

Abstract
As genomic sequencing expands, so does our knowledge of the link between genetic variation and disease. Deeper catalogs of variant frequencies improve identification of benign variants, while sequencing affected individuals reveals disease-associated variation. Accumulation of human genetic data thus makes reanalysis a means to maximize benefits of clinical sequencing. We implemented pipelines to systematically reassess sequencing data from 494 individuals with developmental disability. Reanalysis yielded pathogenic or likely pathogenic (P/LP) variants that were not initially reported in 23 individuals, 6 described here, comprising a 16% increase in P/LP yield. We also downgraded three LP and six variants of uncertain significance (VUS) due to updated population frequency data. The likelihood of identifying a new P/LP variant increased over time, as ~22% of individuals who did not receive a P/LP variant at their original analysis subsequently did after three years. We show here that reanalysis and data sharing increase the diagnostic yield and accuracy of clinical sequencing.
Keywords: Reanalysis, clinical sequencing, developmental delay, intellectual disability, data sharing, VUS, CSER
Graphical abstract
INTRODUCTION
Whole genome sequencing (WGS) and whole exome sequencing (WES) are increasingly used clinically, particularly for rare disease diagnosis. WGS/WES uncovers pathogenic or likely pathogenic (P/LP) variants in 20–60% of sequenced patients1–4, leaving many patients without a relevant genetic finding. Although these individuals may have diseases that are non-genetic or result from complex genetic effects, incomplete knowledge of genetic variation likely prevents identification of many P/LP variants. Systematically reanalyzing data over time may prove useful, as accumulated knowledge—i.e., new publications, updated population frequencies, improved clinical variant databases, and data sharing among researchers—may facilitate new discoveries5–7.
We sought to systematically reanalyze WES/WGS data from probands with developmental delay and/or intellectual disability (DD/ID) enrolled in the Clinical Sequencing Exploratory Research (CSER) project at HudsonAlpha8. An initial reanalysis effort was described in an analysis of the first 371 affected probands8, but an expanded and improved reanalysis pipeline, including new findings, approaches, and implications, is presented here.
MATERIALS AND METHODS
Study overview
Enrollment, sequencing, variant calling and Sanger confirmation were performed as previously described8, although we have now included an additional 123 affected probands, increasing the cohort to 494 affected individuals (see Supplemental Table 1 and Supplemental Materials and Methods). For reanalysis, original joint-called VCFs were reannotated to include updated versions of ClinVar5, ExAC/gnomAD6, CADD9, and DDG2P10. Filtering and curation were performed in light of new data. Reanalysis is performed on a rolling schedule so that all cases are reviewed at least once every 12 months. Genes of uncertain disease significance that harbored candidate variants were submitted to GeneMatcher11. Our study began before publication of the American College of Medical Genetics and Genomics (ACMG)12 standards, but our original classification criteria were conceptually similar and are available on the ClinVar Submitters page (https://submit.ncbi.nlm.nih.gov/ft/byid/yR2NSzwW/HA_assertions_20161101.pdf). For reanalysis, however, ACMG criteria12 were used and evidence codes for reinterpreted variants are provided. All returned variants were submitted to ClinVar5. Sequence data for consenting participants is available through dbGAP13. Supplemental Materials and Methods provide additional details.
Uniparental disomy (UPD) analysis
UPD was called using UPDio14. CNV calls (Supplemental Materials and Methods), were masked and not considered by UPDio.
SLC1A4 Analysis
DNA and RNA isolation, cDNA synthesis, PCR, and qPCR were conducted with standard protocols (Supplemental Materials and Methods). Graphpad Prism version 7.0c was used for graphing and statistics.
RESULTS
New Findings
Based on the success of our initial reanalysis efforts8, we sought to improve and expand our strategy. We have subsequently identified P/LP variation in six additional probands (Table 1). Two of these “upgrades” resulted from recent publications and two from GeneMatcher11 collaborations (Table 1). One of these variants (FGF12, NM_004113.5:c.145G>A, (p.Arg114His)) is a recurrent de novo variant now seen in two unrelated probands in our cohort8,15. In each of these four cases, the variation was in a gene not previously associated with disease and was originally classified as a non-returnable VUS. After data supporting gene-disease associations became available, application of ACMG criteria led to classification of these variants as P/LP (Table 1) and return to families.
Table 1.
Upgraded variants.
| Individual ID |
Gene | Variant(s) | Reason for Update | Original Score† | Updated Score (with ACMG Criteria) |
|---|---|---|---|---|---|
| 00231-C | DHX30 | NM_138615.2:c.2353C>T, (p.Arg785Cys) | Publication20,21 | VUS | Pathogenic (PS2, PS3, PS4, PM2, PP3) |
| 00144-C | SLC1A4 | NM_003038.4:c.766G>A, (p.Glu256Lys); NC_000002.11:g.65244824-65245606del, (p.Ser345Arg, Ser346_Leu410del) | Publication16 | VUS; VUS | Pathogenic (PS3, PS4, PM2, PP1, PP4); Likely Pathogenic (PM2, PM3, PM4, PP4) |
| 00012-C | FGF12 | NM_021032.4:c.341G>A, (p.Arg114His)‡ | GeneMatcher15 | VUS | Pathogenic (PS2, PS4, PM2, PP3) |
| 00303-C | NTRK2 | NM_006180.4:c.1301A>G, (p.Tyr434Cys) | GeneMatcher22 | VUS | Pathogenic (PS2, PS4, PM2, PP3) |
| 00130-C | NA | Paternal isodisomy of chr15 | UPD | NA | Pathogenic§ |
| 00346-C | NA | Maternal heterodisomy of chr15 | UPD | NA | Pathogenic§ |
ACMG, American College of Medical Genetics and Genomics; VUS, Variant of Uncertain Significance; NA, not applicable; UPD, uniparental disomy.
Original scores for SNV/CNVs based on retroactive scoring using ACMG criteria for a gene of uncertain significance (GUS). None of the variants in this table were returned to probands at time of the first analysis, nor was ACMG scoring or criteria explicitly used at that time.
Note that this variant was previously reported with our initial study findings8, although the variant was recently found in one additional unrelated proband in our study, represented here.
ACMG scoring criteria do not apply to UPD.
In one case, we identified a single nucleotide variant (SNV) and a copy number variant (CNV) within SLC1A4, associated with an autosomal recessive neurodevelopmental disorder (MIM:616657)16. Initially (June 2015), no returnable variation was found in this proband. However, upon reanalysis, a paternally-inherited pathogenic missense variant16 was found using updated ClinVar information. A targeted search for a second variant in SLC1A4 revealed a maternally-inherited 782-bp deletion (Table 1). We confirmed the presence of this deletion in genomic DNA from the proband and mother (Supplemental Figures 1, 2). While this variant does not change SLC1A4 transcript levels, it does result in skipping of exon 6 (Supplemental Figures 1, 3) and is predicted to lead to an in-frame deletion of 65 amino acids encompassing two transmembrane domains. This CNV was only identified in the proband by one of the four algorithms in our pipeline and was completely missed in the mother; curation of unfiltered CNVs coupled to manual inspection of reads in both samples was required. Had we not found the previously reported missense variant, or lacked phenotype data suggesting the relevance of SLC1A4, this CNV would have been missed.
In addition to manual reanalysis in light of new data, we also implemented methods to detect uniparental disomy (UPD). While UPD, especially heterodisomy, often goes unnoticed, it can cause DD/ID when imprinted regions are affected17–19. We found two cases of disease-associated UPD, both affecting chromosome 15 (Table 1). Clinical methylation analyses confirmed one result. Clinical methylation analyses were recommended for the second but not performed.
Downgrades
In addition to searching for new P/LP variation, we also reanalyzed all previously returned variants, leading to downgrades of nine variants in seven individuals (Table 2). Six of these variants were originally classified as VUSs and three were considered LP. All downgrades resulted from addition of data in the ExAC/gnomAD databases6. Variants present at non-trivial frequencies in these databases are unlikely to be dominant, highly penetrant DD/ID variants6. Similarly, variation that is homozygous or hemizygous at non-trivial frequencies are unlikely to cause recessive or X-linked disease. Though reanalysis did uncover new P/LP variation in three of these seven probands, upgrade and downgrade decisions were made independently of one another.
Table 2.
Downgraded variants.
| Allele Frequency at time of Original Analysis |
||||||||
|---|---|---|---|---|---|---|---|---|
|
|
||||||||
| Individual ID |
Gene | Suspected Mode of Inheritance |
Variant | Original Score† |
Updated Score (with ACMG criteria) |
1KG | EVS | Allele Frequency at time of Reanalysis (gnomAD) |
| 00026-C | ULK4 | Rec | NM_017886.3:c.2584C>T, (p.Arg862Ter) | VUS | Likely Benign (BS2) | 0.05% | 38/11762 (0 hom), 0.3% | 777/276908 (4 hom), 0.002806% |
| 00026-C | ULK4 | Rec | NM_017886.3:c.2887G>A, (p.Val963Met) | VUS | Likely Benign (BS2) | 0.32% | 45/12217 (0 hom), 0.4% | 942/276788 (4 hom), 0.003403% |
| 00074-C | NRXN1 | Rec | NM_001135659.2:c.3485-110131T>G | VUS | Likely Benign (BS2) | 0.37% | NR | 85/30792 (2 hom), 0.002760% |
| 00074-C | NRXN1 | Rec | NM_001135659.2:c.11405C>T, (p.Pro469Ser) | VUS | Likely Benign (BS2) | 0.09% | 34/12262 (0 hom), 0.3% | 1048/273312 (6 hom), 0.003834% |
| 00130-C | GRIA3 | XLR | NM_007325.4:c.580G>A, (p.Gly194Arg) | Likely Pathogenic | Likely Benign (BS2) | 0.06% | 1/10562 (0 hemi), 0.0095% | 12/200235 (3 hemi), 0.00005993% |
| 00053-C | GRIA3 | XLR | NM_007325.4:c.466T>C, (p.Tyr156His) | Likely Pathogenic | Likely Benign (BS2) | NR | NR | 9/199897 (3 hemi), 0.00004502% |
| 00059-C | PTPN11 | Dom | NM_002834.4:c.1174G>A, (p.Ala392Thr) | Likely Pathogenic | Likely Benign (PS2, BS2) | NR | NR | 8/246234 (0 hom), 0.00003249% |
| 00111-C | CSMD1 | Dom | NM_033225.5:c.1642A>G, (p.Thr548Ala) | VUS | Likely Benign (PS2, BS2) | NR | NR | 7/240916 (0 hom), 0.00002906% |
| 00007-C | SCN1A | Dom | NM_001165963.1:c.4547 C>T, (p.Ser1516Leu) | VUS | Likely Benign (BS2) | NR | NR | 5/245496 (0 hom), 0.00002037% |
1KG, 1000 Genomes Project; EVS, Exome Variant Server; Rec, Recessive; XLR, X-linked Recessive; Dom, Dominant; VUS, Variant of Uncertain Significance; hom, homozygotes; hemi, hemizygotes; NR, Not Reported
ACMG criteria and scoring were not explicitly used at the time of first analysis.
Likelihood of Variant Upgrade over Time
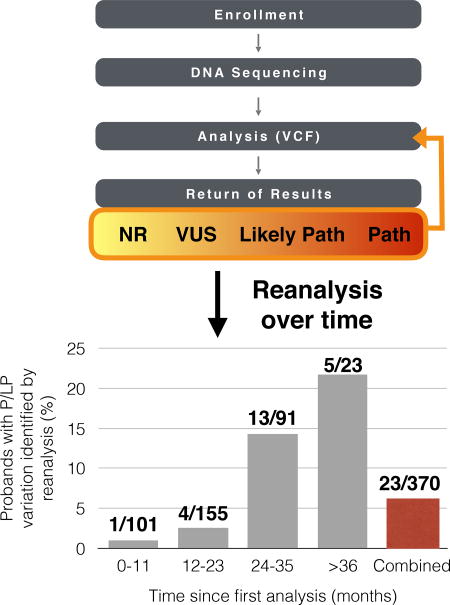
We conducted WES on the first 127 probands of this study and switched to WGS for all subsequent probands (Supplemental Table 1). We measured P/LP rates separately for WES and WGS, both before and after reanalysis (Supplemental Table 2). Although the initial P/LP rate for WGS (26.2%) was greater than that of WES (22.0%), reanalysis yielded an 11.0% increase in the P/LP rate for WES and only 1.6% increase for WGS. To further refine this comparison, since all WES cases were trios, we also restricted calculations for WGS to trios, and found the P/LP rate in WGS-trios increased by 2.2% (28.1% to 30.3%). The larger relative gain in WES yield is likely due to time since initial analysis. WES was initially performed from November 2013 to May 2015, while WGS was performed from June 2015 onward. Indeed, while probands had only a 1% likelihood of upgrade in the first year following analysis, this rate increased over time. ~22% of cases over three years old with a VUS or no returnables were eventually found to have P/LP variation (Table 3, Supplemental Materials and Methods).
Table 3.
Likelihood of variant upgrade over time.
| Time since first analysis (months) |
Number of DD/ID-affected individuals with P/LP variation identified by reanalysis (%) |
|---|---|
| ≥36 | 5/23 (21.7%) |
| 24–35 | 12/91 (13.2%) |
| 12–23 | 4/155 (2.6%) |
| 0–11 | 1/101 (1.0%) |
| Overall | 22/370 (5.9%) |
DISCUSSION
Accumulation of genetic knowledge suggests that reanalysis of sequencing data may lead to discovery of novel medically relevant variants and to refinement of initial variant interpretations. Our reanalysis efforts thus far have led to identification of 22 P/LP variants in 23 probands in a cohort of 494 total patients. These affect ~6% of probands who originally received either a VUS or no returnables, and represents a 16% increase in total P/LP yield. Other groups have also reported success with reanalysis, with upgrade rates from 10–36%7,20.
The ACMG has published guidelines for the interpretation of sequence variants12, and it is important to note that the changed interpretations in our study did not result from altered application of criteria, but rather new evidence supporting (or refuting) pathogenicity.
Three of the downgrades resulted in a change from LP to likely benign. The ACMG suggests that an LP designation represents pathogenicity with 90% confidence12. In our case, these three downgrades represent 9% of all variants initially determined to be LP, suggesting an empirical error rate similar to the conceptual target established by the ACMG. As population variant frequency databases expand, reanalysis of previously reported variants will continue to be necessary.
Based on our experience with reanalysis, we suggest the following framework:
If reanalysis has never been conducted on WES/WGS data, it should be performed, even in the absence of annotation or pipeline updates. New P/LP variation may be discovered simply by reviewing new literature during manual curation.
WES data lacking P/LP variation should be reanalyzed before performing WGS, especially for data over two years old.
If reanalysis of all data cannot be conducted regularly, an automated process to flag variants in genes recently linked to disease can be more easily implemented.
Improvements to bioinformatics pipelines are beneficial, especially updates from population and clinical genetic databases. Additionally, algorithms for detection of non-SNV/indel variants, such as CNVs, UPD, etc., are continually improving and worth updating. While any individual variant type may be a small fraction of P/LP variation, such additions can make a large cumulative difference.
Data sharing through GeneMatcher, ClinVar, and related resources is a key component of reanalysis. These platforms help to establish gene-disease relationships among research groups with small cohorts, in many cases well before formal publication (which can take months or years from the time that a robust disease association has been established). Most of our GeneMatcher submissions (45 of 52 probands) represented one gene of interest within one individual, with de novo variants present in ~50% of these. We expect the benefits of data sharing to increase, as 12% (43/350) of the individuals in our cohort that lack a P/LP variant harbor a variant within a gene that has been submitted to GeneMatcher; 79% (34/43) of these genes have at least preliminary matches.
Time since initial analysis should inform the reanalysis strategy. If WGS/WES data was analyzed within the past year, reanalysis yield will be small – in our case, only 1%. However, datasets first analyzed over two years ago should be prioritized for reanalysis, consistent with observations from others7,20. We also note that timing of analysis is a major factor to consider when evaluating overall yield rates, particularly when focused on comparisons of technologies that have changed over time. For example, while we believe a number of factors support the benefits of WGS over WES for rare disease diagnosis (e.g., improved CNV detection, more uniform depth of coverage, etc.), the tendency for WGS-based analyses to be more recent than WES-based analyses must be accounted for when comparing P/LP variant yields.
In summary, our data strongly support the benefits of systematic reanalysis of WES/WGS data. Such efforts lead to substantial increases in P/LP variant discoveries while simultaneously reducing false positive P/LP rates. Thus, reanalysis can substantially improve the accuracy and benefits of clinical sequencing.
Supplementary Material
Acknowledgments
We thank all the participating families, the staff at North Alabama Children’s Specialists, and the informatics teams, Genomic Services Lab, and Clinical Services Lab at HudsonAlpha. This work was supported by an NHGRI grant (UM1HG007301).
Footnotes
The authors declare no conflicts of interest.
References
- 1.Ankala A, da Silva C, Gualandi F, et al. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol. 2015;77(2):206–214. doi: 10.1002/ana.24303. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor JC, Martin HC, Lise S, et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet. 2015;47(7):717–726. doi: 10.1038/ng.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chong JX, Buckingham KJ, Jhangiani SN, et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am J Hum Genet. 2015;97(2):199–215. doi: 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2017;19(2):209–214. doi: 10.1038/gim.2016.88. [DOI] [PubMed] [Google Scholar]
- 8.Bowling KM, Thompson ML, Amaral MD, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017;9(1):43. doi: 10.1186/s13073-017-0433-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wellcome Trust Sanger Institute. [Accessed November 18, 2014];The Development Disorder Genotype - Phenotype Database (DDG2P) 2015 Dec 3; https://decipher.sanger.ac.uk/ddd - ddgenes.
- 11.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tryka KA, Hao L, Sturcke A, et al. NCBI's Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2014;42(Database issue):D975–979. doi: 10.1093/nar/gkt1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King DA, Fitzgerald TW, Miller R, et al. A novel method for detecting uniparental disomy from trio genotypes identifies a significant excess in children with developmental disorders. Genome Res. 2014;24(4):673–687. doi: 10.1101/gr.160465.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guella I, Huh L, McKenzie MB, et al. De novo FGF12 mutation in 2 patients with neonatal-onset epilepsy. Neurol Genet. 2016;2(6):e120. doi: 10.1212/NXG.0000000000000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srour M, Hamdan FF, Gan-Or Z, et al. A homozygous mutation in SLC1A4 in siblings with severe intellectual disability and microcephaly. Clin Genet. 2015;88(1):e1–4. doi: 10.1111/cge.12605. [DOI] [PubMed] [Google Scholar]
- 17.Driscoll DJ, Miller JL, Schwartz S, Cassidy SB. Prader-Willi Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 18.Dagli AI, Mueller J, Williams CA. Angelman Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 19.Shuman C, Beckwith JB, Weksberg R. Beckwith-Wiedemann Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 20.Eldomery MK, Coban-Akdemir Z, Harel T, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9(1):26. doi: 10.1186/s13073-017-0412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542(7642):433–438. doi: 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamdan FF, Myers CT, Cosette P, et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am J Hum Genet. 2017;101(5):664–685. doi: 10.1016/j.ajhg.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.