

Abstract
Chloroquine-containing 2-(dimethylamino)ethyl methacrylate (DMAEMA) copolymers (PDCs) were synthesized by reversible addition-fragmentation chain-transfer (RAFT) polymerization. Systematic evaluation was performed to test the hypothesis that presence of chloroquine (CQ) in the PDC structure will improve miRNA delivery due to enhanced endosomal escape while simultaneously contribute to anticancer activity of PCD/miRNA polyplexes through inhibition of cancer cell migration. Our results showed that miRNA delivery efficiency was dependent both on the molecular weight and CQ. The best performing PDC/miRNA polyplexes showed effective endosomal escape of miRNA. PDC polyplexes with therapeutic miR-210 showed promising anticancer activity in human breast cancer cells. PDC/miRNA polyplexes showed excellent ability to inhibit migration of cancer cells. Overall, this study supports the use of PDC as a promising polymeric drug platform for use in combination antimetastatic and anticancer miRNA therapeutic strategies.
Keywords: Chloroquine, miRNA delivery, endosomal escape, polymeric drug, polyplex
1. Introduction
MicroRNAs (miRNAs) are small noncoding RNAs that post-transcriptionally regulate various signaling pathways and it is estimated that they are involved in the expression of more than 30% of all human proteins.[1, 2] Dysregulation of miRNA is prominently displayed in cancer due to the important role of miRNA in processes involved in tumorigenesis, tumor growth, angiogenesis, and metastasis.[3–8] Anticancer treatments that use miRNA show great promise because single miRNA typically targets multiple genes simultaneously and is thus capable of regulating multiple biological pathways responsible for the cancer cell phenotype. For example, miR-210 is a major hypoxia-inducible miRNA found overexpressed in multiple types of cancers.[9–11] Oncogenic activity of miR-210 includes a wide range of processes, including cell proliferation, apoptosis, metastasis, differentiation, DNA repair, cell metabolism, and antitumor immune responses.[12–15] Hence, miR-210 inhibition with anti-miR-210 containing sequence complementary to miR-210 mature strand provides a promising target for the treatment of cancer.
Similar to other potential nucleic acid therapeutics, the clinical translation of miRNA in oncology applications has been hindered by significant challenges in achieving efficient and safe systemic delivery. Polycation/nucleic acid complexes (polyplexes) have been among the most widely investigated classes of nucleic acid delivery vectors. Polycations bind with negatively charged nucleic acids to form nanosized particles. Polyplexes based on poly(ethylenimine) (PEI), poly(2-dimethylaminoethyl)methacrylate (PD), poly-L-lysine, and polyarginine, have been successfully used for miRNA delivery.[16–22] However, polyplexes are usually taken up by cells via endocytosis, which leads to entrapment of miRNA in the endosomes and lysosomes where it is subject to enzymatic degradation. Hence, strategies to enhance endosomal escape and ensure effective release of therapeutic miRNA into the cytosol are urgently needed.[23–26] Among the various endosomal escape strategies, pH-sensitive polycations such as PEI and virus-derived or bacteria-derived proteins and peptides have been widely employed as methods to disrupt endosomal membrane and to facility cytoplasmic delivery.[27] Similarly, chloroquine (CQ) has been used as a chemical endosomal agent to improve in vitro transfection of polyplexes. CQ has lipophilic properties in its unprotonated form and can readily penetrate cellular membranes. Upon entering an acidic environment, however, CQ becomes protonated and too polar to penetrate through the membranes.[28] Simple co-transfection of polyplexes with free CQ can enhance cytoplasmic delivery.[29] Loading of sufficient amounts of CQ into nanoparticles can also improve cytoplasmic localization.[30] However, CQ co-delivery has not been used successfully in vivo because in order to achieve the functional levels, toxic doses of CQ are required. To overcome the in vivo limitations of CQ as endosomal agent, it has been reported that covalent conjugation to the gold nanoparticles can improve siRNA delivery in vivo by enhancing endosomal escape.[31] Hence, conjugation of CQ to polycations is expected to improve endosomal escape of polyplexes.
Besides its endosomolytic properties, CQ is a widely used antimalarial drug that has been clinically used for decades. Over the years, a broad spectrum of pharmacologic activities of CQ has been described, including its anticancer activity due to its effects on autophagy and cholesterol metabolism.[32–35] The capability of CQ to inhibit chemokine receptor CXCR4 has also been recognized and successfully employed in the treatment of several types of solid tumors.[36] CQ inhibited CXCR4-mediated invasion and proliferation of pancreatic cancer cells and improved overall survival of tumor-bearing mice when combined with gemcitabine.[37] More recently, our group developed CQ-containing HPMA copolymers, which achieved greatly enhanced inhibition activity against cancer cell migration and tumor metastasis in vivo.[38, 39]
Taken together, CQ is a promising multi-functional agent that is well-suited for the development of novel combination anticancer strategies. Conjugation of CQ to polycations has not been reported before and provides an attractive approach to improve anticancer activity of polyplexes. In this study, we report the development and systematic evaluation of CQ-containing DMAEMA copolymers as vectors for delivery of therapeutic miRNA in combination cancer therapy. We hypothesized that the CQ-containing polycations will improve endosomal escape of miRNA and inhibit cancer cell migration. Such dual functionality is expected to contribute to enhanced combinational anticancer activity.
2. Experimental Section
2.1. Materials
2-Cyano-2-propyl dodecyl trithiocarbonate (CPDT) and 2,2′-azobis(2-methyl-propionitrile) (AIBN) were purchased from Sigma-Aldrich (St. Louis, MO). 2-(Dimethylamino)ethyl methacrylate (DMAEMA) was obtained from Fisher (Fair Lawn, NJ) and distilled before usage. Hydroxychloroquine sulfate was purchased from Acros (Fair Lawn, NJ). Solvents (certified ACS) were purchased from Fisher (Fair Lawn, NJ) and used without further purification. Eagle’s Minimum Essential Medium (EMEM), Dulbecco’s phosphate buffered saline (PBS), and fetal bovine serum (FBS) were from Thermo Scientific (Waltham, MA). Hsa-miR-210-3p-Hairpin Inhibitor (anti-miR-210, mature miRNA sequence: 5′-CUGUGCGUGUGACAGCGGCUGA-3′), negative control miR-NC inhibitor (anti-miR-NC, mature miRNA sequence: 5′-UCACAACCUCCUAGAAAGAGUAGA-3′) and fluorescently labeled FAM-miRNA were purchased from Dharmacon (Lafayette, CO). RT-PCR primers were purchased from Invitrogen (Carlsbad, CA). Protease and phosphatase inhibitor cocktail, Pierce bicinchoninic acid (BCA) protein assay, RIPA buffer and Pierce ECL Western Blotting Substrate were purchased from Thermo Scientific (Waltham,MA). LC3B antibody, GAPDH rabbit antibody and anti-rabbit IgG HRP-linked antibody were purchased from Cell Signaling Technology (Beverly, MA). All other reagents were from Fisher Scientific and used as received unless otherwise noted.
2.2. Synthesis and characterization of poly(DMAEMA-co-MACQ) (PDC)
PDC were synthesized by reversible addition-fragmentation chain-transfer (RAFT) copolymerization of DMAEMA with methacryloyl-hydroxychloroquine (MACQ). MACQ was synthesized as reported in our previous publication.[38] The polymerization was carried out in mixed solvent 1,4-dioxane/DMSO (8/2, v/v) with freshly recrystallized AIBN used as the initiator and CPDT as the RAFT chain transfer agent. In an example synthesis, DMAEMA (0.636 mmol, 100.0 mg), MACQ (0.071 mmol, 28.6 mg), CPDT (0.0088 mmol, 3.05 mg, 3.07 μL), and AIBN (0.003 mmol, 0.5 mg) were loaded into a glass ampule containing 1 mL of 1,4-dioxane/DMSO mixture. After purging with nitrogen for 30 min, the ampule was flame-sealed and immersed into an oil bath at 70 °C. After 24 h, the reaction was stopped by cooling in liquid nitrogen. The mixture was acidified by the addition of HCl. The resulting product was dialyzed (3500 MWCO) against deionized water with HCl (pH=3) for 2 days and against deionized water for another day. The final PDC product was obtained as a hydrochloride salt through lyophilization. Typical yield was around 78%. DMAEMA homopolymers (PD) with no CQ were also synthesized.
The molecular weight of the copolymers was determined by gel permeation chromatography (GPC), using Agilent 1260 Infinity LC system equipped with a miniDAWN TREOS multi-angle light scattering (MALS) detector and a Optilab T-rEX refractive index detector (Wyatt Technology, Santa Barbara, CA). The TSKgel G3000PWXL-CP (Tosoh Bioscience LLC, King of Prussia, PA) column was operated with 0.1 M sodium acetate buffer (pH 5.0) as eluent at a flow rate of 0.5 mL/min at 25 °C. Results were analyzed using Astra 6.1 software from Wyatt Technology. The composition of each polymer was characterized by 1H-NMR (Bruker Avance-III HD 500 MHz) and the data analyzed by Topspin 3.5pl6 software.
2.3. Preparation and physicochemical characterization of polyplexes
The ability of PDC to condense miRNA was determined by electrophoresis in a 2% agarose gel containing 0.5 μg/mL ethidium bromide. Polyplexes were formed by adding predetermined volume of polymer to a miRNA solution (20 μM anti-miR-210 in 30 mM sodium acetate pH 5.0 buffer) to achieve the desired amine-to-phosphate (N:P) ratio and vigorously vortexed for 10 s. Polyplexes were kept at room temperature for 30 min before further use. Polyplexes prepared at different N:P ratios were loaded (20 μL of the sample containing 1.0 μg of miRNA) and run for 30 min at 100 V in 0.5×Tris/Borate/EDTA buffer. The gels were visualized under UV illumination on a KODAK Gel Logic 100 imaging system. Hydrodynamic diameter and zeta potential of the polyplexes were determined by dynamic light scattering (DLS) using a ZEN3600 Zetasizer Nano-ZS (Malvern Instruments Ltd., Massachusetts, United States).
2.4. Cytotoxicity
MDA-MB-231 human mammary gland adenocarcinoma cell line expressing luciferase was used in all the studies (PerkinElmer, Waltham, MA, product No. BW124319). The cells were grown in EMEM supplemented with 10% FBS at 37°C with 5% CO2 in a humidified chamber. Cytotoxicity of the copolymers was evaluated by Cell Titer Blue assay. Briefly, the cells were plated in 96-well microplates at a density of 5,000 cells/well. After 24 h, the medium was replaced by 100 μL of serial dilutions of polymer in complete cell culture medium and the cells were incubated for another 24 h prior to measuring cell viability. The medium was then removed and replaced with a mixture of 100 μL serum-free media and 20 μL of CellTiter-Blue reagent (CellTiter-Blue® Cell Viability Assay, Promega). After a 2-h incubation, the fluorescence intensity [I] (560/590nm) was measured on a SpectraMax iD3 Multi-Mode Microplate Reader (Molecular Devices, CA). The relative cell viability (%) was calculated as [I]treated / [I]untreated × 100%.
2.5. Cellular uptake and intracellular trafficking of polyplexes
Flow cytometry analysis was performed to study the cellular uptake of the polyplexes. MDA-MB-231 cells (5 × 104) were seeded in 24-well plates. After 24 h growth, the cells were incubated for 3 h with polyplexes prepared with FAM-miRNA (100 nM FAM-miRNA). The cells were then trypsinized, washed with cold PBS and subjected to analysis using a BD FACSCalibur flow cytometer (BD Bioscience, Bedford, MA). The results were processed using FlowJo software. Intracellular localization was observed by confocal laser scanning microscopy. Cells were cultured on a 20-mm glass-bottom cell culture dish (Nest) using 1 × 105 cells/dish. After 24 h, the medium was replaced with fresh medium and FAM-miRNA polyplexes were added (100 nM FAM-miRNA). After incubation for 4 h, the cells were washed twice with PBS, stained with LysoTracker® Red DND-99 (Life Technology, USA) for 30 min. The cells were rinsed 3 times with PBS and visualized live using a LSM 710 Laser Scanning Microscope (Zeiss, Jena, Germany).
2.6. Quantitative real-time PCR (qRT-PCR)
Cells were seeded in 24-well plates at 5 × 104 cells/well 24 h prior to transfection. Then, the cells were incubated with the polyplexes containing miRNA (100 nM) in 500 μL of medium. The expression levels of miR-210 were evaluated by TaqMan qRT-PCR. The mirVana™ miRNA Isolation Kit (Ambion™, USA) was used for total RNA extraction from cultured cells. 10 ng of total RNA was converted into cDNA using specific primers for miR-210 (or the internal control Z30 (Applied Biosystems, Foster City, CA)) and the TaqMan microRNA reverse transcription kit (Applied Biosystems). qRT-PCR was performed using TaqMan Universal Master Mix II, No AmpErase UNG (2×) and specific primers for miR-210 or Z30 (Applied Biosystems, Foster City, CA) on a Rotor-Gene Q instrument (QIAGEN) according to the manufacturer’s instructions. MiRNA expression levels were expressed relative to the internal control according to the comparative threshold cycle (Ct) method.
2.7. Cell killing activity
Cell killing ability of anti-miR-210 polyplexes was evaluated by Cell Titer Blue assay in MDA-MB-231 cells. The cells were plated in 96-well microplates at a density of 5,000 cells/well. After 24 h, the cells were incubated as above with PBS, anti-miR-NC polyplexes, or anti-miR-210 polyplexes prepared at N:P ratio of 10 at miRNA concentration of 100 nM for 48 h prior to measuring cell viability. The medium was then removed and replaced with a mixture of 100 μL serum-free media and 20 μL of CellTiter-Blue reagent (CellTiter-Blue® Cell Viability Assay, Promega). After a 2-h incubation, fluorescence (560/590nm) was measured on a SpectraMax iD3 Multi-Mode Microplate Reader (Molecular Devices, CA). The relative cell killing mediated by the nanoparticles was normalized to the viability of untreated cells and expressed as the means ± SD of triplicate samples.
2.8. Transwell cell migration
MDA-MB-231 cells were seeded in 24-well plates at 2 × 105 cells/well 24 h prior to treatment. Then, cells were incubated with HCQ (20 μM), PDC17-H containing 20 μM CQ, or PD-H for 24 h. Then cells were trypsinized, washed with PBS, and suspended in serum-free medium before adding to the transwell inserts at a final concentration of 50,000 cells in 300 μL medium. Inserts were then placed in a 24-well companion plate containing medium with 10% FBS in each well. The cells were then incubated at 37 °C and allowed to migrate through the insert membrane for 8 h. The non-migrated cells on the upper side of the membrane were removed by cotton swabs and the migrated cells attached on the bottom surface were fixed in 100% methanol and stained with 0.2% Crystal Violet solution for 15 min at room temperature. The images were taken by EVOS xl microscope. Three 20× visual fields were randomly selected for each insert and each group was conducted in triplicate.
2.9. Inhibition of autophagy
Western blot was used to measure the effect of polymer on autophagy in MDA-MB 231 cells. Cells were treated with HCQ (20 μM), PD-H, PDC17-H for 24 h, and then washed with cold PBS, and lysed in ice-cold lysis buffer containing protease and phosphatase inhibitors. The lysate was centrifuged at 15,000 rpm for 10 min at 4 °C to collect the supernatant. Total protein was extracted with Laemmli lysis buffer according to the suggested protocol and the protein concentration was quantified by the BCA assay. The samples were then loaded and separated on SDS/PAGE gel, transferred to nitrocellulose membranes followed by probing with LC3B antibody and incubation with anti-rabbit IgG HRP-linked antibody. GAPDH was used as a housekeeping control. Quantification of LC3B levels was performed by ImageJ.
2.10. Statistical analysis
Data are presented as the means ± SD. The statistical significance was determined using ANOVA followed by Bonferroni post hoc correction with p < 0.05 as the minimal level of significance.
3. Results and Discussion
3.1. Polymer synthesis and characterization
To test our hypothesis that presence of CQ moieties in the polycation structure will increase endosomal escape of polyplexes and provide anti-invasive properties, we have synthesized a panel of PDC copolymers of DMAEMA with different molecular weight and increasing content of the MACQ units (CQ content) (Table 1). All copolymers were synthesized by RAFT polymerization (Scheme 1), which was well-controlled as suggested by the polydispersity index (Mw/Mn) values ≤ 1.2 (Table 1). Structural analysis by 1H-NMR showed the characteristic quinoline peaks at δ 6.80, 7.62, 8.01, 8.50 and 8.84 (peaks p, o, n, l and m in Figure 1) indicative of the presence of CQ in the copolymers. In addition, signals at δ 0.82, 1.92, 2.72, 3.26 and 4.26 (d′, e′, a′, b′ and c′ in Figure 1) confirmed the presence of DMAEMA. The CQ content in PDC was calculated from the integration ratio of the quinoline peaks (p and o) to the ethylene peaks of DMAEMA (b′ and c′). PDC copolymers with low (L), medium (M), and high (H) molecular weight and with four different CQ contents of 0, ~5, ~10, and ~17 mol% were synthesized and studied.
Table 1.
Polymer characterization.
| Polymer | Mw | Mw/Mn | Pn | CQ content
|
|
|---|---|---|---|---|---|
| 103 Da | mol % | wt % | |||
| PD-L | 10.9 | 1.1 | 61 | 0 | 0 |
| PD-M | 29.9 | 1.1 | 175 | 0 | 0 |
| PD-H | 39.1 | 1.1 | 233 | 0 | 0 |
| PDC5-L | 7.8 | 1.1 | 45 | 4.3 | 10.4 |
| PDC5-M | 16.8 | 1.04 | 95 | 5.3 | 12.5 |
| PDC5-H | 34.7 | 1.1 | 191 | 4.0 | 9.7 |
| PDC10-L | 8.8 | 1.1 | 43 | 9.7 | 21.6 |
| PDC10-M | 16.7 | 1.03 | 89 | 10.4 | 23.0 |
| PDC10-H | 32.2 | 1.1 | 164 | 9.2 | 20.6 |
| PDC17-L | 9.5 | 1.17 | 40 | 17.8 | 35.8 |
| PDC17-M | 16.8 | 1.1 | 79 | 16.8 | 34.2 |
| PDC17-H | 27.6 | 1.1 | 125 | 17.5 | 35.3 |
Mw...weight-average molecular weight, Pn...number-average degree of polymerization
Scheme 1.

Synthesis of PDC copolymers.
Figure 1.

Representative 1H-NMR of PD-H and PDC17-H.
3.2. Preparation and physicochemical characterization of PDC/miRNA polyplexes
Condensation of nucleic acids by polycations is the first requirement for their successful use as delivery vectors. The ability of PDC to form polyplexes with miRNA was first tested using agarose gel electrophoresis. Polyplexes were prepared by adding PDC to miRNA solution at increasing N/P ratios. As shown in Figure 2A, molecular weight of the polycations was the dominant factor in determining the miRNA condensation. For example, the high molecular weight PD-H was able to fully condense miRNA above N:P ratio of 2.5. Partial miRNA condensation was observed at N:P ratios as low as 1, as indicated by the smear of the miRNA in the gel. In contrast, N:P ratios above 5 were required for PD-M to fully condensed miRNA and the low molecular weight PD-L failed to condense miRNA even at N:P ratio of 10. Presence of CQ in the copolymers had no significant effect on miRNA condensing ability. These results confirmed the well-established understanding that higher molecular weight polycations have better capability to condense nucleic acids and form polyplexes.
Figure 2.

Physicochemical characterization of miRNA polyplexes. (A) miRNA condensation by PD and PDC using agarose gel electrophoresis. (B) Hydrodynamic size and (C) ζ potential of polyplexes at N:P 10 (n=3).
Hydrodynamic size and ζ-potential of the miRNA polyplexes prepared at N:P ratio of 10 were then studied by DLS (Figure 2B and 2C). The PD/miRNA polyplexes had sizes of ~90–110 nm. For the CQ containing PDC copolymers, high molecular weight polycations formed larger sized polyplexes than low molecular weight polycations. For example, the size of PDC17-H polyplexes (~173 nm) was larger than that of PDC17-L polyplexes (~80 nm). As expected, the ζ potential of all tested polyplexes was positive and ranged from 8 to 17 mV. High molecular weight polyplexes had higher ζ potential than low molecular weight polycations.
3.3. Cytotoxicity
Toxicity associated with the use of polycations remains a major hindrance for their use as miRNA delivery systems. Cytotoxicity of all synthesized polycations was evaluated by CellTiterBlue assay in the MDA-MB-231 cells (Figure 3A). The IC50 value of each polymer was calculated from the concentration dependence of cell viability (Figure 3B). For both PD and PDC, the toxicity was highly correlated with the molecular weight. High molecular weight polymers exhibited higher cytotoxicity than lower molecular weight polymers. For example, the IC50 of PD-H (16.5 μg/mL) was lower than IC50 of PD-M (23.8 μg/mL) and PD-L (83.5 μg/mL). Importantly, the cytotoxicity of PDC17 with the highest CQ content was lower than other PDCs with similar molecular weights. For example, IC50 of PDC17-H (42.4 μg/mL) was higher than IC50 of PD-H (16.5 μg/mL), PDC5-H (19.5 μg/mL) and PDC10-H (19.1 μg/mL). This observation may be related to the lower degree of polymerization (Pn) of PDC17-H when compared with the comparable high molecular weight polymers. These results also suggest that high CQ content in the PDC polycations may impart benefits in terms of reduced cytotoxicity and may ultimately improve safety of the polymers in vivo.
Figure 3.

Cytotoxicity of polymers. (A) Cell viability of the polymers determined using Cell Titer-Blue Assay in MDA-MB-231 cells. Results are shown as mean cell viability ± SD (n = 3). (B) IC50 values of the polymers calculated from the dose-response curves.
3.4. Cellular uptake and intracellular trafficking of polymer/microRNA polyplexes
To study cellular uptake and intracellular trafficking, we prepared the polyplexes using a fluorescently labeled miRNA (FAM-miRNA, 100 nM) at N:P ratio of 10. MDA-MB-231 cells were incubated with the FAM-miRNA polyplexes for 3 h and cellular uptake was determined by flow cytometry. As shown in Figure 4A, all miRNA polyplexes were readily taken up by the cells as indicated by the enhanced fluorescence intensity compared with control PBS-treated cells and cells treated with free FAM-miRNA. Increasing the polymer molecular weight in preparation of the polyplexes resulted in enhanced cellular uptake both in terms of the mean fluorescence intensity per cell (Figure 4B) and the percentage of the cells that had internalized the polyplexes (Figure 4C). Over 85% of the cells internalized the polyplexes when prepared using the high molecular weight PDCs and PD. Based on these uptake results, polyplexes prepared using high molecular weight PDC were selected for subsequent studies.
Figure 4.

Cellular uptake of polyplexes in MDA-MB-231 cells. (A) Overlayed histogram of flow cytometry analysis of cells treated with polyplexes at N:P ratio of 10 (100 nM FAM-miRNA) after a 3-h incubation. Quantification of cellular uptake shown as (B) mean fluorescence intensity (MFI) and (C) % of cells that had taken up the polyplexes. Data are shown as the mean ± SD (n = 3).
It is important that miRNA escapes endosomes and is delivered to its site of action in the cytoplasm. Here, we observed and compared the endosomal escape activity of the parent PD homopolymer (PD-H) and PDC17-H. Intracellular distribution and trafficking of these polyplexes prepared with FAM-miRNA at N:P 10 was studied using confocal microscopy. MDA-MB-231 cells were incubated with the polyplexes for 3 h and 6 h and then acidic endosomes/lysosomes were visualized with LysoTrackerRed. As shown in Figure 5, after incubation for 3 h, the fluorescence of the FAM-miRNA (green) in cells treated with PD-H polyplexes was highly colocalized with LysoTracker signal (red), suggesting high entrapment of miRNA in the endosomes and lysosomes. The PDC17-H/miRNA polyplexes also showed obvious endosomal localization after 3 h incubation. However, after incubation for 6 h, the CQ-containing polyplexes achieved highly efficient endosomal escape as suggested by only a limited co-localization with the LysoTrackerRed remaining. The control PD-H polyplexes remained trapped in the endosomes and lysosomes even at 6 h post-incubation, suggesting poor endosomal escape. This result confirmed that high content of CQ in PDC facilitates the endosomal escape and may promote cytoplasmic delivery of miRNA.
Figure 5.

Intracellular trafficking of miRNA polyplexes in MDA-MB 231 cells by CLSM after a 3-h and 6-h incubation.
3.5. Improved miRNA delivery by PDC
Anti-miR-210 and its negative control (anti-miR-NC) were used to evaluate the ability of PDC to deliver functional miRNA into cells. Polyplexes were formed as above and the miR-210 levels in MDA-MB-231 cells were measured using qRT-PCR. As shown in Figure 6, PD-H/anti-miR-210 polyplexes did not reduce cellular miR-210 levels, most likely due to their poor endosomal escape ability. Similarly, anti-miR-210 polyplexes prepared with PDC5-H with the lowest CQ content lacked the ability to reduce miR-210 levels, indicating insufficient delivery of the miRNA to the cytoplasm. In contrast, both PDC with the higher CQ content (PDC10-H and PDC17-H) showed efficient miRNA delivery. The PDC10-H/anti-miR-210 polyplexes reduced miR-210 level by ~37%. Further improvement in the downregulation of miR-210 (~50%) was observed with PDC17-H/anti-miR-210 polyplexes, which was even better than the efficiency of a commercial lipid transfection agent OligoFectamine (~45%). The improved delivery activity of PDC17-H polyplexes was the result of the effective endosomal escape (Figure 5). This result indicated that covalent conjugation of CQ to polycations is effective way of enhancing functional miRNA delivery by improving endosomal escape. Based on these results, PDC17-H polyplexes were selected for subsequent anti-cancer study.
Figure 6.
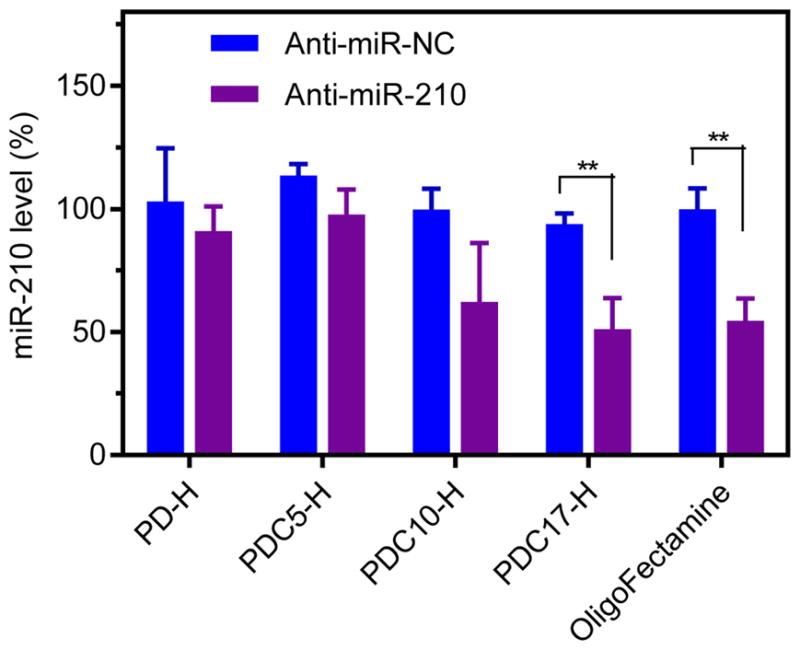
Transfection activity of polyplexes (N:P=10, anti-miRNA 100 nM). MiR-210 level was detected by TaqMan qRT-PCR in MDA-MB 231 cells. Data are shown as mean ± SD (n = 3). **P < 0.01.
3.6. Anticancer activity of PDC/anti-miR-210 polyplexes in vitro
The above results confirm that PDC17-H effectively delivered anti-miR-210 to MDA-MB 231 cells and downregulated cellular miR-210 levels. Previous reports showed that downregulation of miR-210 inhibits proliferation and induces apoptosis in cancer cells.[40, 41] We thus hypothesized that PDC17-H/anti-miR-210 polyplexes will show improved cell killing activity in cancer cells. To evaluate this hypothesis, PDC17-H/anti-miR-210 polyplexes were prepared and their cell killing activity was determined by a cell viability assay. Treatment of the MDA-MB-231 cells with control PD-H/anti-miR-210 polyplexes did not induce any significant cancer cell killing when compared with PD-H/anti-miR-NC polyplexes. In contrast, significant cell killing effect (~43%) was observed in cells treated with PDC17-H/anti-miR-210 polyplexes. The fact that no decrease in cell viability was observed with corresponding anti-miR-NC polyplexes confirmed safety of PDC17-H. The cell killing activity of PDC17-H/anti-miR-210 was even higher than that of OligoFectamine/anti-miR-210 (~29%). These results confirmed that PDC17-H polyplexes improved functional miRNA delivery and induced higher cancer cell killing effect by anti-miRNA in cancer cells.
3.7. Inhibition of cancer cell migration by PDC
Based on our previous work, we proposed that besides being effective miRNA delivery vector, PDC17-H may also function as a polymeric drug with antimetastatic effect. Cancer cell migration and invasion is a vital step in metastatic spread of cancer and strategies to prevent or minimize cancer cell spread are highly desired. Here, transwell migration assay was performed to study the inhibition activity of the synthesized polymers on migration of metastatic MDA-MB-231 cells. The cells were treated with PD-H, PDC17-H, or equivalent free HCQ (20 μM) and then 10% FBS was applied to the lower chamber as the chemoattractant to induce cell migration (Figure 8A and B). Treatment with HCQ and PD-H showed no inhibition of FBS-induced cell migration. In contrast, treatment with PDC17-H decreased cell migration by ~86%. This outstanding anti-migration activity of PDC is a confirmation of our recent work with other CQ-containing polymers in different metastatic breast cancer cells. The anti-migration ability of PDC is believed to be mediated by a broad effects on multiple signaling pathways involved in cancer cell motility, including the CXCR4 axis.[39] Overall, we demonstrated the potential of PDC as both miRNA delivery vectors and effective anti-migration polymeric drug, which represents a promising approach for combination therapy of metastatic cancer.
Figure 8.

Inhibition of cancer cell migration. (A) MDA-MB 231 cells were treated with polymer or equivalent HCQ (20 μM) for 24 h and then allowed to migrate through transwell upon stimulation with 10% FBS for 8 h. (B) Three 20× imaging areas were randomly selected for each insert and each group was conducted in triplicate. Data are shown as mean ± SD (n = 3). ***p < 0.001 vs. PBS.
3.8. Inhibition of autophagy
CQ is commonly believed to inhibit autophagy in cancer cells by preventing the fusion of autophagosomes and lysosomes. To evaluate the mechanism of action of PDC, Western blot was performed to quantify the levels of autophagy marker LC3 (microtubule-associated protein 1A/1B-light chain 3) in cells treated with HCQ, PD-H, and PDC17-H (Figure 9A). The relative expression of LC3-II and total LC3 expression (i.e., LC3-I + LC3-II) were quantified from the Western blots (Figure 9B). The cytosolic form of LC3 (LC3-I) is converted into LC3-II, which is bound to the autophagosomal membrane, indicating autophagic activity. Monitoring degradation of LC3-II is a convenient assay of autophagic activity.[42] The LC3-II degradation is inhibited when cells are treated with CQ, which inhibits lysosomal acidification and leads to the accumulation of LC3-II in the cells. Our results show that HCQ treatment resulted in effective inhibitory activity indicated by the elevated levels of LC3-II (1.6-fold increase). In contrast, PDC17-H showed only a modest autophagy inhibition (1.3-fold increase). PD-H showed no autophagy inhibition. These results clearly suggest that conjugation of CQ to the polycation results in a significant loss of autophagy inhibitory activity. Similar observation was also reported in our previous study.[39]
Figure 9.
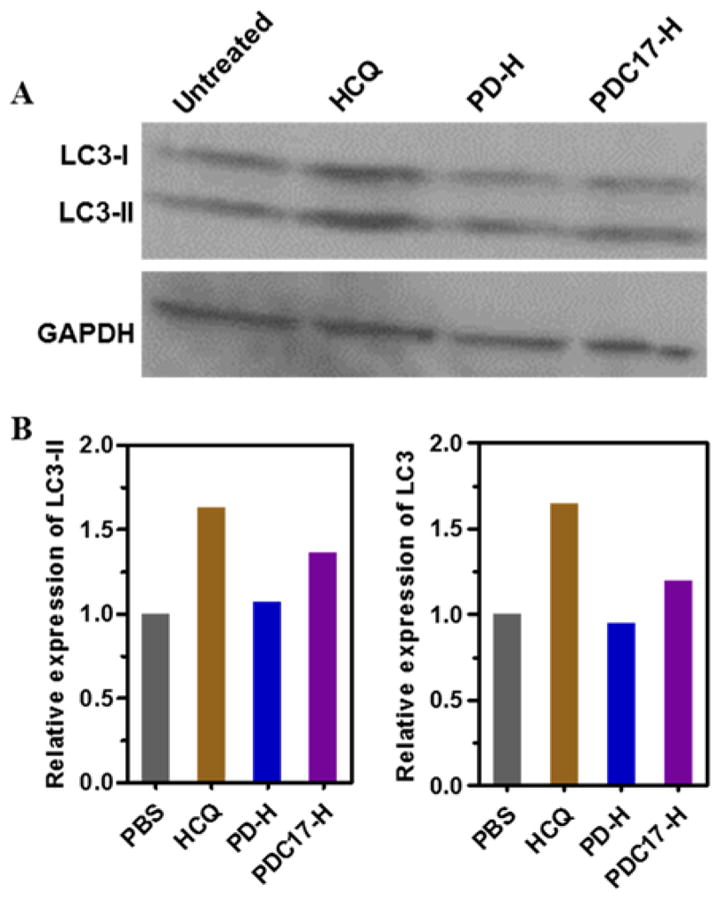
Inhibition of autophagy. (A) MDA-MB 231 cells were treated with HCQ (20 μM), PD-H, PDC17-H for 24 h before Western blot analysis. (B) The band intensities were quantified by ImageJ.
Conclusion
In this study, we developed chloroquine-containing polycations with improved endosomal escape for miRNA therapy and anti-migration activity in metastatic cancer cells. Systematic evaluation was performed to study the effect of the copolymer molecular weight and CQ content on miRNA delivery efficiency. Our results showed that PDC17-H effectively improved endosomal escape and cytoplasmic release of miRNA in cancer cells. Successful delivery of functional anti-miR-210 was confirmed by the downregulation of intracellular levels of miR-210 and induction of effective cell killing in cancer cells. More importantly, PDC17-H also exhibited activity as a polymeric drug with the ability to efficiently inhibit migration of cancer cells. The dual-function chloroquine-containing DMAEMA copolymers provide a promising delivery platform for antimetastatic combination cancer therapies. Our future studies will focus on the synthesis of improved PDC copolymers suitable for systemic targeted delivery of miRNA for the treatment of metastatic cancers.
Figure 7.
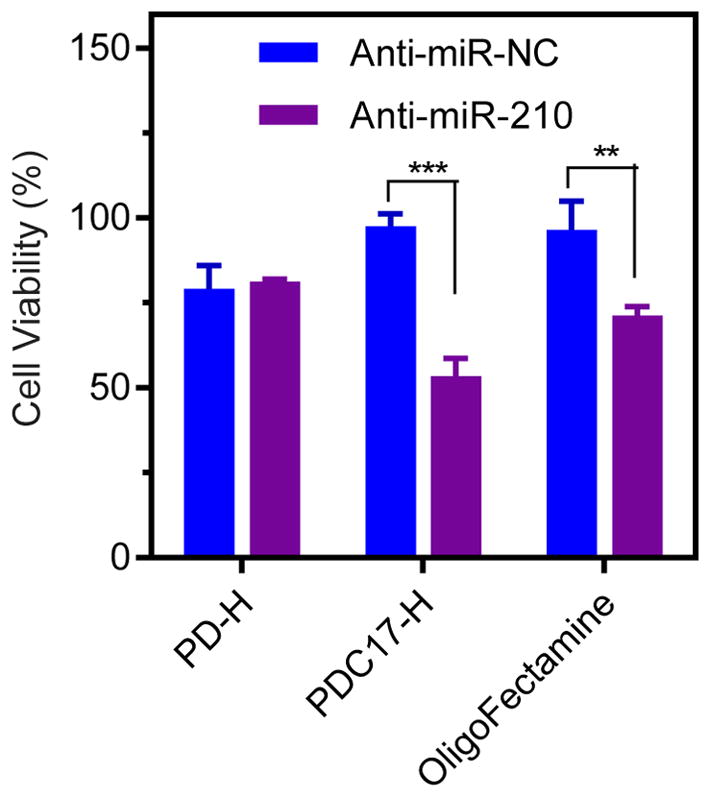
Anticancer activity of polymer/miRNA polyplexes in MDA-MB 231 cells. Cells were treated for 48 h with polymer/anti-miR-NC or polymer/anti-miR-210 prepared at N:P ratio of 10 (anti-miRNA 100 nM). Cell viability was measured by Cell Titer Blue assay. Data are shown as mean ± SD (n = 3). **P < 0.01, ***P < 0.001.
Acknowledgments
This work was supported by the University of Nebraska Medical Center and in part by the National Institutes of Health (EB015216, EB020308, EB019175). China Scholarship Council (YX, YW) is gratefully acknowledged.
Footnotes
Dedication: To my mentor Prof. Karel Ulbrich on the occasion of his 70th birthday.
References
- 1.Bartel DP. Cell. 2004;116:281. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Lewis BP, Burge CB, Bartel DP. Cell. 2005;120:15. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 3.Calin GA, Croce CM. Nature Rev Cancer. 2006;6:857. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 4.Croce CM. Nature Rev Genetics. 2009;10:704. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie Y, Murray-Stewart T, Wang Y, Yu F, Li J, Marton LJ, Casero RA, Oupický D. J Controlled Release. 2017;246:110. doi: 10.1016/j.jconrel.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT. Nature Genetics. 2006;38:1060. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma L, Teruya-Feldstein J, Weinberg RA. Nature. 2007;449:682. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 8.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Nature Genetics. 2008;40:43. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang X, Le QT, Giaccia AJ. Trends Mol Med. 2010;16:230. doi: 10.1016/j.molmed.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang X, Ding L, Bennewith KL, Tong RT, Welford SM, Ang KK, Story M, Le QT, Giaccia AJ. Mol Cell. 2009;35:856. doi: 10.1016/j.molcel.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pulkkinen K, Malm T, Turunen M, Koistinaho J, Ylä-Herttuala S. FEBS Lett. 2008;582:2397. doi: 10.1016/j.febslet.2008.05.048. [DOI] [PubMed] [Google Scholar]
- 12.Chan SY, Loscalzo J. Cell Cycle. 2010;9:1072. doi: 10.4161/cc.9.6.11006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan YC, Banerjee J, Choi SY, Sen CK. Microcirculation. 2012;19:215. doi: 10.1111/j.1549-8719.2011.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noman MZ, Buart S, Romero P, Ketari S, Janji B, Mari B, Mami-Chouaib F, Chouaib S. Cancer Res. 2012;72:4629. doi: 10.1158/0008-5472.CAN-12-1383. [DOI] [PubMed] [Google Scholar]
- 15.Ying Q, Liang L, Guo W, Zha R, Tian Q, Huang S, Yao J, Ding J, Bao M, Ge C. Hepatology. 2011;54:2064. doi: 10.1002/hep.24614. [DOI] [PubMed] [Google Scholar]
- 16.Oupický D, Li J. Macromol Biosci. 2014;14:908. doi: 10.1002/mabi.201400061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Wang Y, Zhu Y, Oupický D. J Controlled Release. 2013;172:589. doi: 10.1016/j.jconrel.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S, Cheng SX, Zhuo RX. Macromol Biosci. 2011;11:576. doi: 10.1002/mabi.201000427. [DOI] [PubMed] [Google Scholar]
- 19.Yang YP, Chien Y, Chiou GY, Cherng JY, Wang ML, Lo WL, Chang YL, Huang PI, Chen YW, Shih YH. Biomaterials. 2012;33:1462. doi: 10.1016/j.biomaterials.2011.10.071. [DOI] [PubMed] [Google Scholar]
- 20.Qian X, Long L, Shi Z, Liu C, Qiu M, Sheng J, Pu P, Yuan X, Ren Y, Kang C. Biomaterials. 2014;35:2322. doi: 10.1016/j.biomaterials.2013.11.039. [DOI] [PubMed] [Google Scholar]
- 21.Gao S, Tian H, Guo Y, Li Y, Guo Z, Zhu X, Chen X. Acta Biomater. 2015;25:184. doi: 10.1016/j.actbio.2015.07.020. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Buhrman JS, Liu Y, Rayahin JE, Gemeinhart RA. Mol Pharmaceutics. 2016;13:1791. doi: 10.1021/acs.molpharmaceut.5b00933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Gao DY, Huang L. Adv Drug Deliv Rev. 2015;81:128. doi: 10.1016/j.addr.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Nature Rev Genetics. 2014;15:541. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 25.He D, Wagner E. Macromol Biosci. 2015;15:600. doi: 10.1002/mabi.201400524. [DOI] [PubMed] [Google Scholar]
- 26.Reinhard S, Wagner E. Macromol Biosci. 2016. [DOI] [PubMed] [Google Scholar]
- 27.Varkouhi AK, Scholte M, Storm G, Haisma HJ. J Controlled Release. 2011;151:220. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Mellman I, Fuchs R, Helenius A. Annual Rev Biochem. 1986;55:663. doi: 10.1146/annurev.bi.55.070186.003311. [DOI] [PubMed] [Google Scholar]
- 29.Pack DW, Hoffman AS, Pun S, Stayton PS. Nature Rev Drug Discov. 2005;4:581. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 30.Bhattarai SR, Muthuswamy E, Wani A, Brichacek M, Castañeda AL, Brock SL, Oupicky D. Pharm Res. 2010;27:2556. doi: 10.1007/s11095-010-0245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perche F, Yi Y, Hespel L, Mi P, Dirisala A, Cabral H, Miyata K, Kataoka K. Biomaterials. 2016;90:62. doi: 10.1016/j.biomaterials.2016.02.027. [DOI] [PubMed] [Google Scholar]
- 32.Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, Fitzgerald SL, George E, Frias E, Cochran N, De Jesus R, McAllister G, Hoffman GR, Bray K, Lemon L, Lucas J, Fantin VR, Abraham RT, Murphy LO, Nyfeler B. Proc Natl Acad Sci. 2016;113:182. doi: 10.1073/pnas.1515617113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King MA, Ganley IG, Flemington V. Oncogene. 2016;35:4518. doi: 10.1038/onc.2015.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solomon VR, Lee H. Eur J Pharmacol. 2009;625:220. doi: 10.1016/j.ejphar.2009.06.063. [DOI] [PubMed] [Google Scholar]
- 35.Janku F, McConkey DJ, Hong DS, Kurzrock R. Nature Rev Clin Oncol. 2011;8:528. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- 36.Kim J, Yip MR, Shen X, Li H, Hsin LYC, Labarge S, Heinrich EL, Lee W, Lu J, Vaidehi N. PLoS One. 2012;7:e31004. doi: 10.1371/journal.pone.0031004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balic A, Sørensen MD, Trabulo SM, Sainz B, Cioffi M, Vieira CR, Miranda-Lorenzo I, Hidalgo M, Kleeff J, Erkan M. Mol Cancer Ther. 2014;13:1758. doi: 10.1158/1535-7163.MCT-13-0948. [DOI] [PubMed] [Google Scholar]
- 38.Yu F, Xie Y, Wang Y, Peng Z-H, Li J, Oupický D. ACS Macro Lett. 2016;5:342. doi: 10.1021/acsmacrolett.5b00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu F, Li J, Xie Y, Sleightholm RL, Oupický D. J Controlled Release. 2016;244:347. doi: 10.1016/j.jconrel.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang W, Sun T, Cao J, Liu F, Tian Y, Zhu W. Exp Cell Res. 2012;318:944. doi: 10.1016/j.yexcr.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 41.Costales MG, Haga CL, Velagapudi SP, Childs-Disney JL, Phinney DG, Disney MD. J Am Chem Soc. 2017;139:3446. doi: 10.1021/jacs.6b11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mizushima N, Yoshimori T, Levine B. Cell. 2010;140:313. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]