

Abstract
C-terminal binding protein 1 (CtBP1) and CtBP2 are transcriptional coregulators that repress numerous cellular processes, such as apoptosis, by binding transcription factors and recruiting chromatin-remodeling enzymes to gene promoters. The NAD(H)-linked oligomerization of human CtBP is coupled to its co-transcriptional activity, which is implicated in cancer progression. However, the biologically relevant level of CtBP assembly has not been firmly established; nor has the stereochemical arrangement of the subunits above that of a dimer. Here, multi-angle light scattering (MALS) data established the NAD+- and NADH-dependent assembly of CtBP1 and CtBP2 into tetramers. An examination of subunit interactions within CtBP1 and CtBP2 crystal lattices revealed that both share a very similar tetrameric arrangement resulting from assembly of two dimeric pairs, with specific interactions probably being sensitive to NAD(H) binding. Creating a series of mutants of both CtBP1 and CtBP2, we tested the hypothesis that the crystallographically observed interdimer pairing stabilizes the solution tetramer. MALS data confirmed that these mutants disrupt both CtBP1 and CtBP2 tetramers, with the dimer generally remaining intact, providing the first stereochemical models for tetrameric assemblies of CtBP1 and CtBP2. The crystal structure of a subtle destabilizing mutant suggested that small structural perturbations of the hinge region linking the substrate- and NAD-binding domains are sufficient to weaken the CtBP1 tetramer. These results strongly suggest that the tetramer is important in CtBP function, and the series of CtBP mutants reported here can be used to investigate the physiological role of the tetramer.
Keywords: transcription coregulator, dehydrogenase, cancer, crystallography, structural biology, CtBP, MALS, NAD(H), tetrameric assembly, cancer target
Introduction
C-terminal binding proteins 1 and 2 (CtBP13 and CtBP2) are paralogous transcriptional co-regulators that modulate numerous cellular processes by binding transcription factors and recruiting chromatin remodeling enzymes such as histone deacetylases, methyl transferases, and demethylases to targeted promoters (1–3). CtBP1 was first identified through interactions with the C-terminal region of the adenovirus E1A oncoprotein and the ability to modulate E1A-transforming activities (4, 5). CtBP co-transcriptional function is important in normal embryogenesis, as it is a regulator of the epithelial-to-mesenchymal transition and is integral in proper fetal cell differentiation. Knockout experiments in mice reveal distinct roles for CtBP1 and CtBP2 in development, with the loss of CtBP2 embryonically lethal, whereas CtBP1-null mice are small but the majority survive (6). Alternate splice forms of CtBP1 and -2 also have nonnuclear roles, including membrane trafficking (7).
Both CtBP paralogues have been implicated as global repressors of the epithelial phenotype and of apoptotic pathways (1), and numerous lines of evidence implicate human CtBP in cancer progression. CtBP is a corepressor of genes including tumor suppressive pro-apoptotic factors (Bik and Noxa), cytoskeletal/cell adhesion molecules (keratin-8 and E-cadherin), and cell-cycle inhibitors (2, 8). CtBP has also been found to act as a coactivator of growth and metastasis-related genes (Tiam1 and MDR1 and certain Wnt target genes), which facilitate the epithelial-to-mesenchymal transition (9–11). Consistent with its role in repression of apoptotic pathways and activation of growth and metastasis, CtBP is up-regulated in a number of cancer tissues, including colorectal cancer (12), melanoma (13), metastatic prostate cancer (14), esophageal squamous cell carcinoma (15), ovarian cancer (16), and breast cancer (17, 18). Strikingly, elevated levels of CtBP in tumor tissue have been correlated with poorer survival in breast cancer (19), ovarian cancer (16), and hepatocellular carcinoma (20). Recent results add to evidence of a link between CtBP and cancer progression by showing increased survival of APCmin/+ mice when CtBP2 levels are lowered by CtBP2+/− heterozygosity (21).
CtBP is unique among transcription factors in the incorporation of a d-isomer–specific 2-hydroxyacid dehydrogenase domain, which reduces or oxidizes substrates using the coenzyme NAD(P)+/NAD(P)H (22, 23). The best substrate identified to date for CtBP is 4-methylthio-2-oxobutyric acid (also known as MTOB or 2-keto-4-methylthiobutyrate) (24), although whether this is a physiologically relevant substrate is unknown. Whereas evidence indicates that catalytic activity is not required for some CtBP activities (8, 25), mutant studies suggest that catalytic activity of CtBP can be important for Drosophila melanogaster development (26).
Regulation of gene expression through the oligomerization of transcriptional factors is an important paradigm (27, 28). In the case of CtBP, substantial evidence exists that oligomerization is linked with NAD(H) binding (3, 25, 29–32), and dimer-destabilizing mutants have been found to inhibit transcriptional function (33–36). Assembly of CtBP has primarily been considered in terms of dimers, as NADH-bound CtBP crystal structures reveal a predominant dimer with extensive interactions between subunit pairs (23, 37, 38). There is, however, evidence for assembly of CtBP into tetramers (29–31) at least when the full C terminus is present. Here, we present multi-angle light scattering (MALS) data showing that the predominant form of CtBP1 and CtBP2 when bound to NAD(H) is tetrameric, with tetramers forming even in the absence of the full C terminus. Moreover, our mutant data provide strong evidence that the solution tetramer is very similar to the tetrameric assembly observed within crystals of the minimal dehydrogenase domains for both CtBP1 and CtBP2. Furthermore, the crystal structure of the CtBP1 A123V mutant reported here suggests that small perturbations in the flexible hinge region are capable of destabilizing the CtBP tetramer.
Results
MALS shows that both CtBP1 and CtBP2 assemble into tetramers in the presence of NADH and NAD+
We investigated the oligomeric state of CtBP1 and CtBP2 using MALS linked with size-exclusion chromatography (SEC). Our initial experiments were carried out using the minimal dehydrogenase domain constructs that we had previously used for crystallization, CtBP1(28–353) and CtBP2(31–364) (37). In contrast with a previous report (30), our SEC-MALS experiments on CtBP lacking the full C terminus showed molecular weights considerably larger than dimers in the presence of sufficient NADH (Fig. 1A). SEC-MALS results for CtBP oligomerization as a function of NADH concentration are shown in Fig. 2A and Table 1. Fitting of the molecular mass dependence on NADH concentration (see “Experimental procedures”) yields an EC50 value for the effect of NADH promoting tetramer formation of about 275 nm for CtBP1(28–353) and 180 nm for CtBP2(33–364) (Table 1). The EC50 value will probably be similar to the dissociation constant for NADH binding; however, the linkage between NADH binding and tetramer assembly suggests that the dimeric and tetrameric forms of CtBP will bind NADH with different affinities, precluding accurate estimates of dissociation constants with the present data.
Figure 1.

SEC trace and MALS molecular masses for the previously crystallized CtBP2(31–364) construct (A) and long (residues 31–445) construct (B) at various levels of NADH. The continuous lines show the light-scattering Rayleigh ratio (arbitrary units) for protein elution from the SEC column, and the small diamonds show the molecular mass measured by MALS measurements for the elution peaks. A, the addition of NADH to 10 μm results in earlier elution from the SEC column and an increase in molecular mass from 106 to 132 kDa (Table 1). B, assembly as a function of NADH concentration is more pronounced for the CtBP construct including the full C terminus, with molecular masses of 142 kDa in the absence of NADH increasing to 182 kDa at 10 μm NADH (Table 2). (Multiple measurements demonstrate that these results are fully reproducible.)
Figure 2.

Dependence of MALS-determined CtBP molecular masses as a function of NAD(H) concentration (see Tables 1 and 2). The approximate molecular masses expected for a pure dimer (left) and tetramer (right) are indicated. The data were fit with Prism version 7 (see “Experimental procedures”). A, MALS-measured Mw values for the crystallized constructs of CtBP1 and CtBP2 as a function of NADH concentration. In the absence of NADH, the Mw of CtBP1(28–353) of 62 kDa indicates a mixture of monomer and dimer, and this value rises to 128 kDa, consistent with about 46% tetramer, in the presence of 10 μm NADH. For CtBP2(31–364), the Mw of 106 kDa is consistent with a mixture of mostly dimer and about 18% tetramer, which rises to about 50% tetramer in the presence of 10 μm NADH. B, MALS-determined Mw values for the constructs CtBP1(28–440) and CtBP2(31–445) with the full C terminus as a function of NADH and NAD+. (These experiments were carried out in triplicate; error bars are shown but are often smaller than the data point shown.) In the absence of coenzyme, CtBP1 has an Mw, consistent with about 15% tetramer, whereas CtBP2 has an Mw, consistent with about 30% tetramer. These values rise to 61 and 66% tetramer in the presence of 10 μm NAD+ for CtBP1 and CtBP2, respectively, and to 79 and 75% tetramer in the presence of 10 μm NADH for CtBP1 and CtBP2, respectively.
Table 1.
Molecular mass values as a function of NAD(H) concentration for crystallized constructs

The MALS molecular mass estimates indicate heterogeneous mixtures, corresponding primarily to dimers and tetramers of CtBP, but also monomers in some cases. Given that light scattering provides weight average molecular mass estimates (Mw) (39), one can calculate approximate fractions of dimers and tetramers, assuming those are the only species contributing to the light scattering (see “Experimental procedures”). Based on these assumptions, CtBP2(31–364) is ∼18% tetramer in the absence of NADH and rises to about 47% tetramer in the presence of 10 μm NADH. In contrast, CtBP1(28–353) appears to be a mixture of monomer and dimer in the absence of NADH but is about 46% tetramer in the presence of 10 μm NADH. Our findings of significant tetramer formation in the absence the full C terminus contrast with an earlier report (30); this probably results from the higher protein concentration used in our experiments. (The loading concentration of 2 μm in the Madison et al. experiments (30) is substantially lower than our loading concentrations of ∼20 μm and will become even more diluted during the ∼50-ml run through the SEC column before CtBP elution compared with the ∼10-ml run before CtBP elution in our SEC-MALS experiments.)
Given previous findings suggesting the importance of the C-terminal residues for tetrameric assembly (30), we investigated constructs that included the full C terminus: CtBP1(28–440) and CtBP2(31–445) (Fig. 1B). Our results show that both assemble with masses indicative of predominantly tetramers in the presence of micromolar levels of NADH (Fig. 2B and Table 2). Assuming a simple mixture of dimers and tetramers, CtBP2(31–445) in the absence of added NADH is about 29% tetramer and plateaus to 75% tetramer at micromolar levels of NADH. CtBP1(28–440) shows only 15% tetramer in the absence of added NADH, but this plateaus to 79% tetramer with sufficient NADH. Thus, in agreement with Madison et al. (30), our experiments suggest that significantly stronger tetramers form in the presence of the full C terminus. Our results also suggest that the affinity of CtBP1 for NADH is slightly higher in constructs with the C terminus (EC50 ∼50 nm) compared with those lacking the final ∼85 residues (EC50 ∼275 nm); however, for CtBP2, the differences are smaller (EC50 ∼180 nm versus ∼110 nm).
Table 2.
Molecular mass values as a function of NAD(H) concentration for full C-terminal constructs (experiments carried out in triplicate)

We also investigated the effect of NAD+ on CtBP tetramer formation (Fig. 2B). For CtBP1(28–440), we observed an EC50 for NAD+ of 140 nm compared with NADH of 50 nm. For CtBP2(31–445), the EC50 values are almost identical for NAD+ (98 nm) and NADH (107 nm). Notable from the curves in Fig. 2B are steeper slopes for NADH-dependent tetramer assembly compared with the slopes for NAD+ assembly, which is reflected in the fitted Hill coefficients (Table 2). This suggests potentially greater cooperativity in NADH-linked assembly of CtBP, which could be relevant for the response of CtBP to coenzyme binding. Although the fitted EC50 values are indirect measurements of binding affinity, our results are consistent with similar affinities for NAD+ and NADH as reported by Madison et al. (30). Our apparent affinities for NADH are similar to that reported by Goodman and colleagues (40–42), but our results are inconsistent with the much lower affinity reported for NAD+ (40).
Tetramer models based on crystal lattices of CtBP1 and CtBP2
Observation of tetramer formation even for the crystallized minimal dehydrogenase domain suggested that the crystals grown from high CtBP concentrations might show assemblies related to the tetramers detected in our SEC-MALS experiments. Examination of the CtBP crystal lattices reveals strikingly similar tetrameric arrangements for both CtBP1 and CtBP2, despite very different crystal lattices (Fig. 3). CtBP1 crystallizes with one subunit in the asymmetric unit of hexagonal space group P6422 (23, 43). Three mutually perpendicular intersecting crystallographic 2-fold axes generate the D2 symmetric tetramer shown in Fig. 3A. The CtBP2 crystal lattice has eight subunits per asymmetric unit (37). These eight subunits are assembled into two tetramers, as shown in Fig. 3 (B and C). The remarkable similarity of these three tetramers is evident, which is confirmed by structural alignment of the full tetramers. The two CtBP2 tetramers align with a root mean square (r.m.s.) deviation of 0.441 Å (for 8083 atom pairs), and the CtBP1 tetramer aligns to each CtBP2 tetramer with r.m.s. values of 0.733 Å (for 7925 atom pairs) and 0.711 Å (for 7776 atom pairs). By far the most extensive interface occurs between two subunits that form the dimer first identified in CtBP1 by Kumar et al. (23), which buries surface areas of 2715–2924 Å2 in CtBP1 and CtBP2. Contacts between two dimers are more limited, with pairs of subunits burying surface areas of 806–857 Å2 (buried surface area calculated with PDBePISA (44)). Thus, the tetramer is a dimer of dimers, encompassing an extensive intradimer interface and a less extensive interdimer interface.
Figure 3.

Crystallographic structures of tetramers for CtBP1 (43) and CtBP2 (37). A, arrangement of four subunits of CtBP1 (PDB code 4U6Q), related by crystallographic symmetry in the space group P6422, forming a tetramer. The main-chain trace is shown in magenta for two subunits and cyan for two subunits, except for the C-terminal residues, which are displayed in orange. A stick model of the NAD(H) is shown with yellow carbon atoms. B and C, two crystallographically independent tetramers in the asymmetric unit of CtBP2 (PDB code 4LCJ). Note the remarkably similar arrangement of subunits in these three crystallographically independent tetramers, each subunit of which projects its C terminus toward a partner subunit across the interdimer interface. D–F, enlarged regions of the interdimer contact for CtBP2 tetramer 1. Each of these regions was investigated by site-directed mutagenesis (Fig. 4).
CtBP subunits comprise an NAD-binding domain (residues 125–319 for CtBP1 and 131–325 for CtBP2) and a discontinuous substrate-binding domain (residues 28–120 and 320–353 for CtBP1; 34–126 and 333–359 for CtBP2). These two domains are connected at residues 320–321 and also by a hinge region including residues 121–124 (CtBP1) and 127–130 (CtBP2), which contributes to the interdimer contacts.
Evident upon examining the CtBP subunit arrangement is a short helix in the NAD binding domain (helix αE, as defined by Kumar et al. (23)), whose projection into the solvent in dimeric CtBP would appear to be unfavorable. The tetramer buries this short helix in a pocket between αD and αE helices of a partner subunit (Fig. 3), potentially providing a more favorable environment.
The arrangement of subunits in the crystallographically observed CtBP tetramers has intriguing features as a potential model for the observed solution tetramer. Interdimer contacts are formed from the NAD-binding domain along with the hinge region between the NAD-binding and substrate-binding domains. Strikingly, each subunit projects its C terminus toward the interdimer interface, suggesting that the presence of an additional ∼85 C-terminal residues may provide interactions to stabilize the tetrameric form, consistent with our MALS results and the results of Madison et al. (30). Additionally, bound NAD(H) coenzymes are in closer proximity across the interdimer interface than across the intradimer interface, suggesting that the interdimer interface may be sensitive to NAD(H) binding. Finally, an Arg side chain (Fig. 3E) provides a direct link between an NAD(H) bound in one subunit with a subunit across the interdimer interface, as discussed below. Thus, the crystallographically observed tetramer provides a detailed and plausible model for the solution tetramer.
Mutational investigation of tetramer model
Based on the tetramer model described above, we designed a series of site-directed mutants for both CtBP1 and CtBP2 to test the hypothesis that the observed interdimer interface stabilizes the solution tetramer. Three groups of residues were investigated, all of which should primarily impact the interdimer interface. 1) A nonpolar interaction along the 2-fold axis relating two dimers involves Leu221 (215 in CtBP1) from the short αE helix (23) of each subunit (Fig. 3D). The atoms from the leucine side chains come within 3.6 Å of each other in CtBP1 and are 3.8–4.8 Å from each other in the lower-resolution CtBP2 structure. We mutated this leucine to the larger Tyr. 2) A particularly intriguing interaction involves Arg190 (Arg184 in CtBP1) (from the amino end of αD (23)), which forms ionic hydrogen bonds with the NAD(H) phosphate in its own subunit and the main-chain carbonyl of Asp215 (Asp209 in CtBP1) across the interdimer interface (Fig. 3E). To test the need for a charged Arg that can donate hydrogen bonds to both groups, we mutated this residue to a Gln. 3) The third group includes residues in the hinge between the NAD and substrate-binding domains of CtBP (Fig. 3F). The contacts involving the hinge regions were interrogated with mutations of Ser128 (Ala122 in CtBP1) to Thr, to test the contact itself and of Ala129 (Ala123 in CtBP1) to the larger Val and Leu, to test whether bulkier residues at this position could alter the hinge conformation and destabilize the interdimer interface. Mutants for both CtBP1 and CtBP2 were constructed in the context of the full C terminus, purified, and subjected to SEC-MALS to investigate the effect on tetramer formation.
Results of SEC-MALS experiments, run with 10 μm NADH, on the CtBP2 mutants are shown in Fig. 4A and Table 3. The dimeric molecular mass for the CtBP2(31–445) construct is 98.0 kDa. WT CtBP2 ran as mostly tetrameric in these experiments, with a MALS molecular mass measured to be 185 kDa. All six putative interdimer-destabilizing mutants eluted at a similar volume (Fig. 4A), with MALS-determined molecular masses of 94–103 kDa. Thus, our results are fully consistent with these mutants destabilizing the tetramer but maintaining their dimeric assemblages, as predicted from the crystallographic tetramer model.
Figure 4.

SEC trace and MALS molecular masses for mutants of CtBP2(31–445) (A) and CtBP1(28–440) (B) in the presence of 10 μm NADH (see also Table 3). The continuous lines show the light scattering Rayleigh ratio (arbitrary units) for protein elution from the SEC column, and the small diamonds show the molecular mass measured by MALS measurements for the elution peaks. The MALS-determined Mw is shown in parentheses for WT and each mutant. A, the six CtBP2 mutants show similar elution volumes and molecular masses that range from 94 to 103 kDa, consistent with primarily dimeric species. B, the equivalent six CtBP1 mutants show a greater range of elution volumes that may result from partial proteolysis within the C-terminal region. Molecular masses for the four mutants whose WT side chains point directly into the interdimer interface, ranging from 84 to 100 kDa, are also consistent with primarily dimeric species. The two hinge region mutants that point into the subunit, A123V and A123L, have molecular masses of 126–132 kDa, suggesting some tetrameric species (20–25%), but with the majority of molecules as dimeric. (MALS measurements on all mutants have been carried out multiple times and are fully reproducible.)
Table 3.
Molecular mass values for CtBP mutants
| Mutant/WT form | M̄w | Elution peak concentration | Tetramer |
|---|---|---|---|
| kDa | μm | % | |
| CtBP2(31–445) | |||
| WT | 185 | 2.8 | 80 |
| A129L | 100 | 3.3 | 4 |
| A129V | 105 | 3.5 | 1 |
| L221Y | 102 | 3.5 | 2 |
| S128T | 99 | 3.4 | 0.5 |
| G216N | 94 | 2.9 | 0 |
| R190Q | 94 | 2.0 | 0 |
| CtBP1(28–440) | |||
| WT | 181 | 2.3 | 86 |
| A123L | 132 | 3.3 | 25 |
| A123V | 125 | 2.3 | 20 |
| L215Y | 90 | 1.8 | 0 |
| A122T | 100 | 3.6 | 3 |
| G210N | 86.8 | 1.4 | 0 |
| R184Q | 83.6 | 2.2 | 0 |
The SEC traces for the CtBP1 mutants are more complex than those for CtBP2 (Fig. 4B). SDS-polyacrylamide gels demonstrate that the C-terminal region of CtBP1 is more susceptible to proteolytic cleavage than that of CtBP2. An SDS-polyacrylamide gel of the samples used in this experiment indicated that all mutants showed some proteolysis in the C-terminal region. All mutants except for R184Q displayed a major band at a full-length size of 47 kDa, whereas the major band for R184Q was at a molecular mass of about 41 kDa, consistent with a sequence ending at approximately residue 378. (The CtBP1 crystals discussed below, grown from constructs expressing the full C terminus, were found to have been proteolyzed just after residue 378.) The greater C-terminal proteolysis for CtBP1, compared with CtBP2, suggests that the C terminus may be more flexible in CtBP1. Such increased flexibility, along with partial proteolysis, may contribute to the observed greater variation in elution volumes for CtBP1 mutants, as the SEC elution depends upon overall protein shape rather than strictly on the molecular mass. (The C-terminal 86 amino acids are the least conserved, with only a 51% sequence identity between human CtBP1 and CtBP2, compared with 89% identity in the crystallized minimal dehydrogenase domain.) The MALS molecular mass values show smaller variations, providing more accurate estimates of the oligomeric state. The four CtBP1 mutants of residues projecting into the interdimer interface show MALS molecular masses of 84–100 kDa (Table 3). These are in the range expected for dimers of CtBP1(28–440) of 94 kDa and those for partially proteolyzed species (residues 28–378) of 82 kDa. These results, thus, support those from CtBP2 indicating that the interdimer interface is similar to that observed in the crystallographic tetramer.
The most subtle mutants designed to destabilize the tetramer were of Ala123/129 (CtBP1/CtBP2), residues that are in the hinge region but project their β-carbon into its subunit rather than toward the interdimeric interface. In this case, the results between CtBP1 and CtBP2 show some interesting differences. Whereas CtBP2 A129V and A129L were found to have molecular masses close to that expected for dimers, the equivalent CtBP1 A123V and A123L show molecular masses between that of dimers and tetramers, suggesting a more subtle destabilization of the CtBP1 interdimer interface (Table 3). These results indicate that subtle conformational changes in the hinge region connecting the two domains can substantially impact tetramer stability.
Crystal structures of CtBP1 with extended C terminus
Our results, complementing those of Madison et al. (30), highlight the contribution of the final ∼85 residues to stabilization of the CtBP tetramer. In an attempt to investigate the C-terminal residues and also the conformational changes induced by mutation of hinge residues, we grew crystals from CtBP1 expressed with the full C terminus for WT and a subtle tetramer-destabilizing mutant (A123V), both in the presence of NADH. (Attempts to grow crystals of A123L, with its slightly larger side chain, were unsuccessful.)
Crystallization of expressed CtBP1(28–440) was successful only intermittently; SDS-polyacrylamide gels revealed that those samples that successfully crystallized were intermediate in size between CtBP1(28–440) and CtBP1(28–353). Mass spectrometry measurements confirmed that these samples were shorter than full-length, with a predominant species encompassing residues 28–378. We therefore consider these crystal structures to be CtBP1(28–378).
All CtBP crystals described to date are built from tetrameric assemblies, although they have not previously been described as such. Thus, it is not surprising that only a subtle tetramer-destabilizing mutant (A123V) successfully crystallized. The crystal structures of both WT and A123V mutants were determined by molecular replacement using the CtBP1-HIPP structure (residues 28–353) (43) and refined to resolutions of 2.6 and 2.4 Å, respectively. (The resolution cutoffs were conservatively chosen.) Final crystallographic statistics are provided in Table 4.
Table 4.
Crystallographic data collection and refinement statistics
| CtBP1(28–378) WT | CtBP1(28–378) A123V | |
|---|---|---|
| PDB code | 6CDF | 6CDR |
| Data collection | ||
| Space group | P6422 | P6422 |
| Unit cell parameters | ||
| a, b, c (Å) | 88.66, 88.6, 163.91 | 89.22, 89.22, 164.23 |
| α, β, γ (degrees) | 90, 90, 120 | 90, 90, 120 |
| Bragg spacings (Å)a | 50. to 2.6 (2.693–2.6) | 50. to 2.4 (2.485–2.4) |
| Rmerge | 0.0453 (0.2695) | 0.0655 (0.4668) |
| I/σI | 30.5 (6.3) | 27.9 (5.3) |
| Completeness (%) | 95.8 (88.7) | 93.3 (99.8) |
| Redundancy | 7.8 (6.8) | 12.8 (10.4) |
| Total reflections | 93,207 (7207) | 193,860 (15932) |
| Unique reflections | 11,944 (1056) | 15,090 (1537) |
| Refinement | ||
| Rwork/Rfree | 0.209/0.243 | 0.235/0.258 |
| No. of non-hydrogen atoms | ||
| Protein | 2578 | 2580 |
| Ligands | 48 | 65 |
| Solvent | 115 | 101 |
| Mean B-factors (Å2) | ||
| Protein | 46.4 | 44.2 |
| Ligands | 36.4 | 38.1 |
| Solvent | 40.3 | 42.1 |
| r.m.s. deviations | ||
| Bond lengths (Å) | 0.003 | 0.003 |
| Bond angles (degrees) | 0.67 | 0.61 |
| Ramachandran (%) | ||
| Favored | 95.2 | 95.8 |
| Allowed | 4.8 | 4.2 |
| Outliers | 0 | 0 |
a Highest-resolution shell shown in parenthesis.
Despite an additional 25 residues in the WT and A123V CtBP1(28–378), only four additional residues are evident in the electron density maps and modeled in our final structures (Fig. 5). These results are consistent with findings on rat CtBP that the C terminus is largely disordered (31).
Figure 5.
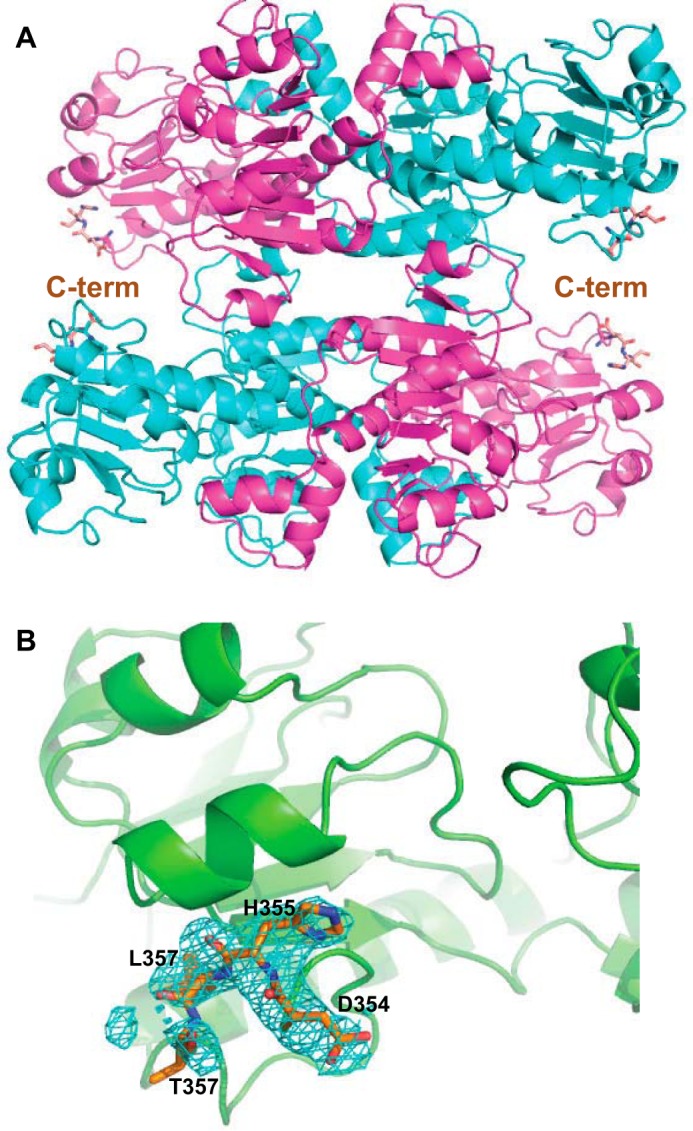
Crystal structure of CtBP1(28–378). A, trace of the CtBP1 tetramer, with two subunits in magenta and two in cyan. Shown with orange carbon atoms is a stick rendition of the additional four residues observed compared with the previous CtBP1(28–353) structures. B, Fo − Fc difference electron density map shown in cyan at a +2σ level for the four additional residues (Asp354–Thr357) observed.
The isomorphous crystal lattices for WT and A123V CtBP1(28–378) permit direct comparison of the structural differences using difference maps (Fo(A123V) − Fo(WT)). As is evident from Fig. 6, the mutation-induced changes largely localize to the mutant site. Substitution of Val for Ala at position 123 induces movements of Glu326 and the α5 helix (23) (which includes Glu326) deeper into the subunit. Interestingly, the maps do not suggest significant movement of residues in the partner subunit across the tetrameric interface. This observation suggests that the tetrameric subunit arrangement is sufficiently stabilized by the CtBP1 crystal lattice that any strain induced by Val123 is absorbed within a subunit. Our MALS results, however, clearly show that the tetramer is destabilized by the mutation, such that in solution there may be significant effects on the interaction between the partner subunits with this mutation. The observation that the homologous mutation (A129V) in CtBP2 is almost entirely dimeric suggests that the CtBP2 subunit structure is unable to accommodate the strain induced by the presence of the Val129.
Figure 6.
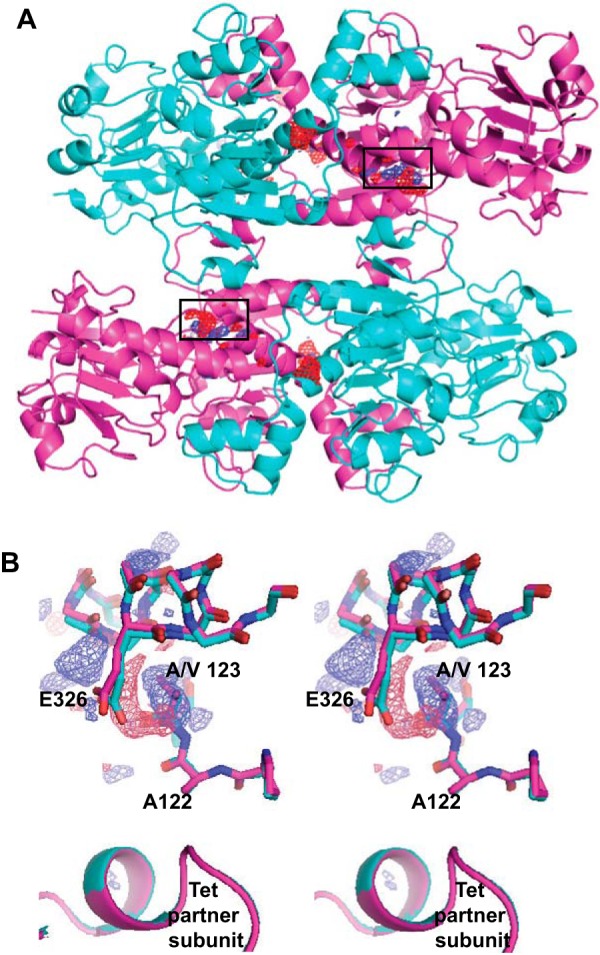
Crystal structure of CtBP1(28–378) A123V compared with the structure of WT CtBP1(28–378). A, trace of the CtBP1 tetramer with the Fo(A123V) − Fo(WT) map contoured at +4σ (blue) and −4σ (red). The electron density is localized to the mutant region (black rectangles) with the exception of a negative density peak (red) near the center, which appears to be an unidentified solvent molecule apparently present in the WT but not in the A123V structure. B, stereoview of the Fo(A123V) − Fo(WT) electron density (blue, +4σ; red, −4σ) in the region of the mutation. Note the positive density for the two methyl carbon atoms of the mutant Val, which induces movement of Glu326 toward the left and upward, taking with it the main chain of helix α5, to which it is attached. There is no evidence of significant movement in the partner subunit, despite the MALS data showing a weakening of the tetramer with the A123V mutation.
Discussion
Assembly of protein components is an important theme in transcriptional regulation (45–47). A central element of this process is often the oligomerization of one or more individual protein components contributing to transcriptional complex assembly (27, 28). In the case of CtBP, strong evidence exists for the role of NAD(H) in both co-transcriptional activity and oligomerization (3, 23, 25, 29, 32, 33). Previously published results have provided evidence that CtBP can assemble to dimers and larger species (3, 25, 29–32) but have not provided a quantitative relationship for the NAD(H) dependence of tetramer assembly nor a stereochemical model of the CtBP tetramer.
The results presented here demonstrate a clear relationship between NAD(H) binding (with an affinity in the 100 nm range) and CtBP assembly into tetramers. Our mutant data provide strong evidence that subunits in the solution tetramer are arranged similarly to the tetrameric assemblages present in previously determined crystal structures of human CtBP1 and CtBP2 (23, 37) and rat CtBP/BARS (38). This tetramer is assembled from the pairing of two dimers to form NAD(H)-sensitive interdimer contacts primarily using the NAD-binding domain of CtBP. In this tetramer, the closest approach of NADH molecules across the intradimer interface is over 30 Å but less than 15 Å across the interdimer interface (see Fig. 3); this closer approach supports the idea that the interdimer interface would be more sensitive to NADH binding than the intradimer interface. Moreover, the interaction between NADH across the interdimer interface to the main-chain carbonyl of residue 209/215 (CtBP1/CtBP2) mediated by Arg184/190 (Fig. 3E) provides an ionically mediated contact across the interdimer interface that should be directly sensitive to NAD(H) binding.
Although NAD(H) stabilizes the CtBP tetramer, some tetramer formation is evident even in the absence of NAD(H) from our experiments. The ability to form tetramers in the absence of NAD(H) allowed Nardini et al. (48) to grow crystals and determine the structure of the NAD(H) binding–impaired G172E mutant rat CtBP1/BARS protein by using seeding with NADH-bound CtBP crystals. These crystals, which are isomorphous to the human CtBP1 crystals described above, have a tetramer as their fundamental unit despite no bound coenzyme. This result shows the ability of NAD(H)-free CtBP to form tetramers, which can be stabilized by lattice interactions triggered by the crystal seeding. The strength of lattice interactions to stabilize the tetramer is also evident in our A123V crystal structure, which, despite having a tetramer-destabilizing mutant, maintains tetramer contacts, by absorbing mutant-induced strain within a subunit.
Previous investigations into the role of assembly in CtBP have investigated the activity of mutants specifically designed to disrupt the CtBP dimer (33–36). Our results strongly suggest that the CtBP dimer is required for the assembly of a CtBP tetramer formed from the pairing of two CtBP dimers. Therefore, it is likely that dimer-destabilizing mutants will also destabilize CtBP tetramers. Thus, previous work showing the importance of dimers is fully consistent with an important role for tetramers.
Our results suggest that the tetramer, assembled as observed in crystal lattices, may be important for CtBP co-transcriptional activity. Importantly, there is an observed correlation between elevated levels of CtBP and poor prognosis in breast cancer (19), ovarian cancer (16), and hepatocellular carcinoma (20) as well as the observation of elevated CtBP levels in other cancers (12–15). Increased levels of CtBP will not only raise the concentration of co-transcriptional factor but also increase the proportion of CtBP that assembles into tetramers, thus potentially increasing its transcriptional activity. Tetramer-destabilizing mutants identified here provide important tools for dissecting the role of CtBP tetramers in co-transcriptional function and cancer progression. Moreover, our model of the tetrameric assembly provides stereochemical details, for the first time, that can be used with structure-based drug design approaches for development of novel CtBP tetrameric destabilizing inhibitors.
Experimental procedures
Expression and mutagenesis of CtBP1 and CtBP2
The expression and purification procedures were adapted and optimized from earlier studies (37, 43). The ligated, purified plasmid containing the desired CtBP construct was transformed into Z-competent BL21(DE3)RIL Escherichia coli cells. A single clonal colony was then grown in a starter culture of lysogeny broth overnight at 37 °C. The starter culture was used to inoculate between three and six 1-liter cultures grown in Research Products International Terrific Broth using 50 ml of starter/liter. Cultures were grown at 37 °C while shaking at 150 rpm and induced with isopropyl 1-thio-β-d-galactopyranoside, at a final concentration of 0.2 mm, after reaching A600 between 0.800 and 1.00. The temperature was reduced to 30 °C at the time of induction, and the cells were harvested 4 h later. The cells were pelleted by centrifuging for 20 min at 4700 rpm and resuspended in 10 ml of harvesting buffer (pH 7.6; 0.1 m NaCl, 0.05 m Tris-HCl, 0.2 mm EDTA) per liter of culture. One tablet of EDTA-free complete Mini (Roche Diagnostics) protease inhibitor mixture was added per liter of culture, and the cells were frozen at −80 °C. Site-directed mutants were created using the QuikChange protocol (Stratagene) using the modified approach of Liu and Naismith (49).
Purification of CtBP1 and CtBP2
Cells were thawed slowly on ice and then lysed in a Microfluidics Corp. model 1109 cell disrupter. 35 mg of Roche Diagnostics DNase I, 500 μl of 2 m MgCl2, and 500 μl of 40 mm CaCl2 were added per 100 ml of lysate. The lysate was then gently stirred at 4 °C for 30 min before the insoluble fraction was pelleted by centrifuging at 19,000 rpm for 45 min. The supernatant was then mixed with 8 ml of HisPurTM nickel-nitrilotriacetic acid resin (Thermo Scientific) and gently stirred at 4 °C for 2 h to allow CtBP to bind to the resin.
The bead/supernatant mixture was placed in a Bio-Rad Econo-Column® at 4 °C, and the soluble fraction was allowed to flow through. The beads were then cleaned with 5 column volumes of wash buffer (0.0625 m Tris-HCl, pH 7.4, 0.375 m NaCl, 0.05 m imidazole, 0.625 mm EDTA, 1.0 mm DTT), followed by 6 column volumes of wash buffer supplemented with an additional 1.7 m NaCl. Another 2 column volumes of wash buffer was passed over the beads before 6 column volumes of wash buffer supplemented with 0.5% Triton X-100 was added. An additional 2 column volumes of wash buffer again followed. CtBP was eluted from the beads using 3 column volumes of wash buffer supplemented with 250 mm imidazole. The eluent was collected and dialyzed overnight in SnakeSkin® dialysis tubing (Thermo Scientific) to remove imidazole. The dialysis buffer consisted of 50 mm Tris-HCl, pH 7.7, 300 mm NaCl, 5 mm EDTA, 2 mm DTT, and 10% glycerol. The protein was then concentrated by centrifuging at 5000 rpm in an Amicon® Ultra-15 10K centrifugation column (Millipore). Protein concentration was measured by UV absorbance at 280 nm using an Ultraspec 2100 pro by Amersham Biosciences.
The protein sample was further purified by size-exclusion chromatography. The FPLC (ÄTKAprime plus by GE Healthcare) and size-exclusion column (HighloadTM 16/60 SuperdexTM200 preparation grade) were equilibrated with “FPLC buffer” (50 mm Tris-HCl, pH 7.7, 300 mm NaCl, 5 mm EDTA, 2 mm DTT). The sample was prepared by adding 1.5 mm NADH to the concentrated protein solution; the solution was then centrifuged at 8000 rpm for 6 min at 4 °C to remove any small insoluble fraction. The flow rate was set to 1 ml/min, 62 fractions of 2 ml each were collected, and the appropriate fractions were concentrated in an Amicon® Ultra-15 10K centrifugation column.
SEC-MALS for NAD(H)-dependent oligomerization studies
Four different CtBP constructs were tested in NAD(H)-dependent oligomerization MALS experiments: CtBP1(28–353), CtBP1(28–440), CtBP2(31–364), and CtBP2(31–445) were expressed and purified as described above. Protein samples were prepared for SEC-MALS by diluting CtBP stocks to 1.0 mg/ml in FPLC buffer supplemented by the desired concentration of NAD(H). The protein samples were filtered through a Costar 0.22-μm Spin-X column at room temperature. 100-μl protein samples were injected into the SEC-MALS instrument. The MALS system consisted of a Dawn Helios-II MALS detector (Wyatt), an Optilab T-rEX differential refractive index detector (Wyatt), and the 1260 Infinity HPLC system (Agilent) with a Wyatt Corp. 0.78 × 30-cm HPLC column with 500-Å pore size. In later experiments, a TSKgel G3000SWxl column (Tosoh Bioscience) was substituted, but the effect of the column on the quality of the data obtained was negligible. For the WT constructs with the full C terminus, measurements at given NADH and NAD+ concentrations were carried out in triplicate (Table 2 and Fig. 2B).
The data for the dependence on molecular mass as a function of NAD(H) concentration (Fig. 2) were fit with Prism version 7 (GraphPad Software, Inc.) to the equation, Y = L + (U − L)/(1 + 10log(EC50 − x)n), where L and U are the lower and upper Mw plateaus, respectively, x is the concentration of NAD(H) in nm, and n is the Hill coefficient.
The molecular mass obtained from light scattering from a heterogeneous mixture of protein molecules is the Mw: Mw = ΣNiM2i/ΣNiMi, where Ni is the molar or fractional concentration (39). Assuming a mixture of just CtBP dimers and tetramers allows simplification to Mw = (FT(2MD)2 + (1 − FT)iMD2)/(FT(2MD) + (1 − FT)MD), where MD is the dimeric molecular weight and FT is the fraction of CtBP in the tetrameric form. Rearranging this equation, we obtained estimates of the fraction tetramer from FT = (Mw − MD)/(3MD − Mw).
SEC-MALS for mutant studies
The mutant SEC studies were carried out as described above using a TSKgel G3000SWxl column (Tosoh Bioscience) working in concert. The instruments were equilibrated in FPLC buffer supplemented with 10 μm NADH. Protein samples consisted of 1 mg/ml CtBP in FPLC buffer. The sample was filtered using 0.22 μm cellulose acetate Costar® Spin-X centrifuge tube filters at 8000 rpm for 1 min at room temperature. 100 μl of sample solution was injected into the MALS system, with a flow rate of 1.0 ml/min. Data were analyzed using the ASTRA software package by Wyatt.
Crystallization and X-ray diffraction
Purified CtBP1(28–378) WT protein was diluted to 18.0 mg/ml from 27.0 mg/ml stock with dH2O before being supplemented with 10% 15 mm NADH and 2% 100 mm tris(2-carboxyethyl)phosphine in dH2O. The sample was then filtered via a Costar 0.22-μm Spin-X column at room temperature. Hanging vapor diffusion drops were set up in a 1:1 ratio of protein to mother liquor with a total volume of 4 μl and incubated at 20 °C. Crystals formed within 24 h after the drops were set up but allowed to grow for several days. A single crystal with hexagonal bipyramidal morphology was observed in 100 mm HEPES buffer, pH 7.5, containing 140 mm CaCl2 and 5% PEG 400. The observable dimensions along its longest axes were 260 × 400 μm. The crystal was harvested for data collection by dipping in a cryoprotectant solution consisting of mother liquor supplemented with 20% ethylene glycol. The crystal was then flash-frozen at 100 K. Data were collected on a MicroMax-007-HF/Saturn 944 CCD X-ray diffraction system (Rigaku) and then processed with HKL-3000R. The structure was solved by molecular replacement, using the CtBP1(28–353) HIPP structure (PDB code 4U6Q (43)) as the search molecule and refined in PHENIX version 1.11.1-2575-000. Model building between rounds of refinement was completed with Coot version 0.8.8.EL. Data were refined to a maximum resolution of 2.60 Å.
For the CtBP1 28–378 A123V mutant, protein was diluted to 17.9 mg/ml from a 33.5 mg/ml stock with dH2O before being supplemented with 10% 15 mm NADH and 2% tris(2-carboxyethyl)phosphine in dH2O. The protein sample was filtered via a Costar 0.22-μm Spin-X column at room temperature. Hanging vapor diffusion drops were set up in a 1:1 ratio of protein to mother liquor with a total volume of 4 μl and incubated at 15 °C. Crystals formed between 3 and 8 days after the drops were set up. A single crystal with hexagonal bipyramidal morphology was observed in 100 mm HEPES, pH 7.5, buffer containing 80 mm MgCl2 and 4% PEG 400. The observable dimensions along its longest axes were 160 × 300 μm. The crystal was harvested for data collection after 3.5 μl of cryoprotectant solution consisting of mother liquor supplemented with 20% ethylene glycol was added to the hanging drop and left to soak for 5 min. The crystal was then flash-frozen at 100 K. Data were collected, solved, and refined as described above. Data were refined to a maximum resolution of 2.40 Å.
LC/MS
LC/MS samples were prepared by diluting CtBP1 28–440 WT stock and CtBP1 28–440 A123V stock to 2.5 mg/ml in 16.7 mm Tris-HCl, pH 7.7, buffer supplemented with 100 mm NaCl and 1.67 mm EDTA. Each sample was spin-filtered in a Costar 0.22-μm Spin-X column at room temperature. LC/MS experiments were carried out by Dr. John Leszyk and Dr. Khaja Muneeruddin of the Proteomics and Mass Spectrometry core at University of Massachusetts Medical School (Worcester, MA).
Author contributions
A. G. B. and W. E. R. conceptualization; A. G. B., A. M. J., and W. E. R. data curation; A. G. B., A. M. J., J. A. H., and W. E. R. formal analysis; A. G. B. and W. E. R. investigation; A. G. B., J. A. H., and W. E. R. methodology; A. G. B. and W. E. R. writing-original draft; A. G. B., A. M. J., J. A. H., C. S., and W. E. R. writing-review and editing; J. A. H. resources; J. A. H. and W. E. R. validation; C. S. and W. E. R. funding acquisition; C. S. and W. E. R. project administration; W. E. R. supervision.
Acknowledgments
We thank Drs. Brian Kelch and C. Robert Matthews for acquiring the Wyatt SEC-MALS instrument; Dr. Brendan Hilbert, Dr. Steven Grossman, and Gordon Lockbaum for helpful discussions; and Drs. John Leszyk and Khaja Muneeruddin for the LC/MS experiments. The diffraction data were collected on a Rigaku CCD system obtained through National Institutes of Health Shared Instrumentation Grant S10 OD012028 (to C. A. S.).
This work was supported by National Institutes of Health Grant R01 GM119014 (to W. E. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 6CDF and 6CDR) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CtBP
- C-terminal binding protein
- MALS
- multi-angle light scattering
- SEC
- size-exclusion chromatography
- r.m.s.
- root mean square
- dH2O
- distilled H2O.
References
- 1. Bergman L. M., and Blaydes J. P. (2006) C-terminal binding proteins: emerging roles in cell survival and tumorigenesis. Apoptosis 11, 879–888 10.1007/s10495-006-6651-4 [DOI] [PubMed] [Google Scholar]
- 2. Chinnadurai G. (2009) The transcriptional corepressor CtBP: a foe of multiple tumor suppressors. Cancer Res. 69, 731–734 10.1158/0008-5472.CAN-08-3349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kuppuswamy M., Vijayalingam S., Zhao L. J., Zhou Y., Subramanian T., Ryerse J., and Chinnadurai G. (2008) Role of the PLDLS-binding cleft region of CtBP1 in recruitment of core and auxiliary components of the corepressor complex. Mol. Cell. Biol. 28, 269–281 10.1128/MCB.01077-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boyd J. M., Subramanian T., Schaeper U., La Regina M., Bayley S., and Chinnadurai G. (1993) A region in the C-terminus of adenovirus 2/5 E1a protein is required for association with a cellular phosphoprotein and important for the negative modulation of T24-ras mediated transformation, tumorigenesis and metastasis. EMBO J. 12, 469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schaeper U., Boyd J. M., Verma S., Uhlmann E., Subramanian T., and Chinnadurai G. (1995) Molecular cloning and characterization of a cellular phosphoprotein that interacts with a conserved C-terminal domain of adenovirus E1A involved in negative modulation of oncogenic transformation. Proc. Natl. Acad. Sci. U.S.A. 92, 10467–10471 10.1073/pnas.92.23.10467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hildebrand J. D., and Soriano P. (2002) Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol. Cell. Biol. 22, 5296–5307 10.1128/MCB.22.15.5296-5307.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corda D., Colanzi A., and Luini A. (2006) The multiple activities of CtBP/BARS proteins: the Golgi view. Trends Cell Biol. 16, 167–173 10.1016/j.tcb.2006.01.007 [DOI] [PubMed] [Google Scholar]
- 8. Grooteclaes M., Deveraux Q., Hildebrand J., Zhang Q., Goodman R. H., and Frisch S. M. (2003) C-terminal-binding protein corepresses epithelial and proapoptotic gene expression programs. Proc. Natl. Acad. Sci. U.S.A. 100, 4568–4573 10.1073/pnas.0830998100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fang M., Li J., Blauwkamp T., Bhambhani C., Campbell N., and Cadigan K. M. (2006) C-terminal-binding protein directly activates and represses Wnt transcriptional targets in Drosophila. EMBO J. 25, 2735–2745 10.1038/sj.emboj.7601153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin W., Scotto K. W., Hait W. N., and Yang J. M. (2007) Involvement of CtBP1 in the transcriptional activation of the MDR1 gene in human multidrug resistant cancer cells. Biochem. Pharmacol. 74, 851–859 10.1016/j.bcp.2007.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paliwal S., Ho N., Parker D., and Grossman S. R. (2012) CtBP2 promotes human cancer cell migration by transcriptional activation of Tiam1. Genes Cancer 3, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nadauld L. D., Phelps R., Moore B. C., Eisinger A., Sandoval I. T., Chidester S., Peterson P. W., Manos E. J., Sklow B., Burt R. W., and Jones D. A. (2006) Adenomatous polyposis coli control of C-terminal binding protein-1 stability regulates expression of intestinal retinol dehydrogenases. J. Biol. Chem. 281, 37828–37835 10.1074/jbc.M602119200 [DOI] [PubMed] [Google Scholar]
- 13. Deng H., Liu J., Deng Y., Han G., Shellman Y. G., Robinson S. E., Tentler J. J., Robinson W. A., Norris D. A., Wang X. J., and Zhang Q. (2013) CtBP1 is expressed in melanoma and represses the transcription of p16INK4a and Brca1. J. Invest. Dermatol. 133, 1294–1301 10.1038/jid.2012.487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang R., Asangani I. A., Chakravarthi B. V., Ateeq B., Lonigro R. J., Cao Q., Mani R. S., Camacho D. F., McGregor N., Schumann T. E., Jing X., Menawat R., Tomlins S. A., Zheng H., Otte A. P., et al. (2012) Role of transcriptional corepressor CtBP1 in prostate cancer progression. Neoplasia 14, 905–914 10.1593/neo.121192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guan C., Shi H., Wang H., Zhang J., Ni W., Chen B., Hou S., Yang X., Shen A., and Ni R. (2013) CtBP2 contributes to malignant development of human esophageal squamous cell carcinoma by regulation of p16INK4A. J. Cell. Biochem. 114, 1343–1354 10.1002/jcb.24475 [DOI] [PubMed] [Google Scholar]
- 16. Barroilhet L., Yang J., Hasselblatt K., Paranal R. M., Ng S. K., Rauh-Hain J. A., Welch W. R., Bradner J. E., Berkowitz R. S., and Ng S. W. (2013) C-terminal binding protein-2 regulates response of epithelial ovarian cancer cells to histone deacetylase inhibitors. Oncogene 32, 3896–3903 10.1038/onc.2012.380 [DOI] [PubMed] [Google Scholar]
- 17. Birts C. N., Harding R., Soosaipillai G., Halder T., Azim-Araghi A., Darley M., Cutress R. I., Bateman A. C., and Blaydes J. P. (2010) Expression of CtBP family protein isoforms in breast cancer and their role in chemoresistance. Biol. Cell 103, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deng Y., Deng H., Liu J., Han G., Malkoski S., Liu B., Zhao R., Wang X. J., and Zhang Q. (2012) Transcriptional down-regulation of Brca1 and E-cadherin by CtBP1 in breast cancer. Mol. Carcinog. 51, 500–507 10.1002/mc.20813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Di L. J., Byun J. S., Wong M. M., Wakano C., Taylor T., Bilke S., Baek S., Hunter K., Yang H., Lee M., Zvosec C., Khramtsova G., Cheng F., Perou C. M., Miller C. R., et al. (2013) Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat. Commun. 4, 1449 10.1038/ncomms2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zheng X., Song T., Dou C., Jia Y., and Liu Q. (2015) CtBP2 is an independent prognostic marker that promotes GLI1 induced epithelial-mesenchymal transition in hepatocellular carcinoma. Oncotarget 6, 3752–3769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sumner E. T., Chawla A. T., Cororaton A. D., Koblinski J. E., Kovi R. C., Love I. M., Szomju B. B., Korwar S., Ellis K. C., and Grossman S. R. (2017) Transforming activity and therapeutic targeting of C-terminal-binding protein 2 in Apc-mutated neoplasia. Oncogene 36, 4810–4816 10.1038/onc.2017.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chinnadurai G. (2002) CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol. Cell 9, 213–224 10.1016/S1097-2765(02)00443-4 [DOI] [PubMed] [Google Scholar]
- 23. Kumar V., Carlson J. E., Ohgi K. A., Edwards T. A., Rose D. W., Escalante C. R., Rosenfeld M. G., and Aggarwal A. K. (2002) Transcription corepressor CtBP is an NAD+-regulated dehydrogenase. Mol. Cell 10, 857–869 10.1016/S1097-2765(02)00650-0 [DOI] [PubMed] [Google Scholar]
- 24. Achouri Y., Noël G., and Van Schaftingen E. (2007) 2-Keto-4-methylthiobutyrate, an intermediate in the methionine salvage pathway, is a good substrate for CtBP1. Biochem. Biophys. Res. Commun. 352, 903–906 10.1016/j.bbrc.2006.11.111 [DOI] [PubMed] [Google Scholar]
- 25. Mani-Telang P., Sutrias-Grau M., Williams G., and Arnosti D. N. (2007) Role of NAD binding and catalytic residues in the C-terminal binding protein corepressor. FEBS Lett. 581, 5241–5246 10.1016/j.febslet.2007.10.011 [DOI] [PubMed] [Google Scholar]
- 26. Zhang Y. W., and Arnosti D. N. (2011) Conserved catalytic and C-terminal regulatory domains of the C-terminal binding protein corepressor fine-tune the transcriptional response in development. Mol. Cell. Biol. 31, 375–384 10.1128/MCB.00772-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen W., Lam S. S., Srinath H., Jiang Z., Correia J. J., Schiffer C. A., Fitzgerald K. A., Lin K., and Royer W. E. Jr. (2008) Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nat. Struct. Mol. Biol. 15, 1213–1220 10.1038/nsmb.1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cubillos-Rojas M., Schneider T., Bartrons R., Ventura F., and Rosa J. L. (2017) NEURL4 regulates the transcriptional activity of tumor suppressor protein p53 by modulating its oligomerization. Oncotarget 8, 61824–61836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Balasubramanian P., Zhao L. J., and Chinnadurai G. (2003) Nicotinamide adenine dinucleotide stimulates oligomerization, interaction with adenovirus E1A and an intrinsic dehydrogenase activity of CtBP. FEBS Lett. 537, 157–160 10.1016/S0014-5793(03)00119-4 [DOI] [PubMed] [Google Scholar]
- 30. Madison D. L., Wirz J. A., Siess D., and Lundblad J. R. (2013) Nicotinamide adenine dinucleotide-induced multimerization of the co-repressor CtBP1 relies on a switching tryptophan. J. Biol. Chem. 288, 27836–27848 10.1074/jbc.M113.493569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nardini M., Svergun D., Konarev P. V., Spanò S., Fasano M., Bracco C., Pesce A., Donadini A., Cericola C., Secundo F., Luini A., Corda D., and Bolognesi M. (2006) The C-terminal domain of the transcriptional corepressor CtBP is intrinsically unstructured. Protein Sci. 15, 1042–1050 10.1110/ps.062115406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thio S. S., Bonventre J. V., and Hsu S. I. (2004) The CtBP2 co-repressor is regulated by NADH-dependent dimerization and possesses a novel N-terminal repression domain. Nucleic Acids Res. 32, 1836–1847 10.1093/nar/gkh344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhambhani C., Chang J. L., Akey D. L., and Cadigan K. M. (2011) The oligomeric state of CtBP determines its role as a transcriptional co-activator and co-repressor of Wingless targets. EMBO J. 30, 2031–2043 10.1038/emboj.2011.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bi C., Meng F., Yang L., Cheng L., Wang P., Chen M., Fang M., and Xie H. (2018) CtBP represses Dpp signaling as a dimer. Biochem. Biophys. Res. Commun. 495, 1980–1985 10.1016/j.bbrc.2017.12.018 [DOI] [PubMed] [Google Scholar]
- 35. Chinnadurai G. (2007) Transcriptional regulation by C-terminal binding proteins. Int. J. Biochem. Cell Biol. 39, 1593–1607 10.1016/j.biocel.2007.01.025 [DOI] [PubMed] [Google Scholar]
- 36. Ray S. K., Li H. J., and Leiter A. B. (2017) Oligomeric form of C-terminal-binding protein coactivates NeuroD1-mediated transcription. FEBS Lett. 591, 205–212 10.1002/1873-3468.12501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hilbert B. J., Grossman S. R., Schiffer C. A., and Royer W. E. Jr. (2014) Crystal structures of human CtBP in complex with substrate MTOB reveal active site features useful for inhibitor design. FEBS Lett. 588, 1743–1748 10.1016/j.febslet.2014.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nardini M., Spanò S., Cericola C., Pesce A., Massaro A., Millo E., Luini A., Corda D., and Bolognesi M. (2003) CtBP/BARS: a dual-function protein involved in transcription co-repression and Golgi membrane fission. EMBO J. 22, 3122–3130 10.1093/emboj/cdg283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Holde K. E. (1971) Physical Biochemistry, pp. 43 and 191, Prentice-Hall, Englewood Cliffs, NJ [Google Scholar]
- 40. Zhang Q., Piston D. W., and Goodman R. H. (2002) Regulation of corepressor function by nuclear NADH. Science 295, 1895–1897 [DOI] [PubMed] [Google Scholar]
- 41. Zhang Q., Wang S. Y., Nottke A. C., Rocheleau J. V., Piston D. W., and Goodman R. H. (2006) Redox sensor CtBP mediates hypoxia-induced tumor cell migration. Proc. Natl. Acad. Sci. U.S.A. 103, 9029–9033 10.1073/pnas.0603269103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fjeld C. C., Birdsong W. T., and Goodman R. H. (2003) Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc. Natl. Acad. Sci. U.S.A. 100, 9202–9207 10.1073/pnas.1633591100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hilbert B. J., Morris B. L., Ellis K. C., Paulsen J. L., Schiffer C. A., Grossman S. R., and Royer W. E. Jr. (2015) Structure-guided design of a high affinity inhibitor to human CtBP. ACS Chem. Biol. 10, 1118–1127 10.1021/cb500820b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 45. Panne D., Maniatis T., and Harrison S. C. (2007) An atomic model of the interferon-β enhanceosome. Cell 129, 1111–1123 10.1016/j.cell.2007.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Payankaulam S., Li L. M., and Arnosti D. N. (2010) Transcriptional repression: conserved and evolved features. Curr. Biol. 20, R764–R771 10.1016/j.cub.2010.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sun L., and Fang J. (2016) Epigenetic regulation of epithelial-mesenchymal transition. Cell. Mol. Life Sci. 73, 4493–4515 10.1007/s00018-016-2303-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nardini M., Valente C., Ricagno S., Luini A., Corda D., and Bolognesi M. (2009) CtBP1/BARS Gly172 → Glu mutant structure: impairing NAD(H)-binding and dimerization. Biochem. Biophys. Res. Commun. 381, 70–74 10.1016/j.bbrc.2009.02.010 [DOI] [PubMed] [Google Scholar]
- 49. Liu H., and Naismith J. H. (2008) An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91 10.1186/1472-6750-8-91 [DOI] [PMC free article] [PubMed] [Google Scholar]