

Abstract
The objective of this study was to investigate whether males who were born preterm took longer to receive a Duchenne muscular dystrophy (DMD) diagnosis than term males. Data for males with DMD identified through a population-based surveillance system were analyzed using Kaplan Meier estimator. The first signs and symptoms (SS) were noted at a median age of two years in both groups. Median age when first SS prompted medical evaluation was 2.59 years among preterm and 4.01 years among term males. Median age at definitive diagnosis was 4.25 years and 4.92 years for preterm and term males, respectively. Neither difference was statistically significant. Preterm males tended to be seen for their initial medical evaluation earlier than term males, though they were not diagnosed significantly earlier. It may take clinicians longer after the initial evaluation of preterm males to arrive at a DMD diagnosis.
Keywords: Duchenne muscular dystrophy, preterm, pediatric, children, epidemiology
Introduction
Duchenne muscular dystrophy is an X-linked recessive disorder resulting from the absence of dystrophin in the dystrophin-glycoprotein complex.1 The disease affects one in 3,600–6,000 live male births and is the most common form of muscular dystrophy in children.2 Signs and symptoms of Duchenne muscular dystrophy are usually noticed before five years of age and consist of mildly delayed motor milestones, inability to run and jump properly, and difficulties rising from the floor.2,3 In cases where there is no family history of dystrophinopathy, the diagnosis is suspected in the presence of delayed development that includes motor difficulties; however, cognitive, behavioral, and language abnormalities are also observed in 30% of cases.4
Early diagnosis of Duchenne muscular dystrophy allows families to make choices about family planning and also allows early therapeutic interventions that can substantially improve prognosis.2,5,6 However, studies that evaluated the diagnostic odyssey of males with Duchenne muscular dystrophy showed a lag between the time of the first symptoms and the time of definitive diagnosis that varied between 2.25 years,7 2.50 years,3 3.20 years,8 and 5 years.9 This lag in diagnosis has not improved in the past 20 years and may well continue, given that some professional societies have still not included early creatine kinase screening in their recommendations for males presenting with motor delay.3,10
Preterm birth, defined as delivery before 37 completed weeks of gestation, is a leading contributor to infant death.11 According to the Centers for Disease Control and Prevention, about one of every 10 infants born in the United States were born preterm in 2015.12 Despite advancements in treatments that have helped improve premature infants’ survival, these babies are vulnerable to a wide array of long-term conditions such as neurodevelopmental disabilities (cerebral palsy, impaired coordination, and motor planning problems), cognitive impairment, visual and hearing impairments, behavioral and socio-emotional disorders, and poor health and growth.13
We hypothesized that preterm males with no family history of Duchenne muscular dystrophy at birth may have their diagnosis delayed beyond the lag from identification of first signs and symptoms to diagnosis observed in term males with no family history of Duchenne muscular dystrophy at birth. This delay could be the result of health care providers attributing the developmental and neurologic symptoms to prematurity rather than considering other diagnoses, such as Duchenne muscular dystrophy. To address these hypotheses, our study compared males with Duchenne muscular dystrophy who were born prematurely to those born at term regarding: 1) the timeline of diagnostic steps; 2) the appearance of first signs and symptoms; and 3) the medical evaluation steps that led to the definitive diagnosis.
Materials and Methods
Study design
We conducted a retrospective cohort study to evaluate and compare the characteristics and timing of diagnostic and medical evaluation steps between males with Duchenne muscular dystrophy with no family history of dystrophinopathy at birth who were born premature to those born at term.
Study population and data sources
Males with Duchenne muscular dystrophy were identified through the Muscular Dystrophy Surveillance Tracking and Research Network. The surveillance methodology has been described by Miller et al.14 Briefly, the Muscular Dystrophy Surveillance Tracking and Research Network is a multisite surveillance system that includes individuals with Duchenne or Becker muscular dystrophy born on or after January 1, 1982, and on or before December 31, 2011, and who resided in the states of Arizona, Colorado, Iowa, Georgia, Hawaii, and a 12-county area in western New York State during any part of that time period.
The males with Duchenne or Becker muscular dystrophy in the Muscular Dystrophy Surveillance Tracking and Research Network cohort were identified both prospectively and retrospectively as having a childhood-onset dystrophinopathy and were diagnosed before their 21st birthday through neuromuscular clinics, hospitals and hospital discharge databases, private physicians, service sites for children with special care needs, and birth defects surveillance programs. Data abstraction began in 2004 in Arizona, Colorado, Iowa, and western New York State, in 2005 in Georgia, and in 2008 in Hawaii.
Trained personnel reviewed and abstracted the birth certificates and medical records of individuals and conducted annual follow-up abstraction until December 2011, time of death, or the time they moved outside of the site for cases identified before September 1st, 2011. Individuals identified between September 2011 and December 2011 were followed through December 2012 to ensure at least one year of follow-up for all cases. Abstractors collected data about demographic characteristics including date and place of birth, gestational age at birth, residential history, diagnostic characteristics and medical history (including earliest signs and symptoms), signs and symptoms that prompted first evaluation, diagnostic tests such as creatine kinase, muscle biopsy, genetic test (DNA), and family history of muscular dystrophy. Supplemental Table 1 displays the distribution of the genetic test methods as reported in the medical records. For more than 50% of cases, Polymerase Chain Reaction and/or Southern Blot were used for diagnosis. Supplemental Table 2 shows the distribution of DNA mutation types reported in the medical records grouped by preterm/term status and definite/probable diagnosis of Duchenne muscular dystrophy. For more than half of the definite Duchenne muscular dystrophy cases (term and preterm) genetic mutation consisted of deletions.
In addition, abstractors identified the person who noticed the earliest signs and symptoms, the provider who evaluated the child and the action taken as a result of the evaluation. Data collection was approved by the Institutional Review Board at University of Arizona and the Hawaii Department of Health and through public health authority in Colorado, Georgia, Iowa and western New York. Clinical data for each case were reviewed by a neuromuscular physician and assigned a case definition: definite, probable, possible, asymptomatic, or female.
We included in our analysis males with a definite or probable dystrophinopathy diagnosis.15 A case was categorized as definite if clinical symptoms referable to a childhood-onset dystrophinopathy were documented, and one or more of the following criteria were met: a positive genetic test for dystrophin mutation; a muscle biopsy demonstrating abnormal dystrophin; or an elevated creatine kinase, family history of an X-linked dystrophinopathy, and an affected family member with a positive muscle biopsy or a dystrophin mutation. Cases with documented clinical symptoms referable to a dystrophinopathy, elevated creatine kinase, and a family history of X-linked dystrophinopathy but no confirmatory genetic testing were classified as probable. The identification of a definite case of muscular dystrophy does not necessarily imply a Duchenne muscular dystrophy case phenotype. For example, a definite case in the Muscular Dystrophy Surveillance Tracking and Research Network data could have a Becker muscular dystrophy phenotype. To further identify definite Duchenne muscular dystrophy cases, individuals were considered to have the condition if they met one or more of the following criteria:1) ambulation ceased prior to age 12, or prior to age 16 and two years of continuous steroid treatment prior to the cease in ambulation; 2) a dystrophin quantity at 5% or less on a Western Blot test, or a confirmed out-of-frame genetic mutation; or 3) onset of motor symptoms prior to age five.
Figure 1 shows the exclusion criteria used to derive the study cohort. We excluded females and individuals whose sex was not known, individuals with Becker muscular dystrophy, pregnancy terminations, non-classifiable muscular dystrophy phenotypes, cases classified as asymptomatic or possible, cases with missing data on birth characteristics, residents of Hawaii due to incomplete case ascertainment and follow-up, cases with family history of dystrophinopathy at birth, siblings of first born males with Duchenne muscular dystrophy, and cases not born in Muscular Dystrophy Surveillance Tracking and Research Network site catchment areas. We excluded the siblings of older males with Duchenne muscular dystrophy because we were interested in males with no known family history of dystrophinopathy at birth. Also, we excluded males not born in Muscular Dystrophy Surveillance Tracking and Research Network site catchment areas because these cases had incomplete medical history data. The final analytic sample consisted of 325 males with Duchenne muscular dystrophy (living or deceased as of December 31, 2011), of which seven were very preterm (28–31 weeks gestation), 23 were moderate preterm (32–36 weeks gestation) and 295 were born at term. While this is a small total number of very preterm births, it comprises 23.33% of all preterm births included in the study sample. Nationally, very preterm births (28–31 weeks gestation) comprise 10.02% of all preterm births (28–36 weeks gestation).16 Because of small number of very preterm males with Duchenne muscular dystrophy in our analytic sample, and consequently imprecise estimates, for analysis we combined the very preterm and moderate preterm cases in one group.
Figure 1.
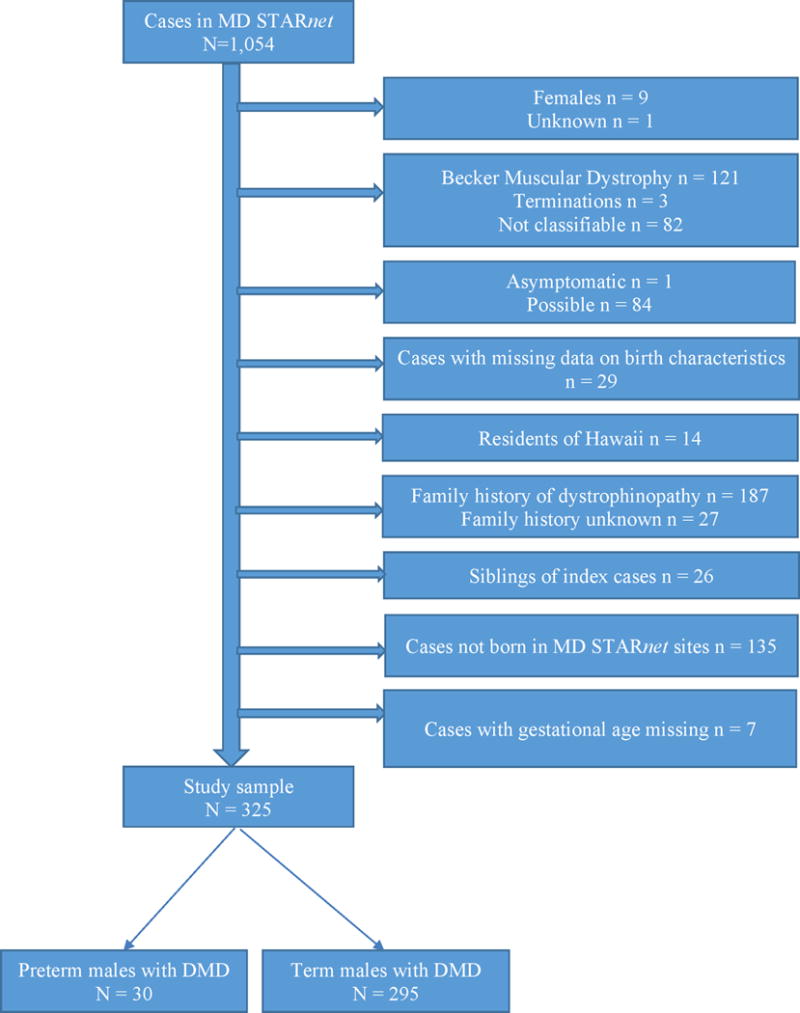
Sample exclusion criteria for males from Muscular Dystrophy Surveillance Tracking and Research Network cohort
Variables
We analyzed individual’s ages at the following events: earliest mobility signs and symptoms, medical evaluation prompted by the first signs and symptoms, first visit to a neuromuscular clinic, and first abnormal diagnostic tests (creatine kinase, muscular biopsy, and DNA test). We determined the age at definitive diagnosis to be the minimum value of the age at first abnormal creatine kinase, age at muscle biopsy and age at DNA test. Also, we calculated the overall time elapsed between age at earliest signs and symptoms and age at definitive diagnosis, age at first signs and symptoms and age at initial medical evaluation, and age at initial medical evaluation and age at definitive diagnosis. To assess the medical evaluation steps, we analyzed motor/mobility and non-motor related symptoms (gait problems, gross motor delay, muscle symptoms, non-motor/neurologic), the individual who noticed the early signs and symptoms, the provider who evaluated the child, and actions taken as a result of the evaluation.
Statistical analysis
None of the age distributions for preterm males with Duchenne muscular dystrophy at various diagnostic steps were normally distributed. Therefore, we assessed medians and interquartile ranges (IQR) and used the Wilcoxon rank-sum test to compare medians between preterm and term males with Duchenne muscular dystrophy. We computed frequency counts and percentages for categorical variables and used Fisher’s exact test to compare the distributions of categorical variables between preterm and term males.
We used Kaplan-Meier survival curves to compare the time elapsed between the following events among preterm and term males: a) age at first signs and symptoms and age at definitive diagnosis; b) age at first signs and symptoms and age at first clinical evaluation; and c) age at first clinical evaluation and age at definitive diagnosis. We used the log-rank test to assess differences between the survival curves for preterm and term males. The significance level for all statistical tests was set to α=0.05. Data management and analyses were conducted using SAS 9.3 software.
Results
Table 1 displays the distribution of race/ethnicity and state of residence for preterm and term males. Most of the preterm and term males with Duchenne muscular dystrophy were White. Preterm and term males with Duchenne muscular dystrophy showed no statistically significant difference in the distribution of race/ethnicity or state of residence.
Table 1.
Sociodemographic characteristics of preterm and term males with Duchenne muscular dystrophy and no family history of dystrophinopathy at birth, MD STARnet
| Sociodemographic Characteristics | Preterm (N=30) | Term (N=295) | * P value | ||
|---|---|---|---|---|---|
|
| |||||
| Race/ethnicity | n | % | n | % | |
| White non-Hispanic | 17 | 56.67 | 197 | 66.78 | 0.08 |
| Black non-Hispanic | 3 | 10.00 | 24 | 8.14 | |
| Hispanic or Latino | 3 | 10.00 | 49 | 16.61 | |
| Other | 7 | 23.33 | 25 | 8.47 | |
| State | |||||
| AZ | 10 | 33.33 | 60 | 20.34 | 0.20 |
| CO | 4 | 13.33 | 59 | 20.00 | |
| GA | 9 | 30.00 | 85 | 28.81 | |
| IA | 1 | 3.33 | 46 | 15.59 | |
| wNYS | 6 | 20.00 | 45 | 15.25 | |
Fisher’s exact test; MD STARnet = Muscular Dystrophy Surveillance Tracking and Research Network; AZ = Arizona; CO = Colorado; GA = Georgia; IA = Iowa; wNYS = western New York State
Medians and IQRs for age at various diagnostic steps are shown in Table 2. Among preterm males with Duchenne muscular dystrophy, median age was under five years at all diagnostic steps, whereas among term males, median age at first visit to a neuromuscular clinic, and median age at DNA test and muscle biopsy was above five years. We did not detect any statistically significant difference in median age at various diagnostics steps between preterm and term males with Duchenne muscular dystrophy.
Table 2.
Median age (years) and interquartile range (IQR) at various diagnostic steps for term and preterm males with Duchenne muscular dystrophy and no family history of dystrophinopathy at birth, MD STARnet
| Preterm births | Term births | P value* | |||
|---|---|---|---|---|---|
| N | Median age (IQR) | N | Median age (IQR) | ||
| Earliest mobility signs/symptoms | 30 | 2.00 (2.16) | 294 | 2.01 (2.67) | 0.84 |
| Initial medical evaluation | 30 | 2.59 (2.83) | 293 | 4.01 (3.08) | 0.07 |
| First neuromuscular visit | 30 | 4.25 (2.80) | 295 | 5.00 (3.00) | 0.23 |
| First abnormal CK test | 25 | 4.17 (2.42) | 275 | 4.92 (3.08) | 0.19 |
| DNA test | 29 | 4.87 (3.28) | 276 | 5.65 (3.47) | 0.23 |
| Muscle biopsy test | 12 | 3.79 (1.99) | 136 | 5.23 (3.24) | 0.18 |
| Definitive diagnosis** | 30 | 4.25 (3.42) | 295 | 4.92 (3.00) | 0.26 |
Wilcoxon rank-sum test; IQR = interquartile range; MD STARnet = Muscular Dystrophy Surveillance Tracking and Research Network;
CK = creatine kinase
We determined the age at definitive diagnosis to be the minimum value among the age at first abnormal CK, age at MB and age at DNA test
We observed an overall time lag between the median age at first signs and symptoms and median age at the definitive diagnosis (2.25 years among preterm and 2.91 years among term males with Duchenne muscular dystrophy); however, the Kaplan-Meier survival curves were not statistically different from each other (Figure 2). Also, we observed a time lag between median age at first signs and symptoms and median age at first medical evaluation (0.59 years among preterm and 2.00 years among term males with Duchenne muscular dystrophy), and between median age at first medical evaluation and median age at definitive diagnosis (1.66 years among preterm and 0.91 years among term males with Duchenne muscular dystrophy). The Kaplan-Meier curves were not statistically significantly different for these outcomes (Figures 3 and 4).
Figure 2.
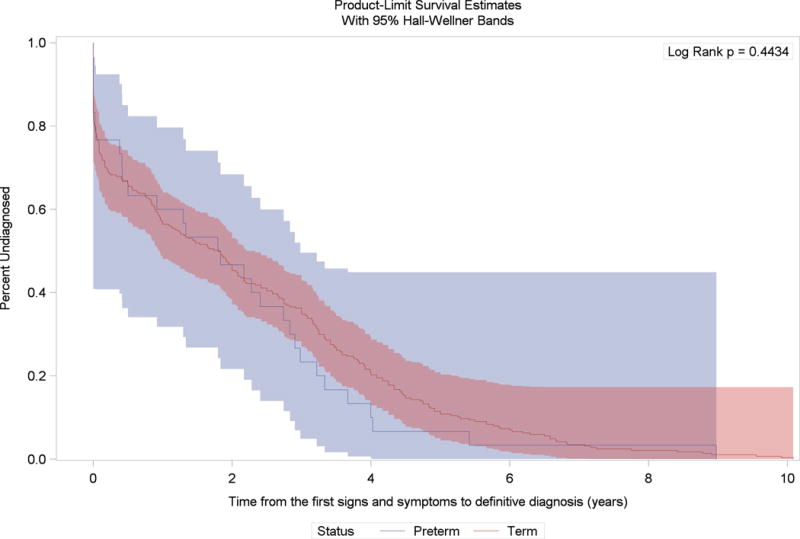
Kaplan-Meier survival curves for the time between age at first signs and symptoms and age at definitive diagnosis among preterm and term born males with Duchene muscular dystrophy and no family history of dystrophinopathy at birth, Muscular Dystrophy Surveillance Tracking and Research Network
Figure 3.
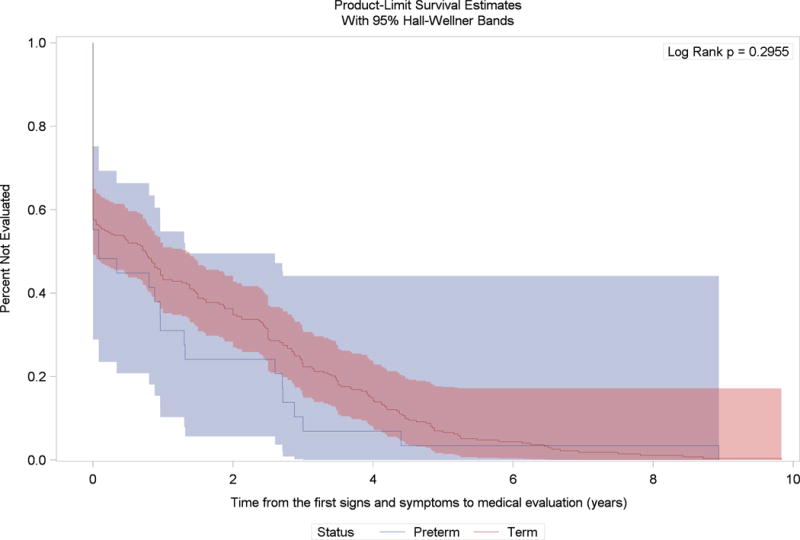
Kaplan-Meier survival curves for the time between age at first signs and symptoms and age at medical evaluation for first signs and symptoms among preterm and term born males with Duchene muscular dystrophy and no family history of dystrophinopathy at birth, Muscular Dystrophy Surveillance Tracking and Research Network
Figure 4.
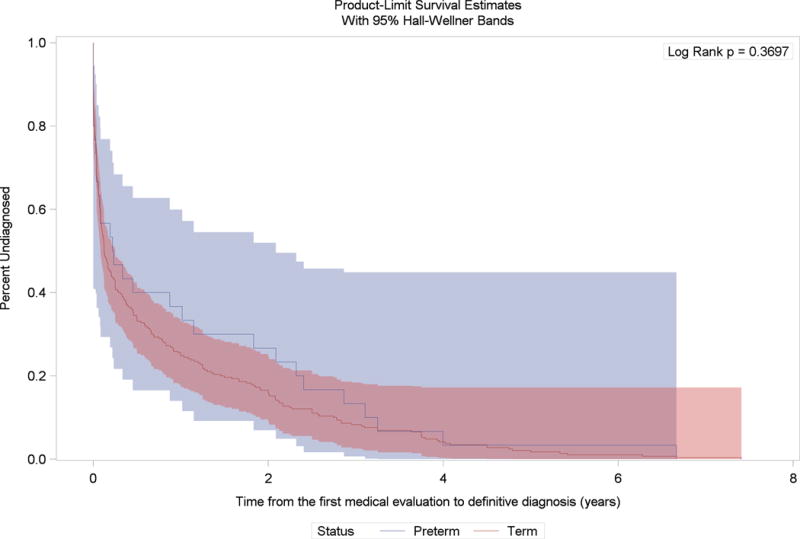
Kaplan-Meier survival curves for the time between age at medical evaluation for first signs and symptoms and age at definitive diagnosis among preterm and term born males with Duchene muscular dystrophy and no family history of dystrophinopathy at birth, Muscular Dystrophy Surveillance Tracking and Research Network
Table 3 displays the distribution of the first signs and symptoms as abstracted from the medical records and the distribution of the variables that describe the medical evaluation process for concerns about first signs and symptoms. Among preterm males with Duchenne muscular dystrophy, gross motor delay and muscle symptoms were documented at a slightly lower percentage than among term males (93% vs 98%), the family or the primary caregiver was the person who noted the first concern at a lower percentage than among term males (43% vs 60%), and a pediatrician or a therapist (speech, occupational or physical) was generally first to evaluate these infants. With respect to referral action taken as the result of the first evaluation, preterm males were seen by physicians of various specialties at a higher percentage than term males (40% vs. 13%), and a lower percentage of preterm males were referred for laboratory tests (27% vs. 36%) or various therapies (17% vs. 25%). However, differences in the medical evaluation process between preterm and term males with Duchenne muscular dystrophy were not statistically significant.
Table 3.
Signs and symptoms first reported to care providers and first medical evaluation of males with Duchenne muscular dystrophy and no family history of dystrophinopathy at birth, MD STARnet
| Preterm births | Term births | P value* | |||
|---|---|---|---|---|---|
|
| |||||
| Signs and symptoms first reported | N | % | N | % | |
| Gait problems1 | 29 | 96.67 | 294 | 99.66 | 0.18 |
| Gross motor delay2 | 28 | 93.33 | 287 | 97.29 | 0.23 |
| Muscle symptoms3 | 28 | 93.33 | 288 | 97.63 | 0.20 |
| Non-motor, neurologic4 | 21 | 70.00 | 198 | 67.12 | 0.84 |
| Other5 | 16 | 53.33 | 130 | 44.07 | 0.34 |
| First concern that prompted initial evaluation | |||||
| Gait problems1 | 16 | 53.33 | 162 | 54.92 | 1.00 |
| Gross motor delay2 | 19 | 63.33 | 175 | 59.32 | 0.70 |
| Muscle symptoms3 | 7 | 23.33 | 106 | 35.93 | 0.23 |
| Non-motor, neurologic4 | 7 | 23.33 | 80 | 27.12 | 0.83 |
| Other5 | 14 | 46.67 | 80 | 27.12 | 0.03 |
| First concern noted by | |||||
| Family, primary caregiver | 13 | 43.33 | 171 | 59.97 | 0.18 |
| Child physician | 6 | 20.00 | 51 | 17.29 | |
| Day care personnel | 1 | 3.33 | 2 | 0.68 | |
| School | 5 | 16.67 | 44 | 14.92 | |
| Other | 3 | 10.00 | 12 | 4.06 | |
| Unknown | 2 | 6.67 | 15 | 5.08 | |
| Health provider that evaluated the child for the first concern | |||||
| Pediatrician | 13 | 43.33 | 110 | 37.29 | 0.81 |
| Family practitioner | 2 | 6.67 | 29 | 9.83 | |
| Orthopedist | 2 | 6.67 | 22 | 7.46 | |
| Neurologist/Neuromuscular specialist | 4 | 13.33 | 63 | 21.36 | |
| Therapist (Speech, Occupational, Physical) | 4 | 13.33 | 16 | 5.42 | |
| Other | 3 | 10.00 | 33 | 11.19 | |
| Unknown | 2 | 6.67 | 18 | 6.10 | |
| Type of referral action as a result of the first evaluation | |||||
| Laboratory testsa | 8 | 26.67 | 107 | 36.27 | 0.57 |
| Consultationsb | 12 | 40.00 | 102 | 13.08 | |
| Therapiesc | 5 | 16.67 | 52 | 25.42 | |
| Otherd | 4 | 13.33 | 30 | 25.30 | |
| None | 1 | 3.33 | 4 | 1.36 | |
Fisher’s exact test; MD STARnet = Muscular Dystrophy Surveillance Tracking and Research Network;
includes: Abnormal gait, Frequent falling/clumsy, Toe-walking, Trouble walking or running, Trouble climbing, Trouble rising or getting up
includes: Loss of motor skills, Muscle hypotonia, Gross motor delay, Inability to keep up with peers in motor;
includes: Muscle pain, Calf hypertrophy, Muscle weakness;
includes: Speech delay or articulation difficulties, Behavioral issues, Cognitive delay, Autism, Other neurology diagnosis
includes: Failure to thrive/poor weight gain, Abnormal lab test, Other factors
includes: Creatine kinase, DNA, Muscle biopsy, and Liver tests
includes: Orthopedics, Pediatrician, Neurology/Neuromuscular, Gastroenterology
includes: Speech Therapy, Occupational Therapy, Physical Therapy, Rehabilitation medicine
includes: Follow up in less than 6 months, Other, Unknown
Discussion
We conducted a descriptive study to investigate whether a history of preterm birth would be associated with additional delays in diagnosing Duchenne muscular dystrophy. This was done by investigating males with no family history of Duchenne muscular dystrophy in the Muscular Dystrophy Surveillance Tracking and Research Network surveillance project. We found that the median age at first signs and symptoms was not significantly different between preterm (2.00 years) and term males (2.01 years) with Duchenne muscular dystrophy, and was similar to the median age observed by Araujo et al. among 78 patients with confirmed diagnosis of Duchenne muscular dystrophy, (2 years, range 0.5–8).9 In a study conducted on 68 males with Duchenne muscular dystrophy born in Eastern Austria, Hauser et al. reported a mean age at first signs and symptoms of 3.1 years, standard deviation (SD)=1.1.17 In our study, the first signs and symptoms consisted of a combination of gait problems, gross motor delay and muscle symptoms. A lower percentage of males had non-motor or neurologic symptoms; however, the distribution of these symptoms was similar between preterm and term males with Duchenne muscular dystrophy. Similarly, Araujo et al. reported that late onset of walking and walking difficulties were the first abnormalities recognized by family members in their study population.9
For preterm infants, these signs and symptoms led to initial medical evaluations at a younger median age (2.59 years) than term males (4.01 years), but this difference was not statistically significant (p=0.07). Our estimates differ from and could not be directly compared to findings by other authors as these studies reported mean age and range or SD. For instance, in the study by Mohamed et al. conducted on 21 boys, the mean age at first specialist (non-general practitioner) visit was 1.9 years, range 1.3–4.2.18 Crisp et al. reported that 51% of 31 patients with Duchenne muscular dystrophy sought medical evaluation before the age of 3 years.8 In the study by Hauser et al., the mean age at first presentation to a physician was 4.8 years, SD=2.0.17
In the present study, gross motor delay and gait problems were the two major symptoms that prompted initial medical evaluation among both preterm and term males. Similar findings were reported by Marshall et al. who observed that 42% of their patients reported an inability to match motor achievements of peers, while 30% reported gait problems.19 The higher percentages observed in our study are likely due to the fact that these signs and symptoms were not recorded in the medical records as being mutually exclusive. In our study, the first concern was noted by family/primary caregiver in a slightly higher proportion among term males with Duchenne muscular dystrophy than among preterm males. Mohamed et al. and Bushby et al. also reported that in most of the cases, family members and/or teachers, not health professionals, expressed the initial concern.7,18
In the current study, it was predominantly pediatricians or various therapists (speech, occupational, physical, and rehabilitation medicine) who evaluated preterm males for the first concern. The initial medical evaluation among preterm males resulted in referral to a specialist (orthopedic, pediatrician, gastroenterology, neurology) who may not have ordered a creatine kinase screening test as the first step in definitive diagnosis, whereas a higher percentage of term males were referred to laboratory tests compared to preterm males, though this difference was not statistically significant.
The time elapsed from first signs and symptoms to initial evaluation was 0.59 years among preterm and 2.00 years among full-term males. The length of the time lag did not differ significantly between the two groups, but it appears that preterm males with Duchenne muscular dystrophy were evaluated sooner, suggesting that they may have been under closer medical scrutiny because of their preterm status. We identified two other studies who reported a slightly longer delay in diagnosis after initial medical evaluation. Marshall et al. observed a mean time lag between presentation to a medical practitioner and definitive diagnosis of 1.92 years, while Read et al. reported that the average time from first presentation to a doctor to definitive diagnosis was 2.0 years, range 0–6.19,20
In our study, the suggestion of an earlier evaluation in the preterm group did not result in earlier diagnosis of Duchenne muscular dystrophy. We found that the median age at definitive diagnosis was similar between preterm and full-term males with Duchenne muscular dystrophy, 4.25 years and 4.92 years, respectively. The time to diagnosis after initial evaluation was longer for the preterm males (1.66 years) than it was for term males (0.91 years). Although these differences were not statistically significant, they suggest that motor and muscle symptoms were more likely attributed to prematurity, with other diagnoses not being pursued as aggressively. A higher median age at diagnosis of 7 years, range 1.75–12, was reported by Araujo et al.9 Other studies reported mean ages at diagnosis that varied as follows: 3.2 years, range 0.4–6.8,18 4.7 years, range 2–8,8 4.8 years, range 1.33–8.25,7 5.1 years (± 2),21 5.2 years, range 1.5–9,20 and 5.5 years, SD=1.7.17
Strengths
To our knowledge, this is the first study that investigates potential diagnosis delays among preterm males with Duchenne muscular dystrophy. These findings supplement the results reported by Ciafaloni et al. using the same source of data3. Muscular Dystrophy Surveillance Tracking and Research Network is a population-based cohort identified through neuromuscular clinics, hospitals and hospital discharge databases, private physicians, service sites for children with special care needs, and birth defects surveillance programs. Data were obtained through abstraction of medical records and therefore are not subject to recall bias. Clinical data were reviewed by neuromuscular physicians who assigned case definition consistently across all participant sites.
Limitations
Since the participant sites in the Muscular Dystrophy Surveillance Tracking and Research Network cohort were not randomly selected, our findings may not be representative of the entire Duchenne muscular dystrophy population in the U.S. To evaluate whether our study sample obtained by exclusion of cases not born in the Muscular Dystrophy Surveillance Tracking and Research Network sites is representative for the Muscular Dystrophy Surveillance Tracking and Research Network cohort, we compared the distribution of sociodemographic characteristics between selected study subjects and the cohort that included males not born in Muscular Dystrophy Surveillance Tracking and Research Network sites. We observed similar distribution of race/ethnicity (p=0.20) and state of residence (p=0.62) (Supplemental Table 3). Due to the small number of preterm males with Duchenne muscular dystrophy in our study, we did not have enough statistical power to reduce uncertainty in the estimates while testing for differences in median age at various diagnostic steps or in length of the delay between preterm and term males with Duchenne muscular dystrophy.
Conclusions
Preterm males with no family history of Duchenne muscular dystrophy may be slightly older at diagnosis than term males with no family history of Duchenne muscular dystrophy, but the difference in these median ages is not statistically significant. However, it appears that preterm males with Duchenne muscular dystrophy are evaluated sooner after the onset of the clinical symptoms, but that it may take the physicians longer to arrive at a Duchenne muscular dystrophy diagnosis compared to term males. The length of time to reach an MD diagnosis has not improved in the past 20 years, and may well continue, given that some professional societies have still not included early creatine kinase screening in their recommendations for males presenting with motor delay.3,10 The timing of Duchenne muscular dystrophy diagnosis has implications for both the child and the family. Early diagnosis allows timely genetic counseling and assessment of carrier status. In addition, early diagnosis helps with avoiding unnecessary diagnostic procedures and tests. Given the progressive decline in muscle function, early diagnosis allows timely initiation of appropriate treatment and implementation of multidisciplinary strategies known to maximize positive outcomes.
Supplementary Material
Acknowledgments
We thank study coordinators and data managers for assistance with data collection and management.
Financial disclosure: This research was supported by Cooperative Agreement numbers DD000187, DD000189, DD000190, DD000191, and DD001117 funded by the Centers for Disease Control and Prevention. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Diseases Control and Prevention or the Department of Health and Human Services.
Footnotes
Author contributions
Aida Soim, PhD, MD conceptualized and designed the study, carried out all data analyses, drafted the initial manuscript, and reviewed and revised the manuscript.
Michael G. Smith Dr.PH replicated all data analyses (as per Muscular Dystrophy Surveillance Tracking and Research Network policies), assisted with study design, and reviewed and revised the manuscript.
Jennifer M. Kwon MD, Joshua R. Mann MD, MPH, Shiny Thomas MPH, MBBS and Emma Ciafaloni MD approved study design, assisted with data analyses, and reviewed and revised the manuscript.
All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Declarations of conflicting interests: The authors declare that there is not conflict of interest.
Ethical approval: Data collection was approved by the institutional review board at University of Arizona and the Hawaii Department of Health and through public health authority in Colorado, Georgia, Iowa and western New York.
Reference list
- 1.Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–17. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 2.Bushby K, Finkel R, Birnkrant DJ, Case LE, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 3.Ciafaloni E, Fox DJ, Pandya S, Westfield CP, et al. Delayed diagnosis in duchenne muscular dystrophy: data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) J Pediatr. 2009;155(3):380–5. doi: 10.1016/j.jpeds.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cyrulnik SE, Fee RJ, De Vivo DC, Goldstein E, et al. Delayed developmental language milestones in children with Duchenne’s muscular dystrophy. J Pediatr. 2007;150(5):474–8. doi: 10.1016/j.jpeds.2006.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bushby K, Finkel R, Birnkrant DJ, Case LE, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9(2):177–89. doi: 10.1016/S1474-4422(09)70272-8. [DOI] [PubMed] [Google Scholar]
- 6.Kakulas BA. Problems and potential for gene therapy in Duchenne muscular dystrophy. Neuromuscul Disord. 1997;7(5):319–24. doi: 10.1016/s0960-8966(97)00056-4. [DOI] [PubMed] [Google Scholar]
- 7.Bushby KM, Hill A, Steele JG. Failure of early diagnosis in symptomatic Duchenne muscular dystrophy. Lancet. 1999;353(9152):557–8. doi: 10.1016/s0140-6736(98)05279-9. [DOI] [PubMed] [Google Scholar]
- 8.Crisp DE, Ziter FA, Bray PF. Diagnostic delay in Duchenne’s muscular dystrophy. JAMA. 1982;247(4):478–80. [PubMed] [Google Scholar]
- 9.Araujo APdQC, Deco MCd, Kloh BdS, Costa MRd, et al. Diagnosis delay of Duchenne Muscular Dystrophy. Brazilian Journal of Mother and Child Health. 2004;4:179–83. [Google Scholar]
- 10.Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, et al. Evidence report: Genetic and metabolic testing on children with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2011;77(17):1629–35. doi: 10.1212/WNL.0b013e3182345896. [DOI] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention. National Center for Health Statistics. Available at: https://www.cdc.gov/nchs/products/databriefs/db279.htm. Accessed September 1, 2017.
- 12.Centers for Disease Control and Prevention. Reproductive Health, Maternal and Infant Health. Available at: https://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Accessed September 1, 2017.
- 13.Behrman RE, Stith Butler A. Preterm Birth: Causes, Consequences, and Prevention. Washington (DC): National Academies Press; 2007. [PubMed] [Google Scholar]
- 14.Miller LA, Romitti PA, Cunniff C, Druschel C, et al. The muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): surveillance methodology. Birth Defects Res A Clin Mol Teratol. 2006;76(11):793–7. doi: 10.1002/bdra.20279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathews KD, Cunniff C, Kantamneni JR, Ciafaloni E, et al. Muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): case definition in surveillance for childhood-onset Duchenne/Becker muscular dystrophy. J Child Neurol. 2010;25(9):1098–102. doi: 10.1177/0883073810371001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Centers for Disease Control and Prevention. National Center for Health Statistics, Division of Vital Statistics, Natality public-use data 2007–2016. Available at: http://wonder.cdc.gov/natality-current.html Accessed March 19, 2018.
- 17.Hauser E, Toifl K, Mad A, Bittner R. The incidence of Duchenne muscular dystrophy in eastern Austria. The controversy regarding CK screening. Wien Klin Wochenschr. 1993;105(15):433–6. [PubMed] [Google Scholar]
- 18.Mohamed K, Appleton R, Nicolaides P. Delayed diagnosis of Duchenne muscular dystrophy. Eur J Paediatr Neurol. 2000;4(5):219–23. doi: 10.1053/ejpn.2000.0309. [DOI] [PubMed] [Google Scholar]
- 19.Marshall PD, Galasko CS. No improvement in delay in diagnosis of Duchenne muscular dystrophy. Lancet. 1995;345(8949):590–1. doi: 10.1016/s0140-6736(95)90503-0. [DOI] [PubMed] [Google Scholar]
- 20.Read L, Galasko CS. Delay in diagnosing Duchenne muscular dystrophy in orthopaedic clinics. J Bone Joint Surg Br. 1986;68(3):481–2. doi: 10.1302/0301-620X.68B3.3733820. [DOI] [PubMed] [Google Scholar]
- 21.Rao VK, Kuntz NL. Delay in Diagnosis of Duchenne Muscular Dystrophy. Pediatr Neurol Briefs. 2015;29(1):5–1. doi: 10.15844/pedneurbriefs-29-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.