

Abstract
Background
Corticosteroids are routinely utilized to alleviate edema in patients with intracranial lesions and are first-line agents to combat immune-related adverse events (irAEs) that arise with immune checkpoint blockade treatment. However, it is not known if or when corticosteroids can be administered without abrogating the efforts of immunotherapy. The purpose of this study was to evaluate the impact of dexamethasone on lymphocyte activation and proliferation during checkpoint blockade to provide guidance for corticosteroid use while immunotherapy is being implemented as a cancer treatment.
Methods
Lymphocyte proliferation, differentiation, and cytokine production were evaluated during dexamethasone exposure. Human T cells were stimulated through CD3 ligation and co-stimulated either directly by CD28 ligation or by providing CD80, a shared ligand for CD28 and CTLA-4. CTLA-4 signaling was inhibited by antibody blockade using ipilimumab which has been approved for the treatment of several solid tumors. The in vivo effects of dexamethasone during checkpoint blockade were evaluated using the GL261 syngeneic mouse intracranial model, and immune populations were profiled by flow cytometry.
Results
Dexamethasone upregulated CTLA-4 mRNA and protein in CD4 and CD8 T cells and blocked CD28-mediated cell cycle entry and differentiation. Naïve T cells were most sensitive, leading to a decrease of the development of more differentiated subsets. Resistance to dexamethasone was conferred by blocking CTLA-4 or providing strong CD28 co-stimulation prior to dexamethasone exposure. CTLA-4 blockade increased IFNγ expression, but not IL-2, in stimulated human peripheral blood T cells exposed to dexamethasone. Finally, we found that CTLA-4 blockade partially rescued T cell numbers in mice bearing intracranial gliomas. CTLA-4 blockade was associated with increased IFNγ-producing tumor-infiltrating T cells and extended survival of dexamethasone-treated mice.
Conclusions
Dexamethasone-mediated T cell suppression diminishes naïve T cell proliferation and differentiation by attenuating the CD28 co-stimulatory pathway. However, CTLA-4, but not PD-1 blockade can partially prevent some of the inhibitory effects of dexamethasone on the immune response.
Electronic supplementary material
The online version of this article (10.1186/s40425-018-0371-5) contains supplementary material, which is available to authorized users.
Keywords: Corticosteroids, Immunotherapy, Glioma, Checkpoint blockade, Dexamethasone
Background
Immunotherapy is emerging as a promising anti-cancer treatment and is now part of the standard of care for certain advanced cancers including melanoma and non-small cell lung carcinoma [1]. Encouraging results from recent studies suggest that intracranial lesions located beyond the blood-brain barrier may also be targeted by the immune system [2–5]. However, patients with intracranial lesions are frequently provided corticosteroids before commencing immunotherapy to combat cerebral edema and reduce symptom burden. Corticosteroids are also first-line agents against immune-related adverse events (irAEs) that may develop during or following immunotherapy, particularly checkpoint blockade [6, 7]. To date, it remains unclear how steroids impact adaptive anti-tumor immunity [8, 9], and whether the effects of corticosteroids on immune response differs if they are administered prior to initiation of immune therapy or after an immune response has been generated.
Although a subset of patients receiving corticosteroids while undergoing treatment with immunce checkpoint inhibitors have achieved clinical benefit, there are concerns that they exhibit poorer response to checkpoint blockade [10–12]. Other studies found that corticosteroids do not negatively impact overall survival of patients on immunotherapy involving CTLA-4 blockade [13–18]. However, these studies were not powered to specifically address the impact of corticosteroids on immunotherapeutic response. Given the varied reports, the immunosuppressive effects of corticosteroids require further interrogation, particularly understanding the impact of this treatment when administered at the initiation of checkpoint blockade therapy, a situation likely to be common in patients with intracranial malignancies.
Exogenous corticosteroids are toxic to immature T cells, including thymocytes and acute lymphoblastic leukemia blasts [19, 20]. However, less is known about the impact of corticosteroids on mature lymphocytes. Whereas corticosteroids have been shown to suppress IL-2-mediated T cell proliferation and cytokine production; they can also induce expression of the pro-survival receptor IL-7Rα [21, 22]. T cell reactivity against cytomegalovirus, a potential antigen in glioblastoma [23, 24], was not impaired in patients with glioblastoma actively receiving or previously on dexamethasone [25]. Further, pre-operative corticosteroids did not reduce the density of tumor-infiltrating lymphocytes in patients with brain metastases [26]. These observations suggest that antigen-stimulated memory T cells, a critical population for patients receiving immunotherapy, may be resistant to corticosteroid exposure.
Here, we interrogated the impact of dexamethasone on T cell proliferation, differentiation, and cytokine production using human T cells and a murine glioblastoma model. We demonstrated that dexamethasone attenuates the CD28 co-stimulatory pathway by upregulating CTLA-4, thereby severely inhibiting naïve T cell proliferation and differentiation. This inhibition can be overcome in vivo by using a CTLA-4 neutralizing antibody, which extended survival in a murine syngeneic glioblastoma model. These findings have important implications for corticosteroid use with immune checkpoint blockade, particularly in patients with central nervous system tumors where corticosteroids are regularly utilized to mitigate peritumoral edema.
Methods
Preparation of Dynabeads®M-450 Tosylactivated beads
Dynabeads® M-450 Tosylactivated beads (Invitrogen by Life Technologies) were coupled with 50 μg of protein according the manufacturer’s guidelines and as previously shown [27]. Isotype beads were coupled with 10% Purified NA/LE Mouse IgG2a, k isotype control (BD Bioscience), 30% Ultra-LEAF Purified Mouse IgG1, k isotype control (Biolegend) and 60% Ultra-LEAF Purified Human IgG1 isotype control antibody (Biolegend). Stimulatory beads were designed using 5% Purified NA/LE Mouse anti-human CD3 (HIT3a; BD Biosciences), 30% Ultra-LEAF Purified Human IgG1 Isotype control antibody (Biolegend), 15% Ultra-LEAF Purified anti-human CD28 antibody (CD28.2; Biolegend) or Recombinant human B7–1/CD80-Fc Chimera Protein, CF (R&D Systems). Beads were stored in Ca2+ and Mg2+ free PBS with 0.1% BSA and 2 mM EDTA pH 7.4 at 4 °C.
T-cell preparation, culture and treatment
Healthy donor leukapheresis packs were obtained from the NIH blood bank (protocol 99-CC-0168). T cells were negatively selected using an EasySep Human T cell isolation kit (Stemcell Technologies) and cryopreserved in 90% FBS and 10% DMSO until use. Cells were thawed in a 37°C water bath and cultured overnight in RPMI1640 medium containing 10% fetal bovine serum, 1% penicillin-streptomycin-glutamine, 1% MEM non-essential amino acids solution, 15 mM HEPES, 1 mM sodium pyruvate and 55 μM 2-mercaptoethanol. Cells were plated at 1*105/200 μl in 96-well round-bottom plates with M-450 Tosylactivated beads. Dexamethasone was purchased from Sigma Aldrich (D4902) and dissolved in DMSO. Niviolumab and ipilimumab F(ab’)2 were used to block PD-1 and CTLA-4, respectively. Ipilimumab F(ab’)2 was created using a Pierce F(ab’)2 Preparation Kit per the manufacturer’s instructions (ThermoFisher Scientific, MA, USA). Cells were incubated at 37 °C in 20% O2, and 5% CO2 for four days for proliferation analyses and two days for Western blot and qPCR analyses.
Flow cytometry analysis
αCD152-PE (BNI), αCD8α-Pacific-Blue or Alexa 488 (RPA-T8), αCD4-APC (RPA-T4), αCD197-Brilliant Violet 605 (G043H7), and αCD45RO-Brilliant Violet 785(UCHL1) were purchased from BioLegend. Anti-CD4-Brilliant UV 496 (SK3) and anti-CD279-APC (clone EH12.2.2H7) were purchased from eBioscience. Cells were stained for 30 min at room temperature, rinsed with FACS buffer (1% BSA and 0.01% sodium azide in PBS), fixed with 4% paraformaldehyde (Alfa Aesar, WA, USA), and resuspended in FACS buffer for flow cytometry analysis. Cell Cycle analysis was conducted using a Click-iT® Edu Flow Cytometry Assay Kit (Invitrogen, Carlsbad, CA, USA) per the manufacturer’s instructions. Cell apoptosis was investigated using Annexin V-Pacific Blue (Biolegend, San Diego, CA, USA) and propidium iodide (Sigma Aldrich, MO, USA). Data were acquired on a BD LSR Fortessa SORP II or LSR Fortessa X50, analyzed with Flowjo version 9.9.4 and SPICE version 5.35.
Western blot analysis
Isolated human T cells were collected after 48 h of stimulation and lysed in RIPA buffer with EDTA- free protease inhibitor cocktail set III (EMD Millipore, Billerica, Massachusetts, USA). Pierce BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL, USA) was used to determine protein concentration. Samples were separated by SDS-PAGE (Bio-Rad) and transferred onto 0.2 μm pore size polyvinylidene fluoride membranes (PVDF) (Invitrogen, Carlsbad, CA, USA). The following antibodies were purchased from Cell Signaling: cleaved caspase 3 (5A1E), p27kip (2552 s), cyclin D3 (DCS22), CDK4 (D9G3E). Anti-CTLA-4 (EPR1476) was purchased from Abcam. The bands were detected by Super Signal West Pico chemiluminescence reagent (Pierce, Rockford, IL, USA). Antibodies against β-actin (AG74) or GAPDH (FL-335) were used as internal standards.
RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen). Reverse transcription was performed using the Superscript III First-Strand Synthesis System (Invitrogen) with oligo dT. Subsequent RT-PCR was performed using SYBR green reagent from ABI and run on an ABI Quant Studio7. β-actin was used as a control to normalize gene expression. Primers and conditions used for RT-PCR are listed in the table below.
| Gene | Primer Sequence | Tm(°C) |
|---|---|---|
| IFNγ | F- 5’ TCGGTAACTGACTTGAATGTCCA 3′ R- 5’ TCGCTTCCCTGTTTTAGCTGC 3’ |
60 |
| IL-2 | F- 5’ AACTCCTGTCTTGCATTGCAC 3′ R- 5’GCTCCAGTTGTAGCTGTGTTT 3’ |
60 |
| CTLA-4 | F- 5’ CATGATGGGGAATGAGTTGACC 3′ R- 5’ TCAGTCCTTGGATAGTGAGGTTC 3’ |
60 |
| β-actin | F-5’ CCACACTGTGCCCATCTAC 3′ R-5’ CCATCTCTTGCTCGAAGTCC 3’ |
60 |
Murine studies
All experiments were approved by the NCI-Bethesda Animal Care and Use Committee. Six to 8 week-old female albino C57BL/6 mice were purchased from Jackson laboratories (Bar Harbor, ME). For intracranial tumor implantation, mice were injected with 1*103 GL261 cells that were stably transduced with a firefly luciferase-mCherry lentiviral vector. Water-soluble dexamethasone (D2915, Sigma Aldrich) was administered at 1 mg/kg/day by oral gavage. The non-toxic solubilizer, 2-hydroxypropyl-beta-cyclodextrin (H-107, Sigma Aldrich) was dissolved with water and matched in concentration to water-soluble dexamethasone for vehicle control. InvivoMab anti-mouse CTLA-4 (BE0131, BioXCell) or InVivoMab polyclonal Syrian hamster IgG isotype antibody (BE0087, BioXCell) were administered by intraperitoneal injection. Tumor was detected by luminescence imaging and analyzed with LivingImage Software.
Statistical analysis
Statistical Analysis was performed using GraphPad Prism 7.0 software. Data are expressed as the mean +/− standard deviation and statistical significance evaluated by two-tailed Student’s t-test. P-values < 0.05 were considered significant.
Results
Dexamethasone impairs proliferation of mature T cells
T cells require two signals for an effective proliferative response; the first signal arises when a T cell receptor (TCR) binds its cognate peptide:MHC complex, resulting in an intracellular signaling cascade through CD3. The second, co-stimulatory signal, is received when CD28 on T cells binds either CD80 or CD86 on antigen-presenting cells (APCs) [28]. Here, T cells isolated from healthy donor peripheral blood samples were stimulated with an αCD3 antibody for signal 1 and either an αCD28 antibody or recombinant CD80 protein for co-stimulation [27]. Recombinant CD80 permitted us to dissect the role of extracellular molecular interactions between CD80 and its multiple binding partners on T cells. These partners include the positive co-stimulatory molecule, CD28, and negative co-stimulatory molecules, such as CTLA-4 and PD-1.
Human T cells incubated with αCD3 and CD80 underwent multiple rounds of division, demonstrating that recombinant CD80 provided an efficient co-stimulatory signal under normal culture conditions (Fig. 1a). However, dexamethasone exposure caused defects in cell division for both CD4 and CD8 T cells. Cell proliferation statistics, including precursor frequency, expansion index, and proliferation index, were performed on multiple heathy donor T cells isolated from peripheral blood mononuclear cells to assess population dynamics and identify the proliferation defect [29]. Precursor frequency, defined as the cell-intrinsic probability that a cell will undergo at least one division, was diminished when T cells were exposed to dexamethasone (Fig. 1b). Similarly, dexamethasone-exposed T cells also had a reduced expansion index, a cell-extrinsic statistic used to express the fold expansion of the final cell count compared to the initial cell count (including undivided cells). Finally, the proliferation index, which represents the number of divisions that cells in the post-mitotic population have undergone, was modestly decreased by dexamethasone. Thus, cell proliferation statistics demonstrate that dexamethasone impaired the ability of CD80 co-stimulated CD4 and CD8 T cells to divide and restricted the expansion potential of these populations. In contrast, dexamethasone imposed only subtle cell division defects under all conditions in which CD28 was exclusively ligated with an αCD28 antibody (Additional file 1: Figure S1), indicating that direct ligation of CD28 can confer resistance to dexamethasone.
Fig. 1.

T cell proliferation is impaired by dexamethasone. Healthy donor T cells were cultured for four days with αCD3/CD80 microbeads in the presence of vehicle or dexamethasone. a Representative flow cytometry plots of CellTrace violet dilution. Plots were derived from gated CD4 (top row) or CD8 (bottom row) T cells. b Negatively-selected healthy donor T cells were stained and proliferation analyses determined by flow cytometry following four days of culture under the indicated conditions. Precursor Frequency, Expansion Index, and Proliferation Index are shown. Each symbol is the average of duplicate wells, and each paired symbol represents a different donor (n = 5 donors). Statistical significance was determined with a paired two-tailed T test. c Cell cycle analysis was performed on healthy donor T cells cultured with vehicle or dexamethasone and stimulated with αCD3/CD80 microbeads. EdU uptake and DNA content were used to identify G0/G1, S, and G2/M phases. Representative flow images (top) and quantification of duplicate wells are shown (bottom) from two independent experiments. d Lysates from healthy donor T cells incubated with the indicated microbeads and vehicle or dexamethasone were probed for the indicated proteins. GAPDH was used as a loading control and is shown for each individual blot. Data are representative of three independent experiments
Dexamethasone attenuates the CD28 signaling pathway
Increased apoptosis and cell cycle blockade can each manifest as a proliferative defect in vitro. Dexamethasone did not increase the frequency of apoptotic T cells when cultured with αCD3 and either αCD28 or CD80 co-stimulation as demonstrated by flow cytometry and Western analysis of cleaved caspase 3 (Additional file 2: Figure S2), demonstrating that cell death was not responsible for the proliferative defect. Indeed, dexamethasone has been shown to protect T cells from activation-induced cell death [30].
Cell cycle progression of CD80-costimulated T cells was next evaulated in response to dexamethasone exposure. Dexamethasone induced, on average, a 13% increase in the G0/G1 phase along with a 67% reduction of cells in S-phase and a 49% reduction in the G2/M phase (Fig. 1c). Dexamethasone-exposed T cells also contained increased levels of p27 protein, an inhibitor of cell cycle entry, and concurrent reductions in CDK4 and cyclin D3 protein. Together, these data demonstrate that dexamethasone blocks cell cycle entry in T cell stimulated with αCD3 and CD80.
Co-stimulation protects T cells from the anti-proliferative effects of dexamethasone
Following stimulation-induced expansion T cells may advance to a more terminally differentiated state. However, T cells cultured with dexamethasone had a greater proportion of naïve T cells (TN) and central memory T cells (TCM) and fewer effector memory T cells (TEM) than cells cultured with vehicle control after four days of stimulation (Additional file 3: Figure S3). Terminal effector T cells (TTE) were unchanged. These data indicate that dexamethasone blocked the ability of T cells to differentiate in response to αCD3/CD80 stimulation. The loss in TEM numbers was reversed by increasing concentrations of CD80 (Fig. 2 and Additional file 4: Figure S4), suggesting that strong co-stimulation may protect this subset from the inhibitory effects of dexamethasone.
Fig. 2.
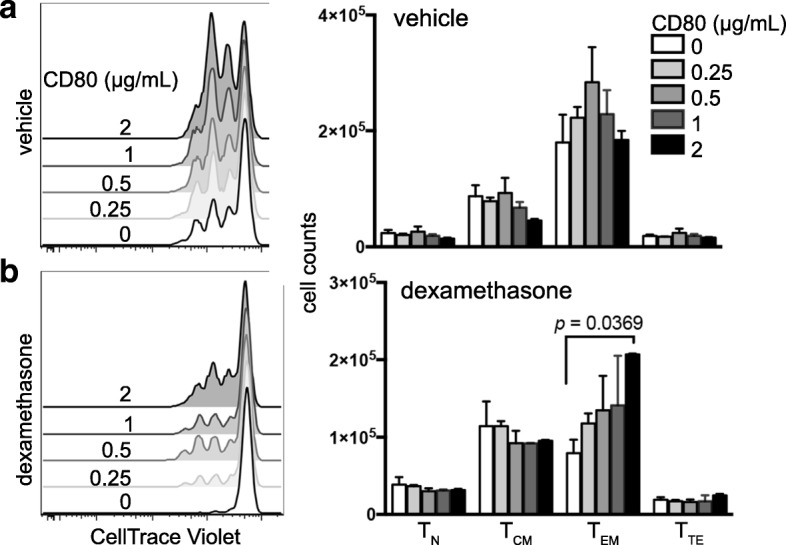
Increased co-stimulation ameliorates the inhibitory effects of dexamethasone. Negatively-selected healthy donor T cells were cultured with 5 μg/mL αCD3 and increasing concentrations of CD80 in the presence of vehicle or dexamethasone. a-b CD4 T cells cultured with vehicle (a) or dexamethasone (b). Flow cytometry plots showing proliferation of cells cultured with the indicated concentration of CD80 (left) and total numbers of naïve (TN), central memory (TCM), effector memory (TEM), and terminal effector (TTE) T cells following four days of culture (right) are shown. Differentiation subsets were assessed by CD45RO and CCR7 staining. Each condition was plated in duplicate, and data are representative of three independent experiments. Data were analyzed with an unpaired, two-tailed T Test
To directly test the impact of dexamethasone on T cell differentiation subsets, circulating T cells from healthy donors were sorted by flow cytometry into TN, TCM, and TEM subsets. The proliferative response of each was assessed in response to stimulation and exposure to vehicle or dexamethasone. Cultures from purified naïve CD4 and CD8 T cells were most severely impaired by dexamethasone, with a diminished precursor frequency, expansion index, and proliferation index (Fig. 3). In contrast, purified TCM response was comparable to vehicle control. TEM demonstrated reduced precursor frequency but greater cell number, suggesting that although dexamethasone impaired cell cycle entry, the cells were not lost due to apoptosis. Also, unlike TN, dexamethasone did not impair the expansion or proliferation index of TEM, demonstrating that this subset was less sensitive to dexamethasone than TN.
Fig. 3.

Naïve and effector memory T cells show sensitivity to dexamethasone. a Healthy donor T cells were sorted into TN, TCM, and TEM subsets by flow cytometry. Sorted subsets were cultured with αCD3/CD80 microbeads in the presence of dexamethasone (red) or vehicle control (black). Total cell numbers, Precursor Frequency, Expansion Index, and Proliferation Index of CD4 T cells (top) and CD8 T cells (bottom) are shown. All samples were plated in duplicate and analyzed with a paired, two-tailed T test. Data are representative of three independent experiments
CTLA-4 blockade protects T cells from the deleterious effects of dexamethasone
CD80 provides a positive co-stimulatory signal to T cells when bound by CD28. However, following TCR signal transduction, CTLA-4 is translocated to the outer membrane of T cells where it can outcompete CD28 to bind CD80 and block co-stimulation [31–34]. Using flow cytometry, we confirmed that T cell stimulation led to increased extracellular CTLA-4 protein levels. In the presence of dexamethasone, however, stimulation caused a fourfold increase in surface CTLA-4 protein compared to vehicle treated (Fig. 4a) as well as an increase in CTLA-4 transcription (Fig. 4b). For these experiments, T cells were stimulated with αCD3 and αCD28 antibodies because surface CTLA-4 is internalized following ligation by CD80 [35], potentially impairing detection by flow cytometry antibodies. CTLA-4 expression was consistently found to be higher in CD4 T cells compared to CD8 T cells during dexamethasone treatment, likely rendering CD4 T cells more susceptible to cell cycle inhibition by checkpoint molecule.
Fig. 4.

CTLA-4 blockade partially restores T cell proliferation in the presence of dexamethasone. a Flow cytometry analysis of CTLA-4 surface expression on CD4 (left) or CD8 (right) T cells stimulated with αCD3/αCD28 microbeads. Unstimulated (dashed line), stimulated in presence of vehicle (solid line), and stimulated in presence of dexamethasone (filled red line) are shown (left) and median fluorescence intensity (MFI) of CTLA-4-expressing T cells is quantified (right). Data are representative of four independent experiments. b Expression of CTLA-4 by qPCR of T cells stimulated in the presence of vehicle or dexamethasone. Data are representative of four independent experiments. c Healthy donor T cells stimulated for four days in the presence of vehicle or dexamethasone and with or without ipilimumab F(ab’)2 antibody. Proliferation analysis of CD4 T cells (top) and CD8 T cells (bottom) was performed, and the ratio of cells stimulated with dexamethasone relative to vehicle control are shown for Precursor Frequency (PF), Expansion Index (EI), and Proliferation Index (PI). All samples were plated in duplicate and the ratios were analyzed with an unpaired, two-tailed T test. Data are representative of 7 healthy donors. d Cells were cultured as in (c). The number of T cells in each differentiation group were quantified by flow cytometry and analyzed by SPICE. e Expression of the indicated cytokines was determined by qPCR. Five healthy donors were assayed for each condition. Each data point represents and average of triplicate wells. Data were analysed with a paired, two-tailed T test
We hypothesized that dexamethasone-induced CTLA-4 upregulation on T cells out-competed CD28 for the shared CD80 ligand, thereby attenuating the CD28 co-stimulatory pathway and inhibiting cell cycle entry. To test this, CTLA-4 was blocked with ipilimumab, a monoclonal anti-CTLA-4 human antibody, thereby enhancing the ability of CD28 to bind CD80. With ipilimumab treatment, dexamethasone-induced cell proliferation defects were partially restored. Precursor frequency and expansion index of dexamethasone-treated CD4 and CD8 T cells were significantly increased (Fig. 4c) as well as the proliferation index of CD4 T cells. In contrast, the proliferation index of CD8 T cells co-stimulated with CD80 was not increased with ipilimumab. T cells exposed to dexamethasone also upregulated of PD-1 during in vitro stimulation, but blocking PD-1 with nivolumab did not significantly change T cell precursor frequency (Additional file 5: Figure S5). These data demonstrate that ipilimumab specifically reversed the proliferation defects caused by dexamethasone, implicating CTLA-4 as a mechanism for the anti-proliferative activities of dexamethasone exposure in T cells.
The relative proportion of TN and TCM were increased in cultures exposed to dexamethasone compared to vehicle control. CTLA-4 blockade with ipilimumab led to an expansion of the TCM subset, and this was recapitulated in T cells cultured with the combination of ipilimumab and dexamethasone (Fig. 4d). Blocking CTLA-4 with ipilimumab increased IFNγ, but not IL-2 transcription during dexamethasone exposure, suggesting that CTLA-4 blockade may rescue IFNγ-producing T cells inhibited by dexamethasone (Fig. 4e).
Checkpoint blockade enhances survival of dexamethasone-treated tumor-bearing mice
To determine if these findings translated to in vivo models, the C57Bl/6 syngeneic glioma tumor line GL261 was implanted intracranially. Consistent with the human in vitro data, dexamethasone increased the percentage of CTLA-4-expressing CD4 T cells of tumor-bearing mice in a dose-dependent manner (Fig. 5a). The percent of CTLA-4-expressing CD8 T cells also significantly increased in mice treated with the highest concentration of dexamethasone (2.5 mg/kg/day). Because dexamethasone upregulated CTLA-4, we hypothesized that CTLA-4 blockade would provide a survival advantage to dexamethasone-treated mice. GL261 tumor cells were implanted one week before commencement of dexamethasone or vehicle treatment to allow for immune surveillance and development of differentiated anti-tumor T cells. CTLA-4 blockade or isotype antibody were administered on days 13, 16, and 19 to vehicle or dexamethasone-treated mice to emulate the clinical scenerios where patients may be off or on corticosteroids before receiving immunotherapy (Fig. 5b). To ensure comparable tumor burden, mice were randomized into cohorts with equivalent tumor luminescence prior to treatment, and luminescence was measured weekly thereafter (Fig. 5c).
Fig. 5.

CTLA-4 blockade enhances survival of dexamethasone-treated mice. a CTLA-4 was measured on circulating CD4 (left) and CD8 (right) T cells 1 h following oral gavage of vehicle or the indicated concentration of dexamethasone. Each cohort contained eight mice with intracranial GL261 tumors. Vehicle and dexamethasone-treated cohorts were statistically analyzed with an unpaired two-tailed student’s T-test. b Schema of treatment cohorts for (c-d). GL261 ffluc-mCherry glioma cells were orthotopically implanted into C57BL/6 mice one week before treatment initiation. Luminescence readings were acquired 6 days following tumor implantation and weekly thereafter. Mice were treated with vehicle or dexamethasone as indicated. CTLA-4 blocking antibody or isotype control were administered on days 13, 16, and 19 following tumor implantation. c Luminescence of tumor-bearing mice at days 13 and 20 following tumor implantation. d Kaplan Meier survival curves of mice receiving the indicated treatments. n = 7 to 8 mice per cohort. Data are representative of two independent experiments
Single-agent treatment with CTLA-4 blocking antibody or dexamethasone did not produce a significant survival benefit compared to vehicle-treated animals in this well-established glioblastoma model. However, mice that received CTLA-4 blockade in addition to dexamethasone treatment survived significantly longer than control animals, with a median survival of 49 versus 39 days, respectively (p = 0.013; Fig. 5d). Here, the immune system surveyed the immunogenic GL261 luciferase-mCherry tumor for one week before dexamethasone treatment was initiated. To determine if immune priming contributed to dexamethasone resistance, this experiment was repeated to include a cohort of mice exposed to dexamethasone one day prior to tumor implantation. In this cohort, CTLA-4 blockade did not provide a survival benefit (Additional file 6: Figure S6). These data indicate that dexamethasone exposure may block a successful anti-tumor immune response if treatment occurs before anti-tumor T cells can differentiate from the TN pool.
The impact of dexamethasone exposure was next evaluated on lymphocyte populations from tumor-bearing mice, including tumor-infiltrating lymphocytes (TILs) and T cells in the tumor-draining cervical lymph nodes (TDLN). Mice exposed to dexamethasone possessed significantly reduced numbers of T cells in TDLNs. However, CTLA-4 blockade increased the total number of CD4 T cells in TDLN and CD8 TILs (Fig. 6a and b). The number of regulatory T cells (Treg) in the tumor-bearing brain were not significantly affected by dexamethasone or CTLA-4 blockade whereas overall numbers were reduced in TDLNs from each dexamethasone-treated cohort (Additional file 7: Figure S7A).
Fig. 6.

CTLA-4 blockade rescues lymphocyte defects induced by dexamethasone. GL261 ffluc-mCherry tumor-bearing mice were randomized into the indicated cohorts. Vehicle or dexamethasone treatment was initiated on day 7, and isotype or CTLA-4 blocking antibody were administered on days 13, 16, and 19 following tumor implantation. Mice were euthanized on day 23 and tissues were harvested for flow cytometry analysis. a-b CD4 (a) and CD8 (b) T cells were quantified along with the indicated differentiation subsets using CD44 and CD62L expression. Brains (n = 8) and cervical lymph nodes (n = 10) were collected. Data are analyzed using a unpaired students T test. c The relative contribution of each differentiation subset is shown for CD4 (top) and CD8 (bottom) TILs. d The total number of IFNγ-producing T cells were quantified from the tumor-bearing hemispheres of mice from the indicated cohorts. Data are analyzed using an unpaired students T test. N = 8 mice/group
Dexamethasone exposure reduced the total number of T cells in the majority of T cell subsets along the differentiation spectrum in the tumor-draining lymph nodes, yet as observed in vitro, this was partially rescued by CTLA-4 blockade (Fig. 6a and b). The number of CD4 TN and TCM TIL subsets were also significantly elevated by CTLA-4 blockade in the dexamethasone-treated group. The relative proportion of TCM TILs was increased by CTLA-4 blockade in both the vehicle and dexamethasone-treated cohorts (Fig. 6c). GBM has been reported to induce high expression of checkpoint molecules on TILs, resulting in a severe exhaustion signature [36]. Dexamethasone treatment increased the percentage of CD8 TILs that expressed Tim-3 from 2 to 12%, although it did not significantly impact other checkpoint molecules (PD-1 or Lag-3) or the frequency of TILs expressing multiple checkpoint molecules (Additional file 7: Figure S7B). Although T cells expressing checkpoint molecules were less frequent in TDLN, dexamethasone decreased the frequency of most checkpoint-expressing T cells with the exception of Tim-3+ CD8 T cells, which increased from 1.5 to 7.5 and 1.3 to 4.7% in isotype and CTLA-4 blockade treated mice, respectively. Finally, the number of IFNγ-producing TILs in dexamethasone-treated mice was significantly increased with CTLA-4 blockade, consistent with in vitro data (Fig. 6d). Together, these data demonstrate that CTLA-4 blockade can partially reverse the inhibitory effects of dexamethasone on T cells in vivo, reduce the proportion of TILs expressing checkpoint molecules, increase IFNγ-expressing TILs and extend survival of dexamethasone-treated mice bearing intracranial gliomas.
Discussion
Corticosteroids, most commonly dexamethasone, are regularly administered to patients with intracranial tumors to combat cerebral edema and provide symptomatic relief [37]. Additionally, corticosteroids are used to treat patients who develop irAEs as a result of immunotherapy. In contrast to the frequent early use of dexamethasone for tumor-associated edema, corticosteroid use for immunotherapy-related toxicity is always given after treatment has commenced. Corticosteroids have been established as causing dose-related immunosuppression, yet the mechanisms behind this impaired immune function, particularly in the context of cancer immunotherapy, have not been defined [38]. Importantly, it is not known if corticosteroids impede the differentiation of freshly stimulated T cells or if they deplete established and already differentiated tumor-reactive lymphocytes.
In this study, the immunosuppressive effects of dexamethasone on individual T cell differentiation subsets were interrogated. TN were identified as being exceptionally sensitive to dexamethasone-mediated cell cycle blockade. TN are a key source of secondary anti-tumor immunity mediated by antigen spread in response to checkpoint blockade [39] and are highly sensitive to anergy imposed by expression of CTLA-4 [40]. Dexamethasone exposure strongly upregulated PD-1 and CTLA-4 transcription and protein, consistent with previous in vitro murine studies [41, 42]. The data presented here extend upon these studies to demonstrate that dexamethasone-induced CTLA-4 upregulation effectively blocked TN proliferation and differentiation in both murine and human T lymphocytes. In tumor-bearing mice, this led to a loss of differentiated T cell subsets in several lymphoid tissues.
In contrast to TN lymphocytes, dexamethasone exposure had much less impact on memory T cell proliferation following flow cytometry sorting. TEM had reduced precursor frequency but increased total numbers. Our data confirm and expand previous studies that demonstrated that the efficacy of TCR transgenic T cells was not impaired by dexamethasone treatment [43]. Here, endogenously generated anti-tumor immunity could be maintained during dexamethasone treatment if CTLA-4 blockade was provided. Blocking CTLA-4, but not PD-1, partially rescued T cell proliferation in the presence of dexamethasone in vitro. These findings may reflect that CTLA-4 blockade acts upon less differentiated T cells, which are most sensitive to dexamethasone. Systemic immunity elicited by CTLA-4 blockade has been previously shown to promote anti-tumor immunity against melanoma metastases within the central nervous system, indicating that CTLA-4 blockade functions outside of the CNS [15, 44]. The GL261 luciferase-mCherry tumor cells provided an immunogenic intracranial tumor model. By waiting one week before dexamethasone treatment, tumor surveillance and immune activation were permitted, potentially leading to differentiation of tumor-reactive T cells before dexamethasone exposure. In this model, CTLA-4 blockade was sufficient to provide a survival advantage to dexamethasone-treated mice. In contrast, mice exposed to dexamethasone prior to tumor implantation and antigen exposure were unresponsive to CTLA-4 blockade. Collectively, these data suggest that tipping the immune response to more differentiated subsets may lesson the immune suppression imposed by dexamethasone.
These results demonstrate that the timing of dexamethasone treatment relative to the development of anti-tumor immunity significantly impacts the efficacy of immunotherapy. Previous work has shown that corticosteroids provided to alleviate irAEs did not impact the overall response rate of patients with melanoma who received nivolumab [45]. For those patients, corticosteroids were provided after immunotherapy was initiated. In contrast, patients with intracranial tumors are routinely provided high-dose corticosteroids from the period of initial diagnosis until chemoradiation completion, a period that can span 8–12 weeks. Corticosteroids are also provided during surgical resection, as they have been shown to extend survival in this context [46]. Thus, for patients with intracranial tumors, corticosteroids provided before initiation of immunotherapy, a time when the immune system is unlikely to be actively proliferating, may blunt the generation of an anti-tumor response.
Similarly, dexamethasone provided to immunologically “cold” tumors or those with insufficient anti-tumor immunity will likely abrogate new priming and differentiation of anti-tumor T cells. However, the concurrent use of CTLA-4 blockade can encourage TN activation, thereby contributing to antigen spread. Once anti-tumor immunity has been initiated, the negative impact of corticosteroids on immune function is markedly reduced. These results may have important implications in designing future immunotherapy strategies helping to optimize clinical trials for patients with brain cancers as well as other diseases where corticosteroid use is common.
Steroid alternatives may need to be considered for patients with intracranial tumors who wish to enroll on immunotherapy trials. For example, blockade of vascular endothelial growth factor by bevacizumab reduces edema by normalizing tumor vasculature. Further, it has been shown to promote lymphocyte infiltration into the tumor and increase circulating memory T cell numbers [47]. Such alternative approaches may be needed to manage symptoms in patients with intracranial tumors while preserving the potential for anti-tumor immunity.
Conclusions
Here, we interrogated the impact of dexamethasone on T cell subsets in the setting of immunotherapy. Dexamethasone blocks naïve T cell proliferation and differentiation by attenuating CD28 co-stimulation. Because co-stimulation is essential for successful T cell priming and expansion, these data suggest that corticosteroids impair response in immunotherapy treatment-naïve patients or those with poorly antigenic tumors. However, T cells may be partially protected or rescued from the immunosuppressive effects of dexamethasone with administration of CTLA-4 blockade. Additionally, negative corticosteroid effects are diminished after developing a successful anti-tumor immune response.
Additional files
Figure S1. T cell stimulated with αCD3/αCD28 microbeads proliferate in the presence of dexamethasone.Healthy donor T cells were cultured for four days with the indicated ratio of αCD3/αCD28 microbeads:total T cells in the presence of vehicle or dexamethasone. A, Representative flow cytometry plots of CellTrace violet dilution. Plots were derived from gated CD4 (top row) or CD8 (bottom row) T cells. B-D, Proliferation analyses of CD4 T cells (top) and CD8 T cells (bottom) performed on the samples shown in (A). Precursor Frequency (B), Expansion Index (C), and Proliferation Index (D) are shown. Samples were plated in duplicate and analyzed with an unpaired students T test. Data are representative of three independent experiments. (PDF 3563 kb)
Figure S2. A, Negatively-selected healthy donor T cells were cultured with the indicated microbeads and vehicle or dexamethasone. The percent of apoptotic CD4 (top) and CD8 (bottom) T cells was assessed by Annexin V/PI. Data are representative of four independent experiments. B, Lysates from healthy donor T cells incubated with the indicated microbeads and vehicle or dexamethasone were probed for the indicated proteins. GAPDH was used as a loading control. (PDF 693 kb)
Figure S3. T cell differentiation subsets formed during in vitro stimulation with αCD3/CD80 stimulation. Negatively-selected healthy donor T cells were cultured with 5 μg/mL αCD3 and the indicated concentration of CD80. T cell differentiation subsets were quantified following four days of culture. A, Flow plot of gating strategy to identify the indicated T cell differentiation subsets. B, Flow plots of CD4 (top) and CD8 (bottom) T cells cultured under the indicated conditions. (PDF 3995 kb)
Figure S4. Increased co-stimulation ameliorates the inhibitory effects of dexamethasone. Negatively-selected healthy donor T cells were cultured with 5 μg/mL αCD3 and increasing concentrations of CD80 in the presence of vehicle or dexamethasone. A-B. CD8 T cells cultured with vehicle (A) or dexamethasone (B). Flow cytometry plots showing proliferation of cells cultured with the indicated concentration of CD80 (left) and total numbers of naïve (TN), central memory (TCM), effector memory (TEM), and terminal effector (TTE) T cells following four days of culture (right) are shown. Differentiation subsets were assessed by CD45RO and CCR7 staining. Each condition was plated in duplicate, and data are representative of three independent experiments. Data were analyzed with an unpaired, two-tailed T Test. (PDF 2573 kb)
Figure S5 PD-1 blockade does not rescue dexamethasone-mediated proliferation defects. A, Flow cytometry analysis of PD-1 surface expression on CD4 (left) or CD8 (right) T cells stimulated with αCD3/αCD28 microbeads. Unstimulated (dashed line), stimulated in presence of vehicle (solid line), and stimulated in presence of dexamethasone (filled red line) are shown. B, Geometric median fluorescence intensity (gMFI) of PD-1 staining on CD4 or CD8 T cells. Cells cultured with vehicle (black bars) and dexamethasone (red bars) are shown. Data are an average of duplicate samples. C, Expression of PD-1 by qPCR of T cells stimulated in the presence of vehicle or dexamethasone. Data are representative of four independent experiments. D-E. Healthy donor T cells were stimulated for four days in the presence of vehicle or dexamethasone and nivolumab or ipilimumab F(ab’)2 antibody as indicated. Precursor frequency of CD4 and CD8 T cells was quantified by FlowJo. The ratio of dexamethasone to vehicle for CD4 (C) and CD8 (D) T cells is shown. All samples were plated in duplicate and the ratios were analyzed with a one-way ANOVA. Data are representative of n = 4 healthy donors. (PDF 2522 kb)
Figure S6 CTLA-4 blockade does not rescue dexamethasone pre-treated mice. A, Schema of survival experiment. Albino C57Bl/6 mice received intracranial implantation of GL261 ffluc-mCherry glioma cells. Dexamethasone was initiated one day prior to tumor implantation (dex (D-1)) or one week following tumor implantation (dex (D7)). CLTA-4 blockade or isotype antibody were injected on days 13, 16, and 19 following tumor implantation. Mice were randomized on day 6 following tumor implantation into groups of equivalent tumor luminescence. B, Kaplan Meier survival curves of mice receiving the indicated treatments. n = 8 to 9 mice per cohort. Data are representative of two independent experiments. (PDF 1525 kb)
Figure S7. Quantification of Treg and checkpoint molecules in tumor-bearing mice. GL261 ffluc-mCherry tumor-bearing mice were randomized into the indicated cohorts based on bioluminescence values from tumor. Vehicle or dexamethasone treatment was initiated on day 7, and isotype or CTLA-4 blocking antibody were administered on days 13, 16, and 19 following tumor implantation. Mice were euthanized on day 23 and tissues were harvested for flow cytometry analysis. A, Treg cell number from tumor-bearing brain hemisphere (left; n = 8) or the cervical tumor-draining lymph nodes (right; n = 10). B, The percentage of CD4 (top two plots) or CD8 (bottom two plots) T cells expressing the indicated checkpoint molecules. Co-expression of molecules was quantified using a Boolean gating strategy. Data were analyzed using a unpaired students T test. (PDF 1891 kb)
Acknowledgements
The authors wish to acknowledge Jonathan D. Ashwell, M.D. for insightful scientific discussions.
Funding
This research was supported in part by the Intramural Research Program of the NIH, NCI.
Availability of data and materials
All data analyzed are included in this article and additional information is available upon request.
Abbreviations
- APC
Antigen-Presenting Cell
- EI
Expansion Index
- irAE
immune-related Adverse Events
- PF
Precursor Frequency
- PI
Proliferation Index
- Tcm
Central Memory T cells
- Tem
Effector Memory T cells
- Tn
naïve T cells
Authors’ contributions
AJG designed research studies, conducted experiments, acquired and analyzed data, and wrote the manuscript. MH designed research studies, conducted experiments, acquired and analyzed data. HMS, JJ, NR, CMR, WZ, HS, RB, and DD conducted experiments, acquired and analyzed data. PEF contributed data and edited the manuscript. DMP designed research studies, provided reagents, and edited the manuscript. MRG designed research studies, analyzed data, and helped write the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable; not a clinical trial.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s40425-018-0371-5) contains supplementary material, which is available to authorized users.
Contributor Information
Amber J. Giles, Email: amber.giles@nih.gov
Marsha-Kay N. D. Hutchinson, Email: marsha-kay.hutchinson@nih.gov
Heather M. Sonnemann, Email: heather.sonnemann@nih.gov
Jinkyu Jung, Email: jinkyu.jung@nih.gov.
Peter E. Fecci, Email: peter.fecci@duke.edu
Nivedita M. Ratnam, Email: nivi.ratnam@nih.gov
Wei Zhang, Email: zhangwe@mail.nih.gov.
Hua Song, Email: hua.song@nih.gov.
Rolanda Bailey, Email: baileyro@mail.nih.gov.
Dionne Davis, Email: ddavis1@mail.nih.gov.
Caitlin M. Reid, Email: reidcaitlin5@gmail.com
Deric M. Park, Email: deric.park@nih.gov
Mark R. Gilbert, Email: mark.gilbert@nih.gov
References
- 1.Hoos A. Development of immuno-oncology drugs - from CTLA4 to PD1 to the next generations. Nat Rev Drug Discov. 2016;15(4):235–247. doi: 10.1038/nrd.2015.35. [DOI] [PubMed] [Google Scholar]
- 2.Farber SH, Tsvankin V, Narloch JL, Kim GJ, Salama AK, Vlahovic G, et al. Embracing rejection: immunologic trends in brain metastasis. Oncoimmunology. 2016;5(7):e1172153. doi: 10.1080/2162402X.2016.1172153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ, et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res. 2017;23(8):1898–1909. doi: 10.1158/1078-0432.CCR-16-2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma eradication following immune checkpoint blockade in an Orthotopic. Immunocompetent Model Cancer Immunol Res. 2016;4(2):124–135. doi: 10.1158/2326-6066.CIR-15-0151. [DOI] [PubMed] [Google Scholar]
- 5.Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13(7):2158–2167. doi: 10.1158/1078-0432.CCR-06-2070. [DOI] [PubMed] [Google Scholar]
- 6.Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(13):1270–1271. doi: 10.1056/NEJMc1509660. [DOI] [PubMed] [Google Scholar]
- 7.Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691–2697. doi: 10.1200/JCO.2012.41.6750. [DOI] [PubMed] [Google Scholar]
- 8.Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–148. doi: 10.1016/j.ejca.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 9.Garant A, Guilbault C, Ekmekjian T, Greenwald Z, Murgoi P, Vuong T. Concomitant use of corticosteroids and immune checkpoint inhibitors in patients with hematologic or solid neoplasms: a systematic review. Crit Rev Oncol Hematol. 2017;120:86–92. doi: 10.1016/j.critrevonc.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Margolin K, Ernstoff MS, Hamid O, Lawrence D, McDermott D, Puzanov I, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459–465. doi: 10.1016/S1470-2045(12)70090-6. [DOI] [PubMed] [Google Scholar]
- 11.Queirolo P, Spagnolo F, Ascierto PA, Simeone E, Marchetti P, Scoppola A, et al. Efficacy and safety of ipilimumab in patients with advanced melanoma and brain metastases. J Neuro-Oncol. 2014;118(1):109–116. doi: 10.1007/s11060-014-1400-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parakh S, Park JJ, Mendis S, Rai R, Xu W, Lo S, et al. Efficacy of anti-PD-1 therapy in patients with melanoma brain metastases. Br J Cancer. 2017;116(12):1558–1563. doi: 10.1038/bjc.2017.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horvat TZ, Adel NG, Dang TO. Momtaz P, Postow MA, Callahan MK, et al. Immune-related adverse events, need for systemic immunosuppression, and effects on survival and time to treatment failure in patients with melanoma treated with Ipilimumab at memorial Sloan Kettering Cancer center. J Clin Oncol. 2015;33(28):3193–3198. doi: 10.1200/JCO.2015.60.8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM, Yang JC, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol. 2005;23(25):6043–6053. doi: 10.1200/JCO.2005.06.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003;100(14):8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harmankaya K, Erasim C, Koelblinger C, Ibrahim R, Hoos A, Pehamberger H, et al. Continuous systemic corticosteroids do not affect the ongoing regression of metastatic melanoma for more than two years following ipilimumab therapy. Med Oncol. 2011;28(4):1140–1144. doi: 10.1007/s12032-010-9606-0. [DOI] [PubMed] [Google Scholar]
- 17.Beck KE, Blansfield JA, Tran KQ, Feldman AL, Hughes MS, Royal RE, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24(15):2283–2289. doi: 10.1200/JCO.2005.04.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Downey SG, Klapper JA, Smith FO, Yang JC, Sherry RM, Royal RE, et al. Prognostic factors related to clinical response in patients with metastatic melanoma treated by CTL-associated antigen-4 blockade. Clin Cancer Res. 2007;13(22 Pt 1):6681–6688. doi: 10.1158/1078-0432.CCR-07-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchetti MC, Di Marco B, Cifone G, Migliorati G, Riccardi C. Dexamethasone-induced apoptosis of thymocytes: role of glucocorticoid receptor-associated Src kinase and caspase-8 activation. Blood. 2003;101(2):585–593. doi: 10.1182/blood-2002-06-1779. [DOI] [PubMed] [Google Scholar]
- 20.Mitchell CD, Richards SM, Kinsey SE, Lilleyman J, Vora A, Eden TO, et al. Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol. 2005;129(6):734–745. doi: 10.1111/j.1365-2141.2005.05509.x. [DOI] [PubMed] [Google Scholar]
- 21.Bianchi M, Meng C, Ivashkiv LB. Inhibition of IL-2-induced Jak-STAT signaling by glucocorticoids. Proc Natl Acad Sci U S A. 2000;97(17):9573–9578. doi: 10.1073/pnas.160099797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franchimont D, Galon J, Vacchio MS, Fan S, Visconti R, Frucht DM, et al. Positive effects of glucocorticoids on T cell function by up-regulation of IL-7 receptor alpha. J Immunol. 2002;168(5):2212–2218. doi: 10.4049/jimmunol.168.5.2212. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro-Oncology. 2008;10(1):10–18. doi: 10.1215/15228517-2007-035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheurer ME, Bondy ML, Aldape KD, Albrecht T, El-Zein R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol. 2008;116(1):79–86. doi: 10.1007/s00401-008-0359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crough T, Beagley L, Smith C, Jones L, Walker DG, Khanna R. Ex vivo functional analysis, expansion and adoptive transfer of cytomegalovirus-specific T-cells in patients with glioblastoma multiforme. Immunol Cell Biol. 2012;90(9):872–880. doi: 10.1038/icb.2012.19. [DOI] [PubMed] [Google Scholar]
- 26.Berghoff AS, Fuchs E, Ricken G, Mlecnik B, Bindea G, Spanberger T, et al. Density of tumor-infiltrating lymphocytes correlates with extent of brain edema and overall survival time in patients with brain metastases. Oncoimmunology. 2016;5(1):e1057388. doi: 10.1080/2162402X.2015.1057388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Broeren CP, Gray GS, Carreno BM, June CH. Costimulation light: activation of CD4+ T cells with CD80 or CD86 rather than anti-CD28 leads to a Th2 cytokine profile. J Immunol. 2000;165(12):6908–6914. doi: 10.4049/jimmunol.165.12.6908. [DOI] [PubMed] [Google Scholar]
- 28.Lanier LL, O'Fallon S, Somoza C, Phillips JH, Linsley PS, Okumura K, et al. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J Immunol. 1995;154(1):97–105. [PubMed] [Google Scholar]
- 29.Roederer M. Interpretation of cellular proliferation data: avoid the panglossian. Cytometry A. 2011;79(2):95–101. doi: 10.1002/cyto.a.21010. [DOI] [PubMed] [Google Scholar]
- 30.Zacharchuk CM, Mercep M, Chakraborti PK, Simons SS, Jr, Ashwell JD. Programmed T lymphocyte death. Cell activation- and steroid-induced pathways are mutually antagonistic. J Immunol. 1990;145(12):4037–4045. [PubMed] [Google Scholar]
- 31.Linsley PS, Nadler SG, Bajorath J, Peach R, Leung HT, Rogers J, et al. Binding stoichiometry of the cytotoxic T lymphocyte-associated molecule-4 (CTLA-4). A disulfide-linked homodimer binds two CD86 molecules. J Biol Chem. 1995;270(25):15417–15424. doi: 10.1074/jbc.270.25.15417. [DOI] [PubMed] [Google Scholar]
- 32.van der Merwe PA, Bodian DL, Daenke S, Linsley P, Davis SJ. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med. 1997;185(3):393–403. doi: 10.1084/jem.185.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11(12):852–863. doi: 10.1038/nri3108. [DOI] [PubMed] [Google Scholar]
- 34.Engelhardt JJ, Sullivan TJ, Allison JP. CTLA-4 overexpression inhibits T cell responses through a CD28-B7-dependent mechanism. J Immunol. 2006;177(2):1052–1061. doi: 10.4049/jimmunol.177.2.1052. [DOI] [PubMed] [Google Scholar]
- 35.Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woroniecka K, Chongsathidkiet P, Rhodin KE, Kemeny HR, Dechant CA, Farber SH, et al. T Cell Exhaustion Signatures Vary with Tumor Type and are Severe in Glioblastoma. Clin Cancer Res. 2018; 10.1158/1078-0432.CCR-17-1846. [DOI] [PMC free article] [PubMed]
- 37.Kaal EC, Vecht CJ. The management of brain edema in brain tumors. Curr Opin Oncol. 2004;16(6):593–600. doi: 10.1097/01.cco.0000142076.52721.b3. [DOI] [PubMed] [Google Scholar]
- 38.Min L, Hodi FS, Kaiser UB. Corticosteroids and immune checkpoint blockade. Aging (Albany NY) 2015;7(8):521–522. doi: 10.18632/aging.100797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gulley JL, Madan RA, Pachynski R, Mulders P, Sheikh NA, Trager J, et al. Role of antigen spread and distinctive characteristics of immunotherapy in Cancer treatment. J Natl Cancer Inst. 2017;109(4) 10.1093/jnci/djw261. [DOI] [PMC free article] [PubMed]
- 40.Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14(2):145–155. doi: 10.1016/S1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 41.Xia M, Gasser J, Feige U. Dexamethasone enhances CTLA-4 expression during T cell activation. Cell Mol Life Sci. 1999;55(12):1649–1656. doi: 10.1007/s000180050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xing K, Gu B, Zhang P, Wu X. Dexamethasone enhances programmed cell death 1 (PD-1) expression during T cell activation: an insight into the optimum application of glucocorticoids in anti-cancer therapy. BMC Immunol. 2015;16(39) 10.1186/s12865-015-0103-2. [DOI] [PMC free article] [PubMed]
- 43.Hinrichs CS, Palmer DC, Rosenberg SA, Restifo NP. Glucocorticoids do not inhibit antitumor activity of activated CD8+ T cells. J Immunother. 2005;28(6):517–524. doi: 10.1097/01.cji.0000177999.95831.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schartz NE, Farges C, Madelaine I, Bruzzoni H, Calvo F, Hoos A, et al. Complete regression of a previously untreated melanoma brain metastasis with ipilimumab. Melanoma Res. 2010;20(3):247–250. doi: 10.1097/CMR.0b013e3283364a37. [DOI] [PubMed] [Google Scholar]
- 45.Weber JS, Hodi FS, Wolchok JD, Topalian SL, Schadendorf D, Larkin J, et al. Safety profile of Nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol. 2017;35(7):785–792. doi: 10.1200/JCO.2015.66.1389. [DOI] [PubMed] [Google Scholar]
- 46.Jelsma R, Bucy PC. The treatment of glioblastoma multiforme of the brain. J Neurosurg. 1967;27(5):388–400. doi: 10.3171/jns.1967.27.5.0388. [DOI] [PubMed] [Google Scholar]
- 47.Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2(7):632–642. doi: 10.1158/2326-6066.CIR-14-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. T cell stimulated with αCD3/αCD28 microbeads proliferate in the presence of dexamethasone.Healthy donor T cells were cultured for four days with the indicated ratio of αCD3/αCD28 microbeads:total T cells in the presence of vehicle or dexamethasone. A, Representative flow cytometry plots of CellTrace violet dilution. Plots were derived from gated CD4 (top row) or CD8 (bottom row) T cells. B-D, Proliferation analyses of CD4 T cells (top) and CD8 T cells (bottom) performed on the samples shown in (A). Precursor Frequency (B), Expansion Index (C), and Proliferation Index (D) are shown. Samples were plated in duplicate and analyzed with an unpaired students T test. Data are representative of three independent experiments. (PDF 3563 kb)
Figure S2. A, Negatively-selected healthy donor T cells were cultured with the indicated microbeads and vehicle or dexamethasone. The percent of apoptotic CD4 (top) and CD8 (bottom) T cells was assessed by Annexin V/PI. Data are representative of four independent experiments. B, Lysates from healthy donor T cells incubated with the indicated microbeads and vehicle or dexamethasone were probed for the indicated proteins. GAPDH was used as a loading control. (PDF 693 kb)
Figure S3. T cell differentiation subsets formed during in vitro stimulation with αCD3/CD80 stimulation. Negatively-selected healthy donor T cells were cultured with 5 μg/mL αCD3 and the indicated concentration of CD80. T cell differentiation subsets were quantified following four days of culture. A, Flow plot of gating strategy to identify the indicated T cell differentiation subsets. B, Flow plots of CD4 (top) and CD8 (bottom) T cells cultured under the indicated conditions. (PDF 3995 kb)
Figure S4. Increased co-stimulation ameliorates the inhibitory effects of dexamethasone. Negatively-selected healthy donor T cells were cultured with 5 μg/mL αCD3 and increasing concentrations of CD80 in the presence of vehicle or dexamethasone. A-B. CD8 T cells cultured with vehicle (A) or dexamethasone (B). Flow cytometry plots showing proliferation of cells cultured with the indicated concentration of CD80 (left) and total numbers of naïve (TN), central memory (TCM), effector memory (TEM), and terminal effector (TTE) T cells following four days of culture (right) are shown. Differentiation subsets were assessed by CD45RO and CCR7 staining. Each condition was plated in duplicate, and data are representative of three independent experiments. Data were analyzed with an unpaired, two-tailed T Test. (PDF 2573 kb)
Figure S5 PD-1 blockade does not rescue dexamethasone-mediated proliferation defects. A, Flow cytometry analysis of PD-1 surface expression on CD4 (left) or CD8 (right) T cells stimulated with αCD3/αCD28 microbeads. Unstimulated (dashed line), stimulated in presence of vehicle (solid line), and stimulated in presence of dexamethasone (filled red line) are shown. B, Geometric median fluorescence intensity (gMFI) of PD-1 staining on CD4 or CD8 T cells. Cells cultured with vehicle (black bars) and dexamethasone (red bars) are shown. Data are an average of duplicate samples. C, Expression of PD-1 by qPCR of T cells stimulated in the presence of vehicle or dexamethasone. Data are representative of four independent experiments. D-E. Healthy donor T cells were stimulated for four days in the presence of vehicle or dexamethasone and nivolumab or ipilimumab F(ab’)2 antibody as indicated. Precursor frequency of CD4 and CD8 T cells was quantified by FlowJo. The ratio of dexamethasone to vehicle for CD4 (C) and CD8 (D) T cells is shown. All samples were plated in duplicate and the ratios were analyzed with a one-way ANOVA. Data are representative of n = 4 healthy donors. (PDF 2522 kb)
Figure S6 CTLA-4 blockade does not rescue dexamethasone pre-treated mice. A, Schema of survival experiment. Albino C57Bl/6 mice received intracranial implantation of GL261 ffluc-mCherry glioma cells. Dexamethasone was initiated one day prior to tumor implantation (dex (D-1)) or one week following tumor implantation (dex (D7)). CLTA-4 blockade or isotype antibody were injected on days 13, 16, and 19 following tumor implantation. Mice were randomized on day 6 following tumor implantation into groups of equivalent tumor luminescence. B, Kaplan Meier survival curves of mice receiving the indicated treatments. n = 8 to 9 mice per cohort. Data are representative of two independent experiments. (PDF 1525 kb)
Figure S7. Quantification of Treg and checkpoint molecules in tumor-bearing mice. GL261 ffluc-mCherry tumor-bearing mice were randomized into the indicated cohorts based on bioluminescence values from tumor. Vehicle or dexamethasone treatment was initiated on day 7, and isotype or CTLA-4 blocking antibody were administered on days 13, 16, and 19 following tumor implantation. Mice were euthanized on day 23 and tissues were harvested for flow cytometry analysis. A, Treg cell number from tumor-bearing brain hemisphere (left; n = 8) or the cervical tumor-draining lymph nodes (right; n = 10). B, The percentage of CD4 (top two plots) or CD8 (bottom two plots) T cells expressing the indicated checkpoint molecules. Co-expression of molecules was quantified using a Boolean gating strategy. Data were analyzed using a unpaired students T test. (PDF 1891 kb)
Data Availability Statement
All data analyzed are included in this article and additional information is available upon request.