

ABSTRACT
Familial chylomicronemia syndrome (FCS) is a rare lipid disorder posing significant clinical burdens on patients. Due to its rarity, variety of presentations, and lack of universal diagnostic criteria, patients see an average of five physicians before diagnosis. We screened adults for a triglyceride level ≥1000 mg/dL from September 2015 to September 2016 and a history of pancreatitis and performed a thorough chart review on those who met the criteria. An adjudication panel used a definition that also called for supportive information including history of hypertriglyceridemia or family history of pancreatitis/hypertriglyceridemia. Among 297,891 adults with laboratory values available, 334 (0.11%) had triglyceride levels ≥1000 mg/dL, and 30 (9%) of those had pancreatitis. Most of these 30 patients were male (73%), had diabetes (90%), were taking a fibrate (93%), and were taking a statin (80%). The average body mass index was 32.5 ± 4.5 kg/m2. Six cases were ruled out, primarily due to substance abuse and/or isolated pancreatitis. Of the 24 suspected FCS cases, the average maximum triglyceride level was 3085 ± 1211 mg/dL. Electronic screening methods based solely on triglycerides ≥1000 mg/dL and pancreatitis eliminated 99.99% of the population, enabling the adjudication panel to focus on 30 cases. In 24 cases, FCS could not be ruled out; hence, the prevalence of FCS may be as high as 1 in 12,413.
KEYWORDS: Chylomicrons, chylomicronemia syndrome, diagnostic tools, familial hypertriglyceridemia, pancreas, pancreatitis, triglycerides
Familial chylomicronemia syndrome (FCS) is a rare inherited lipid disorder that poses significant clinical and psychosocial burdens on patients.1 Though patients have recurrent abdominal pain and fatigue and are prone to episodes of pancreatitis,2 the remaining physical manifestations are heterogeneous; pediatric patients may present with failure to thrive, whereas adolescents and adults may have eruptive xanthomas, lipemia retinalis, and/or hepatomegaly.3,4 A milky “latescent” or “lipemic” appearance of plasma is also a hallmark.1 Due to the rarity of FCS and the variety of presentations, patients see an average of five physicians before receiving the diagnosis, enduring a low quality of life all the while.3,5 The true prevalence of FCS is unknown, with an estimate of 20 per million and suspicion that it is much higher.6,7 Because there are no accepted criteria for FCS diagnosis, our goal was to apply our proposed diagnostic algorithm to efficiently examine a vast number of patients in our health care system to identify suspected FCS cases and obtain an estimate of prevalence.
METHODS
Laboratory records of adult patients in the North Texas division of the Baylor Scott & White Health system were screened from September 2015 to September 2016 for triglyceride values ≥1000 mg/dL. Electronic health records of those with elevated triglycerides were scanned for history of pancreatitis (Figure 1). Those with triglyceride values ≥1000 mg/dL and a history of pancreatitis received a thorough chart review, with data elements selected based on existing FCS literature.2,3
Figure 1.
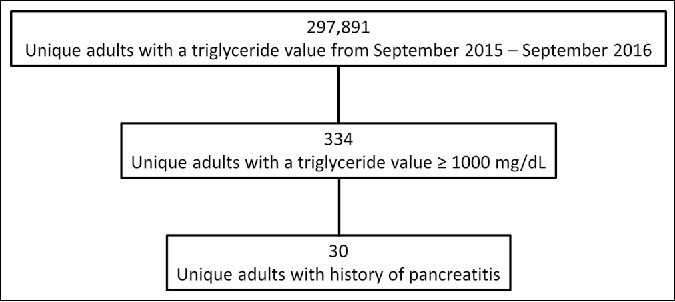
CONSORT diagram detailing the charts eligible for extensive review.
Outpatient clinic notes and documentation from hospitalizations were reviewed to identify xanthomas, a family history of pancreatitis and/or high triglyceride levels, smoking and alcohol behaviors, and medication therapy. Computed tomography (CT) scans and ultrasonography reports were reviewed; the radiologist's interpretation allowed for the determination of hepatic steatosis and splenic vein thrombosis. Maximum and minimum triglyceride values, including inpatient and outpatient records, were recorded. Total cholesterol at the time of the highest triglyceride level was obtained to calculate a triglyceride/total cholesterol ratio. In addition, the most recent results for triglyceride, low-density lipoprotein, high-density lipoprotein, total cholesterol, and hemoglobin levels were recorded.
An adjudication panel, consisting of two physicians, met to discuss the charts and provide a ruling for the cases. The panel used a working definition that called for acute pancreatitis associated with hypertriglyceridemia and at least one piece of supportive information, including a history of hypertriglyceridemia or family history of pancreatitis and/or hypertriglyceridemia. Other clear explanations for the clinical scenario provided the basis for exclusion, such as severe alcoholism.
RESULTS
Thirty patients (0.01%) out of the original 297,891 met criteria for extensive chart review (Table 1). Most of these 30 patients were male (73%), had diabetes (90%), were taking a fibrate (93%), and/or were taking a statin (80%), with 23% of the whole cohort on maximum statin therapy (atorvastatin 40 or 80 mg daily, or rosuvastatin 40 mg daily). The average body mass index was 32.5 ± 4.5 kg/m2. The average age at the onset of pancreatitis was 37.3 ± 10.1 years; however, this was calculated using the first documented date in the patient's chart and not directly from the clinical exam. Forty-three percent of patients had a cholecystectomy. For patients who had available CT scans (n = 21), 29% had evident splenic vein thrombosis. Of those who had CT scans and ultrasonography (n = 23), 87% showed hepatic steatosis. The average maximum triglyceride value was 2864 ± 1255 mg/dL, and the median triglyceride/total cholesterol ratio was 7 (quartile 1 = 5, quartile 3 = 9).
Table 1.
Clinical presentation of the 30 patients who met initial criteria for familial chylomicronemia syndromea
| Variable | Result |
|---|---|
| Male | 22 (73%) |
| Age at pancreatitis onsetb (years) | 37.3 ± 10.1 |
| Age at analysis (years) | 40.2 ± 10.1 |
| Body mass index (kg/m2) | 32.5 ± 4.5 |
| Hispanic | 14 (47%) |
| Family history of high triglycerides | 7 (23%) |
| Xanthoma | 1 (3%) |
| Diabetes mellitus | 27 (90%) |
| Insulin-dependent diabetes mellitus | 21 (70%) |
| Diabetic ketoacidosis | 6 (20%) |
| Cholecystectomy | 13 (43%) |
| Spleen vein thrombosisc | 6 (29%) |
| Hepatic steatosisd | 20 (87%) |
| Alcohol abuse | 6 (20%) |
| Smoker (current) | 9 (30%) |
| Smoker (former) | 15 (50%) |
| Laboratory values | |
| Peak triglyceride in study period (mg/dL) | 2864.3 ± 1254.5 |
| Lowest triglyceride in study period (mg/dL) | 244 [170, 334] |
| High-density lipoprotein (mg/dL) | 30.3 ± 10.7 |
| Low-density lipoprotein (mg/dL) | 80.9 ± 43.9 |
| Total cholesterol (mg/dL) | 213 [164, 286] |
| Triglyceride/total cholesterol (at peak triglyceride) | 7.4 [5.0, 8.7] |
| Triglyceride/total cholesterol (most recent) | 3.3 [1.8, 4.8] |
| Hemoglobin (mg/dL) | 14.1 ± 1.8 |
| Medications | |
| Statin | 24 (80%) |
| Maximum statin | 7 (23%) |
| Fibrate | 28 (93%) |
| Omega-3 | 16 (53%) |
Twenty-four were clinically adjudicated as having familial chylomicronemia syndrome.
This was calculated with the first documented date in a patient's chart; these were not back-dated, even if a patient had pancreatitis as a child.
n = 21.
n = 23.
The adjudication panel ruled out a total of six cases (Figure 2). The remaining 24 patients were chronically very ill, presenting with multiple episodes of abdominal pain and/or pancreatitis throughout their course. Because FCS could not effectively be ruled out, genetic confirmation of FCS would be recommended. Of these patients, 29% had a family history of high triglycerides; 92% had diabetes and 71% required insulin; 21% had a recorded incidence of diabetic ketoacidosis; and 46% had a cholecystectomy. For those with available imaging studies, 5 (29%) had splenic vein thrombosis, and 15 (83%) had hepatic steatosis. Three (13%) had a history of alcohol abuse, 10 (42%) had a history of smoking, and 5 (21%) were currently smokers. Nineteen (79%) patients were on statin therapy and 5 (21%) were on maximal statin therapy; 23 (96%) and 13 (54%) were on a fibrate and omega-3 medication, respectively. The average maximum triglyceride value was 3085 ± 1211 mg/dL, and the median triglyceride/total cholesterol ratio was 7 (quartile 1 = 6, quartile 3 = 9). The average hemoglobin again was within normal range, 14 ± 2 mg/dL.
Figure 2.
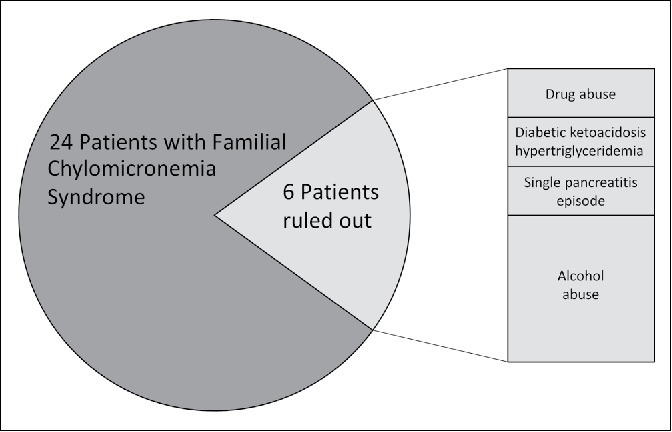
Possible familial chylomicronemia syndrome cases and the subset that was clinically ruled out.
DISCUSSION
Our method of screening triglyceride values ≥1000 mg/dL was exceptionally effective in reducing the pool of potential patients with FCS; only 0.11% of patients met that criterion. Additional screening for pancreatitis further reduced the pool by 91%. Hence, the use of electronic screening methods based solely on those two criteria eliminated 99.99% of the population, enabling the adjudication panel to focus on 30 cases out of the original 297,891. These 30 patients were predominantly male and obese; many had insulin-dependent diabetes, and nearly half had a cholecystectomy. Every patient was taking a statin, fibrate, and/or omega-3. Triglycerides were elevated with a mean >2500 mg/dL and a median triglyceride/total cholesterol ratio >7. Twenty-four cases could not be ruled out and would be advised for genetic confirmation, indicating that this disease may have a prevalence as high as 1 in 12,413.
Excessively high triglycerides (>1000 mg/dL) typically reveal the presence of abnormalities in the lipolysis pathway; determining the etiology of these elevated triglycerides is challenging, however. FCS has a genetic basis resulting from a loss-of-function mutation in genes involved in lipolysis. Five main genes have been implicated in FCS, with over 100 mutations. The most common cause is a mutation of the lipoprotein lipase (LPL) gene itself, present in over 80% of genetically confirmed FCS cases.3,8 The frequency of carriers of LPL deficiency is estimated to be around 1 in 500.9,10 Such mutations can cause a presentation of illness during infancy. Less common mutations of the apolipoprotein C2 (APOC2) gene can cause presentation in childhood. Conversely, patients with glycosylphosphatidylinositol anchored high-density lipoprotein binding protein 1, apolipoprotein A5, and lipase maturation factor 1 mutations typically show signs of chylomicronemia in late adulthood (Figure 3).2
Figure 3.
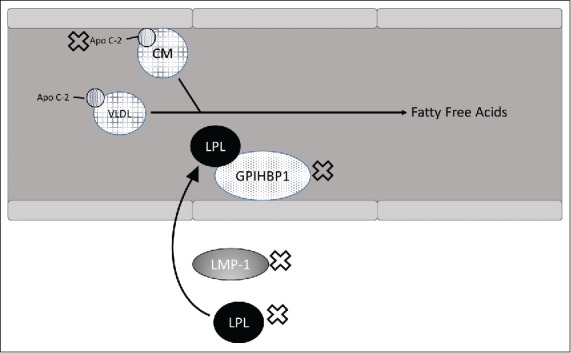
Mechanism of triglyceride breakdown into free fatty acids. Genes with abnormalities that may cause familial chylomicronemia syndrome are labeled with an X. Apo C-2 indicates apolipoprotein C2; CM, chylomicron; GPIHBP1, glycosylphosphatidylinositol anchored high-density lipoprotein binding protein 1; LMF-1, lipase maturation factor 1; LPL, lipoprotein lipase; VLDL, very low-density lipoprotein.
Because the goal of this study was to identify a rare disorder, the adjudication panel focused on ruling out patients, which required clear evidence that pancreatitis or high triglycerides was caused by a secondary factor (e.g., hypothyroidism, medications, alcohol abuse, uncontrolled diabetes).11 Although diabetes has been used to rule out FCS in some algorithms, recurrent pancreatitis may lead to endocrine/exocrine insufficiency, and impaired energy metabolism related to LPL dysfunction may occur from FCS, which could cause diabetes.12 For that reason, the diagnostic algorithm in this study did not rule patients out based on diabetes. Stroes et al introduced the triglyceride/total cholesterol ratio ≥5 as a diagnostic aid, a threshold that approximately 75% of our patients reached.3 Because a variety of lipid disorders yield high triglyceride values, ensuring apolipoprotein B < 120 mg/dL is advised to eliminate the most common diagnosis, polygenic combined hyperlipidemia; however, only one of our cases had an apolipoprotein B measure.3
Burnett and colleagues' review of familial LPL deficiency stated that 25% of children develop symptoms in the first year of life, and the remaining typically present with symptoms before age 10.13 Approximately 50% of patients presented with xanthomas in Burnett and colleagues' work, compared with only approximately 4% of our sample. Controversially, work in the 1970s reported reversible neuropsychiatric changes associated with chylomicronemia; however, no publications in subsequent years confirmed the finding.14 Males and females are thought to be equally affected, with no predilection for a particular race, although certain geographic regions (e.g., Quebec, Canada) exhibit a higher prevalence due to a founder effect.13,15 In our study, 75% of cases of FCS were in men and half were in Hispanics; this ethnic group is known to have a higher rate of hypertriglyceridemia than others.
The median age at FCS diagnosis for 67 genetically confirmed patients in the APPROACH trial was 27 years, and their mean body mass index was 25 ± 6 kg/m2, compared to 32.5 ± 4.5 kg/m2 for our FCS cases. Their median fasting triglycerides of 2209 mg/dL ± 1199 was much lower than our FCS sample's mean of 3085 ± 1211 mg/dL. Eruptive xanthomas were present in 22% of APPROACH cases, with 73% having a documented history of acute pancreatitis.16 Hence, our patient profile was similar but with a lower rate of xanthomas and a higher rate of pancreatitis. A survey of 18 lipidologists from nine countries compiled characteristics and outcomes of 251 FCS patients; the patient profile matched closely with that of those enrolled in the APPROACH trial, further establishing the validity of the FCS diagnosis.17
Fifteen (83%) of our patients with suspected FCS had hepatic steatosis, which indicates mild elevation in liver transaminase and would not be expected to progress to hepatic dysfunction.4 Interestingly, 29% of our suspected FCS patients had splenic vein thrombosis. It is unknown whether this is common in the general FCS population or what the etiology may be, although recurrent episodes of severe inflammatory pancreatitis may be a contributing factor.
Published rates of acute pancreatitis in FCS are between 50% and 80%.3 In some studies, FCS patients have a 360-fold increase in risk of pancreatitis compared to patients with normal triglyceride levels.1,3 For patients with reduced LPL function, pancreatitis may be severe, with complications such as pseudocysts, necrosis, and/or surgical resection, yielding mortality rates from 5% to 30%.1,3 In fact, a simulation model using a cohort of patients with FCS with a mean age of 38 years and mean triglyceride level of 2741 mg/dL yielded an estimated 10 episodes of acute pancreatitis in a lifetime, resulting in 81 inpatient days and a lifetime cost of $154,126.18 Mortality due to acute pancreatitis in this simulation was estimated at 54%.
Almost all of our patients were on fibrate therapy (96%) and/or taking a statin (79%), although lipid-lowering agents such as fibrates and statins have shown limited benefit and the mainstay of therapy remains a stringent fat-restricted diet (20–25 g/day), which is somewhat unrealistic.3 Further, prospective observational studies of patients with autosomal recessive LPL gene deficiency showed that a controlled diet alone is inadequate in preventing pancreatitis.11 Alternative drugs approved for homozygous familial hypercholesterolemia treatment, including a microsomal triglyceride transfer protein inhibitor (lomitapide) and an antisense RNA compound (mipomersen), may be feasible for treating FCS.2,19 Inhibition of diacylglycerol O–acyltransferase 1, which mediates triglyceride synthesis, has also been examined (pradigastat in trial).20 Similarly, volanesorsen, an antisense RNA compound, is in a phase 3 trial.21 ALN-ANG, an antisense oligonucleotide that inhibits translation of angiopoietin-like protein 3 mRNA, has shown efficacy in lowering total cholesterol and triglycerides in preclinical trials.3
This study has limitations inherent to a retrospective chart review. The extent of omissions in patients' charts cannot be fully known. Similarly, though we were able to review current medications, it was not possible to examine medication use corresponding to the time of a particular triglyceride date. Only one patient had a recorded apolipoprotein B value. Additionally, information regarding very low-density lipoprotein was available for only two individuals, so we were unable to distinguish these suspected FCS cases from type V hyperlipidemia. A potential limitation of the proposed methodology is that the definition required a history of pancreatitis, yet it is estimated that only 50% to 80% of patients with FCS have pancreatitis. In this study, the criterion of pancreatitis instantly excluded 304 patients. Additionally, without information regarding uncontrolled diabetes, it is unknown whether some of the individuals with diabetes in this study should be ruled out. Finally, these patients did not undergo genetic confirmation.
Because these chronically ill patients endure psychosocial and medical burdens prior to receiving the proper diagnosis, there is great need to identify FCS early, especially as the possibility of novel pharmaceutical therapies continues to emerge.
Funding Statement
This work was funded in part by Akcea Therapeutics and the Baylor Health Care System Foundation.
References
- 1.Gelrud A, Williams KR, Hsieh A, Gwosdow AR, Gilstrap A, Brown A. The burden of familial chylomicronemia syndrome from the patients' perspective. Expert Rev Cardiovasc Ther. 2017;15:879–887. doi: 10.1080/14779072.2017.1372193. PMID:28847199. [DOI] [PubMed] [Google Scholar]
- 2.Brahm A, Hegele RA. Chylomicronaemia—current diagnosis and future therapies. Nat Rev Endocrinol. 2015;11:352–362. doi: 10.1038/nrendo.2015.26. PMID:25732519. [DOI] [PubMed] [Google Scholar]
- 3.Stroes E, Moulin P, Parhofer KG, Rebours V, Löhr JM, Averna M. Diagnostic algorithm for familial chylomicronemia syndrome. Atheroscler Suppl. 2017;23:1–7. doi: 10.1016/j.atherosclerosissup.2016.10.002. PMID:27998715. [DOI] [PubMed] [Google Scholar]
- 4.Santamarina-Fojo S. The familial chylomicronemia syndrome. Endocrinol Metab Clin North Am. 1998;27:551–567. doi: 10.1016/S0889-8529(05)70025-6. PMID:9785052. [DOI] [PubMed] [Google Scholar]
- 5.Davidson M, Stevenson M, Hsieh A, Ahmad Z, Crowson C, Witztum JL. The burden of familial chylomicronemia syndrome: interim results from the IN-FOCUS study. Expert Rev Cardiovasc Ther. 2017;15:415–423. doi: 10.1080/14779072.2017.1311786. PMID:28338353. [DOI] [PubMed] [Google Scholar]
- 6.Kota S, Kota SK, Jammula S, Modi KD. Familial chylomicronemia syndrome—an uncommon cause of acute pancreatitis with encephalopathy. Indian J Gastroenterol. 2012;31:277–279. doi: 10.1007/s12664-012-0261-6. PMID:23081844. [DOI] [PubMed] [Google Scholar]
- 7.Gagné C, Brun LD, Julien P, Moorjani S, Lupien PJ. Primary lipoprotein-lipase-activity deficiency: clinical investigation of a French Canadian population. CMAJ. 1989;140:405–411. PMID:2914262. [PMC free article] [PubMed] [Google Scholar]
- 8.Rabacchi C, D'Addato S, Palmisano S, et al.. Clinical and genetic features of 3 patients with familial chylomicronemia due to mutations in GPIHBP1 gene. J Clin Lipidol. 2016;10:915–921.e4. doi: 10.1016/j.jacl.2016.03.009. PMID:27578123. [DOI] [PubMed] [Google Scholar]
- 9.Ma Y, Henderson HE, Murthy V, et al.. A mutation in the human lipoprotein lipase gene as the most common cause of familial chylomicronemia in French Canadians. N Engl J Med. 1991;324:1761–1766. doi: 10.1056/NEJM199106203242502. PMID:2038366. [DOI] [PubMed] [Google Scholar]
- 10.Nikkilä E. Familial lipoprotein lipase deficiency and related disorders of chylomicron metabolism In: Stanbury J, Wyngaarden JB, Fredrickson DS, et al, eds. The Metabolic Basis of Inherited Disease. 5th ed. New York, NY: McGraw-Hill; 1983:622–642. [Google Scholar]
- 11.Miller M, Stone NJ, Ballantyne C, et al;. American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Nursing; Council on the Kidney in Cardiovascular Disease. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2292–2333. doi: 10.1161/CIR.0b013e3182160726. PMID:21502576. [DOI] [PubMed] [Google Scholar]
- 12.Gaudet D, de Wal J, Tremblay K, et al;. Review of the clinical development of alipogene tiparvovec gene therapy for lipoprotein lipase deficiency. Atheroscler Suppl. 2010;11:55–60. doi: 10.1016/j.atherosclerosissup.2010.03.004. PMID:20427244. [DOI] [PubMed] [Google Scholar]
- 13.Burnett J, Hooper AJ, Hegele RA. Familial lipoprotein lipase deficiency. In: Adam M, Ardinger HH, Pagon RA, et al, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle. PMID:20301485 Accessed April9, 2018. [PubMed] [Google Scholar]
- 14.Heilman K, Fisher WR. Hyperlipidemic dementia. Arch Neurol. 1974;31:67–68. doi: 10.1001/archneur.1974.00490370093018. PMID:4834546. [DOI] [PubMed] [Google Scholar]
- 15.Normand T, Bergeron J, Fernandez-Margallo T, et al.. Geographic distribution and genealogy of mutation 207 of the lipoprotein lipase gene in the French Canadian population of Québec. Hum Genet. 1992;89:671–675. doi: 10.1007/BF00221960. PMID:1511985. [DOI] [PubMed] [Google Scholar]
- 16.Blom D. Characterizing familial chylomicronemia syndrome: baseline data of the APPROACH study. J Clin Lipidology. 2017;11:816. doi: 10.1016/j.jacl.2017.04.073. [DOI] [PubMed] [Google Scholar]
- 17.Gaudet D. Familial chylomicronemia syndrome (FCS) patients recruited to the APPROACH trial of volanesorsen therapy are representative of subjects with FCS. J Clin Lipidol. 2017;11:817–818. doi: 10.1016/j.jacl.2017.04.076. [DOI] [Google Scholar]
- 18.Lin F, Thomas S, Calado F, Clegg J. Long-term costs and consequences of patients with familial chylomicronemia syndrome—a simulation model approach. Value Health. 2014;17:A400. doi: 10.1016/j.jval.2014.08.908. PMID:27200950. [DOI] [PubMed] [Google Scholar]
- 19.Won J, Zhang J, Tecson KM, McCullough PA. Balancing low-density lipoprotein cholesterol reduction and hepatotoxicity with lomitapide mesylate and mipomersen in patients with homozygous familial hypercholesterolemia. Rev Cardiovasc Med. 2017;18:21–28. PMID:28509890. [DOI] [PubMed] [Google Scholar]
- 20.Meyers C, Tremblay K, Amer A, Chen J, Jiang L, Gaudet D. Effect of the DGAT1 inhibitor pradigastat on triglyceride and apoB48 levels in patients with familial chylomicronemia syndrome. Lipids Health Dis. 2015;14:1–9. doi: 10.1186/s12944-015-0006-5. PMID:25889044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitz J, Gouni-Berthold I. Apoc-III antisense oligonucleotides: a new option for the treatment of hypertriglyceridemia. Curr Med Chem. 2017. doi: 10.2174/0929867324666170609081612. [DOI] [PubMed] [Google Scholar]