

Abstract
Small supernumerary marker chromosomes (sSMC) are chromosomal fragments difficult to characterize genomically. Here we detail a proband with schizoaffective disorder and a mother with bipolar disorder with psychotic features who present with a marker chromosome that segregates with disease. We explored the architecture of this marker and investigated its temporal origin. Array comparative genomic hybridization (aCGH) analysis revealed 3 duplications and 3 triplications that spanned the short arm of chromosome 9, suggestive of a chromoanasynthesis-like event. Segregation of marker genotypes, phased using sSMC mosaicism in the mother, provided evidence that it was generated during a germline-level event in the proband’s maternal grandmother. Whole-genome sequencing (WGS) was performed to resolve the structure and junctions of the chromosomal fragments, revealing further complexities. While structural variations have been previously associated with neuropsychiatric disorders and marker chromosomes, here we detail the precise architecture, human life-cycle genesis, and propose a DNA replicative/repair mechanism underlying formation.
Keywords: Chromosomal abnormalities, Marker chromosome, Structural variation, Microarrays, Psychiatric genetics, SNP analysis
Small supernumerary marker chromosomes (sSMC) are chromosomal fragments that are observed cytogenetically and identified as an addition to the normal complement of 46 human chromosomes, typically during clinical karyotyping studies. Their overall size and genomic content can vary greatly, but generally any additional G-band detected structure smaller than chromosome 20 is considered to be a sSMC. This relatively small size makes it difficult to use traditional cytogenetic G-banding techniques for meaningful characterization at a genomic content level (Liehr, 2011). The chromosomal mechanisms of sSMC formation are heterogeneous; possible explanations include gamete complementation, post-fertilization errors, as well as trisomy and monosomy rescue events (Kotzot, 2002). The lack of detailed resolution for genomic content and architecture that make up a marker chromosome stymies identification of the genomic rearrangement end product. This makes it difficult to surmise which potential DNA recombinant/repair mechanism(s) may apply to specific markers, thereby limiting understanding of the potential mechanisms that may have contributed to the sSMC formation. Although marker chromosomes are found in normal populations with a frequency ranging from 0.044% to 0.075%, there are strong associations between their occurrence and a wide range and severity of disease traits and clinically defined genetic disorders (Liehr, Cirkovic & Lalic, 2013; Liehr, 2011). In some cases, the presence of the sSMC in addition to the normal complement of chromosomes implicates aberrant gene dosage in risk for disease. Alternatively, the presence of the marker, independent of the copy number variation (CNV), may also contribute to altered cellular or developmental processes related to the phenotypic presentation as a result of the cells response to anomalous chromosomal material during division (Crolla, Youings, Ennis & Jacobs, 2005; Liehr et al., 2006).
CNVs has been shown to be associated with a spectrum of neuropsychiatric traits, including autism, schizophrenia, and bipolar disorder (Levy, Xu, Gogos & Karayiorgou, 2012; O’Donovan, Kirov & Owen, 2008). Subsequent analysis of genes located within CNV regions provided evidence for enrichment of genes encoding proteins involved in postsynaptic signaling complexes as well as other neuronal and neurotransmitter specific genes, suggesting a possible mode of disease pathogenesis (Kirov et al., 2012, Pocklington et al., 2015).
A higher incidence of marker chromosomes has been reported among individuals with neuropsychiatric disorders, most notably individuals with autism spectrum disorder and markers containing genomic regions of chromosome 15 and chromosome 9 (Abu-Amero et al., 2010; Gillberg et al., 1991; Schroer et al., 1998). A case report implicated a marker derived from regions spanning 9p24.3q21.11 in a patient with psychotic symptoms, further highlighting the association of a marker chromosome with a neuropsychiatric presentation (Martínez-Jacobo et al., 2015).
Here we detail a proband (BAB3398) and mother (BAB3399) who present with long standing clinical diagnoses of schizoaffective disorder and bipolar disorder with psychotic features, respectively, and a marker chromosome that segregates with disease in this family (Figure 1A). The psychiatric diagnoses of both carriers were based on meeting DSM-IV (1994) criteria, as determined by a review of hospital records and the Structured Clinical Interview for DSM-IV (SCID) (Spitzer et al., 1994). Within a large study of CNVs associated with psychiatric diseases, a proband and mother were shown to carry multiple copy number variants on the short arm of chromosome 9 as revealed by a Nimblegen HD2 2.1 M aCGH platform (data not shown) (Malhotra et al., 2011). Attempts to generate induced pluripotent stem (iPS) cell lines for these individuals revealed that the CNVs presented on a marker chromosome (TCW et al., 2017).
Figure 1.
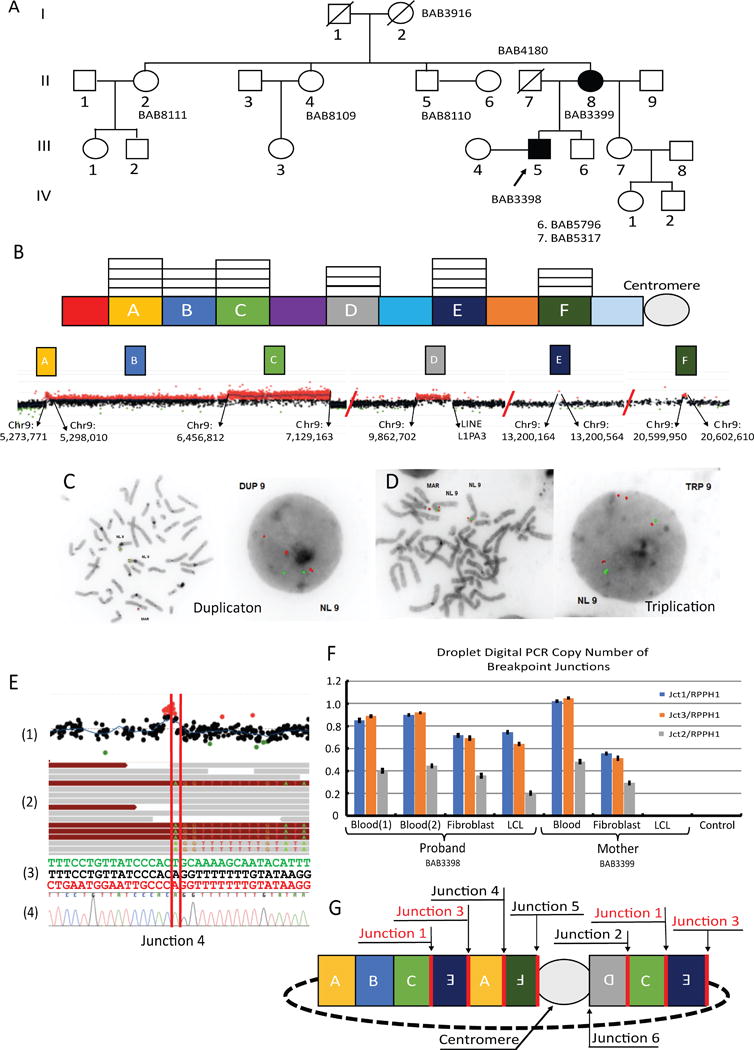
(A) Pedigree showing the extended family with the proband (BAB3398) and mother (BAB3399) (B) The copy number state of each genomic segment with appended high-resolution aCGH detailing the CNV’s presence on the short arm of chromosome 9 with the precise genomic locations for each segment derived from aCGH probe location data. (C & D) FISH studies showing that duplicated segment B, using BAC probe RP11-575C20 (red) (chr9:6,072,717-6,272,364), and triplicated segment C, using BAC probe RP11-106 (red) (chr9:6,576,991-6,744,615) with control BAC probe RP11-362M22 (green) (chr9:12,344,849-12,480,002), are present on the marker. (E) Combination of genomic methods used to map the breakpoint junctions of the marker. (E1) aCGH highlighting the CNV in segment F (E2) Whole genome sequencing (WGS) showing the split reads at the exact point of the junction. (E3) The junction (middle) along with the sequence for the adjoining genomic segments. (E4) Sanger-validation of identified breakpoint junction. (F) Droplet digital PCR of breakpoint junctions showing a duplication of junctions 1 and 3 across cell types in the mother and proband but absent in the LCL of the mother; F refers to fibroblasts. (G) Proposed architectural map of the 9p24 marker chromosome. In red: duplicated junctions 1 and 3 suggest ring formation. All but junction 6 were verified through Sanger sequencing.
Using molecular analyses provided by fluorescence in situ hybridization (FISH), array comparative genomic hybridization (aCGH), breakpoint sequencing, whole genome sequencing (WGS), genome-wide SNP array and droplet digital PCR (ddPCR), we are able to propose a genomic structure for the sSMC, allowing inferences about the DNA mechanism resulting in the genesis of this marker. A custom 4 X 180K Agilent high-resolution tiling-path oligonucleotide microarray spanning the short arm of chromosome 9 confirmed the structural variation, including a duplication approximately 1.15 MB in size (segment B), a triplication 672,351 bp in size (segment C) and a second duplication ranging from 58,512 - 64,535 bp in size (segment D) (Figure 1B), and showed additional CNVs not originally detected that could now be visualized with this higher resolution genome analysis. The newly delineated events included a triplication spanning ~ 24,307 bp (segment A), a triplication of ~ 400 bp (segment E) and an additional duplication of ~ 2,660 bp in size (segment F), bringing the total observed CNV states to 3 duplications and 3 triplications spanning 9p (Figure 1B, Supp. Figure S1). The genes involved in each genomic segment of altered copy number state are highlighted in supplemental figure 1. Subsequent FISH analysis using the bacterial artificial chromosome (BAC) probe RP11-575C20 within the duplicated segment B (Figure 1C), as well as the probe, RP11-106A1, which maps within the triplicated segment C (Figure 1D), and a control probe, RP11-364M22, showed that the regions of copy-number gain identified by the aCGH were adjacent and located on the marker chromosome.
WGS was performed on genomic DNA isolated from the blood of the proband (BAB3398), the mother (BAB3399) and the maternal grandmother (BAB3916) using the Illumina TruSeq PCR-free library and sequenced on the Illumina Hiseq Xten system to identify potential additional structural variants not revealed by aCGH. Data were analyzed using the novoBreak assembly algorithm to detect breakpoint junctions (Figure 1E) (Chong et al., 2017; Liu et al., 2017). In total, 6 junctions were identified through a combination of aCGH and WGS analyses; of those junctions, 5 were confirmed through traditional Sanger-sequencing (Supp. Figure S2). We were unable to resolve the 6th junction at the nucleotide sequence level, but WGS data analysis using novoBreak indicated that it occurred within the pericentromeric repeat region of the long arm of chromosome 9 and involved recombination between two full-length LINEs that share 91% of nucleotide similarity. The first recombinant LINE (L1PA3), spanning 6,024 bp, is located at the proximal break of segment D at 9p23 (chr9:9,921,214 - 9,927,237) whereas the second recombinant LINE (L1PA3), spanning 6,116 bp, is located at 9q12 (chr9:69,101,499-69,107,614). Attempts to confirm this event by PCR amplification were unsuccessful.
In order to further assess the copy number state of each of the junctions using ddPCR in DNA samples from BAB3398 and BAB3399, we used the information obtained from breakpoint junctions (Jct) 1, 2 and 3. Sequencing data identified the distal (in relation to the telomere) break of segment C and the distal break of segment E connected in an inverted orientation (Jct1) followed by the proximal break of segment E connected to the proximal break of segment A (Jct3). Jct 2 is constituted by a proximal break of segment C connected to the proximal break of segment D, the latter also in an inverted orientation. Consistently, results from ddPCR indicate that Jct2 is present once compared with the diploid control region, as expected for junctions formed upon a duplication (Figure 1F). Surprisingly, Jct1 was present at twice the levels observed for Jct2 (Figure 1F), a result confirmed by two sets of primers. Although, this observation is consistent with the dosage indicated in the aCGH for chr9: 7,129,163, double-dosage of junctions occurs only if the initial rearrangement undergoes further non-allelic homologous recombination (NAHR) or if it results from a rolling circle, the latter suggestive of a complex DNA repair aberration (Beck et al., 2015; Gu et al., 2015). In aggregate, the experimental data and information gleaned from the junction copy number, as well as fragment orientation derived through WGS and Sanger-validation, facilitate the derivation of a genome architectural map detailing the content of this sSMC, which is most parsimoniously explained by a specific end product structure for the DNA rearrangement (Figure 1G, Supp. Figure S2). Importantly, the lymphoblastoid cell line (LCL) sample from BAB3399 showed no amplification for any junction tested (Figure 1F), suggesting the marker was lost during EBV transformation, an observation further confirmed by SNP array (Figure 2A). sSMC mosaicism for both BAB3398 and BA3399 was suspected based on ddPCR using distinct samples to confirm copy-number of segments B (DUP) and C (TRP) (Figure 2B). Evidence for mosaicism was further confirmed by FISH in leukocytes, which showed that 13/20 cell nuclei contained the marker in BAB3399 (mother) and 19/20 cell nuclei contained the marker in BAB3398 (proband). Karyotyping studies on fibroblasts from these individuals indicated that the marker element is present in 25% of fibroblasts from BAB3399 and in 56.2% of fibroblasts from BAB3398 (TCW et al., 2017).
Figure 2.
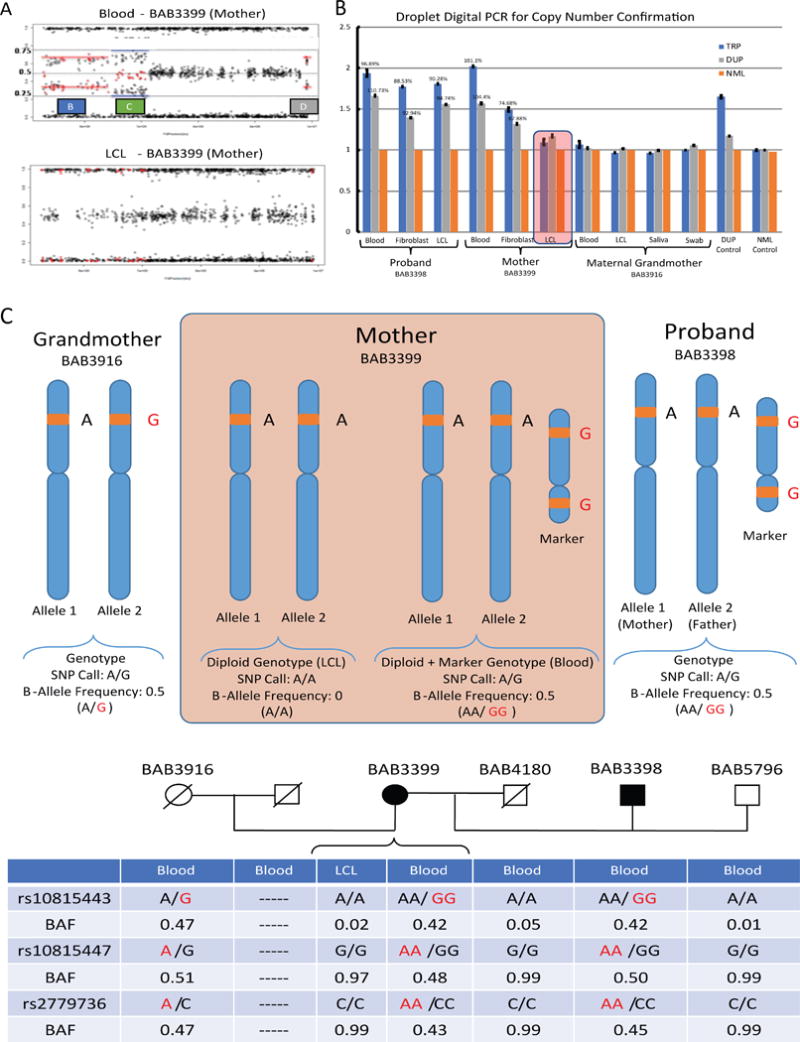
(A) Duplication and triplication events as visualized from SNP array data in BAB3399’s (mother) blood and their absence in lymphoblastoid cell lines (LCL). (B) We designed ddPCR assays to confirm the copy number state of the CNVs identified by aCGH. Primers were designed within the identified duplicated (DUP) and triplicated (TRP) genomic intervals as well as a region of normal (NML) diploid copy number. DNA samples from blood, fibroblasts (F) and LCL were used to confirm the CNVs in the proband (BAB3398), mother (BAB3399) and maternal grandmother (BAB3916). None of the maternal grandmother’s samples, including saliva, showed copy number change, confirming previous aCGH results. The red box highlights the absence of the marker chromosome in maternal LCL. (C) SNPs mapping within the triplicated segments that were homozygous in maternal LCL but heterozygous in maternal blood were classified as informative SNPs and used further for sSMC genotyping and segregation. (D) Examples of informative SNPs used to infer origin of the marker chromosome. Results indicate that sSMC originated in the germline of the maternal grandmother (See Supp. Table S1 for complete SNP data within the CNVs).
The fact that the mother had different subsets of cells with and without the marker provided a unique opportunity to genotype the latter (Figure 2C) and use that information to infer genetic origin. B-allele frequency data within the copy-number variant region was used to estimate the genotype of the marker in BAB3399, which segregates the same way in BAB3398 (Figure 2D); 27 informative SNPs within the triplicated regions (segments A and C) were used to genotype the marker. Importantly, using a Mendelian genomics and genotyping/inheritance strategy, we were able to trace the origin of the marker chromosome to the proband’s maternal grandmother BAB3916 (Supp. Table S1). The absence of the marker in genomic DNA from blood and saliva from BAB3916 suggests that it was generated as a germline-level event. The fact that BAB3399 is the youngest in her sibship is consistent with the higher incidence of marker formation within gametogenesis mutational events in older females (Dalpra et al., 2005; Liehr, 2011).
Here we present a family in which a proband and mother both carry a marker chromosome derived from genomic material on chromosome 9 that segregates with psychotic illness. The lack of resolution afforded by G-banding in marker chromosomes initially made it challenging for cytogeneticists to make informed assumptions about the relation between a marker chromosome and a clinical phenotype, because detailed information about the genomic content located on the marker, its genesis, and transmission status was unclear. The introduction of molecular cytogenetic methods (i.e., FISH and aCGH) and related technologies has allowed for greater insight into and resolution of the genomic content of marker chromosomes, thus allowing for a more genomics data-driven approach (Liehr, 2011; Reddy et al., 2013). While simple segments of fusion could be explained through DNA repair mechanisms such as non-homologous end joining (NHEJ), markers that exhibit cryptic complexity, such as the one we present herein, are likely the result of alternative chromosomal rearrangement mechanisms involving DNA replication/repair events.
Chromoanasynthesis is a phenomenon proposed to explain multiple copy-number gains found on a single or multiple chromosomes along with the seemingly random reorganization of said genomic segments – a recently delineated phenomenon in constitutional genomes that has been proposed to be generated by replicative mechanisms of DNA repair such as microhomology-mediated break-induced replication (MMBIR) and fork stalling and template switching (FoSTeS) (Liu et al., 2011; Maher and Wilson, 2012). These chromosomal catastrophes and genomic phenomena, i.e., chromothripsis, were originally delineated in cancer genome studies (Stephens et al., 2011), were further described in the constitutional genome (Kloosterman et al., 2012; Liu et al., 2011), and now described as a potential mechanism for marker formation (Al-Rikabi, Pekova, Fan, Jančušková & Liehr, 2018). In the individuals reported here, high-resolution aCGH revealed the genomic locations of structural variants that were originally believed to be present in a linear fashion on chromosome 9 (Malhotra et al., 2011). Through additional experimental studies, iPSC generation and subsequent FISH analysis, we determined that the duplication and triplication events mapped to the sSMC, revealing the consequences of a chromoanasynthesis event and delineating a chromosomal rearrangement end product structure, i.e., a sSMC not initially anticipated by the MMBIR replicative repair model (Hastings, Ira & Lupski, 2009). The mechanism of NHEJ was originally proposed to explain multiple CNVs and rearrangements detected on a linear chromosome, but NHEJ may also be a potential causal mechanism in the generation of ring chromosomes (Leibowitz, Zhang & Pellman, 2015). Here we expand the definition of chromoanasynthesis to encompass the formation of a marker chromosome.
In combination with other genomic techniques, WGS facilitated the Sanger validation of 5 out of 6 breakpoints junctions found on the marker (Supp. Figure S2). The 6th junction revealed by WGS proved difficult to discern by aCGH directed breakpoint mapping and Sanger-sequencing, likely due to the occurrence of the breakpoint for this region within a 6.01 kb LINE segment (L1PA3) at 9p23 and with its apparent recombination within the pericentromeric repeat region of another 6.1 kb LINE segment at 9p12, both sharing 91% nucleotide sequence similarity. LINE segments and short interspersed nuclear elements (SINE) are repetitive sequence elements that are often embedded within repeat regions of the genome that are points of genomic instability and mediators of copy number variation as well as sites of rearrangement (Szafranski et al., 2016; Weckselblatt and Rudd, 2015). The repeat nature of this region, the computational and technical limitations of short read WGS sequencing, and a haploid reference devoid of many pericentromeric regions provided limited information regarding the true nature of this specific breakpoint junction.
We hypothesize that the sequence similarity between these two LINE segments was the starting point of break-induced replication (BIR), which may have used regions of homology between the two LINE segments for initiating replicative repair followed by iterative cycles of template-switching (TS) and replication likely by MMBIR, resulting in “patches” of amplified segments copied from the short arm of chromosome 9. Microhomology ranging from 1-3 nucleotides at 4 out of 6 breakpoint recombinant junctions supports the involvement of a replicative-based repair mechanism in the marker generation. Microhomology at all observable breakpoints may have acted as priming events for DNA replication, as found in the MMBIR mechanism for derivation of complex genomic rearrangements (Carvalho and Lupski, 2016). The 4th junction was not only the result of an inversion event but also showed a 62 bp deletion 7 bp upstream of the breakpoint that occurred in the middle of a homopolymeric poly-A stretch (7 bp) implicating the effect of an error-prone polymerase (Carvalho et al., 2013). The discovery that the 1st and 3rd junctions were detected twice is suggestive of a rolling circle-type mechanism during the replicative event itself (Beck et al., 2015; McEachern and Haber, 2006). All together, the data strongly support that an iterative TS model, parsimoniously explained/predicted by MMBIR, underlies formation of the marker chromosome studied in this family (Carvalho and Lupski, 2016, Hastings, Ira & Lupski, 2009). We speculate that a collapsed fork, generated during DNA replication through the highly repetitive pericentromeric region of chromosome 9, resulted in one-ended, double-stranded DNA (oeDNA) that was subject to replicative repair.
SNP array analysis allowed us to infer that the likely life cycle and temporal origin of the maker occurred within the germline of the grandmother. The selection bias against marker chromosomes within sperm is congruent with our finding that this marker is derived from a maternal event (Liehr, 2011).
Although markers are generally seen as unstable genomic events, this marker is stable enough to have been inherited by the mother and subsequently transmitted to the proband. Although karyotyping does not give an accurate representation of the marker itself, presumably it has the required genomic material to allow for transmission and segregation between generations and within cell division; the presence of a centromere and a potential ring structure would aid intergeneration maintenance. Karyotyping studies were unable to resolve this feature, likely because it is below the resolution detectable by that method. The postulated ring structure not only parsimoniously explains the marker’s genomic stability and protection from degradation, but also highlights the hidden architectural complexity that was not visible using older genomic sequencing methods.
The fact that the marker chromosome is found in a mosaic state may have phenotypic consequence since mosaicism for a mutation has been shown to play a role in phenotypic variability (Campbell et al., 2014). Although mosaicism levels have been shown to affect severity of neurodevelopmental disorders, their influence on psychiatric conditions is not well understood (Poduri, Evrony, Cai & Walsh, 2013). For instance, the levels of mosaicism in other chromosomal abnormalities, such as ring 20 [r(20)] syndrome appear to correlate with clinical severity, although this issue has not been extensively studied in patients with marker chromosomes (Daber et al., 2012). It is of interest that the glycine decarboxylase (GLDC) gene is located within the triplicated region of this marker chromosome. GLDC encodes the glycine decarboxylase or glycine cleavage system (GCS) P-protein, which is involved in the catabolism of glycine in glial cells (Takayanagi et al., 2000). Carriers of GLDC triplications would be expected to have increased catabolism of glycine, resulting in low brain levels and NMDA receptor-mediated hypofunction, which has been strongly implicated in the pathophysiology of schizophrenia (Tsai and Coyle, 2002). Consistent with this expectation, mice transgenically modified to overexpress GLDC have significantly reduced extracellular brain glycine levels (Oda et al., 2007). The GLDC gene is also located within a potential locus for autism susceptibility (Abu-Amero et al., 2010), has been found to be fully duplicated in patient with schizophrenia and idiopathic epilepsy (Stewart, Hall, Kang, Shaw & Beaudet, 2011), and has been previously reported as duplicated on a marker chromosome in a patient with psychotic symptoms (Martinez-Jacobo et al. 2015), although the exact biological relevance of dosage changes in this gene remains unknown. Therefore, we postulate that the differing levels of mosaicism in the mother and proband may also contribute to the different clinical severities of their illnesses. Though we can not rule out that the alteration of other dosage-sensitive genes in the region may have contributed to the patient’s phenotype, current evidence supports the role of GLDC as a main candidate gene (Supp. Table S2).
While an increased rare CNV burden is associated with neuropsychiatric disorders, here we detail the precise architecture in the formation of a marker chromosome and delineate a mechanism for formation, transmission genetics and trait manifestation. The final phenotypic outcomes of these patients are likely a combination of the marker chromosome itself, the aberrant gene dosage from genes on the marker as well as the mosaicism levels of the marker in each individual.
Supplementary Material
Supplemental Figure 1: (A) Shows the genomic segments with altered copy number states as denoted by white boxes stacked on top of each segment as well as the genes located within each segment. (B) Table showing each genomic segment that makes up the marker chromosome, their associated copy number state, the genomic coordinates (Hg19) derived from aCGH probe location data, the approximate size of each segment as well as the specific genes involved in each segment.
Supplemental Figure 2: Highlighted are the 5 breakpoint junctions validated through standard Sanger-sequencing and their associated location on the marker chromosome.
Supplemental Table 1: Shows the SNP calls present in the duplicated (red) and triplicated (blue) regions across the family. The genotype of the marker is highlighted in red text across all identified calls.
Supplemental Table 2: Details the other fully duplicated or triplicated genes within the rearrangement and their possible role in neuropsychiatric phenotypes.
Acknowledgments
We would like to thank the patients and their families who generously provided their time and genetic material. We would also like to acknowledge Jesper Eisfeldt who assisted with the initial analysis and file transfer.
Funding Sources: This work was supported in part by the National Institute of Neurological Disorders and Stroke (R35 NS105078) to J.R.L., the National Human Genome Research/National Heart Lung and Blood Institute (NHGRI/NHLBI) grant UM1 HG006542 to the Baylor Hopkins Center for Mendelian Genomics, the National Institute of Health/National Institute of General Medical Sciences (NIH/NIGMS) grant 5 R01 GM106373-04 partially to J.R.L., as well as NIH R01MH071523, R21MH105732, R21MH097470 and to D.L.L., Anonymous Foundation to D.L.L. and J.S., the Ellison Foundation to D.L.L., J.R.L., and C.M.B.C, Team Daniel to D.L.L., the Carmela and Menachem Abraham Fund to D.L.L., J.S. was supported by a grant from the NIH (MH076431) and by a gift to Cold Spring Harbor Laboratory from The Stanley Foundation, NIH R21MH104505 to U.R., the Nilsson-Ehle donations at The Royal Physiographic Society to A.L, the SciLifeLab National Sequencing Project and resources at the Uppmax Project b2015375 to A.L, NIH grant R01 MH101454 to K.B. and the New York Stem Cell Foundation to K.B.
Footnotes
Financial Disclosure:
J.R.L. holds stock ownership in 23andMe Inc. and Lasergen Inc., is a paid consultant for Regeneron Pharmaceuticals, and is a co-inventor on multiple United States and European patents related to molecular diagnostics. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered in the Baylor Medical Genetics Laboratories (BMGL). J.R.L. is on the Scientific Advisory Board of the BMGL.
References
- Abu-Amero KK, Hellani AM, Salih MA, Seidahmed MZ, Elmalik TS, Zidan G, Bosley TM. A de novo marker chromosome derived from 9p in a patient with 9p partial duplication syndrome and autism features: genotype-phenotype correlation. BMC Medical Genetics. 2010;11(1):135. doi: 10.1186/1471-2350-11-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Rikabi ABH, Sona Pekova XF, Jancuskova T, Liehr T. Small Supernumerary Marker Chromosome May Provide Information on Dosage-insensitive Pericentric Regions in Human. Current Genomics. 2018;19(3):192–199. doi: 10.2174/1389202918666170717163830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Carvalho CM, Banser L, Gambin T, Stubbolo D, Yuan B, Lupski JR. Complex genomic rearrangements at the PLP1 locus include triplication and quadruplication. PLoS Genetics. 2015;11(3):e1005050. doi: 10.1371/journal.pgen.1005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Yuan B, Robberecht C, Pfundt R, Szafranski P, McEntagart ME, Stankiewicz P. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. American Journal of Human Genetics. 2014;95(2):173–182. doi: 10.1016/j.ajhg.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nature Reviews Genetics. 2016;17(4):224–238. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Pehlivan D, Ramocki MB, Fang P, Alleva B, Franco LM, Lupski JR. Replicative mechanisms for CNV formation are error prone. Nature Genetics. 2013;45(11):1319–1326. doi: 10.1038/ng.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Z, Ruan J, Gao M, Zhou W, Chen T, Fan X, Chen K. novoBreak: local assembly for breakpoint detection in cancer genomes. Nature Methods. 2017;14(1):65–67. doi: 10.1038/nmeth.4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crolla JA, Youings SA, Ennis S, Jacobs PA. Supernumerary marker chromosomes in man: parental origin, mosaicism and maternal age revisited. European Journal of Human Genetics. 2005;13(2):154–160. doi: 10.1038/sj.ejhg.5201311. [DOI] [PubMed] [Google Scholar]
- Daber RD, Conlin LK, Leonard LD, Canevini MP, Vignoli A, Hosain S, Spinner NB. Ring chromosome 20. European Journal of Medical Genetics. 2012;55(5):381–387. doi: 10.1016/j.ejmg.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Dalpra L, Giardino D, Finelli P, Corti C, Valtorta C, Guerneri S, Larizza L. Cytogenetic and molecular evaluation of 241 small supernumerary marker chromosomes: cooperative study of 19 Italian laboratories. Genetics in Medicine. 2005;7(9):620–625. doi: 10.1097/01.gim.0000182876.57766.2d. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-IV. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- Gillberg C, Steffenburg S, Wahlström J, Gillberg IC, Sjöstedt A, Martinsson T, Eeg-Olofsson O. Case study: autism associated with marker chromosome. Journal of the American Academy of Child & Adolescent Psychiatry. 1991;30(3):489–494. doi: 10.1097/00004583-199105000-00022. [DOI] [PubMed] [Google Scholar]
- Gu S, Posey JE, Yuan B, Carvalho CM, Luk HM, Erikson K, Lupski JR. Mechanisms for the Generation of Two Quadruplications Associated with Split-Hand Malformation. Human Mutation. 2016;37(2):160–164. doi: 10.1002/humu.22929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5(1):e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Owen MJ. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Molecular Psychiatry. 2012;17(2):142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Tavakoli-Yaraki M, van Roosmalen MJ, van Binsbergen E, Renkens I, Duran K, Cuppen E. Constitutional chromothripsis rearrangements involve clustered double-stranded DNA breaks and nonhomologous repair mechanisms. Cell Reports. 2012;1(6):648–655. doi: 10.1016/j.celrep.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Kotzot D. Supernumerary marker chromosomes (SMC) and uniparental disomy (UPD): coincidence or consequence? Journal of Medical Genetics. 2002;39(10):775–778. doi: 10.1136/jmg.39.10.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz ML, Zhang CZ, Pellman D. Chromothripsis: A New Mechanism for Rapid Karyotype Evolution. Annual Review of Genetics. 2015;49:183–211. doi: 10.1146/annurev-genet-120213-092228. [DOI] [PubMed] [Google Scholar]
- Levy RJ, Xu B, Gogos JA, Karayiorgou M. Copy number variation and psychiatric disease risk. Methods in Molecular Biology. 2012;838:97–113. doi: 10.1007/978-1-61779-507-7_4. [DOI] [PubMed] [Google Scholar]
- Liehr T. Small supernumerary marker chromosomes (sSMC): a guide for human geneticists and clinicians. Springer Science & Business Media; 2011. [Google Scholar]
- Liehr T, Cirkovic S, Lalic T. Complex small supernumerary marker chromosomes–an update. Complex small supernumerary marker chromosomes–an update. 2013;6(1):46. doi: 10.1186/1755-8166-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liehr T, Mrasek K, Weise A, Dufke A, Rodríguez L, Guardia NM, Haas OA. Small supernumerary marker chromosomes–progress towards a genotype-phenotype correlation. Cytogenetic and Genome Research. 2006;112(1–2):23–34. doi: 10.1159/000087510. [DOI] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SC, Dhar SU, Kolodziejska KE, Dharmadhikari AV, Bi W. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;146(6):889–903. doi: 10.1016/j.cell.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Yuan B, Carvalho CM, Wuster A, Walter K, Zhang L, Lupski JR. An Organismal CNV Mutator Phenotype Restricted to Early Human Development. Cell. 2017;168(5):830–842. doi: 10.1016/j.cell.2017.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher CA, Wilson RK. Chromothripsis and human disease: piecing together the shattering process. Cell. 2012;148(1–2):29–32. doi: 10.1016/j.cell.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, McCarthy S, Michaelson JJ, Vacic V, Burdick KE, Yoon S, Sebat J. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron. 2011;72(6):951–963. doi: 10.1016/j.neuron.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Jacobo L, Ortiz-Lopez R, Rizo-Mendez A, Garcia-Molina V, Santuario-Facio SK, Rivas F, Rojas-Martinez A. Clinical and molecular delineation of duplication 9p24.3q21.11 in a patient with psychotic behavior. Gene. 2015;560(1):124–127. doi: 10.1016/j.gene.2015.02.010. [DOI] [PubMed] [Google Scholar]
- McEachern MJ, Haber JE. Break-induced replication and recombinational telomere elongation in yeast. Annual Review of Biochemistry. 2006;75:111–135. doi: 10.1146/annurev.biochem.74.082803.133234. [DOI] [PubMed] [Google Scholar]
- Oda M, Kure S, Sugawara T, Yamaguchi S, Kojima K, Shinka T, Omae T. Direct correlation between ischemic injury and extracellular glycine concentration in mice with genetically altered activities of the glycine cleavage multienzyme system. Stroke. 2007;38(7):2157–2164. doi: 10.1161/STROKEAHA.106.477026. [DOI] [PubMed] [Google Scholar]
- O’Donovan MC, Kirov G, Owen MJ. Phenotypic variations on the theme of CNVs. Nature Genetics. 2008;40(12):1392. doi: 10.1038/ng1208-1392. [DOI] [PubMed] [Google Scholar]
- Pocklington AJ, Rees E, Walters JT, Han J, Kavanagh DH, Chambert KD, O’Donovan MC. Novel findings from CNVs implicate inhibitory and excitatory signaling complexes in schizophrenia. Neuron. 2015;86(5):1203–1214. doi: 10.1016/j.neuron.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science. 2013;341(6141):1237758. doi: 10.1126/science.1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy KS, Aradhya S, Meck J, Tiller G, Abboy S, Bass H. A systematic analysis of small supernumerary marker chromosomes using array CGH exposes unexpected complexity. Genetics in Medicine. 2013;15(1):3. doi: 10.1038/gim.2012.78. [DOI] [PubMed] [Google Scholar]
- Schroer RJ, Phelan MC, Michaelis RC, Crawford EC, Skinner SA, Cuccaro M, Stevenson RE. Autism and maternally derived aberrations of chromosome 15q. American Journal of Medical Genetics. 1998;76(4):327–336. doi: 10.1002/(sici)1096-8628(19980401)76:4<327::aid-ajmg8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Spitzer RL, Williams JBW, Gibbon M. Structured Clinical Interview for DSM IV-Patient Edition (SCID-P) American Psychiatric Press; Washington, DC: 1995. [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Campbell PJ. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144(1):27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart LR, Hall AL, Kang SHL, Shaw CA, Beaudet AL. High frequency of known copy number abnormalities and maternal duplication 15q11-q13 in patients with combined schizophrenia and epilepsy. BMC Medical Genetics. 2011;12(1):154. doi: 10.1186/1471-2350-12-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafranski P, Gambin T, Dharmadhikari AV, Akdemir KC, Jhangiani SN, Schuette J, Stankiewicz P. Pathogenetics of alveolar capillary dysplasia with misalignment of pulmonary veins. Human Genetics. 2016;135(5):569–586. doi: 10.1007/s00439-016-1655-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi M, Kure S, Sakata Y, Kurihara Y, Ohya Y, Kajita M, Narisawa K. Human glycine decarboxylase gene (GLDC) and its highly conserved processed pseudogene (Ψ GLDC): their structure and expression, and the identification of a large deletion in a family with nonketotic hyperglycinemia. Human genetics. 2000;106(3):298–305. doi: 10.1007/s004390051041. [DOI] [PubMed] [Google Scholar]
- TCW J, Carvalho CM, Yuan B, Gu S, Altheimer AN, McCarthy S, Brennand KJ. Divergent Levels of Marker Chromosomes in an hiPSC-Based Model of Psychosis. Stem Cell Reports. 2017;8(3):519–528. doi: 10.1016/j.stemcr.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annual Review of Pharmacology and Toxicology. 2002;42(1):165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- Weckselblatt B, Rudd MK. Human Structural Variation: Mechanisms of Chromosome Rearrangements. Trends in Genetics. 2015;31(10):587–599. doi: 10.1016/j.tig.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: (A) Shows the genomic segments with altered copy number states as denoted by white boxes stacked on top of each segment as well as the genes located within each segment. (B) Table showing each genomic segment that makes up the marker chromosome, their associated copy number state, the genomic coordinates (Hg19) derived from aCGH probe location data, the approximate size of each segment as well as the specific genes involved in each segment.
Supplemental Figure 2: Highlighted are the 5 breakpoint junctions validated through standard Sanger-sequencing and their associated location on the marker chromosome.
Supplemental Table 1: Shows the SNP calls present in the duplicated (red) and triplicated (blue) regions across the family. The genotype of the marker is highlighted in red text across all identified calls.
Supplemental Table 2: Details the other fully duplicated or triplicated genes within the rearrangement and their possible role in neuropsychiatric phenotypes.