

ABSTRACT
Like RAS proteins, the aberrant function of RHO family small GTPases has been implicated in driving cancer development and growth. However, unlike the RAS family, where gain-of-function missense mutations are found in ∼25% of all human cancers, missense mutations are relatively rare in RHO proteins. Instead, altered RHO activity in cancer more commonly arises through the aberrant functions of RHO GTPase regulators. In many cancer types, altered expression and/or mutation of RHO-selective guanine nucleotide exchange factors (RHOGEFs) or GTPase-activating proteins (RHOGAPs), which activate or inactivate RHO GTPases, respectively, is observed. For example, deletion or loss of expression of the RHOA GAP DLC1 is well-established to drive cancer growth. Recently, we identified high expression of 2 RHOGAPs, ARHGAP11A and RACGAP1, in the basal-like breast cancer subtype. Unexpectedly, both of these RHOA GAPs exhibited properties of oncoproteins rather than tumor suppressors, in contrast to DLC1. In this commentary, we summarize our findings and speculate that different RHOA GAPs can play distinct roles in cancer depending on their spatial regulation and cancer type context. We also evaluate our results in light of recently-described cancer genome sequencing studies that have identified loss-of-function mutations of RHOA in specific cancer types.
KEYWORDS: ARHGAP11A, basal-like breast cancer, MGCRACGAP, MP-GAP, RACGAP1, RHOA, RHOGAP
RHO family small GTPases are intracellular signaling molecules of the RAS superfamily. Under normal conditions, the activity of these proteins is tightly controlled by 3 different classes of regulatory molecules: guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs) (Fig. 1). GEFs activate RHO GTPases by catalyzing the exchange of a bound GDP molecule for GTP,1 whereas GAPs return these proteins to an inactive, GDP-bound state by stimulating the intrinsic GTPase activity of RHO proteins.2 GDIs are responsible for maintaining a cytosolic pool of stable, inactive RHO GTPases which can readily translocate to membranes, where nucleotide exchange takes place.3 When active, RHO GTPases signal to numerous effectors, through which they regulate cytoskeletal dynamics and a variety of cellular responses such as cell cycle progression, cytokinesis, migration, and polarity.
Figure 1.
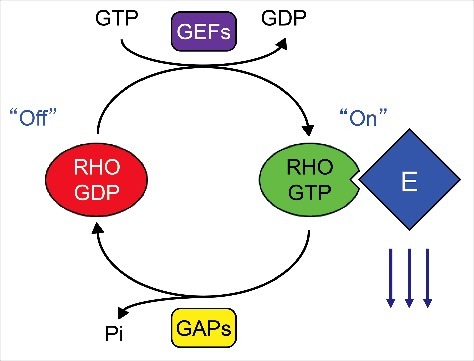
RHO GTPases function as binary GDP-GTP regulated on-off switches. RHOGEFs stimulate nucleotide exchange, resulting in the formation of active GTP-bound RHO GTPases, which preferentially bind to a spectrum of functionally diverse effectors (E; e.g., ROCK serine/threonine kinases). In contrast, RHOGAPs accelerate the intrinsic GTP hydrolysis activity of RHO GTPases, cycling them to their inactive GDP-bound state.
Given the role of RHO GTPases in regulating key aspects of proliferation and invasion, it is unsurprising that these proteins have been strongly implicated in tumorigenesis.4,5 However, unlike RAS GTPases, which are mutated in approximately 27% of all human cancers,6 mutations in RHO proteins are relatively rare (Fig. 2). Indeed, as discussed below, RHO GTPase mutations have only recently been identified in a small set of specific cancer types with the advent of next generation sequencing. Instead, these proteins more commonly contribute to cancer through their dysregulated expression and/or activation.7
Figure 2.
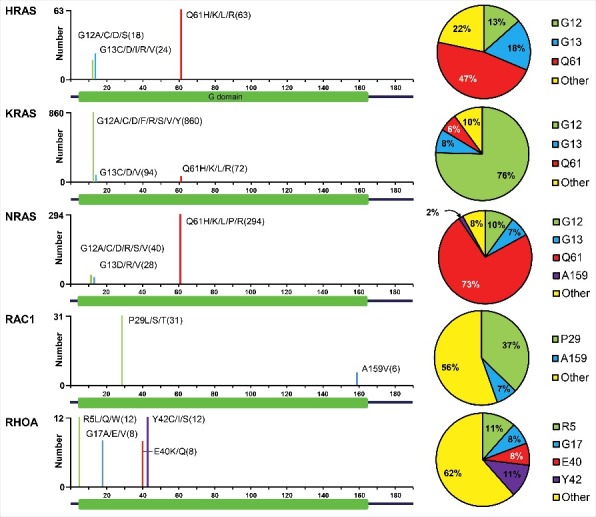
Missense mutations in RAS and RHO GTPases in cancer. Data were compiled for the 3 RAS proteins (HRAS, KRAS, and NRAS), RAC1, and RHOA for all cancers in cBioPortal (http://www.cbioportal.org/). Shown are those residues where 5 or more mutations have been identified. For RAS proteins, there are 3 hotspots for missense mutations (G12, G13, and Q61), leading to impaired intrinsic and RASGAP-stimulated GTP hydrolysis, and/or causing a fast cycling defect, favoring formation of RAS-GTP. For RAC1, the predominant mutation is at P29, causing a fast cycling defect that promotes the GTP-bound state. In contrast, the missense mutations found in RHOA do not favor formation of RHOA-GTP and instead lead to impaired interaction with effectors and/or regulators, resulting in dominant-negative mutant proteins.
In this context, overexpression of several RHOGEFs, including ECT2, PREX1, VAV1, and TIAM1, has been linked to the growth and progression of various cancers.8 GEFs are generally thought to promote cancer by causing hyper-elevated RHO GTPase activity. In contrast, RHOGAPs, which have the opposite biochemical role of RHOGEFs, are generally presumed to suppress tumorigenesis.9 A tumor suppressor role for RAS-specific GAPs is well-established (e.g., the NF1 tumor suppressor).9 The best studied RHOGAP in cancer is the RHOA-selective DLC1 (Fig. 3), which, along with the related RHOGAPs DLC2 and DLC3, form a family of well-validated tumor suppressors that are deleted or transcriptionally silenced in many cancer types.10,11 Little is known about the function of other RHOGAPs in cancer, however.
Figure 3.
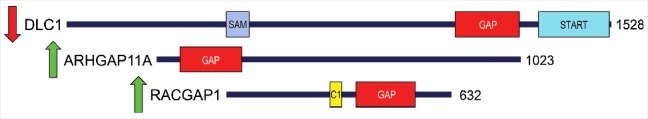
Structural and functional diversity among RHOGAP proteins. Beyond their conserved RHOGAP domains, the 64 human RHOGAPs exhibit significant sequence divergence in their non-catalytic regions. These differences may contribute to the ability of RHOGAPs to act as either tumor supressors (e.g., DLC1) or oncogenes (e.g., ARHGAP11A and RACGAP1). Arrows indicate increased (green) or decreased (red) expression/function of these GAPs in cancer. Domain architecture for each protein was determined in SMART (http://smart.embl-heidelberg.de/). Protein domain abbreviations are: BRCT, breast cancer C-terminal; C1, Protein kinase C conserved region 1 (cysteine-rich domains); DH, Dbl homology (also called RHOGEF); GAP, GTPase-activating protein for RHO-like small GTPases; PH, pleckstrin homology; SAM, Sterile alpha motif; START, in StAR and phosphatidylcholine transfer protein. The total number of amino acids in each full length protein is shown to the right.
In our recent study,12 we aimed to identify potential oncogenic or tumor suppressor roles for the 20 RHO GTPases, 79 Dbl or DOCK family RHOGEFs, 64 RHOGAPs, and 3 RHOGDIs in human breast cancer. Breast tumors can be categorized into one of 5 major subtypes (luminal A, luminal B, HER2-enriched, basal-like, and normal breast-like) depending on their gene expression and, of these, basal-like tumors are particularly aggressive and have a poor prognosis.13-18 The basal-like subtype harbors approximately 80% of the “triple negative” breast cancers (which exhibit low expression of the estrogen receptor, progesterone receptor, and the HER2 receptor tyrosine kinase). As targeted therapies for these cancers are limited, we were particularly interested in identifying novel basal-like breast cancer (BLBC) drivers from among the RHO family of small GTPases and their regulators.
To investigate this, we analyzed The Cancer Genome Atlas (TCGA) Project RNA-Seq data19 from 1,201 human breast tumors of various subtypes for the expression of all of the RHO GTPase, GEF, GAP, and GDI genes. As the general consensus within the field is that RHO GTPases and RHOGEFs are oncogenic, we expected to observe high expression of these genes across breast cancer subtypes. Conversely, we anticipated low expression of RHOGAP genes, given their proposed roles as tumor suppressors. We were therefore greatly surprised when our RNA-Seq data analysis revealed a number of RHOGAP genes that were highly expressed in basal-like tumors relative to normal-like or luminal A tumors, which have a better prognosis.12
This unexpected result prompted us to hypothesize that, in BLBC, certain RHOGAPs can act as oncogenes. To test this hypothesis, we focused on 2 of the RHOGAPs that were most highly upregulated in BLBC, ARHGAP11A and RACGAP1 (Fig. 3), and assessed their function in human BLBC cell lines. Similarly to their mRNA expression in human tumors, protein expression of both of these GAPs was higher in human breast cancer cell lines of the basal-like subtype compared to other subtypes. Strikingly, shRNA-mediated knockdown of ARHGAP11A or RACGAP1 from BLBC cell lines abolished the ability of these cells to proliferate under anchorage-dependent conditions, consistent with an oncogenic role for these GAPs in BLBC.12 We next set about identifying the mechanism(s) through which ARHGAP11A and RACGAP1 supported BLBC proliferation.
RACGAP1 (also known as MGCRACGAP) is a known regulator of cytokinesis,20,21 and ARHGAP11A (also known as MP-GAP) has also recently been identified to control this process.22 Knockdown of RACGAP1 resulted in approximately 30-45% of BLBC cells failing cytokinesis and becoming multinucleated, an effect that is likely to make a substantial contribution to the inability of these cells to proliferate. In contrast, there was no significant cytokinesis defect in BLBC cells depleted of ARHGAP11A. Instead, ARHGAP11A-knockdown caused these cells to arrest in the G1 phase of the cell cycle, as determined using flow cytometry analysis of propidium iodide-stained cells.12 By examining the expression of proteins involved in G1 to S phase progression, we established that the G1 arrest of ARHGAP11A-deficient cells was caused by an induction of the cyclin-dependent kinase (CDK) inhibitor CDKN1B/p27 and the hypophosphorylation of the cell cycle regulator RB1. In RACGAP1-depleted cells, the CDK inhibitor CDKN1A/p21 was upregulated and there was an associated increase in senescence in these cells.12 Hence, ARHGAP11A and RACGAP1 regulate BLBC proliferation via distinct mechanisms: ARHGAP11A-depletion results in p27-mediated cell cycle arrest, whereas knockdown of RACGAP1 causes cytokinesis failure, p21-induction, and the onset of senescence.
In addition to studying the roles of ARHGAP11A and RACGAP1 in promoting BLBC growth, we wanted to identify whether these GAPs were involved in other RHO GTPase-dependent processes, such as cell spreading and migration.23 Depletion of either ARHGAP11A or RACGAP1 caused BLBC cells to spread on fibronectin with an approximately 30–50% larger area than that of control cells, suggesting that both of these GAPs are indeed involved in cytoskeletal remodeling. The random migration velocity of BLBC cells was reduced in the absence of ARHGAP11A, whereas RACGAP1-deficient cells unexpectedly migrated more quickly – again highlighting the divergent functions of these 2 RHOGAPs.12
As the functions that we identified for ARHGAP11A and RACGAP1 in BLBC (proliferation, cytokinesis, cell cycle progression, spreading, and/or migration) can all be regulated by RHO GTPases, we next sought to identify which specific RHO GTPases are regulated by ARHGAP11A and RACGAP1 in BLBC cells. Pulldown experiments for active RHO GTPases revealed that the activity of RHOA, but not that of RAC1 or CDC42, was elevated in cells depleted of either ARHGAP11A or RACGAP1.12 For ARHGAP11A, this result is consistent with in vitro observations that this GAP has GTPase-activating catalytic activity toward RHOA.22,24,25 In contrast, RACGAP1 has previously been demonstrated to act as a GAP for RAC1 and CDC42, but not RHOA.26 The GTPase selectivity of RACGAP1 is somewhat controversial, with a refuted study reporting that this GAP could be converted to a RHOA-specific GAP by aurora kinase B.27 From our results, it is not possible to state whether the effect of RACGAP1 on RHOA activity in BLBC cells arises as a result of a direct interaction between these 2 proteins or through an indirect effect (e.g., via ECT2, a RHOA-specific GEF whose localization is controlled by RACGAP1).21 Regardless of the mechanism through which it occurs, our results suggest that suppression of RHOA activity by ARHGAP11A or RACGAP1 promotes BLBC proliferation. As further evidence for this, inhibition of the RHOA effector ROCK was able to partially rescue the growth defect of ARHGAP11A- or RACGAP1-depleted cells. In addition, BLBC proliferation was reduced by expression of a constitutively active RHOA mutant,12 consistent with previous observations in fibroblasts.28 Hence, our results indicate that increased RHOA activity has a negative impact on BLBC growth.
Having identified that ARHGAP11A and RACGAP1 are required for BLBC survival, we further tested their oncogenic potential by assessing their capacity to induce cellular transformation. Stable overexpression of ARHGAP11A in untransformed immortalized human MCF10A breast myoepithelial cells resulted in an increased proliferation of these cells, supporting the idea that this GAP can act as a cancer driver. Although overexpression of RACGAP1 did not affect the proliferation rate of MCF10A cells, it did cause these cells to grow with a disrupted, less spherical architecture in acinar formation assays, similarly to ARHGAP11A overexpression.12 Hence, the results of our study indicate that ARHGAP11A and RACGAP1 not only support BLBC proliferation but can also induce cancerous phenotypes in untransformed cells. This leads us to conclude that ARHGAP11A and RACGAP1 have oncogenic effects in BLBC.
As RHOGAPs are generally assumed to act as tumor suppressors, our finding that 2 different proteins from this family can support tumorigenesis via distinct mechanisms was unexpected, and illustrates the complex role of RHOGAPs in cancer. On a broader level, our results also raise several intriguing possibilities regarding the role of RHO GTPase signaling networks in cancer.
Our observation that ARHGAP11A and RACGAP1 suppress RHOA activity in BLBC comes at a time when, due to recent developments in the field, the role of RHOA in cancer is under re-assessment. In the last 2 years, several genomic sequencing studies have identified missense RHOA mutations, predominantly in peripheral T cell lymphomas and diffuse-type gastric carcinomas.29-35 Surprisingly, hotspot mutations in RHOA are at sites consistent with loss-of-function (Fig. 2): e.g., G17V, which creates a dominant-negative, GTP-binding-deficient mutant,29-31 and E40Q or Y42C, which are in the effector binding domain of RHOA and impair binding to specific RHO effectors.33-36 Furthermore, another recent study has demonstrated that colorectal cancer growth is enhanced by expression of dominant-negative RHOA.37 Hence, this emerging body of evidence indicates that, at least in certain cancer types, wild type RHOA may act as a tumor suppressor rather than an oncogene.
Our data are consistent with this notion as, if suppressed RHOA activity truly offers a growth advantage to cancer cells, it follows that GAP-mediated inhibition of RHOA would be a means of achieving this. It is notable that, to date, RHOA mutations have only been identified in a relatively restricted subset of cancer types (i.e. gastric cancer or peripheral T cell lymphomas). Indeed, RHOA missense or nonsense mutations and deletions are rare in breast cancer, for example.19 A possible explanation for this is that, in most cancers, an abolition of RHOA activity would make no selective sense, given the vast array of essential functions dependent on this GTPase. Downregulation of RHOA activity by a GAP, however, might allow for such precise spatiotemporal regulation of RHOA activity that this GTPase could still perform other functions within the cell.
An important question arising from our study is whether other RHOGAPs are pro- or anti-tumorigenic, and in which cancer types? Although we have implicated ARHGAP11A and RACGAP1 in promoting BLBC and there is evidence of other RHOGAPs, including ARHGAP35, ARHGAP5, and ARHGAP31,38-41 having pro-tumorigenic functions in breast cancer, it is undeniable that some RHOGAPs, especially DLC1, are bona fide tumor suppressors.10,11 This discrepancy is most likely due to the diverse functions, binding partners, and localization of individual RHOGAP family members. Although the functions of RHOGAP proteins are generally poorly characterized, which has hindered their study in cancer, it is apparent that the 64 human RHOGAP proteins have distinct regulators and functional domains (other than the homologous RHOGAP domain; Fig. 3). Therefore it is of little surprise that certain GAPs can promote tumorigenesis while others suppress this process. It remains to be seen which other RHOGAPs can perform these roles, and in which cancers.
In addition to BLBC,12 there is evidence of increased ARHGAP11A expression in colorectal, brain, and lung cancers.24 Similarly, RACGAP1 overexpression has been reported in several cancers, including breast, colorectal, squamous cell, gastric, uterine, and hepatocellular, and has been linked to increased recurrence and poor prognosis.42-49 Hence, it appears that pro-tumorigenic ARHGAP11A and RACGAP1 functions might be conserved between cancer types. Future work should aim to clarify the mechanisms that control the activity of these GAPs in cancer, particularly for ARHGAP11A, of which little is known regarding its regulation. The possibility that ARHGAP11A and RACGAP1 have additional effects beyond their RHOGAP activity, for example, as a result of a scaffolding role, should also be addressed.
We speculate that the subcellular localization of ARHGAP11A and RACGAP1 throughout the cell cycle is likely to be key to their oncogenic activities. Both proteins contain nuclear localization signals and have previously been shown to localize to both the nucleus and the cytoplasm.22,25,50 Indeed, RACGAP1 has been identified to act as a nuclear chaperone for STAT transcription factors.50 Interestingly, a recent study suggested that colorectal cancer patients had a worse prognosis when RACGAP1 was highly expressed in the nucleus compared to the cytoplasm.49 The ability of ARHGAP11A to regulate the cell cycle is presumably linked to its nuclear localization, and future studies should aim to identify ARHGAP11A binding partners so that the exact mechanism of cell cycle control can be determined.
In summary, in our recent study, we established that ARHGAP11A and RACGAP1 are both drivers of BLBC proliferation, consistent with an oncogenic role for both of these RHOGAP proteins.
Funding Statement
CDL was supported by the US. Army Medical Research and Materiel Command under Award No. W81XWH-14-1-0033. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the US. Army. This work was also supported by National Institutes of Health grants to CJD (CA042978, CA179193, and CA175747).
Abbreviations
- BLBC
basal-like breast cancer
- CDK
cyclin-dependent kinase
- GAP
GTPase-activating protein
- GDI
guanine nucleotide dissociation inhibitor
- GEF
guanine nucleotide exchange factor
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 2005; 6:167-80; PMID:15688002; https://doi.org/ 10.1038/nrm1587 [DOI] [PubMed] [Google Scholar]
- [2].Csepanyi-Komi R, Safar D, Grosz V, Tarjan ZL, Ligeti E. In silico tissue-distribution of human Rho family GTPase activating proteins. Small GTPases 2013; 4:90-101; PMID:23518456; https://doi.org/ 10.4161/sgtp.23708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol 2011; 12:493-504; PMID:21779026; https://doi.org/ 10.1038/nrm3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pajic M, Herrmann D, Vennin C, Conway JR, Chin VT, Johnsson AK, Welch HC, Timpson P. The dynamics of Rho GTPase signaling and implications for targeting cancer and the tumor microenvironment. Small GTPases 2015; 6:123-33; PMID:26103062; https://doi.org/ 10.4161/21541248.2014.973749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Orgaz JL, Herraiz C, Sanz-Moreno V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014; 5:e29019; PMID:25036871; https://doi.org/ 10.4161/sgtp.29019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci 2016; 129:1287-92; PMID:26985062; https://doi.org/ 10.1242/jcs.182873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Porter AP, Papaioannou A, Malliri A. Deregulation of Rho GTPases in cancer. Small GTPases 2016; 1-16; PMID:27104658; https://doi.org/ 10.1080/21541248.2016.1173767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene 2014; 33:4021-35; PMID:24037532; https://doi.org/ 10.1038/onc.2013.362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 2010; 10:842-57; PMID:21102635; https://doi.org/ 10.1038/nrc2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lukasik D, Wilczek E, Wasiutynski A, Gornicka B. Deleted in liver cancer protein family in human malignancies (Review). Oncol Lett 2011; 2:763-8; PMID:22866123; https://doi.org/10.3892/ol.2011.345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Popescu NC, Goodison S. Deleted in liver cancer-1 (DLC1): an emerging metastasis suppressor gene. Mol Diagn Ther 2014; 18:293-302; PMID:24519699; https://doi.org/ 10.1007/s40291-014-0086-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lawson CD, Fan C, Mitin N, Baker NM, George SD, Graham DM, Perou CM, Burridge K, Der CJ, Rossman KL. Rho GTPase transcriptome analysis reveals oncogenic roles for Rho GTPase-activating proteins in basal-like breast cancers. Cancer Res 2016; 76:3826-37; PMID:27216196; https://doi.org/ 10.1158/0008-5472.CAN-15-2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al.. Molecular portraits of human breast tumours. Nature 2000; 406:747-52; PMID:10963602; https://doi.org/ 10.1038/35021093 [DOI] [PubMed] [Google Scholar]
- [14].Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al.. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001; 98:10869-74; PMID:11553815; https://doi.org/ 10.1073/pnas.191367098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol 2011; 5:5-23; PMID:21147047; https://doi.org/ 10.1016/j.molonc.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kassam F, Enright K, Dent R, Dranitsaris G, Myers J, Flynn C, Fralick M, Kumar R, Clemons M. Survival outcomes for patients with metastatic triple-negative breast cancer: implications for clinical practice and trial design. Clin Breast Cancer 2009; 9:29-33; PMID:19299237; https://doi.org/ 10.3816/CBC.2009.n.005 [DOI] [PubMed] [Google Scholar]
- [17].Onitilo AA, Engel JM, Greenlee RT, Mukesh BN. Breast cancer subtypes based on ER/PR and Her2 expression: comparison of clinicopathologic features and survival. Clin Med Res 2009; 7:4-13; PMID:19574486; https://doi.org/ 10.3121/cmr.2008.825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, et al.. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006; 295:2492-502; PMID:16757721; https://doi.org/ 10.1001/jama.295.21.2492 [DOI] [PubMed] [Google Scholar]
- [19].Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al.. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015; 163:506-19; PMID:26451490; https://doi.org/ 10.1016/j.cell.2015.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zuo Y, Oh W, Frost JA. Controlling the switches: Rho GTPase regulation during animal cell mitosis. Cell Signal 2014; 26:2998-3006; PMID:25286227; https://doi.org/ 10.1016/j.cellsig.2014.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhao WM, Fang G. MgcRacGAP controls the assembly of the contractile ring and the initiation of cytokinesis. Proc Natl Acad Sci U S A 2005; 102:13158-63; PMID:16129829; https://doi.org/ 10.1073/pnas.0504145102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zanin E, Desai A, Poser I, Toyoda Y, Andree C, Moebius C, Bickle M, Conradt B, Piekny A, Oegema K. A conserved RhoGAP limits M phase contractility and coordinates with microtubule asters to confine RhoA during cytokinesis. Dev Cell 2013; 26:496-510; PMID:24012485; https://doi.org/ 10.1016/j.devcel.2013.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lawson CD, Burridge K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 2014; 5:e27958; PMID:24607953; https://doi.org/ 10.4161/sgtp.27958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kagawa Y, Matsumoto S, Kamioka Y, Mimori K, Naito Y, Ishii T, Okuzaki D, Nishida N, Maeda S, Naito A, et al.. Cell cycle-dependent Rho GTPase activity dynamically regulates cancer cell motility and invasion in vivo. PloS One 2013; 8:e83629; PMID:24386239; https://doi.org/ 10.1371/journal.pone.0083629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xu J, Zhou X, Wang J, Li Z, Kong X, Qian J, Hu Y, Fang JY. RhoGAPs attenuate cell proliferation by direct interaction with p53 tetramerization domain. Cell Rep 2013; 3:1526-38; PMID:23684608; https://doi.org/ 10.1016/j.celrep.2013.04.017 [DOI] [PubMed] [Google Scholar]
- [26].Bastos RN, Penate X, Bates M, Hammond D, Barr FA. CYK4 inhibits Rac1-dependent PAK1 and ARHGEF7 effector pathways during cytokinesis. J Cell Biol 2012; 198:865-80; PMID:22945935; https://doi.org/ 10.1083/jcb.201204107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Minoshima Y, Kawashima T, Hirose K, Tonozuka Y, Kawajiri A, Bao YC, Deng X, Tatsuka M, Narumiya S, May WS Jr., et al.. Phosphorylation by aurora B converts MgcRacGAP to a RhoGAP during cytokinesis. Dev Cell 2003; 4:549-60; PMID:12689593; https://doi.org/ 10.1016/S1534-5807(03)00089-3 [DOI] [PubMed] [Google Scholar]
- [28].Morin P, Flors C, Olson MF. Constitutively active RhoA inhibits proliferation by retarding G(1) to S phase cell cycle progression and impairing cytokinesis. Eur J Cell Biol 2009; 88:495-507; PMID:19515453; https://doi.org/ 10.1016/j.ejcb.2009.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, Carpenter Z, Abate F, Allegretta M, Haydu JE, et al.. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet 2014; 46:166-70; PMID:24413734; https://doi.org/ 10.1038/ng.2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, Muto H, Tsuyama N, Sato-Otsubo A, Okuno Y, et al.. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet 2014; 46:171-5; PMID:24413737; https://doi.org/ 10.1038/ng.2872 [DOI] [PubMed] [Google Scholar]
- [31].Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, Kim SC, Lee B, Rho K, Lee JE, et al.. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet 2014; 46:371-5; PMID:24584070; https://doi.org/ 10.1038/ng.2916 [DOI] [PubMed] [Google Scholar]
- [32].Nagata Y, Kontani K, Enami T, Kataoka K, Ishii R, Totoki Y, Kataoka TR, Hirata M, Aoki K, Nakano K, et al.. Variegated RHOA mutations in adult T-cell leukemia/lymphoma. Blood 2016; 127:596-604; PMID:26574607; https://doi.org/ 10.1182/blood-2015-06-644948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].The Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513:202-9; PMID:25079317; https://doi.org/ 10.1038/nature13480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kakiuchi M, Nishizawa T, Ueda H, Gotoh K, Tanaka A, Hayashi A, Yamamoto S, Tatsuno K, Katoh H, Watanabe Y, et al.. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet 2014; 46:583-7; PMID:24816255; https://doi.org/ 10.1038/ng.2984 [DOI] [PubMed] [Google Scholar]
- [35].Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, et al.. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet 2014; 46:573-82; PMID:24816253; https://doi.org/ 10.1038/ng.2983 [DOI] [PubMed] [Google Scholar]
- [36].Sahai E, Alberts AS, Treisman R. RhoA effector mutants reveal distinct effector pathways for cytoskeletal reorganization, SRF activation and transformation. EMBO J 1998; 17:1350-61; PMID:9482732; https://doi.org/ 10.1093/emboj/17.5.1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rodrigues P, Macaya I, Bazzocco S, Mazzolini R, Andretta E, Dopeso H, Mateo-Lozano S, Bilic J, Carton-Garcia F, Nieto R, et al.. RHOA inactivation enhances Wnt signalling and promotes colorectal cancer. Nat Commun 2014; 5:5458; PMID:25413277; https://doi.org/ 10.1038/ncomms6458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shen CH, Chen HY, Lin MS, Li FY, Chang CC, Kuo ML, Settleman J, Chen RH. Breast tumor kinase phosphorylates p190RhoGAP to regulate rho and ras and promote breast carcinoma growth, migration, and invasion. Cancer Res 2008; 68:7779-87; PMID:18829532; https://doi.org/ 10.1158/0008-5472.CAN-08-0997 [DOI] [PubMed] [Google Scholar]
- [39].Heckman-Stoddard BM, Vargo-Gogola T, McHenry PR, Jiang V, Herrick MP, Hilsenbeck SG, Settleman J, Rosen JM. Haploinsufficiency for p190B RhoGAP inhibits MMTV-Neu tumor progression. Breast Cancer Res 2009; 11:R61; PMID:19703301; https://doi.org/ 10.1186/bcr2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].McHenry PR, Sears JC, Herrick MP, Chang P, Heckman-Stoddard BM, Rybarczyk M, Chodosh LA, Gunther EJ, Hilsenbeck SG, Rosen JM, et al.. P190B RhoGAP has pro-tumorigenic functions during MMTV-Neu mammary tumorigenesis and metastasis. Breast Cancer Res 2010; 12:R73; PMID:20860838; https://doi.org/ 10.1186/bcr2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].He Y, Northey JJ, Primeau M, Machado RD, Trembath R, Siegel PM, Lamarche-Vane N. CdGAP is required for transforming growth factor beta- and Neu/ErbB-2-induced breast cancer cell motility and invasion. Oncogene 2011; 30:1032-45; PMID:21042277; https://doi.org/ 10.1038/onc.2010.477 [DOI] [PubMed] [Google Scholar]
- [42].Pliarchopoulou K, Kalogeras KT, Kronenwett R, Wirtz RM, Eleftheraki AG, Batistatou A, Bobos M, Soupos N, Polychronidou G, Gogas H, et al.. Prognostic significance of RACGAP1 mRNA expression in high-risk early breast cancer: a study in primary tumors of breast cancer patients participating in a randomized Hellenic Cooperative Oncology Group trial. Cancer Chemother Pharmacol 2013; 71:245-55; PMID:23096218; https://doi.org/ 10.1007/s00280-012-2002-z [DOI] [PubMed] [Google Scholar]
- [43].Imaoka H, Toiyama Y, Saigusa S, Kawamura M, Kawamoto A, Okugawa Y, Hiro J, Tanaka K, Inoue Y, Mohri Y, et al.. RacGAP1 expression, increasing tumor malignant potential, as a predictive biomarker for lymph node metastasis and poor prognosis in colorectal cancer. Carcinogenesis 2015; 36:346-54; PMID:25568185; https://doi.org/ 10.1093/carcin/bgu327 [DOI] [PubMed] [Google Scholar]
- [44].Hazar-Rethinam M, de Long LM, Gannon OM, Boros S, Vargas AC, Dzienis M, Mukhopadhyay P, Saenz-Ponce N, Dantzic DD, Simpson F, et al.. RacGAP1 is a novel downstream effector of E2F7-dependent resistance to doxorubicin and is prognostic for overall survival in squamous cell carcinoma. Mol Cancer Ther 2015; 14:1939-50; PMID:26018753; https://doi.org/ 10.1158/1535-7163.MCT-15-0076 [DOI] [PubMed] [Google Scholar]
- [45].Saigusa S, Tanaka K, Mohri Y, Ohi M, Shimura T, Kitajima T, Kondo S, Okugawa Y, Toiyama Y, Inoue Y, et al.. Clinical significance of RacGAP1 expression at the invasive front of gastric cancer. Gastric Cancer 2015; 18:84-92; PMID:24615626; https://doi.org/ 10.1007/s10120-014-0355-1 [DOI] [PubMed] [Google Scholar]
- [46].Wang SM, Ooi LL, Hui KM. Upregulation of Rac GTPase-activating protein 1 is significantly associated with the early recurrence of human hepatocellular carcinoma. Clin Cancer Res 2011; 17:6040-51; PMID:21825042; https://doi.org/ 10.1158/1078-0432.CCR-11-0557 [DOI] [PubMed] [Google Scholar]
- [47].Ma XJ, Salunga R, Dahiya S, Wang W, Carney E, Durbecq V, Harris A, Goss P, Sotiriou C, Erlander M, et al.. A five-gene molecular grade index and HOXB13:IL17BR are complementary prognostic factors in early stage breast cancer. Clin Cancer Res 2008; 14:2601-8; PMID:18451222; https://doi.org/ 10.1158/1078-0432.CCR-07-5026 [DOI] [PubMed] [Google Scholar]
- [48].Mi S, Lin M, Brouwer-Visser J, Heim J, Smotkin D, Hebert TM, Gunter MJ, Goldberg GL, Zheng D, Huang GS. RNA-seq identification of RACGAP1 as a metastatic driver in uterine carcinosarcoma. Clin Cancer Res 2016; PMID:27121792; https://doi.org/10.1158/1078-0432.CCR-15-2116 [DOI] [PubMed] [Google Scholar]
- [49].Yeh CM, Sung WW, Lai HW, Hsieh MJ, Yen HH, Su TC, Chang WH, Chen CY, Ko JL, Chen CJ. Opposing prognostic roles of nuclear and cytoplasmic RACGAP1 expression in colorectal cancer patients. Hum Pathol 2016; 47:45-51; PMID:26508373; https://doi.org/ 10.1016/j.humpath.2015.09.002 [DOI] [PubMed] [Google Scholar]
- [50].Kawashima T, Bao YC, Minoshima Y, Nomura Y, Hatori T, Hori T, Fukagawa T, Fukada T, Takahashi N, Nosaka T, et al.. A Rac GTPase-activating protein, MgcRacGAP, is a nuclear localizing signal-containing nuclear chaperone in the activation of STAT transcription factors. Mol Cell Biol 2009; 29:1796-813; PMID:19158271; https://doi.org/ 10.1128/MCB.01423-08 [DOI] [PMC free article] [PubMed] [Google Scholar]