD was forming homo-oligomers, segment H1 exhibited higher solvent exposure in the middle of the segment than the two termini with the quenching ratios increasing from PopDL119C-NBD (4.13 ± 0.02) to PopDV129C-NBD (7.05 ± 0.03) and then dropping to low levels for PopDL137C-NBD (1.88 ± 0.07) (Fig. 4D). In PopB and PopD hetero-complexes, however, these NBD-labeled residues showed a similar extent of accessibility to the quenchers (Fig. 4B), suggesting that segment H1 becomes more parallel to the membrane in the presence of PopB. Segment H2 displayed a more tilted angle in homo-oligomers with the N terminus showing a higher solvent exposure than the C terminus (Fig. 4, A and C). Notably, when PopB was present, H2 also became more parallel to the membrane with PopDS68C-NBD and PopDL70C-NBD buried deeper in the lipid bilayer, whereas PopDL78C-NBD became more solvent exposed.
An intact T3S injectisome is required for PopB and PopD insertion into cell membranes at neutral pH
Next, we studied the insertion of the two translocators in P. aeruginosa-infected cell membranes. It is well-known that PopB and PopD can be secreted into the culture media (e.g. by chelating Ca2+ ions with EGTA) (12), and that PopB and PopD are inserted into the plasma membrane when the target cells are incubated with P. aeruginosa at physiological pH (12). Based on our experiments with purified recombinant proteins, we hypothesized that translocators secreted into the media via the injectisome would not be able to efficiently bind to cell membranes, only translocators that are released in close contact to the target membrane will insert and assemble into translocon complexes. To test this hypothesis, we examined PopB and PopD insertion into HeLa cell membranes when the translocators were delivered during the incubation of P. aeruginosa strain PAK (PAK) with HeLa cells, or when using proteins isolated from the bacterial culture media. HeLa cells have proven to be a good model system for P. aeruginosa infection (6, 23). To maximize secretion of translocators, we used a PAK with ΔexsE (lacking T3S system regulator ExsE) (24) and ΔexoSTY (lacking all three T3S system effectors) (25) (Fig. S2).
HeLa cells were infected with PAKΔexsEΔexoSTYΔpopD::popD using a multiplicity of infection of 30 in three different conditions: (i) in PBS, (ii) in Dulbecco's modified Eagle's medium (DMEM), or (iii) in DMEM plus 10% fetal bovine serum (FBS). Both PopB and PopD were largely secreted into the infection media only when FBS was present (Fig. 5A), as reported previously for the Yersinia system (26). Similarly to the poor binding observed with recombinant proteins (Fig. 2A), secreted PopB and PopD isolated from culture media did not efficiently associate with HeLa cell membranes at neutral pH (Fig. 5B). In contrast, PopB and PopD were efficiently detected in HeLa cell membranes at neutral pH in all infection conditions (Fig. 5A), suggesting that secretion and membrane insertion are coupled during translocon assembly at neutral pH.
Figure 5.
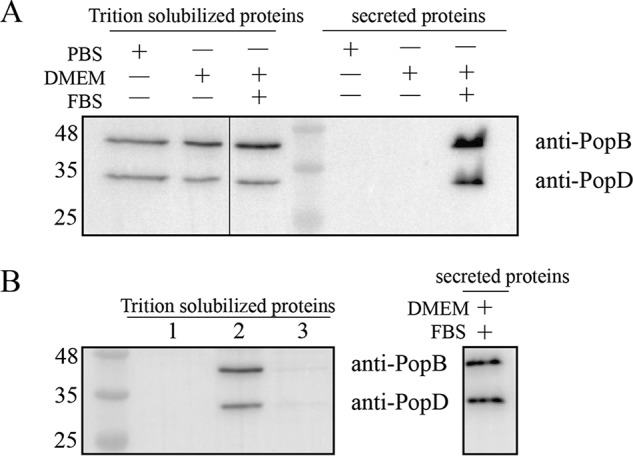
Insertion of PopB and PopD into cell membranes in the presence of P. aeruginosa. A, PopB and PopD were detected on HeLa cell membranes after incubations with PAK at physiological pH. The PAKΔexsEΔexoSTYΔpopD::popD strain was incubated with HeLa cells in the indicated medium for 1 h. After the incubation, secreted proteins in 1 ml of medium were collected by centrifugation, precipitated with TCA, and analyzed by immunoblotting using anti-PopB and anti-PopD antibodies as described under “Experimental procedures.” Infected HeLa cells were lysed with 0.1% Triton X-100, and Triton-solubilized proteins were separated from residual bacteria and insoluble debris using centrifugation. 20 μg of total protein in each Triton-solubilized protein sample was analyzed for the presence of PopB and PopD using immunoblotting. B, translocators secreted to the medium cannot bind to HeLa cells. HeLa cells alone (lane 1), incubated with PAK (lane 2), or incubated with the supernatant containing secreted proteins (lane 3) were collected and analyzed as described in A. The supernatant containing secreted PopB and PopD was obtained by centrifugation after culturing PAK with HeLa cells in DMEM plus FBS for 1 h. Representative blots from two independent experiments are shown.
PopB-assisted PopD insertion into cell membranes
As shown earlier in vitro, hydrophobic segments in PopD displayed different extents of membrane exposure in the presence or absence of PopB (Fig. 4), suggesting that PopB may assist PopD insertion during translocon assembly. To analyze the role of PopB in PopD membrane insertion in vivo, we employed a GSK tag phosphorylation assay that reports on exposure of protein segments to the cytosol of the target cell (27). The GSK tag (MSGRPRTTSFAES) is a 13-amino acid peptide of the N terminus of human glycogen synthase kinase (GSK), where Ser9 is constantly phosphorylated by multiple kinases in the mammalian cell cytosol. Because this tag is not phosphorylated in P. aeruginosa or extracellularly, it constitutes an excellent reporter of accessibility of protein segments to the host cell cytosol.
PopB and PopD are secreted through the T3S needle, and it is possible that a small fraction of these translocators are injected into the cytosol before a switch from translocator secretion to effector secretion can take place (28, 29). Therefore, we introduced a cell permeabilization procedure to selectively collect membrane-associated proteins (Fig. 6A). HeLa cells incubated with P. aeruginosa were washed to remove unbound bacteria, and permeabilized using a Cys-less derivative of perfringolysin O (rPFO). rPFO is a pore-forming toxin that only perforates mammalian cell membranes due to its specificity for cholesterol (30, 31). We used antibodies against glyceraldehyde-phosphate dehydrogenase (GAPDH) and Na+/K+-ATPase to detect cytosolic and plasma membrane fractions, respectively (Fig. 6, B and C). Most of the cytosol was released through the large pores formed by rPFO (∼25–30 nm in diameter), and separated from membranes and insoluble components using centrifugation (PFO sup, Fig. 6B). The presence of some GAPDH in supernatants that were not treated with rPFO indicated that the integrity of some cells was compromised during culture manipulation (Fig. 6B). The majority of the soluble cytosolic components were released after rPFO treatment. Therefore, any translocated PopD will be washed out from the permeabilized membrane fraction and will not interfere with our analysis.
Figure 6.

Insertion of PopD in HeLa cell membranes is promoted by PopB. A, schematic of the membrane protein isolation procedure employed to detect PopD and PopB associated with HeLa cell membranes. Infected cells were first permeabilized with rPFO and centrifuged to separate the released cytosolic components (PFO sup). The pellet containing permeabilized HeLa cells and attached PAK was treated with 0.1% Triton X-100 to selectively solubilize HeLa cell membranes. Insoluble cell debris and bacteria were removed using centrifugation. The supernatant (Triton sup) containing solubilized membrane proteins was precipitated and subjected to immunoblotting as described under “Experimental procedures.” B and C, validation of the membrane protein isolation method. PAKΔexsEΔexoSTYΔpopD::popD-infected HeLa cells were incubated with PBS, 1 μm rPFO, or 2 μm rPFO. PFO sup and Triton sup samples containing 23 μg of total protein were analyzed using immunoblotting for the presence of HeLa cell cytosol or plasma membrane markers using anti-GAPDH (B) and anti-Na+/K+-ATPase (C) antibodies. D, detection of PopD insertion in HeLa cell membranes using a GSK tag phosphorylation assay. PAKΔexsEΔexoSTYΔpopBD or PAKΔexsEΔexoSTYΔpopD strains complemented with pUCP18 plasmid carrying the popB, popD, or GSK-tagged popD genes were incubated with HeLa cells in FBS-free DMEM for 1 h. Proteins associated with HeLa cell membranes were isolated using the membrane protein isolation procedure described in A, and the presence of PopB, PopD, and phospho-GSK PopD was detected by immunoblotting. Representative blots from two independent experiments are shown. The effector translocation ability of used PAK strains into HeLa cells is indicated with a “✓” symbol.
rPFO-permeabilized cell membranes were incubated with 0.1% Triton X-100 to specifically solubilize HeLa cell membrane proteins. Bacterial membranes are not affected by this concentration of detergent (32). Triton X-100-insoluble HeLa cell components plus intact attached bacteria were removed by centrifugation, and the resulting supernatant containing Triton-solubilized proteins was collected and analyzed for the presence of GAPDH and Na+/K+-ATPase (Triton sup, Fig. 6A). When HeLa cells were permeabilized with 2 μm rPFO, the “Triton sup” contained less than 5% of the cytosolic content compared with its “PFO sup.” Therefore, these control experiments corroborated that any PopB or PopD detected in Triton sup fraction (Fig. 6B) was associated with HeLa cell membranes, and not injected into the HeLa cell cytosol.
According to Armentrout and Rietsch (33), the N terminus of PopD is proposed to be located in the target cell cytosol. We chose to insert a single GSK tag after residue Gln40 to avoid potential problems with the PopD secretion signal sequence (34). Proper insertion of PopD is therefore expected to expose the N terminus to the HeLa cell cytosol, where the GSK tag will be phosphorylated. Insertion of the GSK tag did not affect the function of PopD, as determined by complementation of a PAKΔpopD strain with a plasmid encoding PopD–Gln40–GSK (Fig. S3). The effect of PopB on PopD insertion was studied by introducing the plasmid encoding PopD–Gln40–GSK into PAKΔexsEΔexoSTYΔpopD or PAKΔexsEΔexoSTYΔpopBD.
Using the membrane protein isolation procedure described above, we found that PAKΔexsEΔexoSTYΔpopBD strains complemented with plasmids encoding PopB WT, PopD WT, or GSK-tagged PopD were able to insert individual translocators into the target membrane (Fig. 6D), even though strains producing a single translocator were not able to cause perturbation of the actin cytoskeleton as a result of disrupted effectors injection (Fig. S3B) (12). Phosphorylation of the GSK tags was detected using a mAb against phospho-GSK3β(Ser9). The GSK-tagged PopD was not phosphorylated in the absence of PopB. In contrast, phosphorylation of PopD–Gln40–GSK was readily detected when PopB was present. These results clearly showed that PopB assists the insertion of PopD into human cell membranes when forming functional translocons.
Discussion
Combining a series of biophysical and cell-based assays we obtained five important insights into the assembly of the P. aeruginosa T3S translocon, including the interaction of hydrophobic segments of PopD with the membrane and the requirement of PopB for proper PopD assembly into membranes. First, analysis of the enhanced PopD binding observed under acidic conditions revealed a novel membrane-interacting segment that may contribute to anchor the protein into membranes. Second, membrane-assembled PopB and PopD translocators have the properties of integral membrane proteins. Third, the two hydrophobic segments in PopD are buried in the membrane but lie parallel to the membrane surface. Fourth, interaction with PopB modifies the conformation adopted by PopD hydrophobic segments in the membrane. Fifth, PopB promoted the insertion of PopD in P. aeruginosa-infected HeLa cell membranes.
Acidic pH and anionic lipids have been shown to drive the insertion of T3S translocators and other pore-forming toxins like colicin and diphtheria toxin into lipid bilayers (35–39). Acidic pH has been suggested to induce a molten globule intermediate state of proteins, facilitating their interaction with membranes in vitro. Furthermore, an increase of net positive charge of proteins at low pH would decrease protein-protein interactions (i.e. aggregation) during folding in the aqueous solvent, whereas favoring the interaction with membranes containing negatively charged lipids. Therefore, it is not surprising that purified PopB and PopD bind better to model membranes containing anionic lipids at acidic pH. Moreover, it has been shown that increasing concentrations of NaCl inhibit the interaction of PopB and PopD with membranes (36).
The pH-dependent insertion of PopD in the membrane suggests that protonation of acidic residues might play a role in this process, as shown previously for other pore-forming toxins (40, 41). Protonation of acidic residues in PopD revealed a significant increase of hydropathy in one segment (Met63–Phe81, Fig. 1B). The pH-dependent hydrophobicity of this segment is conserved among PopD homologues (Fig. S1), and this unique property is not found in other pore-forming proteins like colicins, diphtheria toxin, or Bcl-like proteins (Fig. S4). Modification of acidic residues in the segment to uncharged amino acids promoted PopD binding to membranes at higher pH values (Fig. 1C), suggesting that protonation of these residues is involved in the pH-dependent insertion of PopD into model membranes.
Protonation of His residues in diphtheria toxin has been suggested to induce a major conformational change in the protein that exposes hydrophobic segments to the membrane (42, 43), whereas protonating the acidic residues in its transmembrane hairpin modifies the membrane insertion (41). Therefore, it is tempting to think that once the pH-sensitive segment in PopD is protonated, it becomes hydrophobic enough to form a transmembrane hairpin with segment H1 (Leu119–Val137) penetrating the membrane and anchoring PopD in the membrane. This hypothesis prompted us to examine the orientation of these segments further in the membrane-inserted PopB–PopD complexes.
Characterization of the structural arrangement of the T3S translocators in model membranes has been difficult given their tendency to form mixtures of homo- and hetero-oligomers (13, 44). However, we found experimental conditions to maximize PopD incorporation into hetero-complexes by mixing PopD with an excess of PopB (13). This method allowed us to study PopD uniformly associated with PopB in hetero-complexes and compare its conformation with the one adopted while forming homo-complexes. PopD was stably associated with membranes, suggesting the presence of one or more transmembrane segments (Fig. 2). Therefore, we examined the conformation of the two hydrophobic segments H1 and H2 when PopD was associated with membranes. Fluorescence measurements on single NBD-labeled PopD derivatives showed that both hydrophobic segments were located in a nonpolar environment (Fig. 3). However, in contrast with the transmembrane orientation assumed in current models for T3S translocons, dual quenching studies positioned the two segments adopting in-plane conformations when PopD was assembled into hetero-complexes (Fig. 4, A and B).
Given that other components of the T3S apparatus (e.g. the needle tip) can affect the translocon assembly, we decided to advance our study in the context of a cellular environment. After being secreted through the needle, the translocators could be (i) released to the media, (ii) inserted directly into the membrane while being secreted, or (iii) translocated into the cytosol if one active translocon is already in place and the switch to effector secretion has not yet occurred. To specifically study T3S-dependent insertion of PopB and PopD in cell membranes, we evaluated these three possibilities. FBS induced secretion of T3S translocators into extracellular medium has been documented in Y. enterocolitica (26) and we found that FBS could induce nonspecific secretion of translocators in PAK cultures. Medium containing PopB and PopD secreted in the presence of FBS was separated from bacteria and incubated with HeLa cells to evaluate post-secretion binding of the translocators. We found that secreted translocators bind to the HeLa cell plasma membrane at very low levels. This observation readily correlates with the little binding observed at neutral pH when using purified recombinant PopB and PopD and model membranes (Fig. 2A). In addition, these results corroborate that no receptor present in the target cell membrane (and absent in model membranes) seems to be sufficient to trigger binding of the translocators. They also emphasize that effective insertion of translocators into cell membranes at physiological pH requires a full T3S system. Eliminating FBS during infection of HeLa cells with PAK strains minimized the secretion of the translocators in the absence of target cells (Fig. 5A) and therefore eliminated any interference from nonspecific binding of translocators secreted to the media.
A source of uncertainty is added when analyzing translocator topology in human cell membranes if translocators are partially delivered into the target cell cytosol. Transient injection of PopD is possible during the time it takes to switch from translocator secretion to effector secretion. Injection of translocators into the target cell cytosol has been noticed in certain T3S systems previously (28, 29). In addition, intracellular functions have been proposed for translocated IpaB, a translocator from the Shigella T3S system (29, 45, 46). Any translocated PopD will interfere with the characterization of membrane-inserted PopD, especially when the GSK tag was used to report on exposure to host cell cytosol. To avoid this problem, we developed a membrane-enrichment procedure that included the permeabilization of the plasma membrane and elimination of water-soluble cytosolic components (e.g. injected translocators). Permeabilization with rPFO allowed the release of soluble cytosol content. Subsequently, proteins in HeLa cell membranes were selectively solubilized with a low concentration of Triton X-100, conditions that do not lyse P. aeruginosa (Fig. 6A). In the end, only translocators associated with the target cell membrane are enriched and analyzed.
Using this approach we demonstrated that a strain lacking both translocators (PAKΔexsEΔexoSTYΔpopBD) complemented with plasmid bearing the popD-GSK gene was able to insert PopD in HeLa cell membranes but no phosphorylation took place on the GSK segment. However, when a strain lacking only PopD (PAKΔexsEΔexoSTYΔpopD) was complemented with the same plasmid, both PopB and PopD inserted into the membrane and the GSK segment was phosphorylated (Fig. 6D). These results clearly indicate that PopB is required to properly insert PopD into the target cell membrane to form functional translocons.
The mechanism of translocator assembly into cell membranes during bacterial infection has been explored previously (12, 33, 47). However, how the membrane-associated translocators transition to a functional translocon has remained elusive. This work provides important insights into the mechanism of translocon assembly. First, PopB and PopD formed a heterodimer on lipid membranes, suggesting that an early PopB and PopD interaction is essential for guiding the assembly of hetero-complexes. Second, the interaction of PopB with PopD is required to properly insert PopD into the target cell membrane and assemble functional translocons.
Experimental procedures
Plasmids and strains construction
Genomic deletion of the genes encoding T3S regulator ExsE, translocator PopD, or translocator PopB were introduced according to a two-step allelic exchange procedure described by Hmelo et al. (48) using the PAKΔexoSTY strain (courtesy of Dr. Stephen Lory (25)) to generate the PAKΔexsEΔexoSTYΔpopD and PAKΔexsEΔexoSTYΔpopBD strains. pUCP18 plasmids containing the DNA fragment coding for PopD WT or PopB WT were generated using Gibson assembly as described previously (13). The expression of the translocators is regulated by the endogenous promoter region that was included in the DNA fragment (13). The plasmid containing the gene encoding PopD–Gln40–GSK was generated using primers: forward, CGCCCTCGCACTACTAGTTTCGCTGAAAGTGTGCCGGCCGCGCGGGCCGATC; reverse, GAAACTAGTAGTGCGAGGGCGACCACTCATCTGCGGCAGGTCCGCAGCCG. Modifications introduced into plasmids used in this work were verified by DNA sequencing. Plasmids carrying the gene encoding the desired protein were introduced into PAKΔexsEΔexoSTYΔpopD or PAKΔexsEΔexoSTYΔpopBD strains using electroporation, and positive clones were identified by their resistance to carbenicillin. The generated strains were evaluated by their ability to restore translocation of effectors into HeLa cells, evidenced by typical cell rounding as described (13).
Protein expression, purification, and fluorescent labeling
PopB and PopD were purified as a complex with the His-tagged chaperone PcrH (hisPcrH) as previously described (37). Single Cys PopD derivatives were generated by site-directed mutagenesis as described previously (37). PopD derivatives were labeled with N,N′-dimethyl-N-(iodoacetyl)-N′-(NBD) ethylenediamine (IANBD amide, Invitrogen) when bound to hisPcrH in 50 mm Hepes, pH 8.0, supplemented with 100 mm NaCl at 20–23 °C. After 2 h, the labeling reaction was stopped by removing the unreacted dye using a Sephadex G-25 size exclusion column. The labeled hisPcrH–PopD complex was bound to an immobilized metal ion affinity chromatography column and PopD was dissociated from hisPcrH using buffer A (20 mm Tris-HCl, pH 8.0, supplemented with 6 m urea and 20 mm glycine) as described previously (37). Purified PopD was kept in buffer A until use. The labeling efficiency was calculated using the molar absorptivities at 280 nm for PopD (13,980 m−1 cm−1) and NBD (25,000 m−1 cm−1) in buffer A. Labeling efficiency was more than 70% for all PopD derivatives. All NBD-labeled PopD derivatives showed a pore-forming activity similar to PopD WT (Fig. S5).
Liposome preparation and reconstitution of homogenous PopB–PopD complexes
Large unilamellar liposomes were prepared using the extrusion procedure with 0.1-μm membranes as reported in detail previously (37). Unless otherwise stated, the lipid composition was 65 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 15 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (POPS), and 20 mol % cholesterol. For quenching experiments with 10-DN, liposomes were prepared as above with 10 mol % of the POPC replaced by 10 mol % 10-DN. For quenching experiments with 12-doxyl-PC, the liposome composition was 20% POPC, 40% 1-palmitoyl-2-stearoyl-sn-glycero-3-phosphocholine (PSPC), 15% POPS, 20% cholesterol, and 5% 12-doxyl-PC. Replacing POPC with a more saturated PSPC did not alter the ability of PopB and PopD to hetero-oligomerize on the membrane (Fig. S6). All lipids were purchased from Avanti Polar Lipids, cholesterol was from Steraloids Inc. PopD WT or NBD-labeled PopD derivatives (30 nm) were mixed with PopB (210–300 nm) in buffer A. The PopB and PopD mixture was added to liposomes suspended in buffer B (50 mm sodium acetate, pH 4.3) or 50 mm Hepes, pH 7.5, supplemented with 100 mm NaCl and incubated at 20–23 °C for 20 min (the range for protein to lipids ratio was maintained at 1:5,000–1:6,000). The final volume for each reaction was 300 μl and when indicated, pH neutralization was achieved with the addition of 2 μl of a 2 m solution of Trizma (Tris base).
Immunoblotting
Protein was separated on a 12.5% SDS-PAGE, transferred to polyvinylidene difluoride membrane (GE Healthcare), and blocked with 5% milk for 1 h at 20–23 °C. Primary antibodies were diluted as indicated using 3% (w/v) BSA in a solution of 25 mm Tris-HCl, pH 7.5, supplemented with 150 mm NaCl, and 0.1% Tween 20, and incubated with the membrane at 4 °C overnight. After washing the membrane three times with the same solution (10 min each), anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody (Sigma) and a chemiluminescent detection reagent (Amersham Biosciences ECL Prime, GE Healthcare) were used to detect targeted primary antibodies as instructed by the manufacturer. Images were analyzed and quantified using ImageJ software. PopD and PopB polyclonal antibodies were raised in rabbits immunized with recombinant full-length proteins extracted from preparative SDS-PAGE gels.
Glutaraldehyde cross-linking
PopD (0.1 μm) or a mixture of PopD (0.1 μm) and PopB (0.7 μm) were incubated with liposomes (4.8 mm total lipids) in a total of 200 μl of buffer B for 20 min at 20–23 °C. Buffer pH was then raised to 7.0 by titration with a solution of 1 m Hepes, pH 8.0 (∼30 μl). A freshly prepared solution of 1% glutaraldehyde (Sigma) in water was added to the reaction to a final concentration of 0.01%, and incubated for 2 min at 20–23 °C. The cross-linking reaction was stopped with the addition of 1 m Tris, pH 8.0, to a final concentration of 150 mm and incubated for 20 min at 20–23 °C. To collect proteoliposomes, samples were centrifuged at 60,000 rpm (average of 128,000 × g) for 30 min in a TLA-120.2 rotor (Beckman Coulter). The resulting pellets were resuspended in 50 μl of SDS-PAGE sample buffer (50 mm Tris-HCl supplemented with 4% β-mercaptoethanol, 10% glycerol, 5 mm EDTA, and, 0.02% bromphenol blue). Protein complexes were resolved in a 4–12% ExpressPlus PAGE gels (GenScript), and probed for PopD by immunoblotting using anti-PopD (diluted 1:4000) polyclonal antibodies.
Protein extraction from liposomal membranes
PopD homo-oligomers or PopB:PopD hetero-oligomers were prepared by adding PopD (to 0.1 μm final concentration) or a mixture of PopB:PopD (0.1:0.7 μm final concentration, respectively) solubilized in buffer A to liposomes (4 mm total lipids final concentration) suspended in 500 μl of buffer B and incubating the sample for 20 min. Proteoliposomes were then spun down as described above. After centrifugation, the pellets were resuspended in 75 μl of a solution of buffer C (control), 6 m urea (buffer A), 0.1 m sodium carbonate, pH 11.5, or 1 m NaCl in 10 mm Hepes, pH 7.5, and samples were incubated on ice for 30 min with occasional mixing. Any unbound protein was separated from liposomes using a floatation assay as previously reported (37). Briefly, samples were mixed with 67% sucrose, and overlaid with 40% sucrose and 4% sucrose. After ultracentrifugation at an average 288,000 × g for 50 min at 4 °C, 300 μl of the top, middle, and bottom fractions were collected and precipitated with 10% TCA. The pellets were resuspended in 80 μl of SDS-PAGE sample buffer, and examined by immunoblotting using anti-PopD (diluted 1:4000) and anti-PopB (diluted 1:200,000) polyclonal antibodies.
Fluorescence measurements
Steady state fluorescence measurements were made with a Fluorolog-3 photon-counting spectrofluorimeter as reported earlier (37). For NBD emission maxima λmax measurements, the wavelength for NBD excitation was set to 475 nm and emission was scanned from 510 to 560 nm, every 1 nm using a 1-s integration time. The bandpass was 2 nm for excitation and 4 nm for emission. Polarizers were in place in the excitation (vertical) and the emission (horizontal) to reduce scattered light and to account for polarization effects in the emission monochromator (49). The spectra were corrected by subtraction of the background emission of a sample containing an identical amount of liposomes.
For collisional quenching by iodide, a set of samples were prepared for each NBD-labeled PopD derivatives. In each set, the samples contained an increasing concentration of KI (0–90 mm) obtained by addition of a solution of 1 m KI and 1 mm Na2S2O3. The ionic strength in each sample was maintained constant by addition of a solution of 1 m KCl and 1 mm Na2S2O3. Initial fluorescence intensity F0 was defined as the intensity of NBD-labeled PopD in 90 mm KCl. Different KI:KCl mixtures were incubated with proteoliposomes for 10 min at 20–23 °C, and NBD intensities were measured as Fiodide. A linear relationship between fluorescence intensities and the concentration of iodide for each NBD-labeled PopD was obtained when data were analyzed using the Stern-Volmer equation (F0/Fiodide) − 1 = Ksv[I−], where Ksv is the Stern-Volmer quenching constant. kq was further calculated from Ksv and τ of NBD-labeled PopD in the absence of quenchers using the equation: kq = KSV/τ. Collisional quenching by membrane-restricted quenchers (12-doxyl-PC or 10-DN) was carried out using two sets of liposomes, one with quencher and one without quencher. F0 represents the fluorescence signal of NBD-labeled PopD incorporated in the liposomes without quenchers. Fdoxyl or F10-DN indicates the fluorescence intensity of NBD-labeled PopD incorporated in the liposomes with quenchers. For the acquisition of NBD emission the wavelength for excitation and emission were 475 and 530 nm, respectively. The bandpass was 5 nm for both excitation and emission. Polarizers were used as described above. The temperature of samples was equilibrated to 12–15 °C before measurements. Quenching ratio was determined using equations kq/(1 − Fdoxyl/F0) or kq/(1 − F10-DN/F0).
Time-resolved fluorescence measurements were taken using a Chronos spectrofluorometer (ISS, Champaign, IL) with the same setups as reported previously (37). The τ of NBD was measured in frequency domain (20 frequencies, 20–200 MHz). A solution of fluorescein (Invitrogen) in 0.1 m NaOH was used as reference with a value of 4.05 ns (50). The emission intensity of the reference sample was matched with that of the measured sample (±10%). An equivalent sample without labeled proteins was used for blank subtraction (51). All τ data were analyzed with Vinci software, fitted to two discrete exponential τ, and the calculated average τ was intensity weighted.
Analysis of translocators associated with HeLa cells
HeLa cells were maintained in DMEM (Hyclone) supplemented with 10% FBS in a 5% CO2 atmosphere at 37 °C. Prior to infection, 2.5–2.8 × 106 cells were washed twice with pre-warmed Dulbecco's PBS (DPBS) (Hyclone) and replenished with 4 ml of DPBS, DMEM, or DMEM supplemented with 10% FBS. PAKΔexsEΔexoSTYΔpopD::popD grown overnight in Miller lysogeny broth at 37 °C was diluted to A600 of 0.15 in fresh broth the next day, and allowed to grow until A600 of 1. Aliquots containing 0.3–0.5 ml of bacterial culture were added to a monolayer of HeLa cells at a multiplicity of infection of 30 and incubated for 1 h at 37 °C in a 5% CO2 atmosphere (Fig. S7). At the end of the incubation, the entire medium containing free bacteria and secreted proteins was removed by aspiration. An aliquot of 1 ml of the medium was centrifuged (twice at 18,000 × g for 10 min at 4 °C), and the supernatant containing secreted proteins was precipitated with TCA, resuspended in SDS-PAGE sample buffer, and analyzed by immunoblotting for the presence of PopB and/or PopD as indicated below. The flasks containing infected cells were washed gently three times with DPBS, cells were scraped in 1 ml of ice-cold DPBS supplemented with protease inhibitor mixture (PIC) (Roche) and 5 mm NaF, and pelleted at 4 °C (2,000 × g, 10 min). Cells were resuspended in 210 μl of lysis buffer (DPBS supplemented with 0.1% Triton X-100, PIC, and 5 mm NaF), and incubated for 30 min at 4 °C with constant mixing. Solubilized plasma membrane and cytosolic proteins were separated from insoluble cell debris and intact bacteria by centrifugation at 4 °C (18,000 × g, 15 min). The supernatant containing Triton-solubilized proteins was precipitated with methanol:chloroform (sample:methanol:chloroform:H2O ratio was 1:4:1:3), and the precipitate was resuspended in 60 μl of buffer C containing 2% SDS. Protein concentrations were measured using bicinchoninic acid assay (Thermo Scientific). An aliquot containing 20 μg of total protein was analyzed by immunoblotting for the presence of PopB and/or PopD using anti-PopB (diluted 1:20,000) and anti-PopD (diluted 1:1000) serums.
To obtain secreted PopB and PopD, PAKΔexsEΔexoSTYΔpopD::popD was incubated with HeLa cells in DMEM plus FBS for 1 h at 37 °C. After infection, the entire medium was removed, and the supernatant containing secreted proteins was clarified by centrifugation (twice at 18,000 × g for 10 min). The presence of PopB and PopD in the supernatant was confirmed by immunoblot. To determine whether secreted PopB and PopD bind to HeLa cell membranes, the supernatant containing secreted proteins was transferred to a flask of HeLa cells that have not been exposed to bacteria, and incubated for 1 h. The binding of secreted PopB and PopB to HeLa cells was analyzed as described above.
HeLa cell membrane protein isolation
PAKΔexsEΔexoSTYΔpopD::popD was incubated with HeLa cells in DMEM free of FBS (to minimize nonspecific secretion of translocators) for 1 h. Infected cells were collected in 1 ml of ice-cold DPBS and spun down at 2,000 × g for 10 min at 4 °C. Cells were washed once with 1 ml of DPBS to remove free bacteria. Cell were resuspended in 230 μl of DPBS (control) or the same volume of a solution containing 1–2 μm rPFO in buffer C (supplemented with PIC and 5 mm NaF) and incubated for 30 min at 20–23 °C. The released cytosolic components were separated from permeabilized cells and attached bacteria by centrifugation at 2,000 × g for 10 min at 4 °C. The released cytosol (PFO sup) was further clarified by centrifugation at 18,000 × g for 20 min, and precipitated with methanol:chloroform as described above. Precipitated proteins were resuspended in 60 μl of buffer C containing 2% SDS. Protein concentration in the PFO sup fraction was measured using a bicinchoninic acid assay. The permeabilized cells were washed with 500 μl of 10 mm Tris, pH 7.4, supplemented with PIC and 5 mm NaF on ice for 5 min, and lysed in 210 μl of lysis buffer for 30 min at 4 °C. Solubilized membrane proteins (Triton sup) were separated from residual bacteria and insoluble debris by centrifugation at 18,000 × g for 15 min at 4 °C. Aliquots containing 23 μg of total protein of each PFO sup or Triton sup fractions were analyzed by immunoblotting using anti-GAPDH antibody (diluted 1:2,000) (Cell Signaling Technology) or anti-Na+/K+-ATPase antibody (diluted 1:1,000) (Cell Signaling Technology).
To detect GSK tag phosphorylation, HeLa cells infected with the indicated P. aeruginosa strains were treated with 2 μm rPFO and analyzed as described above. The resulting Triton sup fraction was analyzed by immunoblotting using anti-PopB, anti-PopD, or anti-phospho-GSK antibodies (1:1,000 dilution as indicated by the manufacturer, Cell Signaling Technology).
Author contributions
Y. T., F. B. R., and A. P. H. conceptualization; Y. T., F. B. R., M. B., and A. P. H. formal analysis; Y. T., F. B. R., M. B., and A. P. H. validation; Y. T., F. B. R., and M. B. investigation; Y. T., F. B. R., and A. P. H. methodology; Y. T. and A. P. H. writing-original draft; Y. T., F. B. R., M. B., and A. P. H. writing-review and editing; A. P. H. supervision; A. P. H. funding acquisition; A. P. H. project administration.
Supplementary Material
Acknowledgment
We thank K. C. Rossi for assistance with the preparation and preliminary characterization of some of the PopD derivatives used in these studies.
This work was supported in part by National Institutes of Health Grant GM097414 (to A. P. H). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S7.
- T3S
- type III secretion
- 10-DN
- 10-doxylnonadecane
- 12-doxyl-PC
- 1-palmitoyl-2-stearoyl-(12-doxyl)-sn-glycero-3-phosphocholine
- τ
- fluorescence lifetime
- DMEM
- Dulbecco's modified Eagle's medium
- DPBS
- Dulbecco's PBS
- GAPDH
- glyceraldehyde-phosphate dehydrogenase
- GSK
- glycogen synthase kinase
- H1/2
- hydrophobic segment 1 or 2
- NBD
- 7-nitrobenz-2-oxa-1,3-diazol-4-yl
- PAK
- Pseudomonas aeruginosa strain PAK
- PIC
- protease inhibitor cocktail
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine
- rPFO
- Cys-less derivative of perfringolysin O
- PSPC
- 1-palmitoyl-2-stearoyl-sn-glycero-3-phosphocholine.
References
- 1. Hauser A. R. (2009) The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 7, 654–665 10.1038/nrmicro2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Howell H. A., Logan L. K., and Hauser A. R. (2013) Type III secretion of ExoU is critical during early Pseudomonas aeruginosa pneumonia. mBio 4, e00032-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pettersson J., Nordfelth R., Dubinina E., Bergman T., Gustafsson M., Magnusson K. E., and Wolf-Watz H. (1996) Modulation of virulence factor expression by pathogen target cell contact. Science 273, 1231–1233 10.1126/science.273.5279.1231 [DOI] [PubMed] [Google Scholar]
- 4. Radics J., Königsmaier L., and Marlovits T. C. (2014) Structure of a pathogenic type 3 secretion system in action. Nat. Struct. Mol. Biol. 21, 82–87 10.1038/nsmb.2722 [DOI] [PubMed] [Google Scholar]
- 5. Engel J., and Balachandran P. (2009) Role of Pseudomonas aeruginosa type III effectors in disease. Curr. Opin. Microb. 12, 61–66 10.1016/j.mib.2008.12.007 [DOI] [PubMed] [Google Scholar]
- 6. Dortet L., Lombardi C., Cretin F., Dessen A., and Filloux A. (2018) Pore-forming activity of the Pseudomonas aeruginosa type III secretion system translocon alters the host epigenome. Nat. Microb. 3, 378–386 10.1038/s41564-018-0109-7 [DOI] [PubMed] [Google Scholar]
- 7. Anantharajah A., Mingeot-Leclercq M.-P., and Van Bambeke F. (2016) Targeting the type three secretion system in Pseudomonas aeruginosa. Trends Pharmacol. Sci. 37, 734–749 10.1016/j.tips.2016.05.011 [DOI] [PubMed] [Google Scholar]
- 8. Deng W., Marshall N. C., Rowland J. L., McCoy J. M., Worrall L. J., Santos A. S., Strynadka N. C. J., and Finlay B. B. (2017) Assembly, structure, function and regulation of type III secretion systems. Nat. Rev. Microb. 15, 323–337 10.1038/nrmicro.2017.20 [DOI] [PubMed] [Google Scholar]
- 9. Kudryashev M., Stenta M., Schmelz S., Amstutz M., Wiesand U., Castaño-Díez D., Degiacomi M. T., Münnich S., Bleck C. K., Kowal J., Diepold A., Heinz D. W., Dal Peraro M., Cornelis G. R., and Stahlberg H. (2013) In situ structural analysis of the Yersinia enterocolitica injectisome. eLife 2, e00792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hu B., Morado D. R., Margolin W., Rohde J. R., Arizmendi O., Picking W. L., Picking W. D., and Liu J. (2015) Visualization of the type III secretion sorting platform of Shigella flexneri. Proc. Natl. Acad. Sci. U.S.A. 112, 1047 10.1073/pnas.1411610112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hu B., Lara-Tejero M., Kong Q., Galán J. E., and Liu J. (2017) In situ molecular architecture of the Salmonella type III secretion machine. Cell 168, 1065–1074.e1010 10.1016/j.cell.2017.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goure J., Pastor A., Faudry E., Chabert J., Dessen A., and Attree I. (2004) The V antigen of Pseudomonas aeruginosa is required for assembly of the functional PopB/PopD translocation pore in host cell membranes. Infect. Immun. 72, 4741–4750 10.1128/IAI.72.8.4741-4750.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Romano F. B., Tang Y., Rossi K. C., Monopoli K. R., Ross J. L., and Heuck A. P. (2016) Type 3 secretion translocators spontaneously assemble a hexadecameric transmembrane complex. J. Biol. Chem. 291, 6304–6315 10.1074/jbc.M115.681031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Snider C., Jayasinghe S., Hristova K., and White S. H. (2009) MPEx: A tool for exploring membrane proteins. Protein Sci. 18, 2624–2628 10.1002/pro.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fujiki Y., Hubbard A. L., Fowler S., and Lazarow P. B. (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93, 97–102 10.1083/jcb.93.1.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ohlendieck K. (1996) Extraction of membrane proteins. in Protein Purification Protocols (Doonan S., ed) pp. 293–304, Humana Press, Totowa, NJ [Google Scholar]
- 17. Heuck A. P., and Johnson A. E. (2002) Pore-forming protein structure analysis in membranes using multiple independent fluorescence techniques. Cell Biochem. Biophys. 36, 89–101 10.1385/CBB:36:1:89 [DOI] [PubMed] [Google Scholar]
- 18. Shatursky O., Heuck A. P., Shepard L. A., Rossjohn J., Parker M. W., Johnson A. E., and Tweten R. K. (1999) The mechanism of membrane insertion for a cholesterol-dependent cytolysin: a novel paradigm for pore-forming toxins. Cell 99, 293–299 10.1016/S0092-8674(00)81660-8 [DOI] [PubMed] [Google Scholar]
- 19. Alder N. N., Jensen R. E., and Johnson A. E. (2008) Fluorescence mapping of mitochondrial TIM23 complex reveals a water-facing, substrate-interacting helix surface. Cell 134, 439–450 10.1016/j.cell.2008.06.007 [DOI] [PubMed] [Google Scholar]
- 20. Johnson A. E. (2005) Fluorescence approaches for determining protein conformations, interactions and mechanisms at membranes. Traffic 6, 1078–1092 10.1111/j.1600-0854.2005.00340.x [DOI] [PubMed] [Google Scholar]
- 21. Chattopadhyay A., and London E. (1987) Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry 26, 39–45 10.1021/bi00375a006 [DOI] [PubMed] [Google Scholar]
- 22. Caputo G. A., and London E. (2003) Using a novel dual fluorescence quenching assay for measurement of tryptophan depth within lipid bilayers to determine hydrophobic α-helix locations within membranes. Biochemistry 42, 3265–3274 10.1021/bi026696l [DOI] [PubMed] [Google Scholar]
- 23. Kaufman M. R., Jia J., Zeng L., Ha U., Chow M., and Jin S. (2000) Pseudomonas aeruginosa mediated apoptosis requires the ADP-ribosylating activity of ExoS. Microbiology 146, 2531–2541 10.1099/00221287-146-10-2531 [DOI] [PubMed] [Google Scholar]
- 24. Rietsch A., Vallet-Gely I., Dove S. L., and Mekalanos J. J. (2005) ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 102, 8006–8011 10.1073/pnas.0503005102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee V. T., Smith R. S., Tümmler B., and Lory S. (2005) Activities of Pseudomonas aeruginosa effectors secreted by the type III secretion system in vitro and during infection. Infect. Immun. 73, 1695–1705 10.1128/IAI.73.3.1695-1705.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee V. T., Mazmanian S. K., and Schneewind O. (2001) A program of Yersinia enterocolitica type III secretion reactions is activated by specific signals. J. Bacteriol. 183, 4970–4978 10.1128/JB.183.17.4970-4978.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garcia J. t., Ferracci F., Jackson M. W., Joseph S. S., Pattis I., Plano L. R., Fischer W., and Plano G. V. (2006) Measurement of effector protein injection by type III and type IV secretion systems by using a 13-residue phosphorylatable glycogen synthase kinase tag. Infect. Immun. 74, 5645–5657 10.1128/IAI.00690-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Francis M. S., and Wolf-Watz H. (1998) YopD of Yersinia pseudotuberculosis is translocated into the cytosol of HeLa epithelial cells: evidence of a structural domain necessary for translocation. Mol. Microbiol. 29, 799–813 10.1046/j.1365-2958.1998.00973.x [DOI] [PubMed] [Google Scholar]
- 29. Iwai H., Kim M., Yoshikawa Y., Ashida H., Ogawa M., Fujita Y., Muller D., Kirikae T., Jackson P. K., Kotani S., and Sasakawa C. (2007) A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell 130, 611–623 10.1016/j.cell.2007.06.043 [DOI] [PubMed] [Google Scholar]
- 30. Johnson B. B., Breña M., Anguita J., and Heuck A. P. (2017) Mechanistic insights into the cholesterol-dependent binding of perfringolysin O-based probes and cell membranes. Sci. Rep. 7, 13793 10.1038/s41598-017-14002-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Savinov S., and Heuck A. (2017) Interaction of cholesterol with perfringolysin O: what have we learned from functional analysis? Toxins 9, 381 10.3390/toxins9120381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Urbanowski M. L., Brutinel E. D., and Yahr T. L. (2007) Translocation of ExsE into Chinese hamster ovary cells is required for transcriptional induction of the Pseudomonas aeruginosa type III secretion system. Infect. Immun. 75, 4432–4439 10.1128/IAI.00664-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Armentrout E. I., and Rietsch A. (2016) The type III secretion translocation pore senses host cell contact. PLoS Pathog. 12, e1005530 10.1371/journal.ppat.1005530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tomalka A. G., Stopford C. M., Lee P.-C., and Rietsch A. (2012) A translocator-specific export signal establishes the translocator–effector secretion hierarchy that is important for type III secretion system function. Mol. Microbiol. 86, 1464–1481 10.1111/mmi.12069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van der Goot F. G., González-Manas J. M., Lakey J. H., and Pattus F. (1991) A “molten-globule” membrane-insertion intermediate of the pore-forming domain of colicin A. Nature 354, 408–410 10.1038/354408a0 [DOI] [PubMed] [Google Scholar]
- 36. Faudry E., Vernier G., Neumann E., Forge V., and Attree I. (2006) Synergistic pore formation by type III toxin translocators of Pseudomonas aeruginosa. Biochemistry 45, 8117–8123 10.1021/bi060452+ [DOI] [PubMed] [Google Scholar]
- 37. Romano F. B., Rossi K. C., Savva C. G., Holzenburg A., Clerico E. M., and Heuck A. P. (2011) Efficient isolation of Pseudomonas aeruginosa type III secretion translocators and assembly of heteromeric transmembrane pores in model membranes. Biochemistry 50, 7117–7131 10.1021/bi200905x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vargas-Uribe M., Rodnin M. V., and Ladokhin A. S. (2013) Comparison of membrane insertion pathways of the apoptotic regulator Bcl-xL and the diphtheria toxin translocation domain. Biochemistry 52, 7901–7909 10.1021/bi400926k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chatterjee A., Caballero-Franco C., Bakker D., Totten S., and Jardim A. (2015) Pore-forming activity of the Escherichia coli type III secretion system protein EspD. J. Biol. Chem. 290, 25579–25594 10.1074/jbc.M115.648204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ladokhin A. (2013) pH-triggered conformational switching along the membrane insertion pathway of the diphtheria toxin T-domain. Toxins 5, 1362–1380 10.3390/toxins5081362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ghatak C., Rodnin M. V., Vargas-Uribe M., McCluskey A., Flores-Canales J. C., Kurnikova M., and Ladokhin A. S. (2015) Role of acidic residues in helices TH8–TH9 in membrane interactions of the diphtheria toxin T domain. Toxins 7, 1303 10.3390/toxins7041303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perier A., Chassaing A., Raffestin S., Pichard S., Masella M., Ménez A., Forge V., Chenal A., and Gillet D. (2007) Concerted protonation of key histidines triggers membrane interaction of the diphtheria toxin T domain. J. Biol. Chem. 282, 24239–24245 10.1074/jbc.M703392200 [DOI] [PubMed] [Google Scholar]
- 43. Kurnikov I. V., Kyrychenko A., Flores-Canales J. C., Rodnin M. V., Simakov N., Vargas-Uribe M., Posokhov Y. O., Kurnikova M., and Ladokhin A. S. (2013) pH-triggered conformational switching of the diphtheria toxin T-domain: the roles of N-terminal histidines. J. Mol. Biol. 425, 2752–2764 10.1016/j.jmb.2013.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schoehn G., Di Guilmi A. M., Lemaire D., Attree I., Weissenhorn W., and Dessen A. (2003) Oligomerization of type III secretion proteins PopB and PopD precedes pore formation in Pseudomonas. EMBO J. 22, 4957–4967 10.1093/emboj/cdg499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Senerovic L., Tsunoda S. P., Goosmann C., Brinkmann V., Zychlinsky A., Meissner F., and Kolbe M. (2012) Spontaneous formation of IpaB ion channels in host cell membranes reveals how Shigella induces pyroptosis in macrophages. Cell Death Dis. 3, e384 10.1038/cddis.2012.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mounier J., Boncompain G., Senerovic L., Lagache T., Chrétien F., Perez F., Kolbe M., Olivo-Marin J.-C., Sansonetti Philippe J., and Sauvonnet N. (2012) Shigella effector IpaB-induced cholesterol relocation disrupts the Golgi complex and recycling network to inhibit host cell secretion. Cell Host Microbe 12, 381–389 10.1016/j.chom.2012.07.010 [DOI] [PubMed] [Google Scholar]
- 47. Broz P., Mueller C. A., Müller S. A., Philippsen A., Sorg I., Engel A., and Cornelis G. R. (2007) Function and molecular architecture of the Yersinia injectisome tip complex. Mol. Microbiol. 65, 1311–1320 10.1111/j.1365-2958.2007.05871.x [DOI] [PubMed] [Google Scholar]
- 48. Hmelo L. R., Borlee B. R., Almblad H., Love M. E., Randall T. E., Tseng B. S., Lin C., Irie Y., Storek K. M., Yang J. J., Siehnel R. J., Howell P. L., Singh P. K., Tolker-Nielsen T., Parsek M. R., Schweizer H. P., and Harrison J. J. (2015) Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat. Protoc. 10, 1820–1841 10.1038/nprot.2015.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jameson D. M., Croney J. C., and Moens P. D. (2003) Fluorescence: basic concepts, practical aspects, and some anecdotes. Methods Enzymol. 360, 1–43 10.1016/S0076-6879(03)60105-9 [DOI] [PubMed] [Google Scholar]
- 50. Boens N., Qin W., Basarić N., Hofkens J., Ameloot M., Pouget J., Lefèvre J. P., Valeur B., Gratton E., vandeVen M., Silva N. D., Engelborghs Y., Willaert K., Sillen A., Rumbles G., et al. (2007) Fluorescence lifetime standards for time and frequency domain fluorescence spectroscopy. Anal. Chem. 79, 2137–2149 10.1021/ac062160k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reinhart G. D., Marzola P., Jameson D. M., and Gratton E. (1991) A method for on-line background subtraction in frequency domain fluorometry. J. Fluoresc. 1, 153–162 10.1007/BF00865362 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.