

Abstract
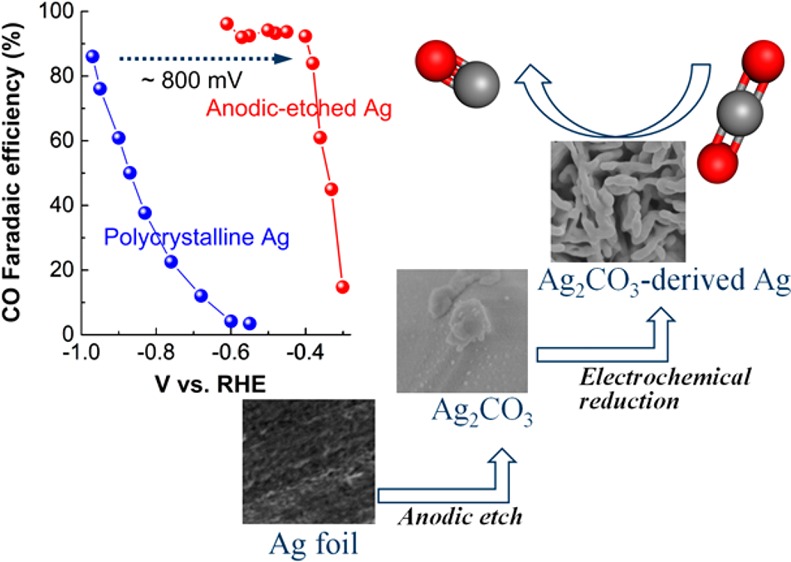
In this work, the highly selective and stable electrocatalytic reduction of CO2 to CO on nanostructured Ag electrocatalysts is presented. The Ag electrocatalysts are synthesized by the electroreduction of Ag2CO3 formed by in situ anodic-etching of Ag foil in a KHCO3 electrolyte. After 3 min of this etching treatment, the Ag2CO3-derived nanostructured Ag electrocatalysts are capable of producing CO with up to 92% Faradaic efficiency at an overpotential as low as 290 mV, which surpasses all of the reported Ag catalysts at identical conditions to date. In addition, the anodic-etched Ag retained ∼90% catalytic selectivity in the electroreduction of CO2 to CO for more than 100 h. The Ag2CO3-derived Ag is able to facilitate the activation of CO2 via reduction of the activation energy barrier of the initial electron transfer and provide an increased number of active sites, resulting in the dramatically improved catalytic activity for the reduction of CO2 to CO.
The electrocatalytic conversion of CO2 into carbon-based fuels and valuable chemicals powered by renewable electricity is an attractive solution to both the utilization of captured CO2 and the storage of renewable energy.1−6 An essential step for achieving this goal is to find a highly efficient and selective electrocatalyst with long-term stability.7,8 Many transition metal catalysts have been evaluated for the selective reduction of CO2 in CO2-saturated aqueous solutions.2,9 Currently, the electrochemical conversion of CO2 into CO provides one of the most promising routes to form a cost-competitive product because syngas (CO and H2) can be employed in Fischer–Tropsch synthesis to produce value-added chemicals and synthetic fuels using already existing industrial technologies.1,4,10 Therefore, significant attention has been focused on finding electrocatalysts that can selectively produce CO from CO2.11−18
While Au is the most active surface for reducing CO2 selectively to CO among the identified metal catalysts, its potential for industrial applications is currently limited by its low abundance and high cost. In this context, Ag has great potential for large-scale applications due to its significantly lower cost than Au and high catalytic selectivity for the reduction of CO2 to CO.9−11,19−24 However, high overpotentials (η) required for driving selective CO2 reduction and rapid catalytic deactivation in favor of H2 evolution on Ag catalysts significantly restrict its practical utilization.20,25
To overcome the limitations of Ag electrocatalysts, many attempts have focused on the development of nanostructured surfaces, which offer mass-transport advantages and contain more low-coordinated sites (edge sites and corner sites) that are more active for CO2 reduction in comparison with a planar metallic surface.7,10,14,22,26,27 It has been demonstrated that nanoporous Ag catalysts prepared by dealloying,10 Ag nanocoral catalysts synthesized via an oxidation–reduction process using chloride anions28 and oxide-derived nanostructured Ag22 are capable of electrochemically reducing CO2 to CO with dramatically enhanced catalytic selectivity at reduced overpotentials, which are ascribed to fast initial electron transfer for CO2 activation on these nanostructured catalysts. In addition, the catalytic stability of the electroreduction of CO2 to CO has been improved on nanostructured Ag catalysts,10,22,28 owing to the enhanced tolerance to heavy metal impurities in the electrolyte.27 While these previous attempts have significantly improved the performance of Ag electrocatalysts, it is still critical to develop a very simple, fast, scalable and low-cost method for preparing and maintaining higher-performance Ag electrocatalysts for practical applications of CO2 electroreduction.
Herein, we demonstrate a simple technique to prepare highly active, stable, and selective Ag electrocatalysts in CO2-saturated KHCO3 electrolyte that is used for CO2 reduction. By first anodic-etching Ag to form Ag2CO3 and then further reducing the Ag2CO3 to metallic Ag with a highly porous scaffold structure, a significantly reduced overpotential for high CO catalytic selectivity was achieved with remarkable catalytic stability, which outcompetes the reported Ag catalysts to date at identical conditions. Thus, the robust performance of the nanostructured catalysts formed via this very easy and low-cost synthesis method may offer a platform for practical applications of CO2 electroreduction.
For the electrochemical synthesis of Ag electrocatalysts used in this work, a polycrystalline Ag foil electrode was immersed in a CO2-saturated 0.1 M KHCO3 electrolyte in a two-compartment cell using a three-electrode configuration (a Pt counter electrode and a Ag/AgCl reference electrode). The two-compartment cell was separated by a Nafion-115 proton exchange membrane to minimize impurity deposition during electrolysis. An anodic potential of 2.6 V vs the reversible hydrogen electrode (RHE) was applied on the Ag foil electrodes for 3 min to synthesize the Ag2CO3 layer, with an estimated thickness of ∼4.9 μm (Table S1). The Ag2CO3 electrodes were then directly utilized for electrocatalytic CO2 reduction in CO2-saturated 0.1 M KHCO3 electrolyte and were electrochemically reduced to metallic Ag in the initial period (<2 min) of CO2 reduction electrolysis (Figure S5d).
In the initial anodic-etching, the Ag electrodes formed short microporous polyhedral rod-like morphologies with smooth surfaces, as presented in Figure 1a, which is consistent with the reported morphologies of Ag2CO3.29Figure 1b shows scanning electron microscope (SEM) images of the same electrodes after the electrolysis (∼0.5 h) of CO2 reduction, revealing that a nanoporous structure was formed via the electroreduction of Ag2CO3. In addition, transmission electron microscopy (TEM) confirmed that the size of the nanostructured materials after the CO2 reduction (Figure 1c), and the selected-area electron diffraction (SAED) pattern in the inset of Figure 1c exhibits the typical nature of crystals. To identify the phase of the prepared materials, X-ray diffraction (XRD) measurements were conducted. The XRD diffractograms in Figure 1d indicate that Ag2CO3 was formed by anodic-etching of Ag foil in KHCO3 solution. After CO2 reduction electrolysis, only Ag diffraction peaks were observed (Figure 1d) without any remaining Ag2CO3, indicating a full transformation from Ag2CO3 to metallic Ag.
Figure 1.

SEM images of anodic-etched Ag (AE-Ag) (4.9 μm) before (a) and after (b) CO2 reduction electrolysis. (c) TEM image of AE-Ag after CO2 reduction electrolysis (the inset is the SAED pattern). (d) XRD patterns and (e) XPS spectra of untreated polycrystalline Ag (blue line) and AE-Ag before (dark yellow line) and after (red line) CO2 reduction electrolysis, respectively.
To verify the surface composition of our samples, X-ray photoelectron spectroscopy (XPS) measurements were performed. As shown in Figure 1e, the Ag 3d5/2 peak at 368.2 eV was observed for polycrystalline Ag. For the anodic-etched Ag (AE-Ag) before CO2 reduction electrolysis, the Ag 3d5/2 peak shifted by about 0.3 eV to the binding energy of 367.9 eV compared to the metal Ag, which is consistent with the value of Ag+ in the synthesized Ag2CO3 according to previous work.29−31 In addition, the binding energy of 288.7 eV in C1s spectra (Figure S3) represents the carbon associated with the carbonate ((CO3)2–),32 which further confirms the formation of Ag2CO3. After electrolysis, the Ag 3d5/2 peak shifted back to 368.2 eV, corresponding to metallic Ag0.29−31 Furthermore, a surface valence band XPS spectrum of AE-Ag after electrolysis is in line with that of metallic Ag foil (Figure S4). All of the above results indicate that the reduction of Ag2CO3 to metallic Ag was complete, implying that only metallic Ag was present on AE-Ag after CO2 reduction electrolysis.
Figure 2a presents a comparison of the electrocatalytic activity of CO2 reduction for untreated polycrystalline Ag (in blue) and 4.9 μm AE-Ag (in red) at various applied potentials (iR-corrected potentials). Both the AE-Ag and untreated Ag experienced a gradually enhanced Faradaic efficiency (FE) for CO production at more negative potentials (Figure 2a), simultaneously accompanying with a decrease in the related FE for H2 formation (Figure S6). Notably, the overpotential required for achieving >90% FE for CO production was shifted toward the positive potential by ∼800 mV on AE-Ag compared to that of untreated Ag. More importantly, a high FE of more than 92% for CO formation was achieved on AE-Ag at a potential of −0.4 V vs RHE, which corresponds to an overpotential (ηco) as low as 0.29 V relative to the CO2/CO equilibrium potential of −0.11 V vs RHE, representing the highest catalytic selectivity for CO2 reduction to CO among the reported Ag catalysts at the same ηco (Table S2). In contrast, no CO production was detected on untreated polycrystalline Ag at identical conditions (ηco = 0.29 V). A plot of the partial current density for CO production (jco) as a function of potential in Figure 2b suggests that the onset potential for the reduction of CO2 to CO on AE-Ag was −0.3 V vs RHE (ηco = 0.19 V), which is a positive shift of ∼250 mV in comparison with that (−0.55 V vs RHE) of untreated polycrystalline Ag. These results show that Ag2CO3-derived nanostructured Ag is a highly selective electrocatalyst for the electrocatalytic reduction of CO2 to CO while inhibiting H2 evolution at significantly reduced overpotentials.
Figure 2.

Comparison of the electrocatalytic activity of polycrystalline Ag and AE-Ag (4.9 μm). (a) FE for CO at various potentials in CO2-saturated 0.1 M KHCO3 (pH 6.8). (b) Current density for CO formation at various potentials. (c) Catalytic stability performance for AE-Ag. The inset shows the catalytic stability of untreated polycrystalline Ag. All of the potentials were iR-corrected.
To test the electrocatalytic stability of AE-Ag catalysts, a long-term CO2 reduction measurement was performed on AE-Ag at a fixed potential of −0.55 V vs RHE (KHCO3 electrolyte without any purification was used). As shown in Figure 2c, AE-Ag exhibited an initially high geometric current density (jtot) at the early stage of electrolysis owing to the electroreduction of Ag2CO3 to Ag and subsequently a stable jtot of ∼1 mA/cm2 with a FE of ∼90% for CO production over ∼37 h. After ∼37 h, slight catalytic deactivation for CO formation was found, which may result from the deposition of impurities on the surface of the catalyst during the electrochemical reduction of CO2.9,27 To overcome this slight deactivation, the same Ag catalyst electrodes were then held at an anodic potential of 2.6 V vs RHE for 3 min in the same CO2-saturated KHCO3 electrolyte, and after returning back to −0.55 V vs RHE, a FE as high as ∼90% for CO formation was recovered and maintained for more than 60 h (no replacement of electrolyte during >100 h electrolysis). This remarkable stability significantly surpasses the currently reported durability for CO2 reduction on single-element catalysts (Table S3) under similar conditions. The in situ reactivation of the catalysts may be attributed to the removal of impurities on the surface of the catalysts by anodic-etching the contaminated surface. In contrast, the polycrystalline Ag electrodes had a very low jtot (∼0.07 mA/cm2) and a very low FE for CO, which decreased from 3.4 to 0% over the course of 2.5 h at −0.55 V vs RHE, which indicates fast catalytic deactivation. Thus, the Ag resulting from AE-Ag exhibited high catalytic selectivity and activity with long-term stability for the electrocatalytic reduction of CO2 to CO.
The electrochemical active surface area (EASA) of nanoporous Ag catalysts reduced from AE-Ag and untreated polycrystalline Ag was measured by forming a monolayer oxide on Ag surface in 0.1 M KOH.20 The charge used for oxidizing the monolayer of the Ag surface was calculated in Figure S7, which shows that the EASA of nanoporous Ag catalysts reduced from AE-Ag (3 min) is more than 10-fold larger than that of untreated Ag, resulting in the discrepancy of jtot between AE-Ag (∼1 mA/cm2) and untreated Ag (∼0.07 mA/cm2), as shown in Figure 2c. Thus, the increased number of active sites (increased EASA) reflects the enhanced catalytic reaction rate. In addition, the normalized jco of AE-Ag by EASA (∼0.08 mA/cm2) is ∼40 times higher in comparison with that (∼0.002 mA/cm2) of untreated Ag, indicating significantly improved intrinsic CO2 reduction activity on nanoporous Ag catalysts reduced from Ag2CO3.
The thickness effect of AE-Ag on the catalytic performance was also evaluated. In this work, the average thickness of AE-Ag was tuned by systematically varying the anodic-etching time (Table S1). As shown in Figure 3a, the thickness of AE-Ag is linearly correlated with the anodic-etching time, and an gradually enhanced FE for CO formation was observed along with decreased FE for H2 evolution at −0.55 V vs RHE with increasing thickness (≤3.5 μm) of AE-Ag. While a high FE of >90% for CO formation was achieved on both 3.5 and 4.9 μm AE-Ag (Figure 3a), the distinct thickness leads to a discrepancy of jtot between the two catalysts (Figure 3b). The EASA was enhanced with increasing thickness of AE-Ag (Figure 3b), which led to the correspondingly increased jtot and jco (Figure S8). These results indicate that the thicker nanoporous Ag is able to provide more active sites for the reduction of CO2 to CO.
Figure 3.
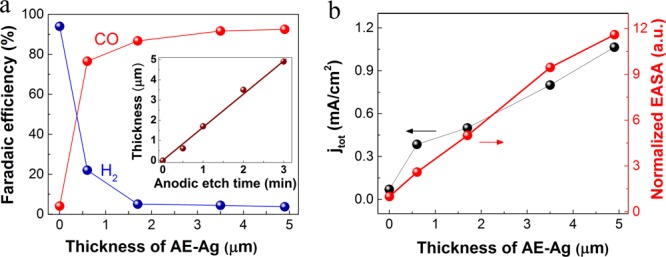
Electrocatalytic activity as a function of the thickness of AE-Ag at −0.55 V vs RHE. (a) FE for CO and H2 on AE-Ag with different thickness in CO2-saturated 0.1 M KHCO3 (pH 6.8). The inset shows the thickness for different anodic-etching times. (b) Geometric current density and normalized EASA.
It has been reported that the surface facets of Ag could significantly influence the catalytic activity of CO2 reduction.20,33 To reveal the variation of Ag surface facets before and after anodic treatment, the adsorption/desorption of OH– was performed on AE-Ag and untreated Ag in argon-purged 0.1 M KOH in the potential range from ∼−0.3 to ∼1 V vs RHE (double layer region) at room temperature.34−36Figure 4a exhibits the difference in peak potentials for the OH– adsorption/desorption processes between AE-Ag (red) and polycrystalline Ag (blue), which correlate with the distinct surface facets of the two catalysts.34−36 In addition, cyclic voltammetry of oxide-derived Ag reported in our previous work22 was also conducted in argon-purged 0.1 M KOH, which shows different peak potentials compared to AE-Ag (Figure S11), reflecting different dominant facets of the Ag surface. While the specific facets of the Ag surface could not be identified, the obvious discrepancy in Ag surface facets may contribute to the difference of the catalytic performance in the reduction of CO2. Therefore, in addition to the increased EASA in the anodic-etched nanostructured Ag compared to polycrystalline Ag, we also provide evidence of a different surface electronic structure that could also influence the catalytic activity and selectivity.
Figure 4.
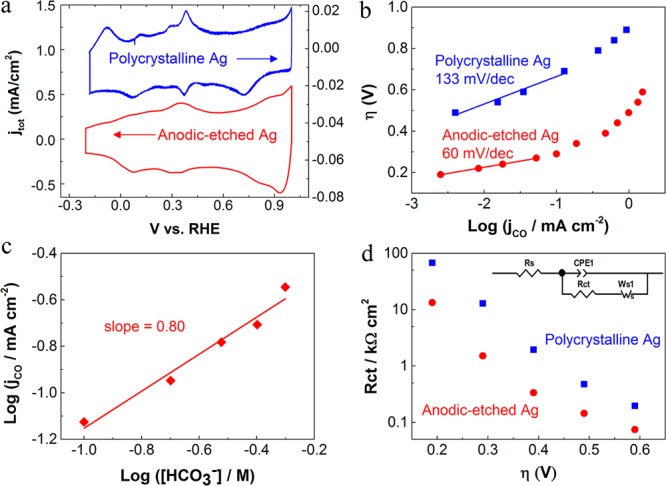
(a) Cyclic voltammetry curves of untreated polycrystalline Ag (blue line) and AE-Ag (4.9 μm) (red line) in argon-purged 0.1 M KOH at room temperature with a sweep rate of 50 mV/s. (b) Tafel plots of the CO partial current density for polycrystalline Ag and AE-Ag (4.9 μm). (c) Bicarbonate concentration at constant potentials of AE-Ag. (d) Charge transfer resistance at various overpotentials. The inset shows the equivalent circuit for the metal/solution interface (Rct, Rs, CPE and Ws are charge transfer resistance, solution resistance, constant phase element, and Warburg-short circuit terminus, respectively).
In order to gain insight into the electrokinetic mechanism of CO2 reduction on AE-Ag and untreated polycrystalline Ag, Tafel analysis was performed. It has been demonstrated that a two-electron transfer is involved for CO2 reduction to CO, and each electron transfer is followed with one proton donation step (or proton-coupled electron transfer steps based on computational studies37−40).7,9,10,22,27 Of particular note, the initial electron transfer for CO2 activation (stabilization of CO2•– or COOH•) is the rate-determining step (RDS) in the whole process due to the much higher activation energy barrier for the first electron transfer compared to the following steps.10,20 In our study, a Tafel plot of untreated Ag (overpotential versus log of the partial current density for CO production) in Figure 4b shows a Tafel slope of 133 mV/dec, which implies that the initial electron transfer for CO2 activation is the RDS for the overall process (Scheme 1).10,16 In contrast, a low Tafel slope of 60 mV/dec was obtained on AE-induced nanostructured Ag catalysts (4.9 μm) at relatively low overpotentials, indicating fast initial electron transfer to a CO2 molecule for CO2 activation (Scheme 1).10,25 In addition, this low Tafel slope is consistent with a fast pre-equilibrium of the initial electron transfer prior to a RDS according to previous work.25 Furthermore, a dramatically increased Tafel slope for nanostructured Ag was observed at relatively high overpotentials, implying that the electrocatalytic CO2 reduction likely reaches a mass transport limitation.
Scheme 1. Proposed Reaction Paths for CO2 Reduction to CO on Untreated Ag and Ag2CO3-Derived Nanostructured Ag.

The grey, red, and white balls represent C, O, and H atoms, respectively. Larger arrows indicate the relatively fast reaction steps.
It has been demonstrated that the initial proton donation is derived from HCO3–.20,25 Thus, to further uncover the reaction mechanism (first proton donation step) on nanostructured Ag, the effect of HCO3– concentration on the CO2 reduction activity was investigated. A plot of log(jco) versus log([HCO3–]) in Figure 4c exhibits a slope of ∼0.8, which corresponds to first-order dependence of the HCO3– concentration on the reaction rate, indicating that proton donation from HCO3– is a RDS for nanostructured Ag in the reduction of CO2 to CO.10,25 Thus, the RDS is switched from the first electron transfer for untreated Ag to the initial proton donation for nanostructured Ag (Scheme 1).
To better understand the charge transfer process at the electrode/electrolyte interface, electrochemical impedance spectroscopy (EIS) was performed at various potentials. The comparison of charge transfer resistance (Rct) between untreated Ag and AE-Ag as a function of overpotential was extracted from EIS (Figure S9) based on the equivalent circuit (Figure 4d). As presented in Figure 4d, AE-Ag exhibited a much lower Rct than that of polycrystalline Ag at identical conditions, suggesting a significantly accelerated charge transfer process on AE-Ag,14 which may reflect the reduced activation energy barrier of electron transfer on nanostructured Ag. This result is consistent with fast initial electron transfer on nanostructured Ag according to Tafel analysis. In addition, a clear mass transport limitation for nanoporous Ag was observed at relatively high overpotentials in Nyquist plots (Figure S10), which is also in line with Tafel analysis (the dramatic increase in the Tafel slope at relatively high overpotentials). These results indicate that, while a mass transport limitation may be reached on nanoporous Ag at high overpotentials, the dramatically improved initial electron transfer for CO2 activation enhances the intrinsic CO2 reduction activity, resulting in high catalytic selectivity and activity for the electrocatalytic reduction of CO2 to CO. EIS has seldom been used in CO2 reduction experiments; thereby, the consistency between the Tafel analysis and EIS shows the potential for this technique to give meaningful information relating to mechanistic charge transfer processes for electrochemical CO2 reduction.
In summary, a simple and fast anodic-etching procedure was used to fabricate highly active, selective, and stable Ag electrocatalysts for the reduction of CO2 to CO. A high FE of >92% for CO was achieved on AE-Ag at a potential of −0.4 V vs RHE (overpotential of 290 mV). Notably, the AE-Ag was capable of maintaining a high catalytic selectivity of ∼90% for CO production for >100 h, which remarkably outcompetes the currently reported durability of single-metal catalysts. The improved CO2 reduction performance is attributed to the increased number of active sites for CO2 reduction and the improved intrinsic CO2 reduction activity by fast initial electron transfer. In this study, after prolonged CO2 reduction, the procedure of anodic-etching can be performed subsequently on the same Ag electrocatalysts in the same KHCO3 electrolyte that is used for CO2 reduction to recover the robust catalytic performance. Thus, the Ag electrocatalysts, prepared by this fast, simple, and cost-effective approach, is capable of reducing CO2 to CO with high catalytic selectivity and excellent stability, offering a very promising platform for industrial applications.
Acknowledgments
This work is supported by an NWO VIDI grant awarded to W.A.S. The authors would like to thank Bartek J. Trześniewski for performing the XPS measurements. We also would like to thank Zerui Zhang for assistance in part of the proton transfer study experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsenergylett.8b00472.
Experimental details of Ag2CO3 fabrication, SEM, TEM, XRD, XPS, and CO2 reduction measurement, thickness calculation of anodic-etched Ag, summarized tables of reported electrocatalysts, EASA measurement, calculation of jCO and normalized jCO, EIS, OH– adsorption/desorption, iR correction, and error bars (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Whipple D. T.; Kenis P. J. A. Prospects of CO2 utilization via direct heterogeneous electrochemical reduction. J. Phys. Chem. Lett. 2010, 1, 3451–3458. 10.1021/jz1012627. [DOI] [Google Scholar]
- Kuhl K. P.; Hatsukade T.; Cave E. R.; Abram D. N.; Kibsgaard J.; Jaramillo T. F. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. 10.1021/ja505791r. [DOI] [PubMed] [Google Scholar]
- Qiao J.; Liu Y.; Hong F.; Zhang J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. 10.1039/C3CS60323G. [DOI] [PubMed] [Google Scholar]
- Ross M. B.; Dinh C. T.; Li Y.; Kim D.; De Luna P.; Sargent E. H.; Yang P. Tunable Cu enrichment enables designer syngas electrosynthesis from CO2. J. Am. Chem. Soc. 2017, 139, 9359–9363. 10.1021/jacs.7b04892. [DOI] [PubMed] [Google Scholar]
- Li C. W.; Ciston J.; Kanan M. W. Electroreduction of carbon monoxide to liquid fuel on oxide-derived nanocrystalline copper. Nature 2014, 508, 504–507. 10.1038/nature13249. [DOI] [PubMed] [Google Scholar]
- Ma M.; Djanashvili K.; Smith W. A. Controllable hydrocarbon formation from the electrochemical reduction of CO2 over Cu nanowire arrays. Angew. Chem., Int. Ed. 2016, 55, 6680–6684. 10.1002/anie.201601282. [DOI] [PubMed] [Google Scholar]
- Li C. W.; Kanan M. W. CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 2012, 134, 7231–7234. 10.1021/ja3010978. [DOI] [PubMed] [Google Scholar]
- Li F.; Chen L.; Knowles G. P.; MacFarlane D. R.; Zhang J. Hierarchical mesoporous SnO2 nanosheets on carbon cloth: a robust and flexible electrocatalyst for CO2 reduction with high efficiency and selectivity. Angew. Chem., Int. Ed. 2017, 56, 505–509. 10.1002/anie.201608279. [DOI] [PubMed] [Google Scholar]
- Hori Y.Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry; Vayenas C. G., White R. E., Gamboa-Aldeco M. E., Eds.; Springer, 2008; pp 89–189. [Google Scholar]
- Lu Q.; Rosen J.; Zhou Y.; Hutchings G. S.; Kimmel Y. C.; Chen J. G.; Jiao F. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 2014, 5, 3242. 10.1038/ncomms4242. [DOI] [PubMed] [Google Scholar]
- Hatsukade T.; Kuhl K. P.; Cave E. R.; Abram D. N.; Jaramillo T. F. Insights into the electrocatalytic reduction of CO2 on metallic silver surfaces. Phys. Chem. Chem. Phys. 2014, 16, 13814–13819. 10.1039/C4CP00692E. [DOI] [PubMed] [Google Scholar]
- Lee H.-E.; Yang K. D.; Yoon S. M.; Ahn H.-Y.; Lee Y. Y.; Chang H.; Jeong D. H.; Lee Y.-S.; Kim M. Y.; Nam K. T. Concave rhombic dodecahedral Au nanocatalyst with multiple high-index facets for CO2 reduction. ACS Nano 2015, 9, 8384–8393. 10.1021/acsnano.5b03065. [DOI] [PubMed] [Google Scholar]
- Hall A. S.; Yoon Y.; Wuttig A.; Surendranath Y. Mesostructure-induced selectivity in CO2 reduction catalysis. J. Am. Chem. Soc. 2015, 137, 14834–14837. 10.1021/jacs.5b08259. [DOI] [PubMed] [Google Scholar]
- Liu M.; Pang Y.; Zhang B.; De Luna P.; Voznyy O.; Xu J.; Zheng X.; Dinh C. T.; Fan F.; Cao C.; et al. Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration. Nature 2016, 537, 382–386. 10.1038/nature19060. [DOI] [PubMed] [Google Scholar]
- Asadi M.; Kumar B.; Behranginia A.; Rosen B. A.; Baskin A.; Repnin N.; Pisasale D.; Phillips P.; Zhu W.; Haasch R.; et al. Robust carbon dioxide reduction on molybdenum disulphide edges. Nat. Commun. 2014, 5, 4470. 10.1038/ncomms5470. [DOI] [PubMed] [Google Scholar]
- Kas R.; Hummadi K. K.; Kortlever R.; De Wit P.; Milbrat A.; Luiten-Olieman M. W. J.; Benes N. E.; Koper M. T. M.; Mul G. Three-dimensional porous hollow fibre copper electrodes for efficient and high-rate electrochemical carbon dioxide reduction. Nat. Commun. 2016, 7, 10748. 10.1038/ncomms10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasileff A.; Zheng Y.; Qiao S. Z. Carbon solving carbon’s problems: recent progress of nanostructured carbon-based catalysts for the electrochemical reduction of CO2. Adv. Energy Mater. 2017, 7, 1700759. 10.1002/aenm.201700759. [DOI] [Google Scholar]
- Cui X.; Pan Z.; Zhang L.; Peng H.; Zheng G. Selective etching of nitrogen-doped carbon by steam for enhanced electrochemical CO2 reduction. Adv. Energy Mater. 2017, 7, 1701456. 10.1002/aenm.201701456. [DOI] [Google Scholar]
- Kim C.; Jeon H. S.; Eom T.; Jee M. S.; Kim H.; Friend C. M.; Min B. K.; Hwang Y. J. Achieving selective and efficient electrocatalytic activity for CO2 reduction using immobilized silver nanoparticles. J. Am. Chem. Soc. 2015, 137, 13844–13850. 10.1021/jacs.5b06568. [DOI] [PubMed] [Google Scholar]
- Rosen J.; Hutchings G. S.; Lu Q.; Rivera S.; Zhou Y.; Vlachos D. G.; Jiao F. Mechanistic insights into the electrochemical reduction of CO2 to CO on nanostructured Ag surfaces. ACS Catal. 2015, 5, 4293–4299. 10.1021/acscatal.5b00840. [DOI] [Google Scholar]
- Qiu J.; Tang J.; Shen J.; Wu C.; Qian M.; He Z.; Chen J.; Shuang S. Preparation of a silver electrode with a three-dimensional surface and its performance in the electrochemical reduction of carbon dioxide. Electrochim. Acta 2016, 203, 99–108. 10.1016/j.electacta.2016.03.182. [DOI] [Google Scholar]
- Ma M.; Trześniewski B. J.; Xie J.; Smith W. A. Selective and efficient reduction of carbon dioxide to carbon monoxide on oxide-derived nanostructured silver electrocatalysts. Angew. Chem., Int. Ed. 2016, 55, 9748–9752. 10.1002/anie.201604654. [DOI] [PubMed] [Google Scholar]
- Yoon Y.; Hall A. S.; Surendranath Y. Tuning of silver catalyst mesostructure promotes selective carbon dioxide conversion into fuels. Angew. Chem., Int. Ed. 2016, 55, 15282–15286. 10.1002/anie.201607942. [DOI] [PubMed] [Google Scholar]
- Vermaas D. A.; Smith W. A. Synergistic electrochemical CO2 reduction and water oxidation with a bipolar membrane. ACS Energy Lett. 2016, 1, 1143–1148. 10.1021/acsenergylett.6b00557. [DOI] [Google Scholar]
- Chen Y.; Li C. W.; Kanan M. W. Aqueous CO2 reduction at very low overpotential on oxide-derived Au nanoparticles. J. Am. Chem. Soc. 2012, 134, 19969–19972. 10.1021/ja309317u. [DOI] [PubMed] [Google Scholar]
- Ma M.; Djanashvili K.; Smith W. A. Selective electrochemical reduction of CO2 to CO on CuO-derived Cu nanowires. Phys. Chem. Chem. Phys. 2015, 17, 20861–20867. 10.1039/C5CP03559G. [DOI] [PubMed] [Google Scholar]
- Lu Q.; Rosen J.; Jiao F. Nanostructured metallic electrocatalysts for carbon dioxide reduction. ChemCatChem 2015, 7, 38–47. 10.1002/cctc.201402669. [DOI] [Google Scholar]
- Hsieh Y.-C.; Senanayake S. D.; Zhang Y.; Xu W.; Polyansky D. E. Effect of chloride anions on the synthesis and enhanced catalytic activity of silver nanocoral electrodes for CO2 electroreduction. ACS Catal. 2015, 5, 5349–5356. 10.1021/acscatal.5b01235. [DOI] [Google Scholar]
- Dong H.; Chen G.; Sun J.; Li C.; Yu Y.; Chen D. A novel high-efficiency visible-light sensitive Ag2CO3 photocatalyst with universal photodegradation performances: Simple synthesis, reaction mechanism and first-principles study. Appl. Catal., B 2013, 134–135, 46–54. 10.1016/j.apcatb.2012.12.041. [DOI] [Google Scholar]
- Murray B. J.; Li Q.; Newberg J. T.; Menke E. J.; Hemminger J. C.; Penner R. M. Shape- and size-selective electrochemical synthesis of dispersed silver(l) oxide colloids. Nano Lett. 2005, 5, 2319–2324. 10.1021/nl051834o. [DOI] [PubMed] [Google Scholar]
- Yu C.; Li G.; Kumar S.; Yang K.; Jin R. Phase transformation synthesis of novel Ag2O/Ag2CO3 heterostructures with high visible light efficiency in photocatalytic degradation of pollutants. Adv. Mater. 2014, 26, 892–898. 10.1002/adma.201304173. [DOI] [PubMed] [Google Scholar]
- Chan C.; Wu J.; Li J.; Cheung Y. Polypropylene/calcium carbonate nanocomposites. Polymer 2002, 43, 2981–2992. 10.1016/S0032-3861(02)00120-9. [DOI] [Google Scholar]
- Hoshi N.; Kato M.; Hori Y. Electrochemical reduction of CO2 on single crystal electrodes of silver Ag(111), Ag(100) and Ag(110). J. Electroanal. Chem. 1997, 440, 283–286. 10.1016/S0022-0728(97)00447-6. [DOI] [Google Scholar]
- Blizanac B. B.; Ross P. N.; Marković N. M. Oxygen reduction on silver low-index single-crystal surfaces in alkaline solution: rotating ring diskAg(hkl) studies. J. Phys. Chem. B 2006, 110, 4735–4741. 10.1021/jp056050d. [DOI] [PubMed] [Google Scholar]
- Jovic B. M.; Jovic V. D.; Stafford G. R. Cyclic voltammetry on Ag(111) and Ag(100) faces in sodium hydroxide solutions. Electrochem. Commun. 1999, 1, 247–251. 10.1016/S1388-2481(99)00049-1. [DOI] [Google Scholar]
- Horswell S. L.; Pinheiro A. L. N.; Savinova E. R.; Danckwerts M.; Pettinger B.; Zei M.-S.; Ertl G. A comparative study of hydroxide adsorption on the (111), (110), and (100) faces of silver with cyclic voltammetry, ex situ electron diffraction, and in situ second harmonic generation. Langmuir 2004, 20, 10970–10981. 10.1021/la0483818. [DOI] [PubMed] [Google Scholar]
- Kortlever R.; Shen J.; Schouten K. J. P.; Calle-Vallejo F.; Koper M. T. M. Catalysts and reaction pathways for the electrochemical reduction of carbon dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. 10.1021/acs.jpclett.5b01559. [DOI] [PubMed] [Google Scholar]
- Calle-Vallejo F.; Koper M. T. M. Theoretical considerations on the electroreduction of CO to C2 species on Cu(100) electrodes. Angew. Chem., Int. Ed. 2013, 52, 7282–7285. 10.1002/anie.201301470. [DOI] [PubMed] [Google Scholar]
- Peterson A. A.; Abild-Pedersen F.; Studt F.; Rossmeisl J.; Nørskov J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311. 10.1039/c0ee00071j. [DOI] [Google Scholar]
- Hansen H. A.; Varley J. B.; Peterson A. A.; Nørskov J. K. Understanding trends in the electrocatalytic activity of metals and enzymes for CO2 reduction to CO. J. Phys. Chem. Lett. 2013, 4, 388–392. 10.1021/jz3021155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.