

Summary
The cellular transcriptional coactivator HCF-1 is required for initiation of herpes simplex virus (HSV) lytic infection and for reactivation from latency in sensory neurons. HCF-1 stabilizes the viral Immediate Early (IE) genes enhancer complex and mediates chromatin transitions to promote IE transcription initiation. In infected cells, HCF-1 was also found to be associated with a network of transcription elongation components including the Super Elongation Complex (SEC). IE genes exhibit characteristics of genes controlled by transcriptional elongation and the SEC-P-TEFb complex is specifically required to drive the levels of productive IE mRNAs. Significantly, compounds that enhance the levels of SEC-P-TEFb also potently stimulated HSV reactivation from latency both in a sensory ganglia model system and in vivo. Thus, transcriptional elongation of HSV IE genes is a key limiting parameter governing both the initiation of HSV infection and reactivation of latent genomes.
Keywords: Herpes simplex virus, transcriptional elongation, latency, host cell factor-1, super elongation complex, P-TEFb
eTOC Blurb
HSV Immediate Early (IE) gene transcription requires the cellular coactivator HCF-1. Alfonso-Dunn et al. demonstrate that during infection, HCF-1 is associated with transcription initiation and elongation components. Both lytic infection and reactivation of latent virus are dependent on elongation factors that mediate a critical checkpoint in viral IE expression.
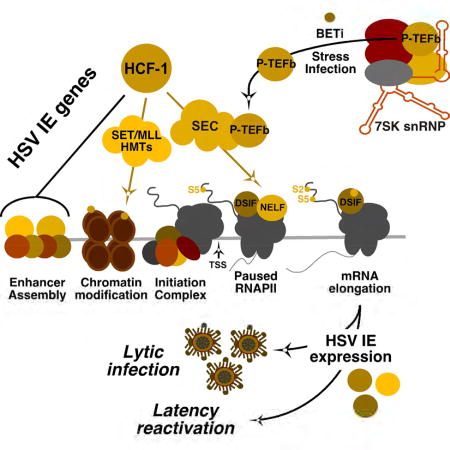
Introduction
Herpes simplex virus (HSV) is a prevalent human pathogen. Following an initial infection, the virus establishes a latent pool in neurons of sensory ganglia that periodically reactivate to produce recurrent disease. Clinical manifestations range from oral and genital lesions to herpetic keratitis, stromal keratitis, and blindness. Additionally, neonatal infections can result in disseminated infection and neurological-developmental issues. Importantly, infection with HSV is correlated with enhanced transmission of human immunodeficiency virus (HIV) (Roizman et al., 2013).
The lytic replication cycle is characterized by ordered and sequential expression of viral Immediate Early (IE), Early (E), and Late (L) genes that are regulated primarily at the level of transcription. Many nuclear DNA viruses like HSV utilize host cell transcriptional machinery, recruiting cellular components to navigate the transcriptional stages to drive the expression of their genes. RNAPII mediated cellular gene expression is regulated at multiple biochemical steps to assure timely cell division, differentiation, and response to both internal and external stimuli. Productive transcription requires the coordination of chromatin modulation machinery, assembly of transcription factor-coactivator complexes, the recruitment of the RNAPII initiation complex, elongation of nascent initiating RNAs, and appropriate RNA processing.
HSV IE gene transcription is mediated by viral (VP16) and cellular transcription factors (i.e. Oct-1, SP1, GABP) that assemble a potent transcription enhancer complex. A primary driver of IE expression is the cellular coactivator HCF-1 that is assembled into IE enhancer complexes via direct interactions with multiple transcription factors, including the viral IE activator, VP16 (Vogel and Kristie, 2013). HCF-1 plays a key role in modulating the chromatin assembled on the IE genes as part of a complex containing histone demethylases (JMJD2/KDM4 and LSD1/KDM1A) and histone H3K4 methyltransferases (SETD1A and MLL1/KMT2A). This complex limits the assembly of heterochromatin at IE promoters and promotes the transition to an active euchromatic chromatin state (Liang et al., 2013; Liang et al., 2009).
Importantly, HCF-1 is also implicated in HSV reactivation from latency in sensory neurons. The protein is rapidly relocalized from the cytoplasm to the nucleus and is recruited to viral IE promoters upon stimuli that promote viral reactivation (Kim et al., 2012; Kristie et al., 1999; Whitlow and Kristie, 2009). Additionally, the HCF-1 associated histone demethylases LSD1 and JMJD2s are required for reactivation (Hill et al., 2014; Liang et al., 2013; Liang et al., 2009), via removing repressive heterochromatin associated with the latent genome. Therefore, this protein is a central regulatory component for both the lytic infectious cycle and for reactivation from latency.
Given the significance of this cellular coactivator, a proteomic analysis of HCF-1 associated complexes was done in uninfected and HSV-infected cells. In addition to transcriptional initiation complexes, this analysis uncovered a striking association of the coactivator with multiple transcription elongation components. Transcriptional elongation has emerged as an important rate-limiting step, particularly for regulating the expression of cellular genes in response to environmental signaling and stress stimuli (Adelman and Lis, 2012; Jonkers and Lis, 2015). Following release from the initiation complex, RNAPII promoter-proximal pausing can prime genes for rapid expression and may also allow for the coordination of chromatin transitions that promote transcription.
Pausing is mediated, at least in part, by association of pausing factors NELF and DSIF with the initiating polymerase (Jonkers and Lis, 2015). Induced elongation of paused polymerase is promoted by recruitment of the P-TEFb complex to specific target genes as part of either the Super Elongation Complex (SEC) or the Bromodomain containing protein 4 (BRD4) adaptors (Jang et al., 2005; Luo et al., 2012b), although other interactions with components such as Mediator have also been described (Takahashi et al., 2011; Wang et al., 2013). P-TEFb, containing the CDK9 kinase, phosphorylates multiple targets including the RNAPII carboxy terminal domain, NELF, and DSIF to stimulate release from pausing.
P-TEFb itself is tightly regulated in a dynamic manner. More than 50% of P-TEFb is sequestered in an inactive form in the 7SK snRNP complex (Nguyen et al., 2001; Yang et al., 2001). Upon stress or growth signaling, P-TEFb is released and associates with the SEC or BRD4 adaptors. Thus, the P-TEFb shuffle and the association with specific adaptors allows for the rapid expression of specific genes primed for transcription.
Investigation of the significance of HCF-1 association with transcriptional elongation components demonstrated that elongation is a limiting stage for HSV IE gene expression and that the SEC-P-TEFb complex is specifically critical to drive IE gene expression and productive infection. Furthermore, BET inhibitors that enhance the levels of active SEC-P-TEFb complexes induced the expression of viral IE genes and promoted reactivation of HSV from latency in sensory neurons. The data supports a model in which the expression of IE genes can be rapidly induced by the SEC-P-TEFb complex upon stress signaling to reactivate a population of latent viral genomes.
Results
HCF-1 is associated with multiple transcription initiation and elongation complexes
While it is clear that HCF-1 functions, in part, to mediate transitions in viral chromatin that promote IE gene expression, the interactions and roles of this essential component have not been fully investigated. Therefore, a proteomic approach was taken to identify HCF-1 interactions in mock infected and HSV-1 infected cells. Endogenous HCF-1 complexes were immunoaffinity purified (IP) from MRC5 cells and the co-isolated proteins were analyzed by mass spectrometry (MS) as described (Diner et al., 2015). Interaction specificity was assessed by comparing HCF-1 IPs against the IgG controls and using the significance analysis of interactome (SAINT) tool (Choi et al., 2011) (Table S1).
A subset of the HCF-1 associated complexes and their interaction networks are depicted in Figure 1 and Table S1. As anticipated, major HCF-1 protein partners (Vogel and Kristie, 2013) were readily detected in the HCF-1 immunoprecipitates. These included the SETD1A, KMT2C/MLL3, KMT2E/MLL5, and KMT2A/MLL1 methyltransferases that install activating histone H3-lysine 4-trimethylation (H3K4me3) marks; OGT (O-GlcNac transferase), a protein that modulates multiple cell pathways but more recently has been implicated in promoting promoter occupancy of RNAPII (Lewis et al., 2016); and BAP1 (BRCA1 Associated Protein 1), a deubiquitinase that stimulates transcription, at least in part, by removing repressive histone H2A-K119 monoubiquitin (Balasubramani et al., 2015).
Figure 1. Transcription related protein complexes associated with HCF-1.

(A) Subunits of complexes identified in MS analyses of HCF-1 immunoisolates from Mock infected and HSV-1 infected (2 hpi) are indicated with black dots. Targets and functions relative to transcription initiation and elongation are depicted. SET complex, KMT2A, KMT2C, KMT2E (histone H3K4 methyltransferases); DSIF and NELF (RNAPII pausing factors); SEC, P-TEFb (phosphorylates RNAPII CTD, DSIF, and NELF), SETD2 (histone H3K36 methyltransferase); FACT (histone chaperone/remodeling complex); TSS (transcription start site). (B) Functional interaction network of HCF-1-associated complexes involved in transcription initiation and elongation. The nodes represent baits identified during HCF-1 immunoaffinity purification and the edges indicate interactions from the Reactome database. See also Table S1.
Most striking was the association of HCF-1 with components of complexes regulating stages of RNAPII transcriptional elongation including the SEC (Super Elongation Complex), PAFc (Polymerase Associated Factor Complex), DSIF (DRB Sensitivity Inducing Factor), SETD2 (histone H3K36 methyltransferase), CDK9 (Cyclin Dependent Kinase 9) and FACT (Facilitates Chromatin Transcription, histone chaperone/remodeler) in both uninfected and HSV-1 infected cells (Table S1). This network of interactions that mediate release of paused polymerase and efficient transcriptional elongation indicated that HCF-1 might also be involved in promoting transcription elongation of its target genes.
A selection of the HCF-1 interactions, identified by mass spectrometry, were confirmed by western blots of HCF-1 immunoprecipitates from mock infected and early (2 h) infected HEK293 cell extracts. As shown in Figure 2A, the HSV IE transactivator VP16 and the cellular Mediator complex (represented by MED12) were detected in HCF-1 immunoprecipitates from infected cells while the HCF-1 associated SETD1A histone methyltransferase complex (represented by SETD1A, RbBP5) was found in both uninfected and infected extracts. Importantly, the interactions of HCF-1 and transcriptional elongation factors (SEC scaffold component AFF4, PAF complex component PAF1, and the FACT subunit SUPT16H) were also confirmed in both uninfected and early 2 h infected cells. Enhanced levels of some components (i.e. AFF4) and the detection of additional components of the SEC complex in the HCF-1 MS and co-IPs from infected cells may reflect other cellular or viral interactions that enhance the stability of the complex (Figure 1 and Table S1). Alternatively, as shown in Figure S1, HSV infection induces the release of P-TEFb from the inactive complex and may thus increase the population of available SEC-P-TEFb complexes.
Figure 2. Coimmunoprecipitation confirmations of HCF-1 MS partner identifications.

(A) HCF-1 and control IgG precipitates from uninfected and HSV infected HEK293 cells were probed for protein partners identified by MS. (B) Schematic of RNAPII preinitiation complex, initiating RNAPII, and elongating RNAPII. CTD S5 phosphorylation by TFIIH promotes promoter clearance and transcription initiation while S2 phosphorylation by CDK9 promotes elongation. (C) HCF-1 and control IgG immunoprecipitates from Mock infected and HSV infected HEK293 cells were probed for forms of RNAPII.
As noted above, subunits of RNAPII were detected in the HCF-1 IP-MS. Given the interactions with protein complexes involved in both transcription initiation and elongation, the association of HCF-1 with specific phospho-forms of RNAPII was investigated. The phosphorylation status of the carboxy-terminal domain (CTD) of the large subunit of RNAPII is a well-established characteristic of the promoter associated (RNAPII-0), initiating (RNAPII-S5p), and elongating (RNAPII-S2p) enzyme (Figure 2B). Coimmunoprecipitation of all forms of RNAPII further indicates that HCF-1 is involved in multiple stages of transcription in both uninfected and infected cells (Figure 2C).
The SEC localizes to early viral transcriptional foci and is required for efficient viral IE gene expression
Association of HCF-1 with the Super Elongation Complex and the P-TEFb CDK9 subunit were of particular interest as CDK9 inhibitors suppress viral gene expression (Durand and Roizman, 2008; Ou and Sandri-Goldin, 2013). Furthermore, the viral IE activator VP16, a partner for HCF-1 in infected cells, was reported to interact with P-TEFb through the CycT1 subunit (Guo et al., 2012; Kurosu and Peterlin, 2004).
The interaction of HCF-1 with the P-TEFb adaptor SEC but not BRD4 in early HSV-1 infected cells (Figures 1, 2A) suggested that the SEC might play a role in mediating expression of the viral IE genes. As shown in Figure 3A, the SEC scaffold component AFF4 accumulated in early viral transcriptional foci as visualized by costaining with ICP4, the major viral transcriptional activator. Furthermore, the levels of viral IE mRNAs in cells depleted of AFF4 were significantly reduced (Figure 3B). In contrast, in cells depleted of the competitive P-TEFb adaptor, BRD4, IE expression was enhanced (Figure 3C). No impact of depletion of the related SEC family member AFF1 or the BET family members BRD2 or BRD3 on viral IE expression was observed (Figure S2). Thus, the data suggest that the SEC-AFF4 complex is specifically important to drive viral IE gene transcription.
Figure 3. AFF4 localizes to early HSV transcriptional foci and is required for efficient IE expression.

(A) MRC5 cells were mock infected or infected with HSV-1 for 2 h. Cells were co-stained with antibodies to the SEC scaffold component AFF4 and the HSV-1 lytic activator ICP4. (B–C) MRC5 cells were transfected with siControl or siRNAs to AFF4 or BRD4. Cells were infected with HSV-1 for 2 h and mRNA levels of the siRNA target (AFF4, BRD4), control cellular gene (GAPDH), and viral IE genes (ICP4, ICP27, ICP22) were determined. Data are levels in siAFF4 or siBRD4 cells relative to control siRNA cells. (Means +/− s.e.m., n > = 6). (D) JQ1+, IBET-762, and HMBA bind to the bromodomains of BRD4 and inhibit its binding to chromatin. These compounds also induce release of 7SK snRNP sequestered P-TEFb. (E–F) MRC5 cells treated with JQ1+ 1 uM, IBET-762 1 uM, or HMBA 5 mM and infected with HSV-1 for 2 h. The mRNA levels of control cellular gene GAPDH and viral IE genes (ICP4, ICP27, ICP22) are relative to those in vehicle treated cells. (Means +/− s.e.m., n > = 6). (G–H) mRNA levels of HSV IE genes and controls (GAPDH, HPRT) in cells treated with the indicated concentrations of CDK9 inhibitors. (Means +/− s.e.m., n = 3). (I–J) MRC5 cells treated with vehicle, JQ1+, or dBET1 and infected with HSV-1 for 2 h. (I) Western blot of ICP4, BRD4, and control GAPDH protein levels. (J) mRNA levels of viral IE and GAPDH are relative to those in vehicle treated cells. (Means +/− s.e.m., n = 3). See also Figures S1–S6.
To confirm these results, cells were infected with HSV-1 and subsequently treated with BET inhibitors (JQ1+, HMBA, IBET-762). These compounds inhibit the binding of BRD4 to its targets but also promote the release of P-TEFb from the inactive 7SK snRNP complex (Bartholomeeusen et al., 2012; Contreras et al., 2007; Filippakopoulos et al., 2010; Mirguet et al., 2013; Nilsson et al., 2016), thereby increasing the pool of SEC containing active P-TEFb (Figure 3D, Figure S3). As shown (Figure 3E–F, Figure S4), treatment of infected cells with JQ1+, HMBA, or IBET-762 results in enhanced IE gene mRNA levels in a manner analogous to depletion of BRD4. In contrast, inhibition of CDK9 activity by treatment of cells with either flavopiridol or the more specific CDK9 inhibitor iCDK9 (Lu et al., 2015) blocked IE expression as expected (Figure 3G–H). Interestingly, in contrast to HSV, depletion of AFF4 and BRD4 or treatment with BET inhibitors did not significantly impact the levels of Human cytomegalovirus IE mRNAs (Figure S5).
Recently, JQ1+ was identified in a compound library screen as an enhancer of HSV lytic infection and gene expression. However, in that study, viral yields and protein accumulation were reduced at a late stage of infection in BRD4-depleted HeLa cells (Ren et al., 2016). Therefore, to further demonstrate that the SEC is the significant P-TEFb adaptor complex that is required for IE gene expression, MRC5 cells were treated with a recently described BET inhibitor (dBET1) that mediates rapid turnover of BRD4 (Winter et al., 2015). As shown in Figure 3I–J and Figure S6, treatment of cells with dBET1 resulted in significant degradation of BRD4 (∼85%) and the anticipated increase in viral IE mRNA levels. Thus, depletion and inhibition studies of BRD4 and the SEC scaffold AFF4 support the hypothesis that the SEC-P-TEFb is the complex that promotes HSV IE expression.
The SEC mediates transcriptional elongation of HSV IE mRNAs
The interaction of HCF-1 with a network of elongation regulatory components and the requirement for SEC-P-TEFb for efficient viral IE expression suggests that transcriptional elongation is an important rate limiting step for IE gene expression. Therefore, small (promoter-proximal initiating) and large (full length) mRNAs (depicted in Figure 4A) were isolated from infected cells depleted of either AFF4 or BRD4. In cells depleted of AFF4, the levels of full length IE mRNAs were suppressed while no significant impact was observed on the levels of small initiating RNAs (Figure 4B, D). As expected, depletion of BRD4 enhanced the levels of full-length mRNAs also without impact on the levels of initiating RNAs (Figure 4C, D).
Figure 4. Depletion of AFF4 or BRD4 differentially impacts elongation of HSV IE gene transcription.

(A) Schematic of small and large IE mRNAs RT-qPCR primer sets (product center). (B–D) The levels of large and small IE RNAs in AFF4 (B) or BRD4 (C) depleted MRC5 cells relative to siControl transfected cells. Cells were infected with HSV-1 for 2 h. BTUB, cellular control gene. The ratios of large to small RNA levels are shown for AFF4 and BRD4 depleted cells (D). (E) The levels of ICP4 large and small RNAs are shown in cells treated with vehicle, JQ1+, or the CDK9 inhibitor flavopiridol (FV) and infected with HSV-1 for 2 h. (B–E) Data are means +/− s.e.m., n = 4.
Consistent with the siRNA results, treatment of infected cells with the CDK9 inhibitor flavopiridol suppressed the accumulation of large RNAs while treatment with the BRD4 inhibitor JQ1+ enhanced this population (Figure 4E). Neither compound affected the levels of small RNAs.
IE genes are regulated by promoter proximal pausing
As RNAPII disengages from the promoter complex, proximal pausing is mediated by factors including the Negative Elongation Factor (NELF). ChIP assays to assess levels of RNAPII and NELF occupancy at viral IE genes illustrate a peak of RNAPII and NELF occupancy proximal to the initiation sites of the IE ICP4 and ICP0 genes (Figure 5A, B). This pattern of NELF and RNAPII occupancy mimics that of the cellular Myc gene whose expression is tightly regulated by elongation (Krumm et al., 1992).
Figure 5. ChIP assays show pausing and elongation factors at viral IE genes.

(A–B) ChIP assays illustrating the occupancy of RNAPII and NELF at viral IE genes (ICP4, ICP0) and the cellular MYC gene. Data are means +/− s.e.m., n = 6. (C) HCF-1, RNAPII-S2P, AFF4, and CDK9 occupancy levels of the ICP4 promoter proximal/TSS region in vehicle and JQ1+ treated cells at 2 hpi. (D) Fold occupancy in JQ1+ treated relative to vehicle treated cells. Data are means +/− s.e.m., n > = 3.
Furthermore, treatment of infected cells with the BRD4 inhibitor JQ1+ enhanced the levels of elongating RNAPII (RNAP-S2p), SEC (AFF4), and P-TEFb (CDK9) associated with the ICP4 viral IE promoter-TSS region (Figure 5C, D). Together the data suggest that expression of HSV IE genes is dependent on release from transcriptional pausing and induced transcriptional elongation.
BRD4-BET inhibitors induce HSV reactivation from latency in a mouse sensory ganglia explant model
Both viral lytic infection and reactivation from latency are dependent on HCF-1-mediated expression of viral IE genes. Given this, the significance of SEC-P-TEFb-mediated transcriptional elongation of viral reactivation was investigated in a sensory ganglia explant model system. Here, reactivation can be stimulated by explant of latently infected mouse trigeminal ganglia into culture. Additionally, signals and mechanisms regulating reactivation can be probed by inclusion of compounds that target critical components of the latency-reactivation dynamic.
Latently infected ganglia were explanted into vehicle, inactive control JQ1− or JQ1+ for 24–48 h. Viral yields were determined as a measure of reactivation (Figure 6A). No significant differences in viral yields were observed between vehicle and control JQ1− treated ganglia. However, treatment with JQ1+ resulted in a strong stimulation of reactivation at both 24 and 48 h timepoints. Similarly, explant in the presence of a second BET inhibitor (IBET-762, Figure S7A) also resulted in enhanced viral reactivation. Strikingly, ganglia explanted with JQ1+ for 3 h, followed by washout of the inhibitor was sufficient (Figure S7B), suggesting that JQ1+ acted at an initial stage in reactivation.
Figure 6. BET inhibitors drive viral reactivation in the mouse ganglia explant model system.

(A) Viral yields from latently infected trigeminal ganglia explanted in the presence of the BRD4-BET inhibitor JQ1+ or control compounds (Vehicle, JQ1−) for 24 or 48 h. Data are yields from individual ganglia, n > = 15. (B) Trigeminal ganglia from latently infected mice were explanted in the presence of JQ1+ or controls (DMSO, JQ1−) for 48 h. Sections were stained for UL29 (HSV lytic ss-DNA binding replication protein, ICP8). (C) The numbers of UL29+ cells per ganglia, n = 12. (D–E) Trigeminal ganglia from latently infected mice were explanted in the presence of acyclovir (ACV) alone or in combination with JQ1+ or JQ1− for 48 h. Ganglia sections were stained for UL29 and the numbers of individual UL29+ cells per ganglia were quantitated, n = 12. (F) Trigeminal ganglia from latently infected mice were explanted in the presence of Vehicle, JQ1+, or JQ1+ and ACV. Viral (ICP27, gC) mRNA levels were determined at 4, 8, and 12 h post explant. (G) Trigeminal ganglia from latently infected mice were explanted in the presence of JQ1+ (1 uM), iCDK9 (0.1 uM), or JQ1+ and iCDK9. Viral mRNA levels were determined at 12 h post explant. (F–G) Samples were normalized based on the levels of cellular GAPDH mRNA. Data are means +/− s.e.m., n = 3 pools of 5 ganglia per group. See also Figure S7.
Enhanced reactivation by JQ1+ treatment was also evident by immunofluorescent staining of ganglia explanted in the presence of vehicle, JQ1+ or JQ1−. Explanted ganglia were sectioned and stained with an antibody to detect the viral lytic DNA replication protein UL29 (ICP8) as a marker of productive reactivation. As shown (Figure 6B, C), JQ1+ treated ganglia exhibited a dramatic increase in the number of cells expressing UL29 relative to control treated ganglia.
Increased numbers of UL29+ cells and viral yields from explanted ganglia in the presence of BET inhibitors suggest that these compounds induce primary reactivation in neurons (initiation) and/or promote the lytic spread of infection throughout the ganglia. To discriminate between these possibilities, ganglia were treated with JQ1+ or JQ1− in the presence of the DNA replication inhibitor ACV to prevent viral spread from the initiating neuron (Figure 6D). Treatment with JQ1+/ACV resulted in ∼8-fold increase in the number of primary reactivation events per ganglia when compared to either ACV alone or JQ1−/ACV (Figure 6E). This increase was also reflected in the number of JQ1+ treated ganglia sections that scored positive for UL29+ cells (Figure S7C).
To define the timecourse of JQ1+ stimulated reactivation, ganglia were explanted in the presence of vehicle or JQ1+ and viral lytic mRNAs levels were determined at points in the reactivation process (0–12 h post explant, Figure 6F). By 8 h post explant, JQ1+ treated ganglia exhibited an induction in the expression of the viral IE ICP27 gene. This was followed by the induction of the representative Late (L, gC) mRNA at 12 h post explant.
In ganglia explanted in the presence of both JQ1+ and ACV to suppress viral DNA replication, the level of the IE gene mRNA (ICP27) was not significantly affected (Figure 6F). In contrast, as gC is a true Late gene and its expression is dependent upon DNA replication, the level of gC mRNA was more potently suppressed in the presence of ACV. Thus, the JQ1+ induction of reactivation proceeded in the characteristically ordered manner seen during lytic infection.
Finally, to demonstrate that CDK9 activity was required for JQ1+ stimulation of viral reactivation, ganglia were explanted in the presence of JQ1+ or JQ1+ and iCDK9 for 12 h. Inclusion of iCDK9 significantly reduced the ability of JQ1+ to stimulate viral gene expression (Figure 6G).
BRD4-BET inhibitors induce HSV reactivation in vivo
To determine if BET inhibitors would stimulate viral reactivation in vivo, latently infected mice were injected intraperitoneal with vehicle or JQ1+ and viral lytic mRNA levels in the trigeminal ganglia were determined at 24 and 48 h post injection. As shown in Figure 7A, IE and L gene mRNAs were induced by 24 h post injection. Continued treatment resulted in an enhanced level of the L gene at 48 h relative to the earlier point.
Figure 7. BET inhibitors drive HSV-1 viral reactivation from latency in vivo.

(A) Latently infected mice were injected twice daily with JQ1+ or control vehicle and mRNA levels of viral lytic genes (ICP27, gC) and control GAPDH were determined at 24 and 48 h post injection. (B) Latently infected mice were injected with JQ1+ or control JQ1− and mRNA levels of viral lytic genes (ICP27, gC) are shown at the indicated times post injection. (A–B) Data are means +/− s.e.m., n = 3 pools of 5 ganglia per group. (C) Viral DNA at 48 h post injection in eyes of latently infected mice injected with vehicle or JQ1+. Data are means +/− s.e.m., n = 8. (A–C) Viral mRNA or DNA levels in samples were normalized based on the levels of cellular GAPDH mRNA or DNA respectively. *p < 0.05; **p < 0.01.
Similar to the induction of these genes in the explant model system, injection of JQ1+ stimulated the detectable expression of the representative IE gene in vivo by 4 h post injection and the accumulation of the L gene mRNA by 8 h post injection (Figure 7B).
Finally, to assess if JQ1+ mediated induction resulted in reactivation as defined by the enhanced detection of viral DNA at the site of the initial infection, latently infected mice were injected once or twice daily with vehicle or JQ1+ and viral DNA levels in the eyes were assessed at 48 h post the initial injection (Figure 7C). In comparison to the control vehicle group, injection of JQ1+ resulted in enhanced detection (>2-fold vehicle maximum value) of viral DNA in 3 of 8 (37.5%) of eye samples. This data does not reach statistical significance, likely due to the small sample size and the expected low level of full reactivation events. However, a clear pattern of induction in JQ1+ treated animals was evident in both experiments and is consistent with the induction of viral lytic mRNAs in the ganglia in vivo (Figure 7A–B).
Discussion
As an essential cellular transcriptional coactivator, HCF-1 plays critical roles in cell cycle dependent transcription (Tyagi et al., 2007) and maintenance of embryonic cell pluripotency (Dejosez et al., 2010) as a component of chromatin modulation complexes including SETD1A and the MLL family of histone methyltransferases. Recently, point mutations in HCF-1 were determined to be a basis for both neurological (X-linked Intellectual Disability) and biochemical (cblX) syndromes (Jolly et al., 2015; Yu et al., 2013).
HCF-1 is also an essential component of the HSV IE gene regulatory program. This coactivator mediates initiation of IE transcription through direct interaction with transcription factors in the IE enhancer complexes and modulates viral chromatin through recruitment of multiple histone modification components. Importantly, the protein and the associated chromatin modulation activities are also components of the regulatory switch mechanism that governs viral reactivation from latency.
The data presented here shows that, in addition to initiation factors, HCF-1 is associated with elongating RNAPII and an extended network of transcriptional elongation complexes including SEC, PAFc, FACT, and SETD2 that play roles in mediating initial release of paused polymerase but also in chromatin modulation during mRNA elongation. Importantly, elongation specifically mediated by the AFF4/SEC-P-TEFb complex, was defined as a critical step for productive viral IE gene expression during lytic infection. The SEC localized to early viral transcriptional foci and depletion of the AFF4 scaffold subunit specifically suppressed viral IE transcriptional elongation while depletion of the major competitor for active P-TEFb (BRD4) resulted in enhanced IE mRNA elongation. Additionally, AFF4 and the P-TEFb-CDK9 kinase occupy IE promoters during productive infection and occupancy increased following treatment with BET inhibitors that increase the levels of available P-TEFb.
It is worth emphasizing that HCF-1 is found in multiple complexes that regulate transcription and chromatin modulation. The associations with both transcription initiation and elongation regulatory complexes shown here may represent distinct HCF-1 complexes or may be networked to couple efficient initiation and elongation. With respect to the identified association with the SEC-P-TEFb complex, the P-TEFb component is regulated at multiple levels (Lu et al., 2013; Mbonye and Karn, 2014) and therefore other cellular or viral regulatory factors are likely to play a role. In addition, while the SEC is clearly important for viral IE expression, multiple P-TEFb complexes can also be recruited to target genes although the functional significance of each is dependent on the target and the regulation context (Lin et al., 2011).
In this study, BET inhibitors that enhance the release of free P-TEFb from the 7SK snRNP complex also stimulated reactivation of HSV from latency in a CDK9 dependent manner. Interestingly, stress stimuli that promote viral reactivation (i.e. UV irradiation) also result in release of P-TEFb (Nguyen et al., 2001). Additionally, compounds that induce reactivation have recently been found to either induce release of P-TEFb (i.e. PKC agonist PMA, HMBA) (Bernstein and Kappes, 1988; Contreras et al., 2007; Fujinaga et al., 2015; Smith et al., 1992) or function as dual kinase-BET inhibitors targeting BRD4 (i.e. PI3K-mTOR inhibitor PP-242 and the PI3K inhibitor LY294002) (Camarena et al., 2010; Ciceri et al., 2014; Dittmann et al., 2014; Kobayashi et al., 2012). It is therefore tempting to speculate that the induction and recruitment of available P-TEFb to viral IE genes is a critical common step in the initiation of reactivation via several distinct stimuli.
The regulated transcriptional elongation, the specific requirement for the SEC-P-TEFb adaptor elongation complex, and the induction of viral reactivation by BET inhibitors share striking similarities to the regulation of HIV transcription and reactivation (Boehm et al., 2013; Cary et al., 2016; Li et al., 2013; Lu et al., 2013; Ott et al., 2011). Importantly, while both BRD4-P-TEFb and the SEC-P-TEFb can mediate HIV basal level expression, the SEC-P-TEFb is required for Tat-mediated induced transcription during reactivation (He et al., 2011; He et al., 2010; Jang et al., 2005; Sobhian et al., 2010; Yang et al., 2005). The use of the SEC for potent transcriptional induction by both HSV and HIV may be reflective of the significant level of RNAPII CTD kinase activity in this complex as compared to that of the BRD4-P-TEFb or the free enzyme (Luo et al., 2012a). In addition, other subunits of this multi-protein complex play roles in promoting highly efficient transcription (Byun et al., 2012; Shilatifard et al., 1996).
Multiple factors impact HSV latency and reactivation including the status of the chromatin structure of the latent viral genome (Bloom et al., 2010; Knipe and Cliffe, 2008; Lieberman, 2016), neuronal antiviral responses (Enquist and Leib, 2016; Maroui et al., 2016; Rosato and Leib, 2015), and microRNAs that suppress lytic gene expression (Flores et al., 2013; Pan et al., 2014). Once considered to be a suppressed static state, the concept of HSV latency has evolved to a more dynamic model (Bloom, 2016) to account for HSV-specific immune surveillance in latently infected ganglia (St Leger and Hendricks, 2011), viral subclinical shedding (Wald and Corey, 2007), and the detection of lytic gene expression in individual latent neurons (Russell and Tscharke, 2016). More recently, a study described a neuronal stress pathway that promotes histone H3S10 phosphorylation at repressed viral lytic genes to stimulate expression in a transient/reversible manner (Cliffe et al., 2015).
The regulation of HSV IE genes by the SEC-P-TEFb complex could provide an opportune manner for HSV to respond rapidly to neuronal stress signals and promote expression of a poised population of viral genomes. This model would suggest that coordinated initiation-elongation control by HCF-1 associated complexes may drive productive viral reactivation.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information concerning these resources and reagents should be directed to the lead author Thomas M. Kristie (tkristie@niaid.nih.gov). Reagents produced by the Kristie Laboratory are the property of the National Institutes of Health, US Health and Human Services and requests may require NIH Material Transfer Agreements.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines, Tissue Culture, and Viruses
MRC5, HFF, HEK293 and Vero cells were grown in high glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 2 mM L-Glutamine, 100 U/ml Penicillin, 100 ug/ml Streptomycin, non-essential amino acids (cDMEM) and 10% fetal bovine serum (FBS). HSV-1(F) or CMV(RC256) high titer stocks were produced using seed stocks.
Mice
Female 6–8 week old BALB/cAnNTac mice were purchased from Taconic Biosciences. Groups of 5 were housed in micro-isolator cages for one week prior to infection with HSV-1(F). All animal care and handling was done in accordance with the National Institutes of Health Animal Care and Use Guidelines and as approved by the NIAID Animal Care and Use Committee.
METHOD DETAILS
Infections of Cultured Cells
Infections were done by incubating cells with HSV-1(F) or CMV(RC256) at the indicated Multiplicity Of Infection (MOI) in cDMEM medium containing 1% fetal bovine serum (FBS) for 1 hour at 37 °C / 5% CO2 with continuous rocking. After virus incubation, the inoculum was removed and fresh cDMEM medium containing 8% FBS was added for 2–5 hrs at 37°C /5% CO2.
Immunoaffinity Purification of HCF-1 Protein Complexes
HCF-1 and control IgG immunoaffinity precipitates were isolated from uninfected and 2 h HSV-1(F)-5 MOI infected MRC5 cells. Cells were harvested and resuspended in buffer A [10 mM Hepes pH 7.9, 10 mM KCl, 0.2% Triton X-100, 1.5 mM MgCl2, 0.34 M sucrose, 10% Glycerol, cOmplete protease inhibitor cocktail (Roche) and phosphatase inhibitors (1 mM NaF, 10 mM β-glycerol phosphate and 0.1 mM sodium orthovanadate)] for 10 m at 4°C. Nuclear pellets were collected by centrifugation and incubated in lysis buffer B (50 mM Tris-HCl pH 7.5, 120 mM NaCl, 0.2% NP-40 1.5 mM MgCl2, 10% Glycerol, cOmplete protease inhibitors and phosphatase inhibitors) supplemented with 100 U/ml Benzonase (Millipore) for 10 m at 20°C and 20 m at 4°C. The supernatant was diluted with buffer B, precleared with Dynabeads Protein G (ThermoFisher Scientific) for 1 h at 4°C, and incubated with magnetic beads pre-conjugated with anti-HCF-1 Ab2124/Ab2130 antibodies or control normal rabbit IgG for 1 h at 4°C. The resulting immunoisolates were washed 4 times with buffer B and once with buffer B containing 250 mM NaCl. Proteins were eluted in 0.5 N NH4OH containing 0.5 mM EDTA.
Preparation of Mass Spectrometry Samples
HCF-1 and control IgG immunoisolates were prepared for in-gel digestion as described in (Joshi et al., 2013) with slight modifications. Samples were resuspended in LDS sample buffer (0.25mM Tris pH 8.5, 2% lithium dodecyl sulfate, 10% glycerol, 0.5mM ethylenediaminetetraacetic acid, and 50mM dithiothreitol), alkylated in 1M iodoacetamide, and separated by SDS-PAGE for 3 cm. Gel lanes with partially resolved protein samples were excised and sliced into 1mm strips and divided into a total of 6 fractions per lane. Proteins were digested with 20 µl of trypsin solution (12.5 ng/µl MS-grade trypsin (Promega) in 50 mM ammonium bicarbonate in water) per fraction for 16 h at 37°C, quenched in 0.5% formic acid, and extracted for 4 h at room temperature. A second extraction was performed in 50% acetonitrile/0.5% formic acid for 2 h. Extracted peptides were concentrated to remove acetonitrile, diluted in 0.5% trifluoracetic acid, and bound to StageTips containing Empore C18 discs (3M Analytical Biotechnologies) (Rappsilber et al., 2007). Bound peptides were washed with 0.5% trifluoracetic acid and eluted from the C18 discs with 80% acetonitrile/0.5% formic acid in water. Peptides were concentrated to remove the acetonitrile and resuspended in 1% formic acid in water for MS analysis.
Mass Spectrometry Analysis of HCF-1 Immunoisolates
Desalted peptides were analyzed by nanoliquid chromatography tandem MS (nLC-MS) using a Dionex Ultimate 3000 nRSLC coupled to an LTQ-Orbitrap XL mass spectrometer (ThermoFisher). Peptides were separated in a 90 m reverse-phase gradient. The mass spectrometer was operated in data-dependent acquisition with dynamic exclusion enabled (repeat count = 1; exclusion duration = 45 s). Full scan range set to 350–1700 m/z with a 60000 resolution at m/z = 400 in the Orbitrap. CID fragmentation was performed on the top 10 most intense precursor ions with minimum signals 1E3 in the dual pressure ion trap. The isolation windows were set to 2.0 Th, normalized collision energy of 30, and activation time of 10 ms. Target values for the Orbitrap full-scan MS and Ion trap MS2 were 1E6 and 5E3, respectively.
Protein Identification
Acquired MS data (RAW files) were searched by SEQUEST (v1.2) in Proteome Discoverer (v1.3, ThermoFisher) with settings allowing up to two missed cleavages, a precursor mass tolerance of 10 ppm, and a fragment mass tolerance of 0.5 Da as described in (Diner et al., 2015). The search was set for dynamic modifications oxidation (M) and phosphorylation (STY), and static modification of carbamidomethylation (C). The database used to perform the search was obtained from Uniprot and contained protein sequences for human, herpesvirus, and common contaminants. The resulting MSF files were then processed using Scaffold (v4.2.1, Proteome Software Inc.) for statistical assessment of peptide/protein identification. The six gel samples per fraction were combined for analysis using the MuDPIT technology in Scaffold. Additional variable modifications were set as part of the X!Tandem algorithm in Scaffold for deamidation (N,Q) and acetylation (n-term). The final list of proteins and spectral counts associated with each protein was obtained at a protein false discovery rate (FDR) of 2%, peptide FDR of 1%, and minimum of 3 peptides sequenced per protein.
SAINT Analysis for Interaction Specificity and Interaction Network Analysis
Interaction specificity was assessed using the SAINTexpress algorithm (Teo et al., 2014) available through the CRAPome server (Mellacheruvu et al., 2013). Protein spectral counts were extracted from Scaffold and formatted in matrix format to be used as input by the SAINT algorithm. For the analysis, control immunoaffinity purifications performed with IgG were used as SAINT controls for non-specific interactions. SAINT probability scores were reported as an average of all replicates. Preys with a SAINT score >0.9 were considered putative specific protein interactions. The functional interaction network was generated using data of Table S1 using the ReactomeFIViz plugin under Cytoscape (v3.4.0) (Shannon et al., 2003).
Co-immunoprecipitation Assays
For co-immunoprecipitation assays, nuclear extracts from mock-infected or 2 h HSV-1 infected (5 MOI) HEK293 cells were generated using the protocol above and incubated with polyclonal antibodies against HCF-1 or control normal rabbit IgG pre-bound to Dynabeads™ Protein G (ThermoFisher Scientific) for 2 h at 4°C with constant rotation. The resulting complex was washed and eluted with non-reducing LDS sample buffer supplemented with DTT, boiled and loaded on 4–15% Criterion™ TGX™ polyacrylamide gels (Bio-Rad). After SDS/PAGE, Western blotting was performed using the indicated antibodies and resolved extracts and immunoprecipitates were developed using WesternBright™ Quantum Western blotting detection Kit (advansta) and quantitated using a Syngene G:BOX Chemi XT4 and GeneTools image analysis software (Syngene).
Inhibitor Treatments
Uninfected or HSV-1 infected MRC5 or HFF cells were treated with the indicated concentrations of Vehicle, ACV, JQ1+, IBET-762, HMBA, inactive analog JQ1−, flavopiridol, or iCDK9 at 1 h post virus absorption. Cells were treated with dBET1 for 2 h prior to infection.
siRNA Depletions
MRC5 cells were transfected with 20–30 mM non-targeting or specific siRNAs using TransIT-X2 (Mirus) or HiPerFect (Qiagen) reagents. RNA and protein were harvested at 48 h post transfection. siRNAs are listed in Table S2.
RNA Isolation and RT-qPCR Quantitation
For quantification of RNA derived from tissue culture cells, cDNA was synthesized from 800 ng of total RNA (ISOLATE II RNA Mini Kit (Bioline)) using a Maxima first-strand cDNA synthesis kit (Thermo Scientific). For isolation of individual small (<200nt) and large RNA (>200nt) fractions, HFF cells were lysed in TriPure Isolation reagent (Roche) and phase separated with the addition of chloroform. After centrifugation, the aqueous layer containing the total RNA fraction was recovered and ethanol was added to a final concentration of 35%. The total RNA fraction was loaded onto RNA isolation columns (Isolate II RNA Mini Kit, Bioline) and centrifuged. The bound fraction representing the large RNA fraction was isolated as per the manufacturer’s protocol. The unbound flow-through (small RNA fraction) was retained and the ethanol concentration was adjusted to 70% followed by loading to a second RNA isolation column. Large mRNAs were reverse transcribed using a Maxima first-strand cDNA synthesis kit (Thermo Scientific). Small-RNA fractions (<200 nucleotide) were reverse transcribed using the Quanta qScript microRNA cDNA Synthesis Kit (Quanta Biosciences). RT-qPCR of cDNAs from small RNAs used a universal reverse primer together with a forward primer corresponding to the 5’ end of the specific nascent RNA. Primers for known small RNAs (miR16 and SNORD44) were used as controls. Amplified cDNA was assessed by RT-qPCR in triplicate using the StepOnePlus™ Real-Time PCR system (ThermoFisher) and FastStart Universal SYBR Green mix (Roche). Viral mRNA levels were normalized between samples based on the levels of cellular GAPDH mRNA. Primer sequences are in Table S2.
Chromatin Immunoprecipitations
MRC5 cells were mock-infected or infected with HSV-1(F) at 5 MOI for 2 h in the presence Vehicle or JQ1+ (1 um). Post infection, cells were washed with cold PBS, fixed in 1% formaldehyde for 10 m at 4°C, and quenched with 125 mM glycine for 5 m at 20°C. Cells were resuspended in 10 ml low-salt buffer (50 mM Hepes-NaOH pH 7.5, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, cOmplete protease inhibitors and phosphatase inhibitors) and incubated for 10 m at 4°C. The isolated nuclei were washed and the chromatin was fragmented in sonication buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH 8.1, cOmplete protease inhibitors and phosphatase inhibitors) to a range of 200–800 bp using a Bioruptor UCD-200 sonicator (Diagonode). Chromatin was diluted with ChIP dilution buffer (2 mM EDTA, 5 mM Tris-HCl pH 8.0, 1% NP-40, 150 mM NaCl, cOmplete protease inhibitors and phosphatase inhibitors), precleared with Dynabeads Protein G for 1 h at 4 °C, and incubated with 3 g of the indicated antibody for 12 h at 4 °C. Immunoprecipitates were washed three times with low salt wash buffer (0.1% SDS, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% Triton X-100), two times with high salt wash buffer (0.1% SDS, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 500 mM NaCl, 1% Triton X-100) and once with Tris-EDTA (TE) buffer (10 mM Tris-HCl pH 8.0, 1.2 mM EDTA). Chromatin was eluted with 100 ul elution buffer (1% SDS, 0.1M NaHCO3) for 30 min at 65 °C, and crosslinking was reversed at 65 °C with NaCl 5M for 12 h. The DNA was purified using the ChIP DNA Clean & Concentrator Kit (Zymo Research) and quantitated by RT-qPCR using a StepOnePlus Real-Time PCR system (ThermoFisher) and FastStart Universal SYBR Green mix (Roche). All reactions were done in triplicates and RT-qPCR signals were normalized to input DNA. Primer sequences are in Table S2.
Immunofluorescence Microscopy
MRC5 cells were grown on cover slips to 40% confluency and mock-infected or HSV-1 infected at MOI 5 for 2 h. After infections, cells were washed with cold PBS, fixed at room temperature in 4% paraformaldehyde-PBS for 30 m, and washed several times with cold PBS. Cells were permeabilized in 0.1% Triton X-100 and stained with the indicated primary and fluorescent secondary antibodies. Cover slips were mounted in Fluoromount-G® containing 4’,6-diamidino-2-phenylindole (DAPI) and visualized using a Leica SP5 confocal microscope with LAS AF software (Leica Microsystems). Images were assembled from sequential Z-sections using Imaris software (version 8.2.0; Bitplane).
Quantitation of Free and 7SK snRNP Bound CDK9
HFF cells were mock-infected or HSV-1 infected for 2 h in the presence of vehicle or BRD4 inhibitors [JQ1+ (1 uM), IBET-762 (2 uM); HMBA (5 mM)]. Differential extractions of free CDK9 and 7SK snRNP bound CDK9 were as described (Biglione et al., 2007). Cells were lysed in buffer A containing low salt concentration (Hepes pH 7.9 10 mM, KCl 10 mM, MgCl2 10 mM, NP-40 0.5%, DTT 1 mM, EDTA 1 mM, cOmplete® protease inhibitor cocktail (Roche) and phosphatase inhibitors) on ice for 10 m. Lysed cells were centrifuged at 5000 X g for 5 m at 4 °C and the supernatant collected and labeled as 7SK snRNP fraction. The remaining pellet was re-suspended and washed two times with buffer A, and extracted with a high salt concentration buffer B (Hepes pH 7.9 20 mM, NaCl 450 mM, MgCl2 1.5 mM, NP-40 0.5%, DTT 1mM, EDTA 0.5 mM, cOmplete ® protease inhibitor cocktail (Roche) and phosphatase inhibitors) for 10 m on ice. Extracts were centrifuged at 10000 × g for 10 m and the resulting supernatant collected and labeled as free P-TEFb fractions. Equivalent amounts of protein from each fraction was loaded on SDS-PAGE gels and Western blots were done using the indicated antibodies. Quantification of CDK9 and loading controls were done using a Syngene G:BOX Chemi XT4 and GeneTools image analysis software (Syngene).
HSV-1 Reactivation in Explanted Trigeminal Ganglia
Balb/c mice were infected with 2×105 pfu HSV-1 (F) via the ocular route. Trigeminal ganglia from latently infected mice (45–60 dpi) were bisected and paired halves were explanted into cDMEM containing 10% FBS and vehicle or inhibitor for the indicated time. Incubated ganglia were homogenized, briefly sonicated in cDMEM containing 1% FBS and viral yields were determined by titering on Vero cell monolayers. To determine the number of UL29+ cells, ganglia explanted for 48 h were fixed in 4% paraformaldehyde/PBS at 4 °C for 8 h and subsequently embedded in paraffin. Sections were rehydrated, treated with citric acid for antigen retrieval, and stained for HSV-1 UL29. UL29+ cells were counted in 3 sections each of 12 ganglia (36 sections). Viral and cellular mRNA levels were determined by RT-qPCR using RNA isolated from 5 ganglia per sample. Ganglia were homogenized in TRIzol® Reagent using lysing matrix D on a FastPrep®24 and RNA was isolated using ISOLATE II RNA Mini Kit (Bioline). Samples were normalized based on the levels of cellular GAPDH mRNA. Primer sequences are in Table S2.
HSV-1 reactivation in vivo
Latently infected mice were injected intraperitoneal with Vehicle, JQ1+, or JQ1− (50 mg/Kg in 2-hydroxypropyl-β-cyclodextrin/PBS) every 12 or 24 h. RNAs were isolated from trigeminal ganglia and analyzed by RT-qPCR. Viral DNA levels were determined by RT-qPCR of genomic DNA from eyes. Samples were normalized based upon the levels of the GAPDH gene. Primer sequences are in Table S2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as means +/− s.e.m. Statistical parameters are reported in the text, figures, and figure legends. Analyses used Mann-Whitney for single comparisons of control vs. treated samples and Wilcoxon matched-pairs signed rank tests for comparisons of paired ganglia samples. n represents the number of independent samples. For ganglia explants, n represents the number of ganglia per group. For in vivo experiments, n represents the number of animals per group. Where indicated, asterisks denote statistical significance as follows: *p < 0.05 and **p < 0.01. Statistical analyses were done using GraphPad Prism 7.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-HCF-1 rabbit polyclonal Ab#2124 | Kristie Laboratory | RRID:AB_2636976 |
| anti-HCF-1 rabbit polyclonal Ab#2130 | Kristie Laboratory | RRID:AB_2636977 |
| anti-MED12 rabbit polyclonal | Bethyl Laboratories | Cat# A300-774A; RRID:AB_669756 |
| anti-AFF4 rabbit polyclonal | Bethyl Laboratories | Cat# A302-538A; RRID:AB_1998985 |
| anti-AFF4 rabbit polyclonal | Bethyl Laboratories | Cat# A302-539A; RRID:AB_1998983 |
| anti-BRD4 rabbit polyclonal | Bethyl Laboratories | Cat# A301-985A; RRID:AB_1576498 |
| anti-CDK9 rabbit polyclonal | Santa Cruz Biotech. | Cat# sc-484; RRID:AB_2275986 |
| anti-PAF1 rabbit polyclonal | Bethyl Laboratories | Cat# A300-172A; RRID:AB_309394 |
| anti-SUPT16H rabbit polyclonal | Santa Cruz Biotech. | Cat# sc-28734; RRID:AB_661341 |
| anti-SETD1A rabbit polyclonal | Abiocode | Cat# R0196-2; RRID:AB_2636972 |
| anti-RbBP5 rabbit polyclonal | Bethyl Laboratories | Cat# A300-109A; RRID: AB_210551 |
| anti-GAPDH mouse monoclonal | Santa Cruz Biotech. | Cat# sc-32233; RRID:AB_627679 |
| anti-VP16 rabbit polyclonal | Sigma-Aldrich | Cat# V4388; RRID:AB_261865 |
| anti-ICP4 mouse monoclonal | Virusys | Cat# H1A021; RRID:AB_2636973 |
| anti-UL29 (ICP8) rabbit polyclonal | Gift of William Ruyechan, University of Buffalo, SUNY | RRID:AB_2636974 |
| anti-NELFA goat polyclonal | Santa Cruz Biotech. | Cat# sc-23599; RRID: AB_2241683 |
| anti-RNAPII-o (RNAPII 8WG16) mouse monoclonal | Covance | Cat# MMS-126R; RRID: AB_10013665 |
| anti-RNAPII-S5p (H14) mouse monoclonal | Covance | Cat# MMS-134R; RRID: AB_10063994 |
| anti-RNAPII-S2p (H5) mouse monoclonal | Covance | Cat# MMS-129R; RRID: AB_10063704 |
| anti-RNAPII-S2p rabbit polyclonal | Abcam | Cat# Ab5095; RRID: AB_304749 |
| anti-POLR2B rabbit polyclonal | Santa Cruz Biotech. | Cat# sc-67318; RRID: AB_2167642 |
| Normal Rabbit IgG | Millipore | Cat# 12-370; RRID: AB_145841 |
| anti-Rabbit Alexa 488 | Jackson ImmunoResearch Labs | Cat# 711-545-152; RRID: AB_2313584 |
| anti-Rabbit Alexa 594 | Jackson ImmunoResearch Labs | Cat# 711-585-152; RRID: AB_2340621 |
| anti-Mouse Alexa 594 | Jackson ImmunoResearch Labs | Cat# 715-585-150; RRID: AB_2340854 |
| Bacterial and Virus Strains | ||
| HSV-1 (strain F) | Gift of B. Roizman, University of Chicago | |
| hCMV (strain RC256) | ATCC | VR-2356 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| HMBA (N, N’-Hexamethylene bis(acetamide)) | Sigma-Aldrich | CAS: 564468-51-5; Cat# 224235 |
| JQ1+ | APExBIO/Cayman Chemical/Biovison | CAS: 1268524-70-4; Cat# A1910/Cat #11187/Cat# 2070 |
| JQ1− | APExBIO/Cayman Chemical/Biovision | CAS: 1268524-71-5; Cat# A8181/Cat# 106/Cat# 2384 |
| dBET1 | Chemietek | CAS: 1799711-21-9; Cat# CT-DBET1 |
| I-BET762 | Cayman Chemical | CAS: 1260907-17-2; Cat# 10676 |
| Flavopiridol | Santa Cruz Biotech. | CAS: 146426-40-6; Cat# sc-202157 |
| i-CDK9 (N2’-(trans-4-aminocyclohexyl)-5’-chloro-N6-(3-fluorobenzyl)-2,4’-bipyridine-2’,6-diamine) | MedChem Express | CAS: 1263369-28-3; Cat# HY-16462 |
| Acyclovir | Sigma-Aldrich | CAS: 59277-89-3; Cat# A4669 |
| Vehicle (DMSO; Dimethyl sulfoxide) | Sigma-Aldrich | CAS: 67-68-5; Cat# D2650 |
| Critical Commercial Assays | ||
| ISOLATE II RNA Mini Kit | Bioline | Cat# BIO-52073 |
| Maxima first-strand cDNA synthesis kit | ThermoFisher Scientific | Cat# K1671 |
| Quanta qScript microRNA cDNA Synthesis Kit | Quanta Biosciences | Cat# 95107 |
| Dynabeads™ Antibody Coupling Kit | ThermoFisher Scientific | Cat# 14311D |
| Sequence-Based Reagents | ||
| siGENOME siRNA SMARTpool targeting BRD2 | GE Dharmacon | M-004935-02 |
| siGENOME siRNA SMARTpool targeting BRD3 | GE Dharmacon | M-004936-01 |
| siGENOME siRNA SMARTpool targeting AFF1 | GE Dharmacon | M-020074-02 |
| siGENOME Individual siRNA targeting AFF4: AFF4-1 | GE Dharmacon | M-020276-02-0001 |
| siGENOME Individual siRNA targeting AFF4: AFF4-2 | GE Dharmacon | M-020276-02-0004 |
| FlexiTube Individual siRNA targeting BRD4: BRD4-1 | Qiagen | SI03190845 |
| FlexiTube Individual siRNA targeting BRD4: BRD4-2 | Qiagen | SI04307009 |
| Non-targeting siControl | GE Dharmacon | D-001206-13, D-001206-14 |
| Experimental Models: Cell Lines | ||
| MRC5; Normal human lung fibroblast | ATCC | CCL-171 |
| HFF; Human Foreskin Fibroblast (TERT-immortalized) | Gift of T. Shenk, Princeton University | |
| HEK-293; Human Embryonic Kidney | ATCC | CRL-1573 |
| Vero | ATCC | CCL-81 |
| Experimental Models: Organisms/Strains | ||
| BALB/c mice, Female, 6–8 weeks old | Taconic | BALB/cAnNTac |
| Oligonucleotides | ||
| Primers used in this study are listed in Table S2 | ||
| Software and Algorithms | ||
| Imaris (v8.2.0) | BITPLANE | |
| SEQUEST (v1.2)/Proteome Discoverer (v1.3) | ThermoFisher | |
| Scaffold (v4.2.1) | Proteome Software Inc.; http://www.proteomesoftware.com/products/scaffold/ | |
| SAINTexpress | http://www.crapome.org | |
| ReactomeFIViz/Cytoscape (v3.4.0) | http://www.cytoscape.org/ | |
| GeneTools image analysis software/G:BOX Chemi XT4 | Syngene;http://www.syngene.com/ | |
| Other | ||
| TriPure Isolation reagent | Roche/Sigma-Aldrich | Cat# 11667157001 |
| Lysing Matrix D Tubes | MP-Biomedicals | Cat# 6913 |
Highlights.
The coactivator HCF-1 is critical for HSV Immediate Early (IE) gene transcription
HCF-1 is associated with a network of both initiation and elongation complexes
The SEC containing AFF4-P-TEFb drives HSV IE transcriptional elongation
BET inhibitors increase available P-TEFb and induce HSV reactivation from latency
Acknowledgments
We thank J. Vogel (Laboratory of Viral Diseases, NIAID) for discussions and review of this manuscript, A. McBride and M.K. Jang (Laboratory of Viral Diseases, NIAID) for BRD reagents, NIAID Bld33 Vivarium staff, and A. Reed (Laboratory of Viral Diseases, NIAID) for excellent technical support. This study was supported by the Intramural Research Program of the NIH, NIAID (T.M.K.), the Mallinckrodt Scholar Award (I.H.C.) and NIGMS (GM114141, I.M.C.). The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
R.A.D. performed MS immunoprecipitations, siRNA and inhibitor studies, ganglia explants and in vivo experiments; A.M.T. performed MS immunoprecipitations and RNA analyses; P.M.J.B. and H.G.B. performed MS; J.H.A. contributed to HSV animal studies; I.M.C. and T.M.K. designed experiments and analyzed data; T.M.K. and R.A.D. wrote the manuscript; all authors commented on the manuscript.
References
- Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nature reviews. 2012;13:720–731. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramani A, Larjo A, Bassein JA, Chang X, Hastie RB, Togher SM, Lahdesmaki H, Rao A. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat Commun. 2015;6:7307. doi: 10.1038/ncomms8307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomeeusen K, Xiang Y, Fujinaga K, Peterlin BM. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J Biol Chem. 2012;287:36609–36616. doi: 10.1074/jbc.M112.410746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein DI, Kappes JC. Enhanced in vitro reactivation of latent herpes simplex virus from neural and peripheral tissues with hexamethylenebisacetamide. Arch Virol. 1988;99:57–65. doi: 10.1007/BF01311023. [DOI] [PubMed] [Google Scholar]
- Biglione S, Byers SA, Price JP, Nguyen VT, Bensaude O, Price DH, Maury W. Inhibition of HIV-1 replication by P-TEFb inhibitors DRB, seliciclib and flavopiridol correlates with release of free P-TEFb from the large, inactive form of the complex. Retrovirology. 2007;4:47. doi: 10.1186/1742-4690-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom DC. Alphaherpesvirus Latency: A Dynamic State of Transcription and Reactivation. Adv Virus Res. 2016;94:53–80. doi: 10.1016/bs.aivir.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Bloom DC, Giordani NV, Kwiatkowski DL. Epigenetic regulation of latent HSV-1 gene expression. Biochim Biophys Acta. 2010;1799:246–256. doi: 10.1016/j.bbagrm.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li PC, Planelles V, Bradner JE, et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle. 2013;12:452–462. doi: 10.4161/cc.23309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun JS, Fufa TD, Wakano C, Fernandez A, Haggerty CM, Sung MH, Gardner K. ELL facilitates RNA polymerase II pause site entry and release. Nat Commun. 2012;3:633. doi: 10.1038/ncomms1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe. 2010;8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary DC, Fujinaga K, Peterlin BM. Molecular mechanisms of HIV latency. J Clin Invest. 2016;126:448–454. doi: 10.1172/JCI80565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Larsen B, Lin ZY, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras AC, Nesvizhskii AI. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Methods. 2011;8:70–73. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciceri P, Muller S, O’Mahony A, Fedorov O, Filippakopoulos P, Hunt JP, Lasater EA, Pallares G, Picaud S, Wells C, et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat Chem Biol. 2014;10:305–312. doi: 10.1038/nchembio.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe. 2015;18:649–658. doi: 10.1016/j.chom.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras X, Barboric M, Lenasi T, Peterlin BM. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 2007;3:1459–1469. doi: 10.1371/journal.ppat.0030146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejosez M, Levine SS, Frampton GM, Whyte WA, Stratton SA, Barton MC, Gunaratne PH, Young RA, Zwaka TP. Ronin/Hcf-1 binds to a hyperconserved enhancer element and regulates genes involved in the growth of embryonic stem cells. Genes Dev. 2010;24:1479–1484. doi: 10.1101/gad.1935210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner BA, Lum KK, Javitt A, Cristea IM. Interactions of the Antiviral Factor Interferon Gamma-Inducible Protein 16 (IFI16) Mediate Immune Signaling and Herpes Simplex Virus-1 Immunosuppression. Mol Cell Proteomics. 2015;14:2341–2356. doi: 10.1074/mcp.M114.047068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmann A, Werner T, Chung CW, Savitski MM, Falth Savitski M, Grandi P, Hopf C, Lindon M, Neubauer G, Prinjha RK, et al. The commonly used PI3-kinase probe LY294002 is an inhibitor of BET bromodomains. ACS Chem Biol. 2014;9:495–502. doi: 10.1021/cb400789e. [DOI] [PubMed] [Google Scholar]
- Durand LO, Roizman B. Role of cdk9 in the optimization of expression of the genes regulated by ICP22 of herpes simplex virus 1. J Virol. 2008;82:10591–10599. doi: 10.1128/JVI.01242-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist LW, Leib DA. Intrinsic and innate defenses of neurons: Detente with the herpesviruses. J Virol. 2016 doi: 10.1128/JVI.01200-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores O, Nakayama S, Whisnant AW, Javanbakht H, Cullen BR, Bloom DC. Mutational inactivation of herpes simplex virus 1 microRNAs identifies viral mRNA targets and reveals phenotypic effects in culture. J Virol. 2013;87:6589–6603. doi: 10.1128/JVI.00504-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Luo Z, Schaufele F, Peterlin BM. Visualization of positive transcription elongation factor b (P-TEFb) activation in living cells. J Biol Chem. 2015;290:1829–1836. doi: 10.1074/jbc.M114.605816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Wu WJ, Liu LD, Wang LC, Zhang Y, Wu LQ, Guan Y, Li QH. Herpes simplex virus 1 ICP22 inhibits the transcription of viral gene promoters by binding to and blocking the recruitment of P-TEFb. PLoS One. 2012;7:e45749. doi: 10.1371/journal.pone.0045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Chan CK, Sobhian B, Chou S, Xue Y, Liu M, Alber T, Benkirane M, Zhou Q. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A. 2011;108:E636–645. doi: 10.1073/pnas.1107107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell. 2010;38:428–438. doi: 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Quenelle DC, Cardin RD, Vogel JL, Clement C, Bravo FJ, Foster TP, Bosch-Marce M, Raja P, Lee JS, et al. Inhibition of LSD1 reduces herpesvirus infection, shedding, and recurrence by promoting epigenetic suppression of viral genomes. Science translational medicine. 2014;6:265ra169. doi: 10.1126/scitranslmed.3010643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Jolly LA, Nguyen LS, Domingo D, Sun Y, Barry S, Hancarova M, Plevova P, Vlckova M, Havlovicova M, Kalscheuer VM, et al. HCFC1 loss-of-function mutations disrupt neuronal and neural progenitor cells of the developing brain. Hum Mol Genet. 2015;24:3335–3347. doi: 10.1093/hmg/ddv083. [DOI] [PubMed] [Google Scholar]
- Jonkers I, Lis JT. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. 2015;16:167–177. doi: 10.1038/nrm3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, Cristea IM. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol. 2013;9:672. doi: 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog. 2012;8:e1002540. doi: 10.1371/journal.ppat.1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol. 2008;6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Wilson AC, Chao MV, Mohr I. Control of viral latency in neurons by axonal mTOR signaling and the 4E–BP translation repressor. Genes Dev. 2012;26:1527–1532. doi: 10.1101/gad.190157.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM, Vogel JL, Sears AE. Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc Natl Acad Sci U S A. 1999;96:1229–1233. doi: 10.1073/pnas.96.4.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm A, Meulia T, Brunvand M, Groudine M. The block to transcriptional elongation within the human c-myc gene is determined in the promoter-proximal region. Genes Dev. 1992;6:2201–2213. doi: 10.1101/gad.6.11.2201. [DOI] [PubMed] [Google Scholar]
- Kurosu T, Peterlin BM. VP16 and ubiquitin; binding of P-TEFb via its activation domain and ubiquitin facilitates elongation of transcription of target genes. Curr Biol. 2004;14:1112–1116. doi: 10.1016/j.cub.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Lewis BA, Burlingame AL, Myers SA. Human RNA Polymerase II Promoter Recruitment in Vitro Is Regulated by O-Linked N-Acetylglucosaminyltransferase (OGT) J Biol Chem. 2016;291:14056–14061. doi: 10.1074/jbc.M115.684365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Guo J, Wu Y, Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013;41:277–287. doi: 10.1093/nar/gks976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Vogel JL, Arbuckle JH, Rai G, Jadhav A, Simeonov A, Maloney DJ, Kristie TM. Targeting the JMJD2 Histone Demethylases to Epigenetically Control Herpesvirus Infection and Reactivation from Latency. Science translational medicine. 2013;5:167ra165. doi: 10.1126/scitranslmed.3005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med. 2009;15:1312–1317. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman PM. Epigenetics and Genetics of Viral Latency. Cell Host Microbe. 2016;19:619–628. doi: 10.1016/j.chom.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Garrett AS, De Kumar B, Smith ER, Gogol M, Seidel C, Krumlauf R, Shilatifard A. Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC) Genes Dev. 2011;25:1486–1498. doi: 10.1101/gad.2059211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Li Z, Xue Y, Zhou Q. Viral-host interactions that control HIV-1 transcriptional elongation. Chem Rev. 2013;113:8567–8582. doi: 10.1021/cr400120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Xue Y, Yu GK, Arias C, Lin J, Fong S, Faure M, Weisburd B, Ji X, Mercier A, et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. Elife. 2015;4:e06535. doi: 10.7554/eLife.06535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Lin C, Guest E, Garrett AS, Mohaghegh N, Swanson S, Marshall S, Florens L, Washburn MP, Shilatifard A. The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol Cell Biol. 2012a;32:2608–2617. doi: 10.1128/MCB.00182-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Lin C, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol. 2012b;13:543–547. doi: 10.1038/nrm3417. [DOI] [PubMed] [Google Scholar]
- Maroui MA, Calle A, Cohen C, Streichenberger N, Texier P, Takissian J, Rousseau A, Poccardi N, Welsch J, Corpet A, et al. Latency Entry of Herpes Simplex Virus 1 Is Determined by the Interaction of Its Genome with the Nuclear Environment. PLoS Pathog. 2016;12:e1005834. doi: 10.1371/journal.ppat.1005834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbonye U, Karn J. Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology. 2014;454–455:328–339. doi: 10.1016/j.virol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirguet O, Gosmini R, Toum J, Clement CA, Barnathan M, Brusq JM, Mordaunt JE, Grimes RM, Crowe M, Pineau O, et al. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J Med Chem. 2013;56:7501–7515. doi: 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- Nguyen VT, Kiss T, Michels AA, Bensaude O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001;414:322–325. doi: 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- Nilsson LM, Green LC, Muralidharan SV, Demir D, Welin M, Bhadury J, Logan DT, Walse B, Nilsson JA. Cancer Differentiating Agent Hexamethylene Bisacetamide Inhibits BET Bromodomain Proteins. Cancer Res. 2016;76:2376–2383. doi: 10.1158/0008-5472.CAN-15-2721. [DOI] [PubMed] [Google Scholar]
- Ott M, Geyer M, Zhou Q. The Control of HIV Transcription: Keeping RNA Polymerase II on Track. Cell Host Microbe. 2011;10:426–435. doi: 10.1016/j.chom.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou M, Sandri-Goldin RM. Inhibition of cdk9 during herpes simplex virus 1 infection impedes viral transcription. PLoS One. 2013;8:e79007. doi: 10.1371/journal.pone.0079007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Flores O, Umbach JL, Pesola JM, Bentley P, Rosato PC, Leib DA, Cullen BR, Coen DM. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe. 2014;15:446–456. doi: 10.1016/j.chom.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Ren K, Zhang W, Chen X, Ma Y, Dai Y, Fan Y, Hou Y, Tan RX, Li E. An Epigenetic Compound Library Screen Identifies BET Inhibitors That Promote HSV-1 and −2 Replication by Bridging P-TEFb to Viral Gene Promoters through BRD4. PLoS Pathog. 2016;12:e1005950. doi: 10.1371/journal.ppat.1005950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizman B, Knipe DM, Whitley RJ. Herpes Simplex Viruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6. Philadelphia: Lippincott Williams & Wilkins; 2013. pp. 2501–2601. [Google Scholar]
- Rosato PC, Leib DA. Neurons versus herpes simplex virus: the innate immune interactions that contribute to a host-pathogen standoff. Future Virol. 2015;10:699–714. doi: 10.2217/fvl.15.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell TA, Tscharke DC. Lytic Promoters Express Protein during Herpes Simplex Virus Latency. PLoS Pathog. 2016;12:e1005729. doi: 10.1371/journal.ppat.1005729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A, Lane WS, Jackson KW, Conaway RC, Conaway JW. An RNA polymerase II elongation factor encoded by the human ELL gene. Science. 1996;271:1873–1876. doi: 10.1126/science.271.5257.1873. [DOI] [PubMed] [Google Scholar]
- Smith RL, Pizer LI, Johnson EM, Jr, Wilcox CL. Activation of second-messenger pathways reactivates latent herpes simplex virus in neuronal cultures. Virology. 1992;188:311–318. doi: 10.1016/0042-6822(92)90760-m. [DOI] [PubMed] [Google Scholar]
- Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell. 2010;38:439–451. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Leger AJ, Hendricks RL. CD8+ T cells patrol HSV-1-infected trigeminal ganglia and prevent viral reactivation. J Neurovirol. 2011;17:528–534. doi: 10.1007/s13365-011-0062-1. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Parmely TJ, Sato S, Tomomori-Sato C, Banks CA, Kong SE, Szutorisz H, Swanson SK, Martin-Brown S, Washburn MP, et al. Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell. 2011;146:92–104. doi: 10.1016/j.cell.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo G, Liu G, Zhang J, Nesvizhskii AI, Gingras AC, Choi H. SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J Proteomics. 2014;100:37–43. doi: 10.1016/j.jprot.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi S, Chabes AL, Wysocka J, Herr W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol Cell. 2007;27:107–119. doi: 10.1016/j.molcel.2007.05.030. [DOI] [PubMed] [Google Scholar]
- Vogel JL, Kristie TM. The dynamics of HCF-1 modulation of herpes simplex virus chromatin during initiation of infection. Viruses. 2013;5:1272–1291. doi: 10.3390/v5051272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald A, Corey L. Persistence in the Population: epidemiology, transmissiion. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore MS, Roizman B, Whitley R, Yamanishi K, editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge UK: Cambridge University Press; 2007. [PubMed] [Google Scholar]
- Wang W, Yao X, Huang Y, Hu X, Liu R, Hou D, Chen R, Wang G. Mediator MED23 regulates basal transcription in vivo via an interaction with P-TEFb. Transcription. 2013;4:39–51. doi: 10.4161/trns.22874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlow Z, Kristie TM. Recruitment of the transcriptional coactivator HCF-1 to viral immediate-early promoters during initiation of reactivation from latency of herpes simplex virus type 1. J Virol. 2009;83:9591–9595. doi: 10.1128/JVI.01115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Yang Z, Zhu Q, Luo K, Zhou Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001;414:317–322. doi: 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- Yu HC, Sloan JL, Scharer G, Brebner A, Quintana AM, Achilly NP, Manoli I, Coughlin CR, Geiger EA, 2nd, Schneck U, et al. An X-linked cobalamin disorder caused by mutations in transcriptional coregulator HCFC1. Am J Hum Genet. 2013;93:506–514. doi: 10.1016/j.ajhg.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.