

Abstract
We describe the use of thioester exchange equilibria to measure the propensities of amino acid residues to participate in helical secondary structure at room temperature in the absence of denaturants. Thermally or chemically induced unfolding has previously been employed to measure α-helix propensities among proteinogenic α-amino acid residues, and quantitative comparison with precedents indicates that the thioester exchange system is reliable for residues that lack side chain charge. This system allows the measurement of α-helix propensities for D-α-amino acid residues and propensities of residues with non-proteinogenic backbones, such as those derived from a β-amino acid, to participate in an α-helix-like secondary structure.
Graphical Abstract
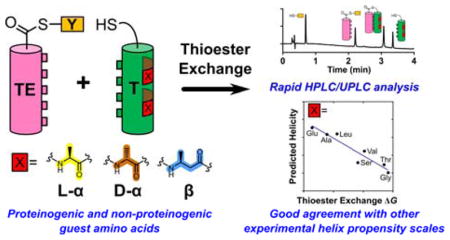
The functional diversity of proteins emerges from structural diversity. Thousands of tertiary folds are known; however, poly-L-α-amino acid backbones appear to favor only a very small number of regular secondary structures, among which the α-helix and β-sheet are dominant. This situation has sparked interest in establishing quantitative scales for secondary structure “propensity” among the proteinogenic amino acid residues,1 because these propensities must be essential to the relationship between a protein’s sequence and its native folding preference. As synthetic advances provide access to proteins containing unnatural side chains and ultimately push the concepts of secondary and tertiary structure beyond L-α-amino acid-derived subunits,2 the challenge of determining subunit secondary structural propensities broadens.
Here we describe an approach that enables the helix propensity of a “guest residue” to be linked quantitatively to a chemical exchange process for which the equilibrium constant can be measured by HPLC or UPLC at room temperature in the absence of denaturants. The resulting α-helix propensity ranking for a set of proteinogenic residues is found to correlate quantitatively with precedents based on more classical experimental designs. In the classical approach, a chemical denaturant is employed to disrupt the folded state, as monitored by circular dichroism or other spectroscopic method, and extrapolation is required to derive thermodynamic parameters in the absence of denaturant.3 Because of the small size of the peptide components required, our approach can be readily employed to evaluate subunits that are not derived from L-α-amino acids, as we illustrate by extension to a D-α-amino acid residue and to β-amino acid residues.
Our measurement strategy is based on thiol-thioester exchange reactions (Figure 1), which rapidly reach equilibrium in aqueous solution at neutral pH.4 In properly designed systems, the thiol-thioester exchange equilibrium constant can be related to the stability of the tertiary structure formed by a long thiodepsipeptide, such as TE−T in Figure 1 (right side). This experimental design has been useful for examining factors that control the packing of α-helices against one another in coiled coils, which are common tertiary structural motifs in proteins.5 Specifically, we have used the thioester exchange strategy to evaluate thermodynamic contributions of side chains at the α-helical interface to coiled-coil stability.6 Here we show that varying residues at sites that are distal to the helix-helix interface provides an accurate measure of α-helical propensity for residues bearing side chains that are not charged.
Figure 1.

Outline of thioester exchange experiment. X indicates positions of guest residues for which helix propensities can be evaluated. [TE−T]u and [TE−T]f indicate folded and unfolded states of the coiled-coil-forming full-length thiodepsipeptide, respectively.
The system used for the current experiments (Figure 2) emerged from a previous study7 and was designed based on the heptad repeat pattern that is characteristic of sequences that form coiled coils.5 In thiodepsipeptide TE−T, Leu and Ile are placed at heptad positions a and d to drive intramolecular packing of the two helical segments via knobs-into-holes interactions. The exterior faces of both helices, defined by heptad positions b, c and f, feature ionic side chains to discourage intermolecular associations. The e and g residues are cationic for the thioester peptide helix (designated TE), and the e and g residues are anionic for the thiol peptide helix (designated T); this Coulombic complementarity should stabilize the intended parallel coiled-coil tertiary structure via interhelical salt bridges.8 Arg or Glu was selected for specific b, c and f positions so as to maximize the number of intrahelical i,i+3 and i,i+4 salt bridges9 and thereby stabilize the individual α-helices and the intramolecular coiled-coil in TE−T. The two α-helix-forming components are linked via a flexible Gly-rich segment in TE−T. The labile thioester bond is located near the center of this linker.
Figure 2.

(a) Helix-wheel diagram of thioester peptide TE (with small molecule Y as the thiol segment, highlighted in orange) and thiol peptide T. (b) Sequence of full-length thiodepsipeptide T−TE. Succ = succinyl.
Factors that affect the stability of an isolated α-helix also affect the stability of tertiary10 or quaternary11 structures that incorporate that helix. We selected f positions as “guest” sites in the T segment for evaluation of α-helical propensities because residues in f positions should not contact the helix formed by the TE segment upon adoption of the helix-loop-helix tertiary structure by TE−T. Therefore, any effect on tertiary stability caused by changing guest residue identity should reflect the impact of the guest on α-helix stability. Two variable sites at f positions, bearing identical guest residues, were employed to maximize the resulting effects on stability. Because both sites reside in the thiol peptide, each new measurement requires the synthesis of only one new T peptide. The TE component is invariant in these studies.
Initial evaluation focused on the version of T with Glu residues at both guest sites, which corresponds to a variant from the previously studied system.7 Thioester exchange measurements indicated ΔGfold = −2.32 kcal/mol for this TE−T variant (ΔGfold = −RT ln(Kfold); Kfold defined in Figure 1; see ref. 4a for calculation of Kfold based on Kex). This value did not change significantly when peptide concentration was varied between 25 and 200 μM, which suggests that Kex measured in this range is not influenced by intermolecular interactions (Figure 3a).12 This concentration-independence is essential if a reliable ΔGfold value is to be derived from the thioester exchange equilibrium constant.4,6
Figure 3.

(a) Thioester exchange-derived ΔGfold at different concentrations of T and TE. Red circles indicate results from individual experiments. Error bars are ± standard deviation. “Asn” in parentheses indicates T variant with Leu→Asn substitution at coiled-coil interfacial residue 13′. (b) ΔGfold measured for T variants with various guest residues and their coiled-coil interfacial Leu→Asn counterparts.
Our experimental design stipulates that tertiary folding is driven by burial of hydrophobic surfaces (a and d side chains) on each helical segment. This hypothesis was tested by replacing one of the d position Leu residues in the T component with Asn, the polar side chain of which is not well accommodated within a hydrophobic coiled-coil interface.6 If the formation of the predicted hydrophobic interface drives tertiary folding in our system, then the Leu→Asn change should be destabilizing. This prediction was borne out: ΔGfold = −0.70 kcal/mol for the Asn-containing T variant with Glu at the back-face guest sites, a decline in tertiary structure stability of ≈1.5 kcal/mol relative to the variant containing Leu at the d position (Figure 3b).
Having validated the thioester exchange system, we examined several variants in which the T component bore identical residues at the two guest sites (Table 1). Precedents11,13 suggest that the guests we employed, Ala, Leu, Ser, Val, Thr and Gly, span a wide range of α-helical propensities among proteinogenic residues. Only proline, which is strongly destabilizing, lies outside this range. Ala and Leu are generally judged to have the strongest α-helical propensities among residues with non-ionizable side chains. Ser, Val and Thr have more moderate α-helical propensities, and the flexible Gly residue has a low propensity. For the cases with Ala or Gly residues in the guest sites, we evaluated the controls in which a d Leu residue was replaced with Asn. In both cases, large declines in ΔGfold result from this interfacial Leu→Asn modification (Figure 3b). These observations support the conclusion that tertiary folding of TE−T requires formation of the expected coiled-coil interface.
Table 1.
ΔGfold measured by thioester exchange for thiol peptides containing various guest residues.a
| Guest Residue | ΔGfold (kcal/mol)b |
|---|---|
| Glu | −2.32 ± 0.14 |
| Glu(Asn)c | −0.70 ± 0.16 |
| Ala | −2.07 ± 0.12 |
| Ala(Asn)c | −0.39 ± 0.04 |
| Leu | −1.91 ± 0.05 |
| Ser | −1.56 ± 0.03 |
| Val | −1.46 ± 0.07 |
| Thr | −1.13 ± 0.10 |
| Gly | −1.06 ± 0.06 |
| Gly(Asn)c | +0.02 ± 0.12 |
| D-Ala | +0.05 ± 0.12 |
| β3hAla | −0.86 ± 0.14 |
| β3hAla(Asn)c | +0.16 ± 0.15 |
| βhGly | −0.47 ± 0.05 |
ΔGfold = −RT ln(Kfold); Kfold is defined in Figure 1. The guest positions are defined in Figure 2. See ref. 4a for calculation of ΔGfold based on Kex. Conditions: 50 μM each T and TE, 50 mM phosphate, pH 7.0, 2 mM TCEP, room temperature.
Mean ± standard deviation of at least five separate experiments.
Leu→Asn substitution at coiled-coil interfacial residue 13′ of T.
α-Helical propensities of proteinogenic residues have been measured based on variations at guest sites in diverse polypeptide hosts,11,13–14 and these precedents offer a basis of comparison for the results in Table 1. Several studies have involved synthetic peptides that autonomously adopt α-helical conformations in solution.11,13c,15 In other systems, α-helix formation has been linked to adoption of higher-order structure.13b,14,15c For comparisons with our results, we focused on two previous studies. O’Neil and DeGrado designed a host sequence that forms a parallel coiled-coil homodimer with the guest site on the back face of each helix.11 Fersht et al. developed a barnase-based system in which the guest site is a solvent-exposed position on an α-helix within the tertiary structure of the protein.14 In both systems, thermodynamic comparisons among variants with different guest residues were conducted in the presence of substantial urea concentrations. Thioester exchange measurements, in contrast, are conducted in the absence of denaturants.
α-Helix propensities derived from thioester exchange measurements for the six proteinogenic residues lacking an ionized side chain show a good quantitative correlation with results from O’Neil and DeGrado (Figure 4a).11 A good correlation is observed also with results from Fersht et al. (Figure 4b)14 and with helix propensity scales described by others.12 However, our system indicates that Glu, a guest residue that bears a charged side chain under the measurement conditions, is substantially more favorable for α-helix formation than is reported by O’Neil and DeGrado11 or by Fersht et al.14 We attribute this difference to the fact that the side chain carboxylates of the guest Glu residues in the thiol peptide can engage in favorable intrahelical Coulombic interactions with nearby Arg residue side chains in the folded state. In contrast, the systems employed by O’Neil and DeGrado11 and by Fersht et al.14 were carefully designed to minimize interactions of guest residue side chains with neighboring sites in the folded state.
Figure 4.

(a) Comparison of thioester exchange-derived ΔΔGfold values (ΔGfold per residue normalized to the value for the variant of T containing Gly at guest sites) with helix propensities measured in the coiled-coil model system of O’Neil and DeGrado (ΔΔGpept).11 The datum for the variant of T containing Glu, indicated in red, was omitted from the linear regression. (b) Comparison of ΔΔGfold with helix propensities measured in the barnase system of Fersht et al. (ΔΔGprot).14 The datum for the variant of T containing Glu, indicated in red, was omitted from the linear regression. (c) Comparison of ΔΔGfold with AGADIR-predicted helix content for the T peptides. (d) Comparison of ΔΔGfold with per-residue molar ellipticity of T peptides at 223 nm (100 μM T variant, 50 mM phosphate, pH 7.0, 2 mM TCEP, 20°C).
Our explanation of Glu behavior is supported by percent helicity predicted with the program AGADIR16 for the T peptides bearing different guest residues (Figure 4c). AGADIR takes into account Coulombic interactions between ionized side chains that are juxtaposed upon helix formation (i,i+3 and i,i+4 sequence relationships). The calculated percent helicity values for T peptides, including the variant with Glu in the guest sites, correlate well with ΔGfold values derived from thioester exchange measurements. Circular dichroism (CD) data for the T peptides lend additional support to our explanation for the high ΔΔGfold value for Glu as guest relative to the experimental scales from O’Neil and DeGrado and from Fersht et al. (Figure 4d). The variant of T containing Glu displays a very strong minimum at 223 nm, which is characteristic of α-helicity, as expected if intrahelical electrostatic interactions contribute to ΔΔGfold for this variant.
There is growing interest in the use of site-specifically incorporated D-α-amino acid residues for the design of bioactive peptides and proteins,2d and we therefore used the thioester exchange system to evaluate the propensity of D-Ala to be incorporated into a right-handed α-helix, i.e., an α-helix composed largely of L-α-amino acid residues. The results indicated that D-Ala is substantially more destabilizing to a right-handed α-helix than is Gly, which is qualitatively consistent with previous reports.17 DeGrado et al.17a used a coiled-coil homodimer to address this question and determined that incorporation of a D-Ala residue into an α-helix is 0.18 kcal/mol less favorable than incorporation of a Gly residue. Krause and coworkers17c found that D-Ala was approximately 0.5 kcal/mol less favorable than Gly in an α-helix, using a monomeric model system. The latter value is consistent with our thioester exchange results, which suggest that D-Ala is 0.55 kcal/mol less favorable than Gly in a right-handed α-helix. Given the quantitative correlation between our system and that of O’Neil and DeGrado for L-α-amino acid residue helix propensities in the absence of side chain charge (Figure 4a), the origin of the discrepancy in terms of D-Ala is unclear. Perhaps the α-helix propensity of D-Ala, and D residues generally, is more sensitive to local environment than are propensities of L residues.
The new system was used to evaluate the propensity of β-amino acid residues to participate in an α-helix-like conformation. These experiments were designed to detect the impact of a side chain on helix stability. β3-Homoalanine (β3hAla) is the homologue of alanine that contains an additional methylene between the side chain-bearing carbon and the carbonyl carbon, and β-homoglycine (βhGly) has no side chain. Thioester exchange measurements indicate ΔGfold = −0.86 kcal/mol for the TE−T variant containing β3hAla at the guest positions. When these two β substitutions are combined with a d position Leu→Asn substitution, ΔGfold = +0.16 kcal/mol. The ≈1 kcal/mol destabilization resulting from this Asn substitution suggests that formation of the coiled-coil interface remains the driving force for tertiary folding of TE−T after the backbone has been altered via two α→β modifications. This conclusion is consistent with previous crystallographic demonstrations that peptides containing combinations of α and β3 residues can adopt an α-helix-like secondary structure.18 Thioester exchange experiments using the T peptide with βhGly at the guest positions gave ΔGfold = −0.47 kcal/mol. Thus, the data suggest an energetic penalty of approximately 0.2 kcal/mol for each β3hAla→βhGly substitution. This trend is consistent with the destabilization of the α-helix that has been generally observed to result from replacement of Ala with Gly.11,13–15 Direct comparisons in the context of our thioester exchange system suggest that the beneficial effect of a methyl side chain may be smaller for β residues than for α residues (Figure 5).
Figure 5.

Thioester exchange-derived ΔGfold for T peptides containing Ala, Gly, β3hAla, or βhGly. Error bars are ± standard deviation.
The thioester exchange measurement involving β3hAla at the guest sites implies that each α→β substitution causes a conformational destabilization of 0.6 kcal/mol per residue, because Ala at the guest site yielded ΔGfold = −2.07 kcal/mol. We propose that the conformational destabilization associated with α→β substitution arises largely from the impact of backbone modification (insertion of a methylene unit) on helical stability, since structural evidence shows that β3 residues are compatible with an α-helix-like secondary structure.18c More specifically, we propose that Ala→β3hAla substitution is destabilizing to the helical state because of the additional backbone bond, which has relatively low barriers to rotation, in the β3 residue. Restriction of rotation upon folding might increase the entropic cost of helix formation for a β3hAla residue relative to an Ala residue; however, experiments by Reinert and Horne have shown that α→β3 substitution in a different tertiary context causes a helix stability penalty that is enthalpic.19 It is possible that the backbone modification in our system causes subtle changes in the arrangement of side chains projected from the other side of the helix20 (positions a and d), which could affect the energetics of knobs-into-holes packing at the coiled-coil interface. Such changes in helix-helix packing energy would contribute to ΔGfold. Whether or not the unfavorable 0.6 kcal/mol increment associated with α→β substitution can be attributed entirely to a change in helical propensity, this measurement provides valuable insight on the energetic consequences of α residue-to-β3 residue substitution in the context of an α-helix-like conformation.
We have shown that thioester exchange equilibria can provide quantitative measurements of α-helical propensity for α-amino acid residues bearing uncharged side chains under “native” conditions (no denaturant; room temperature). Moreover, we have shown that this operationally convenient approach can be extended to non-proteinogenic subunits, including D-α-amino residues and β-amino acid residues. There is growing interest in the structures and functions of polypeptide analogues containing subunits with unnatural backbones,2,21 and this interest is magnified by the prospect that these unnatural subunits may be brought into the realm of ribosomal incorporation.22 The results described above suggest that thioester exchange offers a versatile approach for analyzing the impact of backbone modification on conformational stability.
Supplementary Material
Acknowledgments
This work was supported in part by the National Science Foundation (CHE-1565810). The authors thank Dr. Young-Hee Shin for providing small thiol Y and Prof. Jay Steinkruger for helpful comments.
Footnotes
Notes
The authors declare no competing financial interest.
Experimental details including peptide synthesis, thioester exchange UPLC chromatograms, and comparisons of thioester exchange data with other helix propensity scales. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Chou PY, Fasman GD. Biochemistry. 1974;13:211. doi: 10.1021/bi00699a001. [DOI] [PubMed] [Google Scholar]
- 2.(a) Werner HM, Horne SW. Curr Opin Chem Biol. 2015;28:75. doi: 10.1016/j.cbpa.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Checco JW, Gellman SH. Curr Opin Struct Biol. 2016;39:96. doi: 10.1016/j.sbi.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guichard G, Huc I. Chem Commun. 2011;47:5933. doi: 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]; (d) Simon MD, Maki Y, Vinogradov AA, Zhang C, Yu H, Lin YS, Kajihara Y, Pentelute BL. J Am Chem Soc. 2016;138:12099. doi: 10.1021/jacs.6b03765. [DOI] [PubMed] [Google Scholar]
- 3.(a) Pace NC, Shaw KL. Proteins: Struct, Funct, Bioinf. 2000;41(Suppl 4):1. doi: 10.1002/1097-0134(2000)41:4+<1::aid-prot10>3.3.co;2-u. [DOI] [PubMed] [Google Scholar]; (b) Huyghues-Despointes B, Scholtz MJ, Pace NC. Nat Struct Mol Biol. 1999;6:910. doi: 10.1038/13273. [DOI] [PubMed] [Google Scholar]
- 4.(a) Woll MG, Gellman SH. J Am Chem Soc. 2004;126:11172. doi: 10.1021/ja046891i. [DOI] [PubMed] [Google Scholar]; (b) Woll MG, Hadley EB, Mecozzi S, Gellman SH. J Am Chem Soc. 2006;128:15932. doi: 10.1021/ja0634573. [DOI] [PubMed] [Google Scholar]; (c) Hadley EB, Witek AM, Freire F, Peoples AJ, Gellman SH. Angew Chem Int Ed. 2007;46:7056. doi: 10.1002/anie.200702449. [DOI] [PubMed] [Google Scholar]
- 5.Woolfson DN. Adv Protein Chem. 2005;70:79. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- 6.(a) Hadley EB, Gellman SH. J Am Chem Soc. 2006;128:16444. doi: 10.1021/ja067178r. [DOI] [PubMed] [Google Scholar]; (b) Hadley EB, Testa OD, Woolfson DN, Gellman SH. Proc Natl Acad Sci USA. 2008;105:530. doi: 10.1073/pnas.0709068105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Steinkruger JD, Woolfson DN, Gellman SH. J Am Chem Soc. 2010;132:7586. doi: 10.1021/ja100080q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steinkruger JD, Bartlett GJ, Woolfson DN, Gellman SH. J Am Chem Soc. 2012;134:15652. doi: 10.1021/ja3063088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Shea EK, Lumb KJ, Kim PS. Curr Biol. 1993;3:658. doi: 10.1016/0960-9822(93)90063-t. [DOI] [PubMed] [Google Scholar]
- 9.(a) Burkhard P, Meier M, Lustig A. Protein Sci. 2000;9:2294. doi: 10.1110/ps.9.12.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Burkhard P, Ivaninskii S, Lustig A. J Mol Biol. 2002;318:901. doi: 10.1016/S0022-2836(02)00114-6. [DOI] [PubMed] [Google Scholar]
- 10.Myers JK, Pace CN, Scholtz JM. Biochemistry. 1997;36:10923. doi: 10.1021/bi9707180. [DOI] [PubMed] [Google Scholar]
- 11.O’Neil KT, DeGrado WF. Science. 1990;250:646. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 12.See Supporting Information.
- 13.(a) Muñoz V, Serrano L. Proteins: Struct, Funct, Genet. 1994;20:301. doi: 10.1002/prot.340200403. [DOI] [PubMed] [Google Scholar]; (b) Blaber M, Zhang XJ, Matthews BW. Science. 1993;260:1637. doi: 10.1126/science.8503008. [DOI] [PubMed] [Google Scholar]; (c) Chakrabartty A, Kortemme T, Baldwin RL. Protein Sci. 1994;3:843. doi: 10.1002/pro.5560030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horovitz A, Matthews JM, Fersht AR. J Mol Biol. 1992;227:560. doi: 10.1016/0022-2836(92)90907-2. [DOI] [PubMed] [Google Scholar]
- 15.(a) Lyu PC, Liff MI, Marky LA, Kallenbach NR. Science. 1990;250:669. doi: 10.1126/science.2237416. [DOI] [PubMed] [Google Scholar]; (b) Park SH, Shalongo W, Stellwagen E. Biochemistry. 1993;32:7048. doi: 10.1021/bi00078a033. [DOI] [PubMed] [Google Scholar]; (c) Myers JK, Pace CN, Scholtz JM. Proc Natl Acad Sci USA. 1997;94:2833. doi: 10.1073/pnas.94.7.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muñoz V, Serrano L. J Mol Biol. 1995;245:275. doi: 10.1006/jmbi.1994.0023. [DOI] [PubMed] [Google Scholar]
- 17.(a) Fairman R, Anthony-Cahill SJ, DeGrado WF. J Am Chem Soc. 1992;114:5458. [Google Scholar]; (b) Rothemund S, Krause E, Beyermann M, Dathe M, Bienert M, Hodges RS, Sykes BD, Sönnichsen FD. Pept Res. 1996;9:79. [PubMed] [Google Scholar]; (c) Krause E, Bienert M, Schmieder P, Wenschuh H. J Am Chem Soc. 2000;122:4865. [Google Scholar]; (d) Chen Y, Mant CT, Hodges RS. J Pept Res. 2002;59:18. doi: 10.1046/j.1397-002x.2001.10994.x. [DOI] [PubMed] [Google Scholar]
- 18.(a) Horne WS, Price JL, Keck JL, Gellman SH. J Am Chem Soc. 2007;129:4178. doi: 10.1021/ja070396f. [DOI] [PubMed] [Google Scholar]; (b) Horne SW, Price JL, Gellman SH. Proc Natl Acad Sci USA. 2008;105:9151. doi: 10.1073/pnas.0801135105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Horne WS, Johnson LM, Ketas TJ, Klasse PJ, Lu M, Moore JP, Gellman SH. Proc Natl Acad Sci USA. 2009;106:14751. doi: 10.1073/pnas.0902663106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Patgiri A, Joy ST, Arora PS. J Am Chem Soc. 2012;134:11495. doi: 10.1021/ja301953j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Johnson LM, Gellman SH. Methods Enzymol. 2013;523:407. doi: 10.1016/B978-0-12-394292-0.00019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reinert ZE, Horne WS. Chem Sci. 2014;5:3325. doi: 10.1039/C4SC01094A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee EF, Smith BJ, Horne SW, Mayer KN, Evangelista M, Colman PM, Gellman SH, Fairlie DW. ChemBioChem. 2011;12:2025. doi: 10.1002/cbic.201100314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Wang PSP, Schepartz A. Chem Commun. 2016;52:7420. doi: 10.1039/c6cc01546h. [DOI] [PubMed] [Google Scholar]; (b) Gopalakrishnan R, Frolov AI, Knerr L, Drury WJ, 3rd, Valeur E. J Med Chem. 2016;59:9599. doi: 10.1021/acs.jmedchem.6b00376. [DOI] [PubMed] [Google Scholar]
- 22.(a) Dedkova LM, Fahmi N, Golovine SY, Hecht SM. J Am Chem Soc. 2003;125:6616. doi: 10.1021/ja035141q. [DOI] [PubMed] [Google Scholar]; (b) Dedkova LM, Fahmi N, Paul R, del Rosario M, Zhang L, Chen S, Feder G, Hecht SM. Biochemistry. 2012;51:401. doi: 10.1021/bi2016124. [DOI] [PubMed] [Google Scholar]; (c) Czekster C, Robertson WE, Walker AS, Söll D, Schepartz A. J Am Chem Soc. 2016;138:5194. doi: 10.1021/jacs.6b01023. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fujino T, Goto Y, Suga H, Murakami H. J Am Chem Soc. 2016;138:1962. doi: 10.1021/jacs.5b12482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.