

Pathogen-induced host plasma membrane damage is a recently recognized mechanism used by pathogens to promote their entry into host cells. We identified key transducers activated upon host cell perforation by the pore-forming toxin LLO to promote Listeria entry. This pathway is distinct from the pathway that reseals the toxin-perforated cell.
Abstract
Host cell invasion is an indispensable step for a successful infection by intracellular pathogens. Recent studies identified pathogen-induced host cell plasma membrane perforation as a novel mechanism used by diverse pathogens (Trypanosoma cruzi, Listeria monocytogenes, and adenovirus) to promote their internalization into target cells. It was concluded that T. cruzi and adenovirus damage the host cell plasma membrane to hijack the endocytic-dependent membrane resealing machinery, thereby invading the host cell. We studied L. monocytogenes and its secreted pore-forming toxin listeriolysin O (LLO) to identify key signaling events activated upon plasma membrane perforation that lead to bacterial internalization. Using various approaches, including fluorescence resonance energy transfer imaging, we found that the influx of extracellular Ca2+ subsequent to LLO-mediated plasma membrane perforation is required for the activation of a conventional protein kinase C (cPKC). cPKC is positioned upstream of Rac1 and the Arp2/3 complex, which activation leads to F-actin–dependent bacterial internalization. Inhibition of this pathway did not prevent membrane resealing, revealing that perforation-dependent L. monocytogenes endocytosis is distinct from the resealing machinery. These studies identified the LLO-dependent endocytic pathway of L. monocytogenes and support a novel model for pathogen uptake promoted by plasma membrane injury that is independent of membrane resealing.
INTRODUCTION
Intracellular pathogens use a large repertoire of virulence factors to subvert host cell machineries, thereby ensuring their life cycle and propagation within the infected host. An initial and indispensable step is the internalization of the pathogen into host cells (Cossart and Sansonetti, 2004; Cossart and Helenius, 2014). The present study elucidated a signaling pathway underlying a unique mechanism of pathogen uptake by host cells, in which pathogens damage the host cell plasma membrane to promote their internalization.
The process of pathogen internalization into host cells can be passive from the perspective of the pathogen when a professional phagocyte uses its phagocytic receptors to engulf the pathogen (Brumell and Grinstein, 2003; Groves et al., 2008; Stow and Condon, 2016). Alternatively, the uptake process can be active when pathogens express virulence factors that hijack the host cell endocytic pathways (Cossart and Sansonetti, 2004; Ham et al., 2011; Ribet and Cossart, 2015; Pizarro-Cerda et al., 2016). Although there is a high diversity of pathogens, they evolved common strategies to actively promote their internalization into host cells. Some pathogens use surface invasins that specifically activate a host receptor-mediated endocytic pathway (Isberg, 1990; Seveau et al., 2007; Mercer and Helenius, 2012; Ribet and Cossart, 2015). Other pathogens use a secretion apparatus that injects effectors into the host cell cytosol. These effectors directly subvert signaling pathways that orchestrate the endocytic uptake of the pathogen (Brumell and Grinstein, 2003; Galan, 2009; Velge et al., 2012; Mellouk and Enninga, 2016). Recent studies propose an additional invasion strategy that involves host cell plasma membrane perforation. Indeed, unrelated pathogens such as the eukaryotic parasite Trypanosoma cruzi, the bacterial pathogen Listeria monocytogenes, and the adenovirus HAdV-C2 perforate the host cell plasma membrane to promote their internalization into nonphagocytic cells (Fernandes et al., 2011; Vadia et al., 2011; Vadia and Seveau, 2014; Luisoni et al., 2015). Some pathogens express several invasion factors and use multiple mechanisms to promote their internalization. Collectively, cooperation between these factors may increase the efficiency of host cell invasion. For example, to enter host cells Salmonella uses the surface invasin Rck and injects effectors via a type III secretion machinery (T3SS) (Rosselin et al., 2010; Velge et al., 2012; Wiedemann et al., 2016). Adenoviruses can be internalized into host cells as a result of host receptor-mediated endocytosis and/or of plasma membrane injury (Bergelson et al., 1997; Loustalot et al., 2015; Luisoni et al., 2015).
A particularity of the Gram-positive foodborne pathogen L. monocytogenes is its ability to infect a large diversity of cells including cells that are normally nonphagocytic such as enterocytes, hepatocytes, cytotrophoblasts, and neurons (Vazquez-Boland et al., 2001). Therefore, it is not surprising that this pathogen uses multiple invasins and invasion strategies. L. monocytogenes expresses the surface invasins InlA and InlB to promote its internalization into cells that express the internalins receptors, E-cadherin and c-Met, respectively (Seveau et al., 2004 , 2007). Additionally, the secreted pore-forming toxin listeriolysin O (LLO) perforates the host cell plasma membrane to promote L. monocytogenes internalization into epithelial cells (Vadia et al., 2011; Vadia and Seveau, 2014). LLO is a member of the MACPF/CDC (membrane attack complex perforin/cholesterol-dependent cytolysins) superfamily, which includes numerous eukaryotic pore-forming proteins and the largest family of bacterial pore-forming toxins, the CDCs (Rosado et al., 2008; Seveau, 2014). CDCs form large β-barrel pores across cholesterol-containing membranes and are major virulence factors produced by Gram-positive pathogens. The CDC LLO and intermedilysin O (ILO), which is secreted by Streptococcus intermedius, induce bacterial internalization into human hepatocytes (Sukeno et al., 2005; Vadia et al., 2011).
Although numerous pathways for host receptor- and T3SS effector-mediated pathogen internalization have been extensively studied, less is known about the molecular machineries that link plasma membrane perforation to pathogen internalization. Recent studies have proposed that the pathway involved in resealing the damaged plasma membrane is directly responsible for pathogen uptake (Fernandes et al., 2011; Andrews et al., 2014; Luisoni et al., 2015). Animal cells are frequently wounded due to numerous physical and biological stressors. For example, cells that compose contractile tissues, including those present in skeletal muscles or the intestinal tissue, are subject to mechanical stress. Cells throughout the body may also be exposed to pore-forming toxins produced by pathogens or by membrane-damaging agents released by the immune system. Owing to the frequency of these traumatic events and the inherent fragility of lipid bilayers, animal cells are able to rapidly reseal their plasma membrane to avoid necrosis or programmed cell death pathways. Several resealing mechanisms have been proposed including endocytosis of the damaged membrane (Tam et al., 2010; Jimenez and Perez, 2017). It was proposed that the endocytic machinery that promotes membrane resealing is hijacked by pathogens such as T. cruzi and adenovirus to gain entry into host cells (Fernandes et al., 2011; Luisoni et al., 2015).
The present studies were designed to 1) identify key signaling events activated downstream from LLO-induced host cell plasma membrane perforation that mediate L. monocytogenes internalization and 2) delineate the potential involvement of membrane resealing in this signaling pathway. Together, our studies identified a novel endocytic pathway of L. monocytogenes and support a model for damage-dependent pathogen uptake that is independent of membrane resealing.
RESULTS
Rac1 is required for LLO-mediated internalization of L. monocytogenes
We previously showed that LLO pore formation was sufficient to induce F-actin–dependent L. monocytogenes internalization into epithelial cells (Vadia et al., 2011). Because the Rho GTPases Rac1, Cdc42, and RhoA are master regulators of F-actin remodeling during bacterial internalization, we first established whether they were involved in the LLO-mediated entry pathway (Cox et al., 1997; Etienne-Manneville and Hall, 2002; Sit and Manser, 2011). Expression of Rac1, RhoA, and Cdc42 was knocked down in human hepatocytes (HepG2) via treatment with silencing RNAs (siRNAs) for 48 h. The resulting knockdown efficiencies were assessed in each experiment by Western blot analysis of Rac1, RhoA, or Cdc42 in nontreated and siRNA-treated cells (Figure 1A). For each experiment, Rac1 protein level was decreased by 50–85% relative to negative control cells (NC) treated with NC siRNA, and RhoA and Cdc42 protein levels were decreased by more than 87% relative to NC cells (Figure 1A). Additionally, because these Rho GTPases can have overlapping activities, such as Rac1 and Cdc42 in the formation of lamellipodia (Hall, 2005), we also verified that silencing of one Rho GTPase did not result in a compensatory overexpression of the other two (Figure 1B). Densitometry analysis of the Western blots indicated that cell treatment with Rho GTPase-specific siRNAs does not result in compensatory expression of the two other Rho GTPases when compared with cells treated with NC siRNA (Supplemental Table S1). To determine the importance of these Rho GTPases for LLO-dependent internalization, nontreated and siRNA-treated cells were incubated with wild-type L. monocytogenes (WT, 10403s) or its isogenic LLO-deficient mutant (Δhly) for 30 min at 37°C, and bacterial internalization (entry) efficiency was measured by immunofluorescence microscopy (Haghighat and Seveau, 2010). In all treatment conditions, entry efficiencies of the LLO-deficient bacteria were less than 14% relative to entry of WT bacteria in NC siRNA-treated cells, further verifying that L. monocytogenes internalization is LLO dependent in HepG2 cells (Vadia et al., 2011). Also, there was no significant difference in bacterial entry between the nontreated and NC siRNA-treated cells, showing that cell transfection did not alter bacterial uptake (Figure 2A). Knocking down Rac1 reduced entry of WT bacteria by 51.3 ± 3.2% (p < 0.01), whereas knocking down RhoA or Cdc42 did not significantly affect entry (Figure 2A). To further demonstrate the role of Rac1 in the LLO-mediated entry pathway in the absence of any other virulence factors, we measured the entry of 1-µm polystyrene beads coated with purified recombinant LLO. Beads were covalently coated with bovine serum albumin (BSA) followed by a noncovalent adsorption of LLO to mimic the release of LLO by bacteria (Vadia et al., 2011). BSA-coated beads were used as a negative control. Nontreated and cells treated with NC-, Rac1-, RhoA-, and Cdc42-siRNAs were incubated with BSA- or BSA/LLO-coated beads for 30 min at 37°C and bead entry efficiency was measured by immunofluorescence microscopy. Relative to the NC siRNA treatment, Rac1-siRNA-treated cells exhibited a 61.8 ± 0.1% decrease in LLO-coated bead entry (p < 0.005). There was no statistically significant difference between RhoA and Cdc42 knockdown conditions relative to the NC siRNA-treated cells (Figure 2B).
FIGURE 1:

Rac1, RhoA, and Cdc42 knockdown efficiencies. (A, B) Western blot analysis of Rac1, RhoA, and Cdc42 in nontreated HepG2 cells and in HepG2 cells treated with a negative control siRNA (NC) or with Rac1-, RhoA-, or Cdc42-specific siRNAs. (A) Western blot analysis of cell lysates (nondiluted, 1:2, 1:4, and 1:8 diluted) using anti-Rac1, -RhoA, -Cdc42, and -actin antibodies. (B) Western blot analysis of nondiluted cell lysates to verify the absence of compensatory expression of Rac1 (top), RhoA (middle), or Cdc42 (bottom) in cells treated with Rac1-, RhoA-, and Cdc42-siRNAs. All Western blots are representative of at least three independent experiments.
FIGURE 2:

Role of Rac1 in LLO-dependent entry of L. monocytogenes. (A, B) Nontreated HepG2 cells and HepG2 cells treated with negative control (NC), Rac1-, RhoA-, and Cdc42-siRNAs were incubated with WT (10403s) or the LLO deficient (Δhly) L. monocytogenes (A), or with BSA or BSA/LLO-coated beads (B), at a multiplicity of infection 20 (MOI 20) for 30 min at 37°C. (C) HepG2 cells expressing mCit-Rac1 or dominant negative mCit-Rac1N17 were incubated with BSA/LLO-coated beads at MOI 5 for 30 min at 37°C. Cells were then fixed and bacteria or beads were fluorescently labeled to enumerate the total number of bacteria (Nt) and the number of extracellular bacteria associated with host cells (Ne) (A, B, C). Entry efficiency was measured as 100 × [(Nt)-(Ne)]/(Nt) and results are expressed as the percentage entry ± SEM relative to NC-siRNA-treated cells incubated with WT (A) or BSA/LLO-coated polystyrene beads (B) (n ≥ 3). In C, results are expressed as the percentage entry ± SEM relative to mCit-Rac1 expressing cells incubated with BSA/LLO-coated beads (n = 4). Statistics are as follows: *p < 0.05; **p < 0.01; ***p < 0.005.
On guanosine nucleotide exchange, the Rho GTPases undergo a conformational change into an active GTP-bound state, which permits their reversible association with downstream effectors such as p21 activated kinases (PAK) (Van Aelst and D’Souza-Schorey, 1997). We assessed whether Rac1 activation was required for the LLO-mediated entry pathway by utilizing a Rac1 dominant negative variant (Rac1N17) that is unable to bind GTP. HepG2 cells were transfected to express the fluorescent chimeras (monomeric Citrine) of native Rac1 (mCit-Rac1) or the dominant negative Rac1 variant (mCit-Rac1N17) (Hoppe and Swanson, 2004). Entry of BSA/LLO-coated beads into transfected, fluorescent cells expressing mCit-Rac1N17 was 74.6 ± 0.1% lower than cells expressing the native form of Rac1 (p < 0.05) (Figure 2C). Collectively, these data show that Rac1, but not RhoA or Cdc42, plays a crucial role in the LLO-mediated entry pathway of L. monocytogenes.
Rac1 activation by LLO is pore and Ca2+ influx dependent
To confirm that LLO is sufficient to activate Rac1, we monitored the spatiotemporal dynamics of Rac1 activation in living cells using a fluorescence resonance energy transfer– (FRET) based fluorescence microscopy method (Hoppe et al., 2002; Hoppe and Swanson, 2004). HepG2 cells cotransfected to express the FRET-pair mCit-Rac1 and monomeric cyan fluorescent protein–p21-binding domain (mCFP-PBD) were imaged in an atmosphere-controlled, wide-field fluorescence microscope. In this system, Rac1 activation leads to the formation of the complex mCit-Rac1/mCFP-PBD, resulting in the generation of a FRET signal that can be analyzed quantitatively using the FRET stoichiometry method (Hoppe et al., 2002; Hoppe and Swanson, 2004) (Supplemental Figure S1 and Supplemental Movie S1). Cells that coexpressed mCFP-PBD and mCit-Rac1 were imaged for 5 min before the addition of physiologically relevant concentrations of LLO (0.1 nM). Importantly, this toxin concentration is nonlethal and sufficient to induce the entry of LLO-deficient L. monocytogenes and BSA-coated beads when LLO is added exogenously to the cell culture medium (Vadia, Arnett et al., 2011). LLO-exposed cells exhibited elevated levels of Rac1 activation, as evidenced by transient FRET flashes that paralleled the appearance and disappearance of membrane ruffles in successive waves at different cellular locations (Figure 3A and Supplemental Movie 1). Because Rac1 was activated only within discrete cell regions in a transient manner, we quantified Rac1 FRET in regions of standard size that were positioned where Rac1 activation was observed or positioned arbitrarily if Rac1 activation was absent. Two regions of standard size were positioned on each cell and were analyzed whether Rac1 FRET fluctuations were observed. Data show that Rac1 activation was detected in 76.61% of cells exposed to LLO (Table 1; corresponding statistical analyses in Supplemental Table S2). Rac1 activation was defined as an at least twofold increase in FRET efficiency within the analyzed region (FRET-positive region). In contrast, only 2.43% of control cells (not exposed to LLO) were positive for Rac1 FRET. Rac1 activation kinetics measured in all regions and kinetics of Rac1 activation averaged across all regions are presented in Figure 3, B–D. This analysis showed that at the cell population level, Rac1 was activated ∼40 s after addition of LLO with a maximal amplitude at 80–120 s for a duration of 2 min. Statistical analyses of the FRET kinetics presented in Figure 3 are presented in Supplemental Table S3. To establish the role of LLO pore formation in Rac1 activation, cells were exposed to an LLO variant (listeriolysin O prepore lock, LLOpL) that binds to the host cell plasma membrane and forms a prepore complex but is unable to transition into the transmembrane β-barrel pore (Hotze et al., 2001; Vadia et al., 2011). Indeed, LLOpL was shown to be inactive at concentrations exceeding 104 nM as compared with native LLO (EC50 = 1 nM), but its pore-forming activity was restored to the native LLO level upon treatment with 4 mM dithiothreitol (DTT) (Supplemental Figure S2A) (Seveau, 2014). Treatment with 0.1 and 10 nM LLOpL failed to activate Rac1 in HepG2 cells (Table 1; statistical analyses in Supplemental Table S2). Thus, as reported for LLO-dependent L. monocytogenes entry and membrane ruffling, Rac1 activation requires LLO pore formation (Vadia et al., 2011). Formation of the transmembrane pore leads to an influx of extracellular Ca2+, which is required for LLO-mediated L. monocytogenes entry (Vadia and Seveau, 2014). To prevent Ca2+ influx, cells were analyzed in Ca2+-free medium. Because the influx of Ca2+ is indispensable for membrane resealing, for in the absence of extracellular Ca2+ cell integrity is rapidly compromised upon damage, FRET analysis was limited to time points 0–5 min post–LLO exposure (Vadia and Seveau, 2014). In the absence of extracellular Ca2+, LLO-induced Rac1 activation occurred in 5.68% of cells compared with 74.83% in the presence of extracellular Ca2+ (Table 1; statistical analyses in Supplemental Table S2). Collectively, these data establish that LLO is sufficient to activate Rac1 within membrane ruffles in a pore- and Ca2+-influx–dependent manner.
FIGURE 3:

FRET stoichiometry to monitor Rac1 activation by LLO. HepG2 cells coexpressing mCFP-PBD and mCit-Rac1 were imaged on the microscope stage at 37°C. Phase-contrast (PC), and fluorescence images (IA, ID, and IF) were acquired every 20 s for 20 min. LLO was added after 5 min of imaging (T0). (A) PC, overlay of PC and IA, IA, ID, IF, and FRET (EAVE presented in pseudocolor scale). Scale bar = 10 μm. From top to bottom: time points 0, 3, and 5 min after addition of LLO. (B, C) For each imaged cell, two regions of standard size (305 pixels) were applied at locations where FRET variations were observed. If no FRET variation was observed, then the regions were arbitrarily positioned. The y-axis indicates the fold change in EAVE and the z-axis represents all individual regions of control cells (182 regions, 91 cells) (B), and all FRET-positive regions from the LLO-exposed cells (92 regions, 57 cells) (C), in which FRET positive is defined as a greater than twofold increase in FRET efficiency (n = 3). (D) Kinetics of the averaged EAVE of control (182) and LLO-exposed (92) regions. The black vertical line indicates the time of addition of medium alone (control) or 0.1 nM LLO.
TABLE 1:
LLO induces Rac1 activation in a Ca2+- and pore-dependent manner.
| Toxin | – | LLO | LLOpL (0.1 nM) | LLOpL (10 nM) | – | LLO | – | LLO |
|---|---|---|---|---|---|---|---|---|
| Medium (CaCl2) | + | + | + | + | + | + | – | – |
| Time (min) | 20 | 20 | 20 | 20 | 10 | 10 | 10 | 10 |
| NC | 91 | 75 | 164 | 148 | 91 | 75 | 125 | 173 |
| FRET+ cells | 3 | 57 | 9 | 12 | 0 | 56 | 8 | 10 |
| % FRET | 2.43 | 76.61 | 5.88 | 7.22 | 0 | 74.83 | 4.78 | 5.68 |
Cells were exposed, or not, to 0.1 nM LLO, 0.1 nM LLOpL, and 10 nM LLOpL in cell culture medium with (+) or without (−) CaCl2. Time indicates the total duration of imaging. In Ca2+-free medium (−) cells were imaged for only 10 min due to cell damages caused by LLO. NC is the number of analyzed cells. FRET+ cells is the number of FRET-positive cells (exhibiting at least twofold increase in FRET). % FRET is the percentage of FRET-positive cells (n > 3).
LLO-mediated Rac1 activation results in Arp2/3-dependent F-actin remodeling
The subcortical F-actin cytoskeleton acts as a dynamic scaffold that promotes the formation of membrane ruffles and the formation of the phagocytic cup (Rougerie et al., 2013). We used a live-cell fluorescence imaging approach to visualize F-actin rearrangement in Tomato-β-actin-expressing HepG2 cells. We could clearly visualize the formation of F-actin bundles within membrane ruffles in cell exposed to LLO (Figure 4 and Supplemental Movie 2). We observed by live-cell phase-contrast imaging that 85% of cells exposed to LLO initiated membrane ruffling as early as 84.8 ± 26.7 s (average ± SD; >100 cells, n = 8) following exposure to LLO. Additionally, LLO-induced membrane ruffling was frequently followed by the formation of large endocytic vacuoles reminiscent of macropinosomes (Supplemental Movie 3). F-actin polymerization can involve two types of actin nucleators: formins and the Arp2/3 complex (Chesarone and Goode, 2009). Because Arp2/3 is most commonly involved in remodeling F-actin at bacterial entry sites, we focused on establishing the role of the Arp2/3 complex in HepG2 cells exposed to LLO in the presence or absence of the Arp2/3 inhibitor CK-666 (Hetrick et al., 2013). Inhibition of Arp2/3 abolished LLO-induced membrane ruffling, as assessed by live cell phase-contrast imaging (0% of cells exhibited membrane ruffling; >100 cells, n = 3) as compared with control cells (85%, >100 cells, n = 3; Supplemental Movies 4 and 5). Therefore, LLO induces Arp2/3-dependent de novo F-actin remodeling and subsequent membrane ruffling. To establish whether Rac1 activation occurs upstream from Arp2/3-dependent F-actin remodeling, cells were transfected to express fluorescent chimeras of the native (mCit-Rac1)- or dominant-negative (mCit-Rac1N17)-Rac1 or fluorophore alone (mCit). In cells expressing the fluorophore alone (126 cells, n = 3) or mCit-Rac1 (112 cells, n = 3) 75.4% and 58.04% of cells exhibited membrane ruffling in response to LLO, respectively. However, cells expressing mCit-Rac1N17 (114 cells, n = 3) exhibited substantial decrease in membrane ruffling upon LLO exposure (6.14% ruffling). As expected, nonfluorescent neighboring cells that did not express mCit-Rac1N17 exhibited membrane ruffling, further supporting the hypothesis that Rac1 activation is a prerequisite for LLO-induced membrane ruffling (Supplemental Movie 6). Importantly, we also established that the Arp2/3 complex is required for L. monocytogenes entry into HepG2 cells using the Arp2/3 inhibitors CK-636 and CK-666. Figure 5 shows the dose-dependent inhibition of L. monocytogenes entry by the Arp2/3 inhibitors, indicating that the same machinery that is driving LLO-mediated F-actin–dependent remodeling is also necessary for L. monocytogenes internalization. Collectively, our data indicate that LLO-mediated entry involves Rac1 activation upstream from Arp2/3-dependent F-actin remodeling.
FIGURE 4:
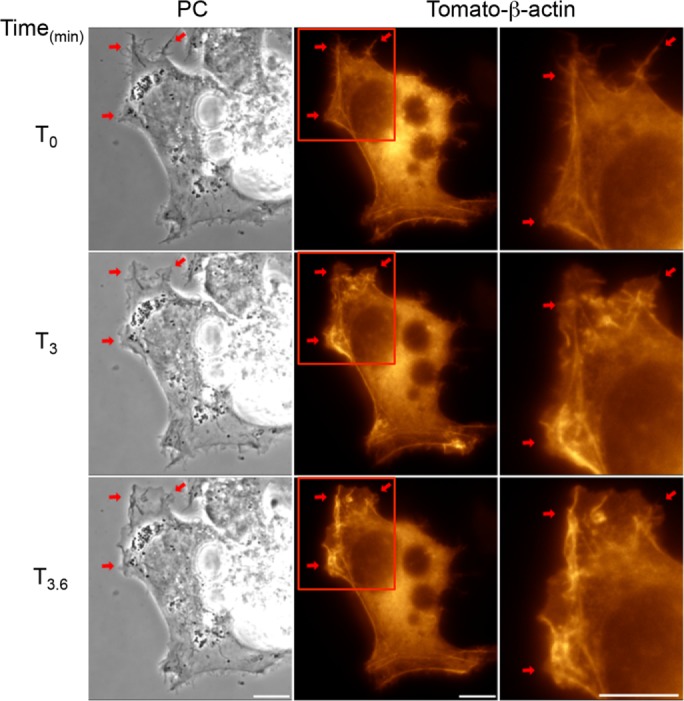
LLO induces F-actin rearrangement. HepG2 cells expressing Tomato-β-actin were imaged on the microscope stage at 37°C. Phase-contrast (PC, column 1), and fluorescence images were acquired every 20 s for 20 min. LLO was added after 5 min of imaging (T0). LLO induces dynamic F-actin rearrangement (red arrows) within membrane ruffles. Regions highlighted in column 2 are shown in column 3. Scale bar = 10 μm.
FIGURE 5:
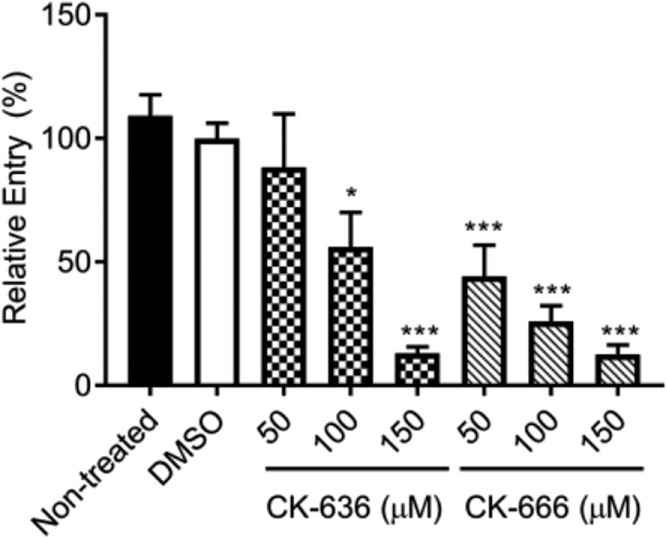
Role of Arp2/3 in LLO-dependent entry of L. monocytogenes. Nontreated and cells treated with 0.2% DMSO or the Arp2/3 inhibitors CK-636 and CK-666 were infected with WT L. monocytogenes at MOI 20 for 30 min at 37°C. Cells were fixed, and bacteria were fluorescently labeled to enumerate the total number of bacteria (Nt) and the number of extracellular bacteria associated with host cells (Ne). Entry efficiency was measured as 100 × [(Nt) - (Ne)]/(Nt), and results are expressed as the mean percentage entry ± SEM relative to the DMSO control. Statistics are as follows: *p < 0.05; ***p < 0.005 (n = 3).
A conventional PKC is required for LLO-mediated entry of L. monocytogenes
The conventional Ca2+-responsive PKC alpha (PKCα) has been shown to be involved in Rac1 translocation to the plasma membrane of cells treated with thapsigargin or ionomycin, which, similarly to LLO, cause an increase in cytosolic Ca2+ (Price et al., 2003). To determine whether a PKC is involved in L. monocytogenes internalization into HepG2 cells, we measured L. monocytogenes entry into cells pretreated with broad spectrum (Gö6983) and conventional (GF109203x) PKC inhibitors. Treatment with 0.5 µM Gö6983 or 1 µM GF109203x resulted in a 59.9 ± 1.5% (p < 0.05) and 61.5 ± 1.9% (p < 0.01) decrease in L. monocytogenes entry, respectively, relative to the vehicle control, indicating a role for a conventional PKC (Figure 6).
FIGURE 6:
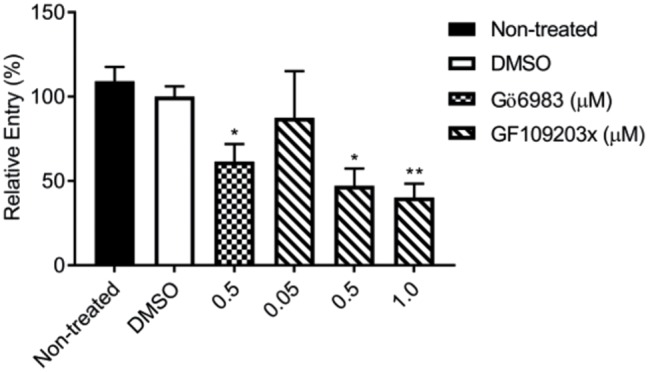
Role of a conventional PKC in LLO-dependent entry of L. monocytogenes. Nontreated and cells treated with 0.2% DMSO, the broad spectrum (Gö6983) or conventional (GF109203x) PKC inhibitors, were infected with WT L. monocytogenes at MOI 20 for 30 min at 37°C. Cells were chemically fixed, and bacteria were fluorescently labeled to enumerate the total number of bacteria (Nt) and the number of extracellular bacteria associated with host cells (Ne). Entry efficiency was measured as 100 × [(Nt) - (Ne)]/(Nt), and results are expressed as the mean percentage entry ± SEM relative to the DMSO control. Statistics are as follows: *p < 0.05; **p < 0.01 (n = 3).
A conventional PKC is activated by the influx of extracellular Ca2+ leading to Rac1 activation
We then focused on demonstrating the activation of a PKC by LLO. We transfected HepG2 cells with fluorescent chimeras of two conventional (α, βII) and two novel (ε, δ) PKC isoforms. HepG2 cells exposed to LLO exhibited dynamic translocation of GFP-PKCα (translocation was observed in 70.8% of transfected cells; 250 cells, n = 7; Figure 7 and Supplemental Movie 7), GFP-PKCε (63.63%; 22 cells, n = 1), and GFP-PKCβII (37.5%; 24 cells, n = 1), but not of GFP-PKCδ (0%; 65 cells, n = 2) or GFP alone (0%; 32 cells, n = 1; Supplemental Movie 7) to regions of plasma membrane ruffles as early as 40 s after addition of LLO. We further established that the influx of extracellular Ca2+ was indispensable for PKC translocation, as observed in cells exposed to LLO in Ca2+-free medium (0% of GFP-PKCα translocation, 23 cells, n = 1; Supplemental Movie 8). Importantly, cells treated with the conventional PKC inhibitor GF109203x did not undergo F-actin–dependent membrane ruffling in response to LLO exposure (3.5% of cells exhibited membrane ruffling, >100 cells, n = 3), suggesting that a Ca2+-responsive conventional PKC was involved in Rac1 activation. To demonstrate that LLO activates a PKC, we used the PKC FRET biosensor myristoylated and palmitoylated c kinase activity reporter (MyrPalm-CKAR), which has a 20% dynamic range and was designed to specifically assess PKC activity at the plasma membrane (Violin et al., 2003; Gallegos and Newton, 2011). In the nonphosphorylated state, MyrPalm-CKAR exists in a constrained conformation that places the N-terminal mCFP in close proximity to the C-terminal mCit leading to FRET, but phosphorylation of the interconnecting sequence by any PKC isoform relaxes the sensor leading to a loss of FRET. We tested whether addition of the Ca2+ ionophore, ionomycin, which is known to activate PKCs, would lead to pronounced phosphorylation of the biosensor. As expected, addition of ionomycin resulted in a sudden drop in FRET efficiency followed by a quick recovery (Supplemental Figure S3). This result is consistent with the notion that Ca2+ influx leads to activation of PKC at the plasma membrane followed by dephosphorylation of the biosensor thereafter, as previously observed in mouse oocytes (Gonzalez-Garcia et al., 2013). HepG2 cells expressing MyrPalm-CKAR and exposed to LLO exhibited an initial drop in FRET, that is, MyrPalm-CKAR phosphorylation, 40 s after addition of LLO, which overlays with the kinetics of PKCα translocation and Rac1 activation (Figure 8, Supplemental Movie 9, and statistical analyses in Supplemental Table S4). The magnitude of change in FRET efficiency in LLO-exposed cells is 65% lower than observed in ionomycin-treated cells, but the duration of PKC activity is prolonged, which may be due to smaller and prolonged Ca2+ fluxes in LLO-exposed cells. Importantly, LLO-induced MyrPalm-CKAR phosphorylation was inhibited by cell pretreatment with the conventional PKC inhibitor GF109203x (Figure 8). We then established whether a conventional PKC was involved in Rac1 activation. Rac1 activation was measured by FRET stoichiometry as presented in Figure 3. We found that Rac1 was no longer activated by LLO in cells pretreated with the conventional PKC inhibitor GF109203x (Table 2; statistical analyses in Supplemental Table S5). Collectively, our data show that a conventional PKC links Ca2+ influx to Rac1 activation and Arp2/3-dependent F-actin remodeling for bacterial internalization.
FIGURE 7:
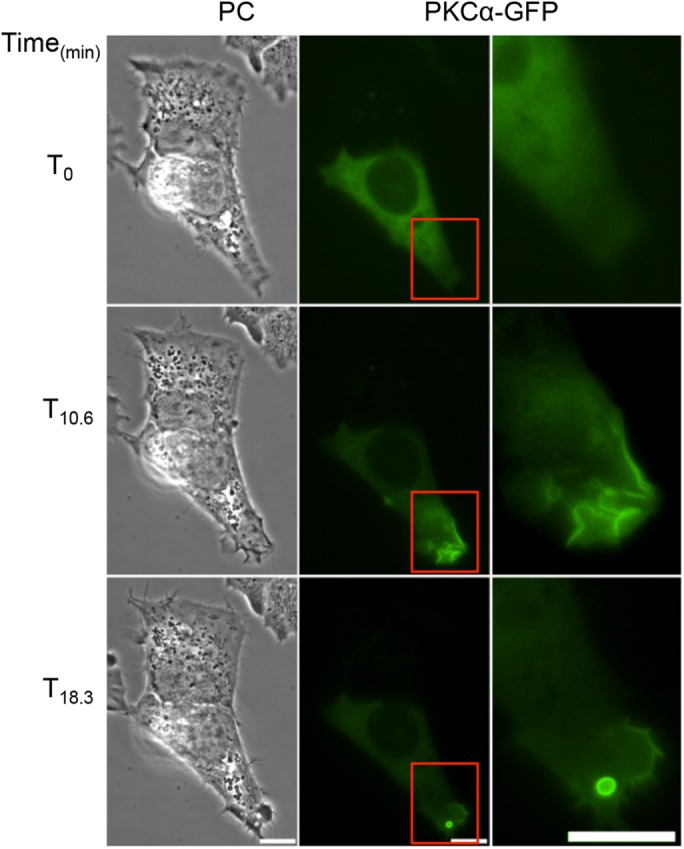
LLO induces PKCα translocation to the plasma membrane. HepG2 cells expressing GFP-PKCα were imaged on the microscope stage at 37°C. Phase-contrast (PC), and fluorescence images were acquired every 20 s for 20 min. LLO was added after 5 min of imaging (T0). LLO induces GFP-PKCα translocation to regions of dynamic plasma membrane ruffles and around newly formed vesicles. Regions highlighted in column 2 are shown in column 3. Scale bar = 10 μm.
FIGURE 8:

A PKC is activated by LLO. HepG2 cells expressing MyrPalm-CKAR were imaged on the microscope stage at 37°C. Phase-contrast (PC) and fluorescence images (IA, ID, and IF) were acquired every 20 s for 20 min. LLO was added after 5 min of imaging (T0 and black vertical line). (A) A representative cell exhibiting LLO-induced MyrPalm-CKAR phosphorylation. From left to right: PC, overlay of PC + IA, IA, ID, and IF, and FRET (in pseudo-color scale). From top to bottom: time points 0, 1, and 5 min after addition of LLO. Scale bar = 10 μm. (B) Data are presented as the average fold change in EAVE ± SEM (n = 3, 24 cells in DMSO control, 26 cells in GF109203x, 28 cells in DMSO +LLO, and 26 cells in GF109203x +LLO).
TABLE 2:
Rac1 is activated downstream of a conventional PKC.
| Toxin | — | LLO | LLO |
|---|---|---|---|
| Medium | DMSO | DMSO | GF109203x |
| Time (min) | 20 | 20 | 20 |
| NC | 115 | 92 | 136 |
| FRET+ cells | 1 | 50 | 26 |
| % FRET | 0.87 | 54.3 | 19.12 |
Cells were exposed to DMSO (0.2%), 0.1 nM LLO, or GF109203x. Time indicates the total duration of imaging. NC is the number of analyzed cells. FRET+ cells is the number of FRET-positive cells (exhibiting at least a twofold increase in FRET). % FRET is the percentage of FRET-positive cells (n > 3).
Inhibition of the LLO-mediated entry pathway facilitates membrane resealing
We previously showed that cells exposed to physiological concentrations of LLO undergo rapid Ca2+-dependent membrane resealing (Vadia and Seveau, 2014). To investigate if LLO-mediated L. monocytogenes entry pathway is part of the membrane resealing machinery, we measured the efficiency of membrane resealing in cells pretreated with the inhibitors that prevent LLO-induced L. monocytogenes entry (Pathak-Sharma et al., 2017). HepG2 cells were incubated with inhibitors of conventional PKC, Arp2/3, or F-actin and exposed to LLO. As a control, inhibition of membrane resealing was achieved by incubating cells in Ca2+-free medium. These experiments were performed in the presence of the plasma membrane-impermeant nucleic acid–binding probe propidium iodide. Propidium iodide flows through damaged cell membranes, leading to a strong, quantifiable fluorescence signal, whereas cells with intact plasma membranes exclude the probe or are minimally labeled. We found that HepG2 cells treated with the conventional PKC inhibitor, the Arp2/3 inhibitor, or the F-actin inhibitor and exposed to LLO, resealed their plasma membrane more efficiently than cells treated with LLO (+ vehicle) in the absence of inhibitors (Figure 9). Indeed, statistical analyses revealed that the presence of the inhibitors improved cell integrity (p < 0.0001). Importantly, LLO pore-forming activity was not altered by any of these inhibitors (Supplemental Figure S2b). In conclusion, LLO pore formation was followed by F-actin–independent membrane resealing and by F-actin–dependent bacterial entry. Both processes were initiated by the influx of extracellular Ca2+, but involved distinct signaling pathways thereafter.
FIGURE 9:
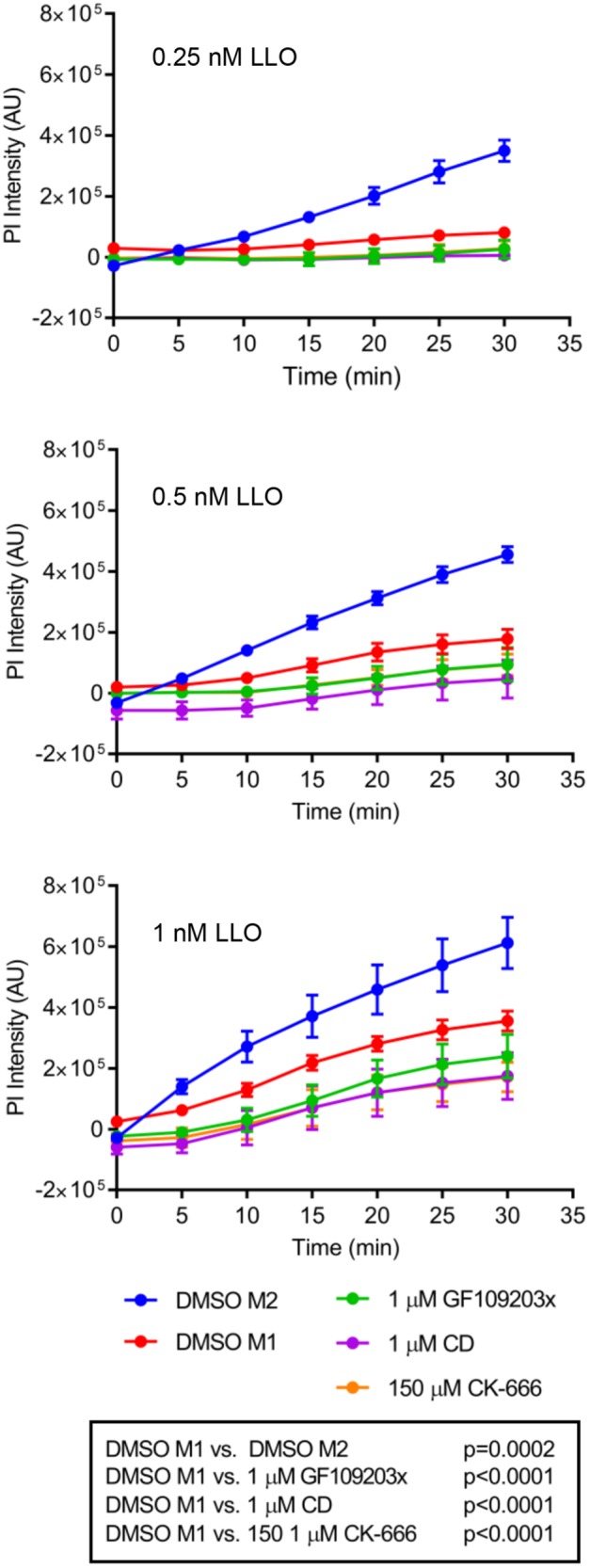
The LLO-dependent entry pathway is dispensable for membrane resealing. HepG2 cells were plated in a 96-well plate and incubated in the presence of DMSO (control), GF109203x, cytochalasin D (CD), or CK-666 in Ca2+-containing medium (M1) or in DMSO in Ca2+-free medium (M2, control for membrane repair). Cells were exposed to 0.25, 0.5, or 1 nM LLO, and the fluorescence intensity of propidium iodide (PI) was measured in the plate reader every 5 min for 30 min at 37°C. Experimental triplicates were performed for each condition, and three independent experiments were performed. Data are presented as the average PI intensity ± SEM. Statistical analyses were performed as described under Materials and Methods and are presented in the text box (n = 3).
DISCUSSION
This study identified a signaling pathway activated by the pore-forming toxin listeriolysin O (LLO) to promote the internalization of the bacterial pathogen L. monocytogenes into human hepatocytes. LLO is an exotoxin that forms large pores across the host cell plasma membrane causing a rapid influx of extracellular Ca2+. Ca2+ influx is then involved in the translocation and activation of conventional protein kinase C (cPKC) at the plasma membrane. Subsequently, the cPKC is required for Rac1 activation upstream of Arp2/3-dependent de novo assembly of the F-actin cytoskeleton for L. monocytogenes internalization (Figure 10). This is the first identification of a signaling pathway activated downstream from plasma membrane perforation by a pore-forming toxin leading to the internalization of a bacterial pathogen. Because pore-forming toxins are virulence factors produced by a large diversity of pathogens, perforation of the host cell plasma membrane is likely a ubiquitous mechanism by which pathogens hijack signaling to enter host cells. Importantly, this work also shows that the identified pathway is dispensable for efficient resealing of the plasma membrane. Although membrane resealing is required for L. monocytogenes internalization, for in its absence cell integrity is lost, the signaling pathway directing F-actin remodeling at the bacterial entry site is distinct from the membrane resealing pathway.
FIGURE 10:

Model for LLO-mediated L. monocytogenes entry. L. monocytogenes secretes the pore-forming toxin listeriolysin O (LLO), which binds to the cholesterol-rich plasma membrane and oligomerizes into a prepore complex. The prepore complex then transitions into a transmembrane pore, which permits the influx of extracellular Ca2+. A rise in cytosolic Ca2+ results in the translocation and activation of a conventional PKC at the plasma membrane. Activated PKC promotes the activation of the Rho GTPase Rac1, which transduces the signal down to induce Arp2/3-mediated F-actin remodeling at the plasma membrane leading to L. monocytogenes entry. The influx of extracellular Ca2+ also induces a membrane resealing pathway.
Link between pathogen internalization and resealing of the injured plasma membrane
Studies on the eukaryotic parasite T. cruzi and the adenovirus HadV-C2 proposed that pathogens damage the host cell plasma membrane and co-opt the subsequent membrane repair machinery to promote their endocytosis (Fernandes et al., 2011; Luisoni et al., 2015). It is thought that flagellar-mediated motility of T. cruzi induces mechanical damages and that the adenovirus perforates the cell by uncoating of the membrane damaging protein-VI. Both studies support the hypothesis that cell injury leads to the influx of extracellular Ca2+, which promotes plasma membrane resealing via the following sequence of events: Ca2+-dependent exocytosis of lysosomes, release of the lysosomal enzyme acid sphingomyelinase (ASM), conversion of sphingomyelin into ceramide by ASM on the outer leaflet of the plasma membrane, and ceramide-mediated endocytosis (Andrews et al., 2014). This pathway leads to the endocytosis of the damaged membrane together with the pathogen (Tam et al., 2010). Furthermore, it was established that resealing is independent of F-actin, dynamin, and clathrin, but requires caveolin-rich membrane microdomains (Corrotte et al., 2013; Andrews et al., 2014). While the evidence supports that membrane resealing is required for pathogen internalization, it is unclear whether the membrane resealing process is directly responsible or whether it is a prerequisite for pathogen uptake. In the latter hypothesis, resealing would be necessary to maintain cell homeostasis and allow for a distinct pathway to promote uptake of the pathogen. Our studies favor the latter hypothesis. Indeed, we identified signaling events required for LLO-dependent L. monocytogenes internalization that were dispensable for membrane resealing. Inhibition of cPKC, Arp2/3, and F-actin assembly improved the efficiency of cell resealing (Figure 9) without altering LLO pore formation (Supplemental Figure S2b). Similar to previous studies, we found that resealing of cells damaged by LLO is independent of F-actin, clathrin, and dynamin (Vadia et al., 2011). Therefore, the resealing machinery downstream from LLO perforation is expected to proceed via the same pathway as described for T. cruzi and adenovirus, but LLO-induced L. monocytogenes endocytosis does not directly involve the resealing machinery. In accordance with our findings, studies have also identified roles for cPKC and F-actin reorganization in entry of adenovirus and T. cruzi into nonprofessional phagocytes (Li et al., 1998; Woolsey and Burleigh, 2004; Maeda et al., 2012; Yousuf et al., 2016). Whether or not T. cruzi and adenovirus utilize the resealing machinery or the signaling pathway described in this work to gain entry into cells, pathogen-mediated host plasma membrane damage represents an invasion mechanism broadly used by unrelated pathogens.
Role of F-actin remodeling in L. monocytogenes uptake and in membrane resealing
Internalization of most pathogens requires F-actin remodeling for the formation of the endosome that contains the pathogen. This was demonstrated for most, if not all, bacterial pathogens, including L. monocytogenes invasion of professional phagocytes and normally nonphagocytic cells as well as for the endocytosis of viruses, such as the adenovirus and for T. cruzi invasion of nonphagocytic cells (Li et al., 1998; Wang et al., 1998; Fernandes et al., 2013). Similarly, the LLO-dependent L. monocytogenes entry pathway is dependent on de novo F-actin assembly (Vadia et al., 2011). A point of convergence is that an intact F-actin cytoskeleton is not required for resealing the plasma membrane during pathogen uptake (Idone, Tam et al., 2008; Fernandes et al., 2011; Vadia and Seveau, 2014). Plasma membrane resealing is thought to require initial disassembly of cortical F-actin to 1) clear the way for docking and fusion of lysosomes with the plasma membrane and 2) decrease surface tension, which prevents enlargement of the plasma membrane wound (Cooper and McNeil, 2015). In accordance with this model, our data show that inhibiting signaling events that favor F-actin assembly improves plasma membrane resealing (Figure 9). Thus, how does one explain that cells undergo de novo F-actin assembly in response to perforation by LLO if F-actin assembly is dispensable for resealing? Similarly to our observation, membrane damage in Xenopus laevis oocytes was followed by Rho GTPase activation and F-actin assembly (Simon et al., 2013). In this example, F-actin assembly was proposed to facilitate repair of very large mechanical wounds. It is possible that injured cells respond to wounding by a sequence of events that include initial disassembly of the actin network followed by its reassembly, regardless of the nature of the wound. We propose that F-actin reassembly 1) repairs large mechanical wounds but is dispensable for repairing smaller injuries caused by toxin pores and 2) restores the cortical F-actin network. Alternatively, one can propose that the reassembly of the F-actin network is due to an additional property of LLO.
LLO is sufficient to activate cPKC-, Rac1-, and Arp2/3-dependent F-actin remodeling
LLO is a multifunctional toxin that affects transcriptional regulation, protein ubiquitination, and mitochondrial dynamics (Hamon et al., 2006; Gonzalez et al., 2011; Seveau, 2014; Osborne and Brumell, 2017). The present studies identified LLO as a regulator of the F-actin cytoskeleton. To monitor the spatiotemporal dynamics of signaling events downstream from LLO pore formation, we used a highly sensitive live-cell FRET-based approach that quantitatively detects small changes in protein activation in single cells (Hoppe et al., 2002; Hoppe and Swanson, 2004). This approach revealed that the kinetics of PKC activation overlay and potentially precede Rac1 activation, a fact that we confirmed with pharmacological inhibitors (Figures 3 and 8). Interestingly, Rac1 activation proceeded in a wavelike pattern, reminiscent of calcium waves. This could be observed because we used a fluorescent chimera of native Rac1. However, the MyrPalm-CKAR biosensor is artificially anchored to the plasma membrane, which allows us to monitor the kinetics of PKC activation at this location, but not native PKC translocation (Violin et al., 2003; Gallegos and Newton, 2011; Antal et al., 2014). To assess PKC translocation, we used cells expressing fluorescent chimeras containing the membrane-binding domains of the PKC isoforms PKCα, PKCβII, and PKCε. Fluorescent cPKC domains were translocated to regions of the membrane where membrane ruffles and macropinosomes were formed similarly to the Rac1 activation pattern (Figure 7 and Supplemental Movie 7). The present work demonstrates for the first time the activation of a conventional PKC by LLO. Previous studies reported the activation of the calcium-independent PKCε (novel PKC) (Shaughnessy et al., 2007). Because activation of conventional and novel PKC requires production of diacylglycerol (DAG), LLO may also activate host phospholipases, as previously suggested (Shaughnessy et al., 2007). Our finding that Ca2+ influx leads to cPKC activation upstream to Rac1 is reminiscent of previous work using the calcium ionophore ionomycin (Price et al., 2003). In that study, a cPKC responsive to Ca2+ influx phosphorylated a RhoGDI facilitating Rac1 activation (Price et al., 2003). Thus, the influx of Ca2+ through LLO pores could suffice to activate Rac1 in a similar manner. This idea is supported by the dependence of PKC and Rac1 activation on Ca2+ influx and the fact that the prepore-locked form of LLO (LLOpL) was inactive. In accordance with our results, the Streptococcus pneumoniae CDC pore-forming toxin pneumolysin induces Ca2+-dependent F-actin remodeling in a Rho GTPase (Rac1, Cdc42, and RhoA)-dependent manner (Iliev et al., 2007). Our FRET studies established that LLO also activates Cdc42 (Supplemental Figure S4), but to a lower extent than Rac1, and we showed that Cdc42 (and RhoA) are both dispensable for the LLO-mediated entry pathway.
Why does L. monocytogenes use multiple invasins and invasion strategies?
Listeria monocytogenes infects multiple nonphagocytic cell types, including enterocytes, hepatocytes, cardiac myocytes, neurons, cytotrophoblasts, endothelial cells, and so on. It is therefore not surprising that this pathogen evolved to express multiple strategies to invade host cells. The major L. monocytogenes invasin, InlA, acts by attaching the bacterium to the cell adhesion molecule E-cadherin but not all cell types infected by L. monocytogenes express E-cadherin (Gaillard et al., 1991; Mengaud et al., 1996; Ortega et al., 2017). The second invasion protein InlB acts via activating c-Met and was shown to cooperate with InlA to potentiate the bacterial internalization pathway (Dramsi et al., 1995; Shen et al., 2000; Pentecost et al., 2010; Gessain et al., 2015). Although the initial stage of activation of bacterial internalization differs for each invasin (InlA binds to E-cadherin, InlB activates c-Met, LLO forms pores) they all converge toward activation of Rho GTPases and the Arp2/3 complex leading to F-actin– and dynamin-dependent internalization (Ribet and Cossart, 2015). More recently it was shown that the InlB internalization pathway involves a PKC similarly to our finding with LLO (Bhalla et al., 2017). InlA, InlB, and LLO are coexpressed and may cooperate to activate a more robust endocytic signaling in normally nonphagocytic cells. Alternatively, the invasins may promote L. monocytogenes invasion in a cell-type–dependent manner.
Overall, this work confirms the novel concept that pathogens use host cell plasma membrane damage as a strategy to induce their endocytic uptake into normally nonphagocytic cells, thereby ensuring the first step of their intracellular life cycle. Unlike previous studies, this work proposes that the plasma membrane resealing pathway is a prerequisite but is not directly responsible for pathogen uptake. We propose that in the example of L. monocytogenes, a Ca2+-dependent pathway that is distinct from plasma membrane resealing is activated downstream from cell injury to restore the cortical actin cytoskeleton, which is hijacked for pathogen internalization.
MATERIALS AND METHODS
Reagents and inhibitors
Cells were pretreated for 1 h with the following pharmacological inhibitors (Sigma-Aldrich): kanamycin (30 μg/ml), ampicillin (100 μg/ml), chloramphenicol (5 μg/ml), CK-636 and CK-666 (50–150 μM), GF109203x (0.05–1.0 μM), and Gö6983 (0.5 μM). Cells were pretreated for 10 min with cytochalasin D (1 μM; Sigma-Aldrich). Antibodies include the following: anti–L. monocytogenes rabbit polyclonal antibodies (GeneTex); goat anti-rabbit secondary antibodies conjugated to Alexa488 and Alexa568 (Molecular Probes); anti-BSA rabbit antiserum (B1520; Sigma-Aldrich); mouse monoclonal antibodies directed against Rac1, RhoA, and Cdc42 (clones ARC03, ARH04, and ACD03, respectively; Cytoskeleton); rabbit anti-actin (clone A2103; Sigma-Aldrich); and secondary goat anti-mouse immunoglobulin G (IgG) and goat anti-rabbit IgG antibodies conjugated to horseradish peroxidase (Cell Signaling). FluoSpheres carboxylate modified microspheres (1 μm) (Molecular Probes), BSA (Santa Cruz Biotechnology), and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (Thermo-Fisher) were used to prepare the beads for LLO coating. Lipofectamine RNAiMax and Lipofectamine 2000 (Life Technologies) were used to transfect siRNA and plasmid DNA, respectively, using the manufacturer’s protocols. Silencer select siRNA for Rac1, RhoA, Cdc42, and NC were used at 60–90 pg per well in a six-well format (Life Technologies).
Listeriolysin O purification
Escherichia coli BL21(DE3) containing pET29b hly, which encodes a C-terminal 6His-tagged listeriolysin O missing the signal sequence (N-terminal 25 amino acids), or pET29b hly (G80C, S215C), which encodes a C-terminal 6His-tagged listeriolysin O missing the signal sequence (N-terminal 25 amino acids) and contains two amino acid substitutions (G80C, S215C) to produce the prepore locked variant LLOpL, were grown on Luria broth (LB) agar containing 30 μg/ml kanamycin at 37°C overnight. Bacterial cultures were grown overnight at 37°C in LB-supplemented kanamycin (30 μg/ml). The overnight culture was subcultured in 0.5 l LB supplemented with kanamycin and grown to OD600 = 0.6 at which point isopropyl β-d-1-thiogalactopyranoside (1 mM final concentration) was added to induce expression of the 6His-tagged listeriolysin O and the culture was agitated at room temperature for 5 h. The culture was pelleted at 6000 × g for 10 min. All buffers used for the LLOpL purification lacked beta-mercaptoethanol (BME) and dithiothreitol (DTT). Cell pellets were suspended in lysis buffer (50 mM phosphate, pH 8.0, 1 M NaCl, 5 mM BME, and protease inhibitor) and lysed using a French press. Cell lysate was centrifuged and the supernatant was applied to Ni-NTA Agarose (Qiagen) for 2 h at 4°C. The agarose was washed with (50 mM phosphate, pH 6, 1 M NaCl, 20 mM imidazole, 5 mM BME, 0.1% Tween-20, 10% glycerol) followed by two washes with (50 mM phosphate, pH 6, 1 M NaCl, 20 mM imidazole, 5 mM BME). The protein was eluted with (50 mM phosphate, pH 6, 1 M NaCl, 500 mM imidazole, 5 mM BME) and dialyzed in 50 mM phosphate, pH 6, 0.5 M NaCl, 5 mM DTT.
Bacterial cultures
Wild-type L. monocytogenes (strain 10403s) and its isogenic hly deletion mutant were gifts from Dan Portnoy (University of California, Berkeley). Bacterial cultures were grown overnight at 37°C under agitation in brain and heart infusion (BHI; BD Biosciences). Escherichia coli XL1blue and DH5α were grown at 37°C under agitation in LB supplemented with kanamycin (30 μg/ml), ampicillin (100 μg/ml), or chloramphenicol (5 μg/ml).
Plasmid construction and purification
Escherichia coli XL1Blue harboring the plasmids pmECFP-C1, pmEYFP-C1, pECFP-YFP, pmECFP-PBD, pmEYFP-Rac1, pmEYFP-Rac1-T17N, and pmEYFP-Rac1-H40Y/Q61L were prepared as described previously (Hoppe et al., 2002, Hoppe and Swanson, 2004). Sanger sequencing with primers A and B was used to verify insert sequences (Supplemental Table S6). To obtain the constitutively active form of Rac1 (pmEYFP-Rac1-Q61L), pmEYFP-Rac1-H40Y/Q61L was mutated via site-directed mutagenesis using primers C and D. Sanger sequencing with primers A and B was used to verify that the proper mutation was made. Escherichia coli DH5α containing pcDNA3-hPKCβII-YFP, pcDNA3-hPKCε-HA1-GFP, and pcDNA3-hPKCδ-HA1-GFP and E. coli XL1Blue containing pEGFP-PKCα were gifts from Alexandra Newton (University of California, San Diego) (Antal et al., 2014). Escherichia coli containing pcDNA3 constructs was grown in LB supplemented with 100 µg/ml ampicillin while E. coli containing pEGFP constructs was grown in LB supplemented with 30 µg/ml kanamycin. Escherichia coli DH5α containing pcDNA3 MyrPalm-CKAR was grown in LB supplemented with 100 µg/ml ampicillin (Addgene) (Violin et al., 2003). Plasmids pEGFPN1 and pEGFP-PKCα were submitted for Sanger sequencing using primers E and G and E, F, and G, respectively (Supplemental Table S6). The GenElute Endotoxin-free Plasmid Midiprep Kit was used to purify endotoxin-free plasmids according to the manufacturer instructions (Sigma-Aldrich).
Mammalian cell culture, silencing, and transfection
The human hepatocyte carcinoma cell line HepG2 (ATCC HB-8065) was grown in MEM with Earle’s salts and 2 mM l-glutamine supplemented with 10% heat-inactivated fetal bovine serum (FBS; Lonza), 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Cells were grown at 37°C in a 5% CO2 humidified cell culture incubator. Cells were seeded at 1 × 104 cells/well in a 24-well tissue culture plate, 4 × 105 cells/dish in No. 1.5, 10-mm glass diameter MatTek dish for live cell imaging, or 5 × 105 cells/well in six-well tissue culture plates containing 22 × 22 mm No. 1.5 cover glass (Gold Seal). To silence the expression of the Rho GTPases, cells were transfected in antibiotic-free medium using Lipofectamine RNAi Max (Invitrogen) with silencer select siRNA for Rac1, RhoA, Cdc42, or negative control siRNA. The media was replaced 24 h after seeding and incubated for an additional 24 h. To monitor Rho GTPase activation by FRET microscopy, cells were seeded at 4 × 105 cells in 35 mm No. 1.5, 10-mm-diameter glass MatTek dishes (MatTek P35GC-1.5-10-C). Twenty-four hours after seeding, cells were transfected with 2500 μg total plasmid DNA in antibiotic- and serum-free medium using Lipofectamine 2000 (ThermoFisher Scientific). Serum was added to a final 10% after 1 h, and then the cell culture media was replaced with antibiotic-free medium after an additional 1 h.
Western blot analysis for Rac1, RhoA, and Cdc42 in HepG2 expression
After 48 h of treatment with, or without (control, nontreated), the siRNAs, HepG2 cells were washed once with PBS, pH 7.4, and lysed in 100 μl boiling reduced sample buffer. Cell lysates were boiled for 5 min and equivalent to 1.5 × 105 (nondiluted), 7.5 × 104 (1:2 dilution), 3.75 × 104 (1:4 dilution), and 1.88 × 105 (1:8 dilution) cells were loaded into a 10% SDS–PAGE. All samples were subjected to SDS–PAGE and Western blot analysis using Rac1, RhoA, Cdc42, and actin antibodies, followed by a goat anti-mouse IgG conjugated to HRP or goat anti-rabbit IgG conjugated to HRP. Western blots were developed using the Amersham ECL Select Detection Reagent (GE Healthcare Life Sciences) and imaged using a ChemiDoc XRS+ system and ImageLab software (Bio-Rad). Once the conditions for knockdown were established, at least three independent experiments were performed to confirm efficient silencing, and Western blot analyses were performed in each experiment to verify knockdown efficiencies.
Densitometry analysis.
Nontreated and siRNA-treated HepG2 cell lysates were processed for Western blot analysis. Densitometry analysis using ChemiDoc XRS+ system and ImageLab software (Bio-Rad) was performed to assess band intensities (verified for nonsaturation), and all values were normalized relative to NC siRNA-treated sample. The ratio of Rho GTPase to actin was then calculated to compare the Rho GTPases expression levels (Supplemental Table S1).
Bacterial invasion assay
Overnight cultures of L. monocytogenes were diluted 1:20 in BHI and grown at 37°C to mid–log phase at an OD600 = 0.8. Bacteria were washed 3 times in phosphate-buffered saline (PBS), pH 7.4, diluted into MEM at a multiplicity of infection (MOI) 20 and added to the mammalian cells that were seeded at 5 × 105 cells/well in a 24-well plate and cultured for 48 h. Cell culture plates were centrifuged at 520 × g for 3 min at room temperature and incubated for 30 min at 37°C. Infected cells were washed with MEM twice followed by two additional washes in PBS and fixed with 4% paraformaldehyde (PFA) in PBS, pH 7.4, for 15 min at room temperature. Cells were washed three times with 0.1 M glycine in PBS and blocked in 0.1 M glycine in PBS/10% FBS for 1 h. For labeling, extracellular bacteria were labeled with the primary anti–L. monocytogenes antibody and secondary anti-rabbit antibody conjugated to Alexafluor-488, and total bacteria were labeled, after cell permeabilization with 0.5% TX-100 (in PBS), with a primary anti–L. monocytogenes rabbit polyclonal antibody and a secondary anti-rabbit antibody conjugated to Alexafluor-568. Cells were preserved in Prolong Gold Antifade Mountant medium containing 4’,6 diamidino-2-phenylindole, dihydrochloride (DAPI). To quantify the numbers of bacteria, 40 sets of images (DAPI, Alexa-488, Alexa-568, and phase-contrast) were randomly acquired using a Metamorph programmed, motorized stage and the 20× objective. Metamorph imaging and analysis software were used to determine the number of extracellular (Ne) and total (Nt) bacteria such that the efficiency of bacterial entry was measured as the percentage of intracellular bacteria (Ni) using the equation Ni = (Nt - Ne)*100/Nt.
Polystyrene beads invasion assay
FluoSpheres carboxylate-modified microspheres (1 µm) (blue fluorescent 365/415) were washed with 15 mM NaoAc, pH 5, three times; incubated with 5 mg/ml BSA in 15 mM NaoAc, pH 5; and treated with 0.008 g 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride and 0.1 N NaOH at 4°C overnight to covalently coat the beads with BSA. The following day, 2.5 M glycine was added for a 30-min incubation followed by two washes in 0.33× PBS, pH 7.4 (Gedde et al., 2000). Beads were washed twice with ice-cold buffer A (20 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM NiCl2), suspended in ice-cold buffer B (20 mM HEPES, pH 7.5, 50 mM NaCl) and 5 μg LLO (or none) to adsorb LLO to the surface, and incubated on ice for 10 min. Two washes with buffer B removed excess LLO, and beads were diluted into ice-cold MEM to obtain MOI 5 or 20. Beads were added to mammalian cells in a six- or 24-well plate format followed by centrifugation at 520 × g for 3 min at room temperature and incubated for 30 min at 37°C. Cells were washed with MEM twice followed by two washes with PBS and fixed with 4% paraformaldehyde (PFA) in PBS, pH 7.4, for 15 min at room temperature. Cells were washed three times with 0.1 M glycine in PBS, pH 7.4, and blocked in 0.1 M glycine in PBS/5% milk for 1 h. Extracellular beads were labeled using an anti-BSA rabbit antiserum followed by a secondary goat anti-rabbit IgG antibody conjugated to Alexa-488. To quantify the number of intracellular beads, 40 sets of images (DAPI, Alexa-488, and phase-contrast) were randomly acquired using a Metamorph programmed, motorized stage, and the 20× objective. Metamorph imaging and analysis software was used to determine the number of extracellular (Alexa-488, Ne) and total (DAPI, Nt) beads such that the efficiency of bead entry was measured as the percentage of intracellular beads (Ni) using the equation Ni = (Nt - Ne)*100/Nt.
Microscope equipment
A motorized, atmosphere-controlled inverted wide-field fluorescence microscope (Axio Observer D1; Carl Zeiss) equipped with a PZ2000 XYZ Series Automated Nano stage (Applied Scientific Instruments); a TempModule S1, Heating Unit XL S, and Objective heater 22.5/34 S1 (D) (Carl Zeiss); 20X Plan Neofluar, 40X Plan Neofluar, and 100× Plan Apo objective lenses (NA = 0.5, 1.3, 1.4, respectively); Cascade II:512 electron-multiplying charge-coupled device (EMCCD) camera (Photometrics); ORCA-FLASH4.0 V2 Digital complementary metal-oxide-semiconductor camera (Hamamatsu); and a Smart Shutter (Sutter Instrument) on the LED lamp. A Lambda 10-3 optical emission filter changer and DG-4 ultra-high-speed wavelength switching illumination system with a 300-W Xenon arc bulb (Sutter Instrument Company) equipped with excitation filters for CFP and FRET (S430/25x) and for YFP (S500/20x), emission filters for CFP (S470/30) and for YFP and FRET (S535/30), and a JP4 beamsplitter for CFP and YFP (86002v2-spr) from Chroma Technology, were controlled with Metamorph Microscopy Automation and Image Analysis Software (Molecular Devices).
FRET stoichiometry imaging
Fluorescence resonance energy transfer or FRET is a biochemical technique that assesses the physical proximity of fluorophores at distances ≤10 nm. On excitation, a donor fluorophore transfers its energy in a nonradiative manner to a lower energy acceptor fluorophore leading to acceptor fluorescence. FRET stoichiometry utilizes donor emission at donor excitation wavelength (ID), acceptor emission at acceptor excitation wavelength (IA), and acceptor emission at donor excitation wavelength (IF) to quantify FRET. FRET stoichiometry relies on four predetermined coefficients (α, β, γ, and ξ) that are characteristic of the microscope settings and fluorophores used (Hoppe et al., 2002, Hoppe and Swanson, 2004). The coefficients were determined using HepG2 cells expressing mCit alone (α = 0.072), mCFP alone (β = 0.584) and mCit-mCFP tandem (γ = 0.058 and ξ = 0.0156).
Phase-contrast (PC), ID, IA, and IF images were acquired with the Cascade II:512 EMCCD camera (Photometrics) at 200-ms exposure every 20 s over 20 min, in which no photobleaching was observed. Images were corrected for camera dark noise and adjusted with a binary mask, and FRET stoichiometry was applied to determine, pixel by pixel, EA and ED using the Metamorph software. The apparent FRET efficiency EA is proportional to the fraction of mCit in complex with mCFP, while ED is proportional to the fraction of mCFP in complex with mCit (Hoppe et al., 2002). The average apparent FRET efficiency, EAVE = (EA+ED)/2, corrects for variations in donor and acceptor expression levels and therefore normalizes FRET signals among different transfected cells (Beemiller et al., 2006).
FRET stoichiometry measurements between mCit-Rac1 and mCFP-PBD
HepG2 cells cotransfected to express mCit-Rac1 and mCFP-PBD, or the mentioned chimeras, were imaged for PC, ID, IA, and IF under 40× magnification. Cells were imaged in microscopy cell culture medium (150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM KCl, 20 mM HEPES, 10 mM D + glucose) or Ca2+-free microscopy cell culture medium. FRET stoichiometry was applied to each image, pixel by pixel, using ID, IA, and IF to create a calculated image corresponding to EAVE = (EA + ED)/2 or FRET. As controls for Rac1 FRET analysis, we quantified the apparent FRET efficiency in cells that coexpressed mCFP-PBD plus the mCit fluorophore alone (FRET negative control), the native mCit-Rac1 (control for basal Rac1 activity), the dominant negative mCit-Rac1N17 (negative control for Rac1 activation), and the constitutively active mCit-Rac1L61 (positive control for Rac1 activation). Additionally, cells separately expressing mCFP and mCit (FRET negative control), or the FRET pair mCit-mCFP tandem (FRET-positive control for the two fluorophores), were analyzed for FRET efficiency. Cells were placed on the microscope stage at 37°C, and image series were collected every 20 s for 20 min (Supplemental Figure S1 and Supplemental Movie S1). In cells that coexpressed mCit-Rac1 and mCFP-PBD and exposed to LLO and/or inhibitors, FRET (EAVE) was calculated within two standard regions (305 pixels) that were positioned on each cell, at each time point. For each region, the average FRET value over the first 5 min of imaging (before the addition of medium or LLO) was calculated to determine the baseline FRET within the analyzed region; all data points were then normalized relative to this value. In cells exposed to LLO and control nontreated cells, the regions were positioned where Rac1 FRET variations were observed. If no FRET variation were observed, then the two regions were arbitrarily positioned at the cell periphery. A FRET-positive cell was determined as a cell that displayed at least a twofold increase in normalized FRET efficiency.
FRET measurements of the MyrPalm-CKAR biosensor
The C kinase activity reporter (CKAR) encoded in pcDNA3.1(+) is a chimeric protein containing an N-terminal mCFP fused to the Rad53p FHA2 domain, followed by an optimal substrate sequence for all PKC isoforms and a C-terminal mYFP (mCit). A 10-residue sequence from Lyn Kinase, which signals for myristoylation and palmitoylation, is located at the N-terminal end of the reporter resulting in the plasma membrane associated reporter MyrPalm-CKAR. In a nonphosphorylated state, the reporter exists in a clamped conformation resulting in FRET between mCFP and mCit. On substrate sequence phosphorylation by a PKC, the FHA2 phosphopeptide domain binds the phosphorylated sequence resulting a conformational change moving mCit away from mCFP resulting in a loss of FRET. Importantly, this PKC biosensor has a dynamic range of 20% (Violin et al., 2003; Gallegos and Newton, 2011). FRET stoichiometry was applied to each image, pixel by pixel, using HepG2 cells transfected to express MyrPalm-CKAR. Cells were washed once with microscopy cell culture medium (150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM KCl, 20 mM HEPES, 10 mM d-glucose) and incubated in the same medium for 1 h at 37°C prior to imaging. The ID, IA, and IF images were acquired with the Cascade II:512 EMCCD camera (Photometrics) at 200-ms exposure time every 20 s for 20 min under 100× magnification. FRET stoichiometry was applied to each image, pixel by pixel, using ID, IA, and IF to create a calculated image corresponding to EAVE = (EA + ED)/2 (or FRET) and EAVE was averaged over the entire cell surface at each time point. The FRET values were normalized relative to EAVE averaged over the first 5 min before the addition of medium or LLO.
Membrane repair assay
HepG2 cells were seeded in a 96-well black-walled and clear bottom cell culture plate at 5 × 104 cells/well in 200 μl and incubated for 24 h. For Ca2+-containing conditions, cells were washed twice with 200 μl M1 (Hank’s balanced salt solution [HBSS] supplemented with 0.5 mM MgCl2, 1.2 mM CaCl2, 10 mM HEPES, 25 mM glucose, pH 7.4) at 37°C. For the Ca2+-free condition, cells were washed once with M2 (HBSS supplemented with 0.5 mM MgCl2, 10 mM HEPES, 25 mM glucose, pH 7.4) containing 5 mM EGTA and twice with M2 at 37°C. M1 or M2 (100 µl) was added to the respective wells. Reagents were added to a separate 96-well plate maintained on ice: 100 μl of 120 μM PI in medium containing Ca2+ (M1) or not (M2), plus 100 μl of 4×-concentrated LLO (M1 or M2). The amount of 100 μl of medium containing the reagents were then transferred to the plate containing the cells. Additionally, as a positive control of plasma membrane damage, 0.1 and 1% Triton X-100 was added in place of LLO. As a negative control, M1 alone and M1 containing PI was added. HepG2 cells were exposed to 0.25, 0.5, and 1 nM LLO (final concentration) in M1 or M2. The kinetics assay was performed on a Spectra Max i3X Multi Mode Detection Platform (Molecular Devices) with the following parameters: 37°C, fluorescence readings every 5 min for 30 min with an excitation/emission filter of 535/617 and 15/15 nm bandwidth (Pathak-Sharma et al., 2017).
Erythrocyte isolation and hemolysis assays
Human peripheral blood was obtained from healthy adult volunteer via venipuncture (Institutional Review Board protocol H0045). All donors provided a written informed consent. Erythrocytes were separated from plasma, leukocytes, and platelets by centrifugation on Polymorphprep (Axis-Shield) and erythrocytes were washed three times with Alsever’s Solution (71.9 mM NaCl, 27 mM C6H5Na3O7⋅2H2O, 2.6 mM C6H5Na3O7⋅H2O, 113.8 d-glucose).
Hemolysis assay (end point).
Erythrocytes (5 × 107 cells/ml) were suspended in PBS or in PBS containing 4 mM DTT. LLO or LLOpL were serially diluted in PBS or PBS containing 4 mM DTT in 10 μl in a 96-well cell culture plate, and 150 μl of erythrocytes in the respective media was added. Erythrocytes were incubated at 37°C for 30 min and then centrifuged at 343 × g for 5 min. Supernatant (100 µl) from each well was transferred to a new 96-well cell culture plate, and OD = 540 nm was measured using the Spectra Max i3X Multi Mode Detection Platform (Molecular Devices).
Hemolysis assay (kinetics).
Erythrocytes were suspended in ice-cold PBS containing DMSO vehicle control, 1 mM cytochalasin D, 1 mM GF109203x, or 150 μM CK-666 to a concentration of 5 × 107 cells/ml. LLO (10 nM) in PBS containing DMSO vehicle control, 1 mM cytochalasin D, 1 mM GF109203x, or 150 μM CK-666 was distributed in 10-μl aliquots in a 96-well cell culture plate that was placed on ice. Erythrocytes (150 μl) were added, and the plate was placed in the Spectra Max i3X Multi Mode Detection Platform (Molecular Devices). The OD = 700 nm was measured every min for 30 min at 37°C.
Statistics
In the entire article, “n” refers to the number of independent experiments. Statistical analyses were performed as follows.
Invasion assays.
A linear mixed effects model was used to take account of the correlation among observations obtained from the same day and Holm’s procedure was used to control for multiple comparisons.
Rac1 FRET and MyrPalm-CKAR FRET.
The area under the FRET curve (AUC) between time points T0 (LLO addition), and T15 min (end of the movies) in cell culture with Ca2+, or T0 and T5 in cell culture without Ca2+, was calculated and analyzed using the Student’s t test for unequal variances and analysis of variance, respectively (Supplemental Tables S2 and S3 for Rac1 FRET and Supplemental Tables S4 and S5 for MyrPalm CKAR FRET). When comparing the percentage of FRET-positive cells for the Rac1 FRET (Supplemental Tables S2 and S3), linear mixed effects models were used to account for repeated experiments. Holm’s procedure was used to adjust the p values (Pathak-Sharma et al., 2017) for all the above multiple comparisons. SAS9.4 was used for the analyses (SAS Institute).
Membrane repair.
Propidium iodide (PI) intensity values were first background corrected, log10-transformed, and averaged for a given condition for each time point for each experiment. Negative values were set to 0 after transformations. The fixed effects of conditions with DMSO or the inhibitors GF109203x, CK-666, and cytochalasin D, time (as a continuous variable), and LLO concentration (as a categorical variable) were tested with a linear mixed model to take into account the correlation among observations from the same independent replicates using the average log-transformed intensity values. A pairwise comparison was made between the different LLO concentrations (0, 0.25, 0.5, and 1 nM) and their differences on log10 (intensity), and the same analysis was performed on the different inhibitor conditions (DMSO, 1 μM cytochalasin D, 1 μM GF109203x, and 150 μM CK-666) (Supplemental Table S5). Holm’s procedure was used to adjust the p values (Pathak-Sharma et al., 2017) for all above multiple comparisons. SAS9.4 was used for the analyses (SAS Institute).
Supplementary Material
Acknowledgments
We acknowledge Alexandra Newton (University of California, San Diego) for providing guidance for the study of PKC activation and providing PKC encoding plasmids. We thank Daniel Wozniak for editing. Research reported in this article was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number RO1AI107250 to Stephanie M. Seveau and award number 1-T32-AI-112542, a NRSA training grant administered by the Center for Microbial Interface Biology at The Ohio State University to Sarika Pathak-Sharma. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations used:
- ASM
acid sphingomyelinase
- CDC
cholesterol-dependent cytolysin
- cPKC
conventional protein kinase C
- FRET
fluorescence resonance energy transfer
- LLO
listeriolysin O
- LLOpL
listeriolysin O prepore lock
- mCFP
monomeric cyan fluorescent protein
- mCit
monomeric Citrine
- MyrPalm-CKAR
myristoylated and palmitoylated c kinase activity reporter
- NC
negative control
- PBD
p21-binding domain of PAK1
- PI
propidium iodide.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-09-0561) on November 29, 2017.
REFERENCES
- Andrews NW, Almeida PE, Corrotte M. Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014;24:734–742. doi: 10.1016/j.tcb.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antal CE, Violin JD, Kunkel MT, Skovso S, Newton AC. Intramolecular conformational changes optimize protein kinase C signaling. Chem Biol. 2014;21:459–469. doi: 10.1016/j.chembiol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beemiller P, Hoppe AD, Swanson JA. A phosphatidylinositol-3-kinase-dependent signal transition regulates ARF1 and ARF6 during Fcgamma receptor-mediated phagocytosis. PLoS Biol. 2006;4:e162. doi: 10.1371/journal.pbio.0040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- Bhalla M, Law D, Dowd GC, Ireton K. Host serine/threonine kinases mTOR and protein kinase C-alpha promote InlB-mediated entry of Listeria monocytogenes. Infect Immun. 2017;85:e00087-17. doi: 10.1128/IAI.00087-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumell JH, Grinstein S. Role of lipid-mediated signal transduction in bacterial internalization. Cell Microbiol. 2003;5:287–297. doi: 10.1046/j.1462-5822.2003.00273.x. [DOI] [PubMed] [Google Scholar]
- Chesarone MA, Goode BL. Actin nucleation and elongation factors: mechanisms and interplay. Curr Opin Cell Biol. 2009;21:28–37. doi: 10.1016/j.ceb.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ST, McNeil PL. Membrane repair: mechanisms and pathophysiology. Physiol Rev. 2015;95:1205–1240. doi: 10.1152/physrev.00037.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrotte M, Almeida PE, Tam C, Castro-Gomes T, Fernandes MC, Millis BA, Cortez M, Miller H, Song W, Maugel TK, Andrews NW. Caveolae internalization repairs wounded cells and muscle fibers. Elife. 2013;2:e00926. doi: 10.7554/eLife.00926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P, Helenius A. Endocytosis of viruses and bacteria. Cold Spring Harb Perspect Biol. 2014;6:a016972. doi: 10.1101/cshperspect.a016972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P, Sansonetti PJ. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science. 2004;304:242–248. doi: 10.1126/science.1090124. [DOI] [PubMed] [Google Scholar]
- Cox D, Chang P, Zhang Q, Reddy PG, Bokoch GM, Greenberg S. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J Exp Med. 1997;186:1487–1494. doi: 10.1084/jem.186.9.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dramsi S, Biswas I, Maguin E, Braun L, Mastroeni P, Cossart P. Entry of Listeria monocytogenes into hepatocytes requires expression of inIB, a surface protein of the internalin multigene family. Mol Microbiol. 1995;16:251–261. doi: 10.1111/j.1365-2958.1995.tb02297.x. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Fernandes MC, Cortez M, Flannery AR, Tam C, Mortara RA, Andrews NW. Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J Exp Med. 2011;208:909–921. doi: 10.1084/jem.20102518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes MC, Flannery AR, Andrews N, Mortara RA. Extracellular amastigotes of Trypanosoma cruzi are potent inducers of phagocytosis in mammalian cells. Cell Microbiol. 2013;15:977–991. doi: 10.1111/cmi.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard JL, Berche P, Frehel C, Gouin E, Cossart P. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell. 1991;65:1127–1141. doi: 10.1016/0092-8674(91)90009-n. [DOI] [PubMed] [Google Scholar]
- Galan JE. Common themes in the design and function of bacterial effectors. Cell Host Microbe. 2009;5:571–579. doi: 10.1016/j.chom.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos LL, Newton AC. Genetically encoded fluorescent reporters to visualize protein kinase C activation in live cells. Methods Mol Biol. 2011;756:295–310. doi: 10.1007/978-1-61779-160-4_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedde MM, Higgins DE, Tilney LG, Portnoy DA. Role of listeriolysin O in cell-to-cell spread of Listeria monocytogenes. Infect Immun. 2000;68:999–1003. doi: 10.1128/iai.68.2.999-1003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessain G, Tsai YH, Travier L, Bonazzi M, Grayo S, Cossart P, Charlier C, Disson O, Lecuit M. PI3-kinase activation is critical for host barrier permissiveness to Listeria monocytogenes. J Exp Med. 2015;212:165–183. doi: 10.1084/jem.20141406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MR, Bischofberger M, Freche B, Ho S, Parton RG, van der Goot FG. Pore-forming toxins induce multiple cellular responses promoting survival. Cell Microbiol. 2011;13:1026–1043. doi: 10.1111/j.1462-5822.2011.01600.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia JR, Machaty Z, Lai FA, Swann K. The dynamics of PKC-induced phosphorylation triggered by Ca2+ oscillations in mouse eggs. J Cell Physiol. 2013;228:110–119. doi: 10.1002/jcp.24110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves E, Dart AE, Covarelli V, Caron E. Molecular mechanisms of phagocytic uptake in mammalian cells. Cell Mol Life Sci. 2008;65:1957–1976. doi: 10.1007/s00018-008-7578-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighat AC, Seveau S. Quantification of host-microbe interactions by automated fluorescence microscopy. J Immunol Methods. 2010;352:186–191. doi: 10.1016/j.jim.2009.11.013. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33(Pt 5):891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- Ham H, Sreelatha A, Orth K. Manipulation of host membranes by bacterial effectors. Nat Rev Microbiol. 2011;9:635–646. doi: 10.1038/nrmicro2602. [DOI] [PubMed] [Google Scholar]
- Hamon M, Bierne H, Cossart P. Listeria monocytogenes: a multifaceted model. Nat Rev Microbiol. 2006;4:423–434. doi: 10.1038/nrmicro1413. [DOI] [PubMed] [Google Scholar]
- Hetrick B, Han MS, Helgeson LA, Nolen BJ. Small molecules CK-666 and CK-869 inhibit actin-related protein 2/3 complex by blocking an activating conformational change. Chem Biol. 2013;20:701–712. doi: 10.1016/j.chembiol.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe A, Christensen K, Swanson JA. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys J. 2002;83:3652–3664. doi: 10.1016/S0006-3495(02)75365-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe AD, Swanson JA. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol Biol Cell. 2004;15:3509–3519. doi: 10.1091/mbc.E03-11-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotze EM, Wilson-Kubalek EM, Rossjohn J, Parker MW, Johnson AE, Tweten RK. Arresting pore formation of a cholesterol-dependent cytolysin by disulfide trapping synchronizes the insertion of the transmembrane beta-sheet from a prepore intermediate. J Biol Chem. 2001;276:8261–8268. doi: 10.1074/jbc.M009865200. [DOI] [PubMed] [Google Scholar]
- Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol. 2008;180:905–914. doi: 10.1083/jcb.200708010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliev AI, Djannatian JR, Nau R, Mitchell TJ, Wouters FS. Cholesterol-dependent actin remodeling via RhoA and Rac1 activation by the Streptococcus pneumoniae toxin pneumolysin. Proc Natl Acad Sci USA. 2007;104:2897–2902. doi: 10.1073/pnas.0608213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg RR. Pathways for the penetration of enteroinvasive Yersinia into mammalian cells. Mol Biol Med. 1990;7:73–82. [PubMed] [Google Scholar]
- Jimenez AJ, Perez F. Plasma membrane repair: the adaptable cell life-insurance. Curr Opin Cell Biol. 2017;47:99–107. doi: 10.1016/j.ceb.2017.03.011. [DOI] [PubMed] [Google Scholar]
- Li E, Stupack D, Bokoch GM, Nemerow GR. Adenovirus endocytosis requires actin cytoskeleton reorganization mediated by Rho family GTPases. J Virol. 1998;72:8806–8812. doi: 10.1128/jvi.72.11.8806-8812.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loustalot F, Kremer EJ, Salinas S. The intracellular domain of the coxsackievirus and adenovirus receptor differentially influences adenovirus entry. J Virol. 2015;89:9417–9426. doi: 10.1128/JVI.01488-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luisoni S, Suomalainen M, Boucke K, Tanner LB, Wenk MR, Guan XL, Grzybek M, Coskun U, Greber UF. Co-option of membrane wounding enables virus penetration into cells. Cell Host Microbe. 2015;18:75–85. doi: 10.1016/j.chom.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Maeda FY, Cortez C, Yoshida N. Cell signaling during Trypanosoma cruzi invasion. Front Immunol. 2012;3:361. doi: 10.3389/fimmu.2012.00361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellouk N, Enninga J. Cytosolic access of intracellular bacterial pathogens: the Shigella paradigm. Front Cell Infect Microbiol. 2016;6:35. doi: 10.3389/fcimb.2016.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud J, Ohayon H, Gounon P, Mege RM, Cossart P. E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell. 1996;84:923–932. doi: 10.1016/s0092-8674(00)81070-3. [DOI] [PubMed] [Google Scholar]
- Mercer J, Helenius A. Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr Opin Microbiol. 2012;15:490–499. doi: 10.1016/j.mib.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Ortega FE, Rengarajan M, Chavez N, Radhakrishnan P, Gloerich M, Bianchini J, Siemers K, Luckett WS, Lauer P, Nelson WJ, Theriot JA. Adhesion to the host cell surface is sufficient to mediate Listeria monocytogenes entry into epithelial cells. Mol Biol Cell. 2017;28:2945–2957. doi: 10.1091/mbc.E16-12-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne SE, Brumell JH. Listeriolysin O: from bazooka to Swiss army knife. Philos Trans R Soc Lond B Biol Sci. 2017;372 doi: 10.1098/rstb.2016.0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak-Sharma S, Zhang X, Lam JGT, Weisleder N, Seveau SM. High-throughput microplate-based assay to monitor plasma membrane wounding and repair. Front Cell Infect Microbiol. 2017;7:305. doi: 10.3389/fcimb.2017.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentecost M, Kumaran J, Ghosh P, Amieva MR. Listeria monocytogenes internalin B activates junctional endocytosis to accelerate intestinal invasion. PLoS Pathog. 2010;6:e1000900. doi: 10.1371/journal.ppat.1000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Cerda J, Charbit A, Enninga J, Lafont F, Cossart P. Manipulation of host membranes by the bacterial pathogens Listeria, Francisella, Shigella and Yersinia. Semin Cell Dev Biol. 2016;60:155–167. doi: 10.1016/j.semcdb.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price LS, Langeslag M, ten Klooster JP, Hordijk PL, Jalink K, Collard JG. Calcium signaling regulates translocation and activation of Rac. J Biol Chem. 2003;278:39413–39421. doi: 10.1074/jbc.M302083200. [DOI] [PubMed] [Google Scholar]
- Ribet D, Cossart P. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect. 2015;17:173–183. doi: 10.1016/j.micinf.2015.01.004. [DOI] [PubMed] [Google Scholar]
- Rosado CJ, Kondos S, Bull TE, Kuiper MJ, Law RH, Buckle AM, Voskoboinik I, Bird PI, Trapani JA, Whisstock JC, Dunstone MA. The MACPF/CDC family of pore-forming toxins. Cell Microbiol. 2008;10:1765–1774. doi: 10.1111/j.1462-5822.2008.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosselin M, Virlogeux-Payant I, Roy C, Bottreau E, Sizaret PY, Mijouin L, Germon P, Caron E, Velge P, Wiedemann A. Rck of Salmonella enterica, subspecies enterica serovar enteritidis, mediates zipper-like internalization. Cell Res. 2010;20:647–664. doi: 10.1038/cr.2010.45. [DOI] [PubMed] [Google Scholar]
- Rougerie P, Miskolci V, Cox D. Generation of membrane structures during phagocytosis and chemotaxis of macrophages: role and regulation of the actin cytoskeleton. Immunol Rev. 2013;256:222–239. doi: 10.1111/imr.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seveau S. Multifaceted activity of listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Subcell Biochem. 2014;80:161–195. doi: 10.1007/978-94-017-8881-6_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seveau S, Bierne H, Giroux S, Prevost MC, Cossart P. Role of lipid rafts in E-cadherin–and HGF-R/Met-mediated entry of Listeria monocytogenes into host cells. J Cell Biol. 2004;166:743–753. doi: 10.1083/jcb.200406078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seveau S, Pizarro-Cerda J, Cossart P. Molecular mechanisms exploited by Listeria monocytogenes during host cell invasion. Microbes Infect. 2007;9:1167–1175. doi: 10.1016/j.micinf.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Shaughnessy LM, Lipp P, Lee KD, Swanson JA. Localization of protein kinase C epsilon to macrophage vacuoles perforated by Listeria monocytogenes cytolysin. Cell Microbiol. 2007;9:1695–1704. doi: 10.1111/j.1462-5822.2007.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Naujokas M, Park M, Ireton K. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell. 2000;103:501–510. doi: 10.1016/s0092-8674(00)00141-0. [DOI] [PubMed] [Google Scholar]
- Simon CM, Vaughan EM, Bement WM, Edelstein-Keshet L. Pattern formation of Rho GTPases in single cell wound healing. Mol Biol Cell. 2013;24:421–432. doi: 10.1091/mbc.E12-08-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sit ST, Manser E. Rho GTPases and their role in organizing the actin cytoskeleton. J Cell Sci. 2011;124(Pt 5):679–683. doi: 10.1242/jcs.064964. [DOI] [PubMed] [Google Scholar]
- Stow JL, Condon ND. The cell surface environment for pathogen recognition and entry. Clin Transl Immunology. 2016;5:e71. doi: 10.1038/cti.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukeno A, Nagamune H, Whiley RA, Jafar SI, Aduse-Opoku J, Ohkura K, Maeda T, Hirota K, Miyake Y, Kourai H. Intermedilysin is essential for the invasion of hepatoma HepG2 cells by Streptococcus intermedius. Microbiol Immunol. 2005;49:681–694. doi: 10.1111/j.1348-0421.2005.tb03647.x. [DOI] [PubMed] [Google Scholar]
- Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, Schuchman E, Tabas I, Andrews NW. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol. 2010;189:1027–1038. doi: 10.1083/jcb.201003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadia S, Arnett E, Haghighat AC, Wilson-Kubalek EM, Tweten RK, Seveau S. The pore-forming toxin listeriolysin O mediates a novel entry pathway of L. monocytogenes into human hepatocytes. PLoS Pathog. 2011;7:e1002356. doi: 10.1371/journal.ppat.1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadia S, Seveau S. Fluxes of Ca2+ and K+ are required for the listeriolysin O-dependent internalization pathway of Listeria monocytogenes. Infect Immun. 2014;82:1084–1091. doi: 10.1128/IAI.01067-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, Goebel W, Gonzalez-Zorn B, Wehland J, Kreft J. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev. 2001;14:584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velge P, Wiedemann A, Rosselin M, Abed N, Boumart Z, Chausse AM, Grepinet O, Namdari F, Roche SM, Rossignol A, Virlogeux-Payant I. Multiplicity of Salmonella entry mechanisms, a new paradigm for Salmonella pathogenesis. Microbiologyopen. 2012;1:243–258. doi: 10.1002/mbo3.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Zhang J, Tsien RY, Newton AC. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol. 2003;161:899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Huang S, Kapoor-Munshi A, Nemerow G. Adenovirus internalization and infection require dynamin. J Virol. 1998;72:3455–3458. doi: 10.1128/jvi.72.4.3455-3458.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann A, Mijouin L, Ayoub MA, Barilleau E, Canepa S, Teixeira-Gomes AP, Le Vern Y Rosselin M, Reiter E, Velge P. Identification of the epidermal growth factor receptor as the receptor for Salmonella Rck-dependent invasion. FASEB J. 2016;30:4180–4191. doi: 10.1096/fj.201600701R. [DOI] [PubMed] [Google Scholar]
- Woolsey AM, Burleigh BA. Host cell actin polymerization is required for cellular retention of Trypanosoma cruzi and early association with endosomal/lysosomal compartments. Cell Microbiol. 2004;6:829–838. doi: 10.1111/j.1462-5822.2004.00405.x. [DOI] [PubMed] [Google Scholar]
- Yousuf MA, Lee JS, Zhou X, Ramke M, Lee JY, Chodosh J, Rajaiya J. Protein kinase C signaling in adenoviral infection. Biochemistry. 2016 doi: 10.1021/acs.biochem.6b00858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.