

Abstract
Trauma remains one of the leading causes of death worldwide. Traumatic injury disrupts immune system homeostasis and may predispose patients to opportunistic infections and inflammatory complications. Prevention of multiple organ dysfunction syndrome due to septic complications following severe trauma is a challenging problem. Following severe injury, the immune system usually tends toward a pro‐inflammatory phenotype and then changes to a counter‐inflammatory phenotype. This immune system homeostasis is believed to be a protective response based on the balance between the innate and adaptive immune systems. We reported that injury activates inflammasomes and primes Toll‐like receptors. The primed innate immune system is prepared for a rapid and strong antimicrobial immune defense. However, trauma can also develop the “two‐hit” response phenotype. We also reported that injury augments regulatory T cell activity, which can control the “two‐hit” response phenotype in trauma. We discuss the current idea that traumatic injury induces a unique type of innate and adaptive immune response that may be triggered by damage‐associated molecular pattern molecules, which are a combination of endogenous danger signal molecules that include alarmins and pathogen‐associated molecular pattern molecules.
Keywords: CARS, injury, macrophage, regulatory T cells (Tregs), SIRS
Introduction
Trauma is one of the leading causes of death worldwide.1 The timing of death following trauma is trimodal.2 The first phase of death after injury represents the immediate effects of trauma that result in death at the scene or within the first hour, and the causes of death are heart rupture, disruption of the cervical spine, or massive exencephaly. The second phase is death occurring within 24 h following injury, which may be due primarily to hemorrhagic shock. The third phase is late, with death usually occurring later than 1 week following the initial insult, and the cause of death in this phase is mainly infectious complications such as sepsis, septic shock, and multiple organ dysfunction syndrome (MODS). Sepsis occurs in approximately 10% of trauma patients and is associated with the severity of injury3 and with a significant increase in mortality compared with that in non‐septic patients.
Why do patients die of sepsis later than 1 week after injury? This is a question of how injury affects the immune system and predisposes the host to opportunistic infections and complications. Obviously, the immune system functions to protect the host from infection and also has a crucial role in controlling tissue injury and cell death in burns and trauma. The immune system is activated by damaged tissues and this can in turn activate specific types of responses that can enhance innate immune reactivity, control excessive pro‐inflammatory responses, and reduce continued tissue damage. Thus, trauma induces dynamic changes in the behavior of the immune system that can be classified as pro‐inflammatory and counter‐inflammatory. Given this general view, the pro‐inflammatory response is driven by the innate immune system, and the counter‐inflammatory response is regulated by the adaptive immune system.4, 5, 6, 7 These responses are clinically called systemic inflammatory response syndrome (SIRS), compensatory anti‐inflammatory response syndrome (CARS), and mixed anti‐inflammatory response syndrome.8
Injury initiates the immune system
The large amount of tissue injury induced by trauma releases various sorts of antigens and mediators. As these endogenous factors alert the immune system to the presence of danger, they are called alarmins. Alarmins interact with immune cells to initiate the inflammatory responses.9, 10, 11 An overview of trauma‐associated alarmins has already been reported.12 Figure 1 shows an overview of the initiation of the innate immune system following trauma. Alarmins are detected by pattern recognition receptors, such as Toll‐like receptors (TLRs).13 Pattern recognition receptors also bind exogenous antigens called pathogen‐associated molecular pattern molecules (PAMPs). This combination of endogenous (alarmins) and exogenous (PAMPs) danger signals comprises what are called the damage‐associated molecular pattern molecules. After trauma, patients are exposed to alarmins that activate the immune system to protect them from tissue injury and microbe invasion. However, a primed immune system could cause excessive inflammation called a “two‐hit” response phenotype, as discussed below (Fig. 2).
Figure 1.
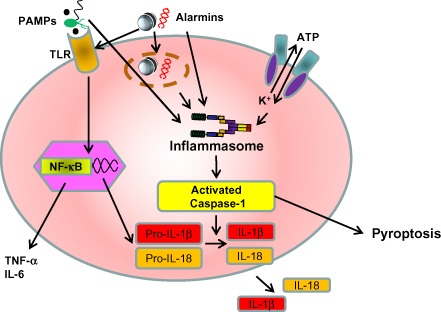
Immune system activation pathway following injury. The inflammasome is a large multiprotein complex that plays a key role in innate immunity by participating in the production of pro‐inflammatory cytokines such as interleukin (IL)‐1β and IL‐18. They are both produced as inactive precursors, pro‐IL‐1β and pro‐IL‐18, and share a common maturation mechanism that requires activated caspase‐1. The inflammasomes sense damage‐associated molecular patterns including pathogen‐associated molecular patterns (PAMPs) and the host‐derived signals known as alarmins. Cell death induced by inflammasome activation is known as pyroptosis. Alarmins also stimulate Toll‐like receptors (TLRs), which produce IL‐6 and tumor necrosis factor (TNF)‐α in response to injury. NF‐κB, nuclear factor‐κB.
Figure 2.
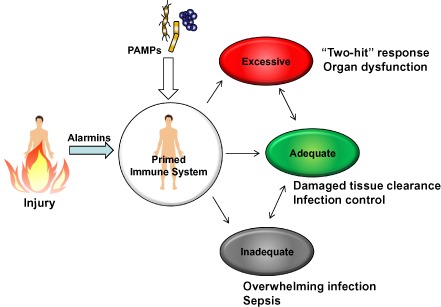
Injury primes immune system. After trauma, patients are exposed to alarmins that prime the immune system to protect them from tissue injury and microbe invasion. The primed immune system could trigger excessive inflammatory cascades that may develop multiple organ failure. This excessive inflammatory cascade is called “two‐hit” response. PAMPs, pathogen‐associated molecular patterns.
Pro‐inflammatory responses to injury
Systemic inflammatory response syndrome commonly follows traumatic injury.14, 15 Our group recently reported that injury activates the inflammasome pathway in injury‐site draining lymph nodes within 2 h, and then the activation spreads systemically. Inflammasome activation is seen predominantly in macrophages, as judged by caspase‐1 activation. Importantly, we found that blocking inflammasome activation worsens prognosis following injury. The cytokine profiles of injured mice with blocked inflammasome activation showed decreased interleukin (IL)‐1β levels and a marked increase in IL‐6.16 Interestingly, it has been described that injured patients who are not able to develop a febrile response after injury—an IL‐1β‐dependent physiological response—show worse outcome than patients who do develop a febrile response post‐injury.17 This clinical observation may be related to differences in the inflammasome activation pathway among these patients. Paterson et al. showed that immune cells display enhanced TLR reactivity following injury as early as 1 day after injury and persists for at least 7 days. This enhanced TLR reactivity is also seen predominantly in macrophages.18 In contrast, Zang et al. showed that T cells demonstrate pro‐inflammatory activity at early time points following injury, but the pro‐inflammatory T cell response is not seen at 7 days after injury.19 In fact, T cells show a reduced pro‐inflammatory phenotype and higher counter‐inflammatory cytokine production phenotype by 7 days after injury. Based on these observations, we contend that the pro‐inflammatory immune response is driven predominantly by macrophages following trauma and the counter‐inflammatory response is mediated by T cells. The beneficial and harmful phenotypes of inflammatory responses to injury are illustrated in Figure 3.
Figure 3.
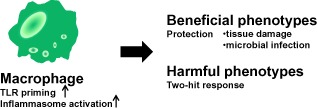
Inflammatory responses to injury. Inflammatory responses to injury are driven predominantly by macrophages. Inflammasomes and Toll‐like receptors (TLRs) are activated by alarmins. The inflammatory responses have crucial roles in the protection of the injured host, such as clearing damaged tissue and eliminating infected microbiomes. Excessive inflammation can cause a two‐hit response.
Beneficial phenotype of pro‐inflammatory responses to trauma
A theoretical reason why pro‐inflammatory responses develop after trauma is that it functions to protect the host from secondary infections by heightening the antimicrobial immunity. Accordingly, Maung et al. showed that trauma induces enhanced resistance to Escherichia coli infections. The enhanced resistance was seen as early as 1 day after injury, and maximal resistance was observed at days 7 and 14.20 Southard et al. also reported enhanced antimicrobial immunity in mice exposed to a pulmonary contusion model.21 These findings support the tenet that heightened innate immune system reactivity after trauma can boost the antimicrobial defense mechanisms. Clinical support for this conclusion comes from a study showing that patients with minor injuries do not require antimicrobial treatments,22 suggesting that the mammalian immune system works to adequately protect the host from microbial invasions.
Harmful phenotype of pro‐inflammatory responses to trauma
The two‐hit response is among one of the serious complications of trauma. The definition of this phenomenon, first defined by Moore et al.,4 is that “traumatic injury can prime the host such that a later, otherwise innocuous, secondary inflammatory insult precipitates an exaggerated systemic inflammatory response.” Research findings that showed that TLR reactivity is enhanced after trauma supports that the two‐hit response phenotype may be mediated by enhanced TLR reactivity by the innate immune system, primarily macrophages and neutrophils. These innate immune cells become hyper‐reactive to bacteria and bacterial toxins to produce high levels of cytokines, which can trigger a secondary SIRS‐like response and MODS.18, 23, 24
Counter‐inflammatory responses to injury
Counter‐inflammatory responses are believed to act as a natural compensatory host response to trauma‐induced inflammation, much like a negative feedback signaling network in cells. Researchers have shown that major injury leads to diminished resistance to infection due to the development of a physiological response to trauma called CARS.7, 25, 26 There are a number of observations to suggest that CARS is mediated primarily by the adaptive immune system and, in particular, by T cells. These include the findings that severely injured patients show reduced delayed‐type hypersensitivity responses, prolonged skin allograft survival, and reduced T cell proliferation to polyclonal and specific recall antigen stimulation.27, 28, 29 Moreover, T helper 1 (Th1)‐type immune responses are reduced after trauma,30, 31, 32 and Th2‐type immune responses are promoted.33, 34 In one study, Kelly et al. showed that Th1 antibody responses to antigen immunization and antigen‐specific Th1‐type cytokines were markedly suppressed in burn‐injured mice.31 Another study by Guo et al. used an adoptive transfer approach to demonstrate that antigen‐specific CD4 T cells showed suppressed antigen‐driven expansion and Th1 cytokine production without an increase in Th2‐type responses.35 We summarize potential beneficial and harmful influences of the T‐cell‐mediated counter‐inflammatory responses to injury in Figure 4.
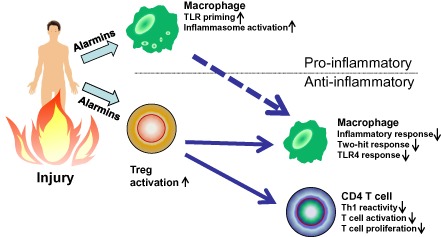
Figure 4.

Counter‐inflammatory responses to injury. Regulatory T cell activation controls Toll‐like receptor (TLR) reactivity and T helper 1 (Th1)‐type responses and T cell proliferation to protect the injured host from the two‐hit response and excessive inflammation as a main regulator. Counter‐inflammatory responses may predispose the injured host to opportunistic infections.
Beneficial phenotype of counter‐inflammatory responses to trauma
If the development of a counter‐inflammatory response to injury is a programmed or evolutionarily conserved response by the immune system, then we should consider what the purpose of this type of an immune response to trauma might be. The answer may be that it acts as a damage control mechanism to limit excessive inflammation following injury. As we found that T cells appear to mediate the counter‐inflammatory response to injury, we focused our research efforts towards a role for a subset of CD4 T cells, called regulatory T cells (Tregs). The reasoning behind this is that Tregs are known to tolerate and control inflammation in autoimmune and inflammatory diseases,36, 37 and Tregs have the ability to produce transforming growth factor‐β1 and IL‐10, both of which can suppress immune responses. Subsequently, Ni Choileain et al. showed that Tregs from injured mice could more effectively block CD4 T cell proliferation than Tregs from sham mice.38 They also found that Tregs could actively suppress Th1‐type reactivity in immunized mice.38, 39 Furthermore, Hanschen et al. provided direct evidence that injury activates CD4+ Tregs as early as 15 min after burn injury in lymph nodes draining the injury site.40 This simple observation suggests that Tregs may specifically react to injury or damaged tissues.
As mentioned above, Zang et al.19 showed that a high‐level of T cell activation induced by the injection of bacterial superantigen into mice at early time points after trauma led to high mortality, whereas Zang et al. and Carretto et al. found that there was no mortality when injured mice were challenged with superantigen at 1 week after the injury.41 These observations provide in vivo evidence that T cell reactivity shows a phenotypic shift towards a counter‐inflammatory and potentially beneficial immune system phenotype. The clinical relevance of these observations is that superantigens could be released by Gram‐positive cocci to trigger toxic shock syndrome in burn patients between 2 and 4 days after injury.42, 43 Considering the time point when Treg numbers are increased and that Tregs can potently suppress CD4 T cell activation, Treg activation might help control toxic shock syndrome following injury. Regulatory T cells may also control innate inflammatory responses to injury. Maung et al. found that Treg‐deficient injured mice were significantly more susceptible to lipopolysaccharide challenge: Treg‐deficient injured mice showed 100% mortality, whereas wild‐type injured mice showed 50% mortality.44 These findings suggest that Tregs may also actively control the severity of the two‐hit response phenotype following trauma.
Harmful phenotype of counter‐inflammatory responses to trauma
A complication of the counter‐inflammatory response to injury is that it can actively suppress antimicrobial immunity and may be responsible for the increased susceptibility of trauma patients to opportunistic infections that often occur during the counter‐inflammatory phase of the injury response. Although there is no known clinical relationship between the development of post‐injury sepsis and altered Treg numbers or function, there is evidence that injury and sepsis can influence Tregs in patients. MacConmara et al.45 reported that circulating Tregs from trauma patients demonstrate enhanced Treg activity by 5–7 days after injury compared with Tregs prepared from patients at 1 day after injury or from normal individuals. Another report showed that septic patients develop higher numbers of circulating Tregs.46 These findings suggest that enhanced Treg presence or responsiveness may also be part of the host response to sepsis and strong inflammatory responses. Further research studies are needed to reveal whether modulating Tregs might be a way to protect trauma patients from post‐injury infections, sepsis, or systemic inflammatory complications.
Conclusions
How the immune system responds to traumatic injuries is a complex host response that has undergone evolutionary changes and adaptation similar to other types of immune system responses. Following injury, cells and mediators of the innate and adaptive immune systems undergo temporal changes that have been categorized into pro‐inflammatory and counter‐inflammatory immune responses. Figure 5 illustrates that the pro‐inflammatory response to injury is driven by the innate immune system such as inflammasome and TLR activation predominantly in macrophages and that the counter‐inflammatory response is regulated by the adaptive immune system, mainly by Tregs. In this review, we presented and discussed that the immune response to injury involves a series of programmed responses that may act to protect the injured host from infections and excessive inflammatory responses. Data support that a compartmentalized infection can be controlled effectively by the innate immune system, but if injuries or infections cannot be controlled, the immune system develops a wider imbalance and further departure from immune system homeostasis (Fig. 6). We believe that restoring immune homeostasis could protect patients from developing opportunistic infections and systemic complications like sepsis syndrome or organ failure. Thus, we propose that future basic and pre‐clinical research should be directed at advancing our understanding of how trauma disrupts immune system homeostasis and at therapeutic approaches with potential to restore immune system homeostasis to help reduce clinical complications in trauma patients.
Figure 5.
Interaction between immune cell subsets in reaction to injury. Injury‐induced tissue damage releases alarmins, which can activate macrophages and regulatory T cells (Tregs). In macrophages, injury triggers a number of changes in phenotype and function, including enhanced Toll‐like receptor 4 (TLR4) reactivity and antimicrobial responses. Regulatory T cells are activated and have been shown to act as “master regulators” of the injury response by suppressing both innate (macrophage) and adaptive (T cell) cellular responses to trauma. Th1, T helper 1.
Figure 6.
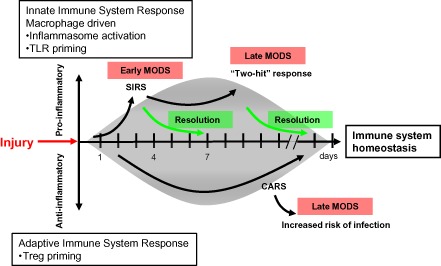
Traumatic injury disrupts normal immune system homeostasis. Injury disrupts immune system homeostasis and leads to the development of systemic inflammatory response syndrome (SIRS) and compensatory anti‐inflammatory response syndrome (CARS) in trauma patients. The pro‐inflammatory response is driven by the innate immune system and the anti‐inflammatory response is regulated by the adaptive immune system. Soon after injury, inflammasomes are activated and Toll‐like receptors (TLRs) are primed predominantly in macrophages, and at the same time, regulatory T cells (Tregs) are also primed. Once a pro‐inflammatory phenotype is overexpressed, the injured host can develop the “two‐hit” response phenotype, which may lead to multiple organ dysfunction syndrome (MODS). However, if the counter‐inflammatory response is overexpressed, the injured host will be at high risk of developing trauma‐associated complications such as sepsis, septic shock, or MODS.
Conflict of Interest
None.
References
- 1. World Health Organization . The global burden of disease [database on the Internet]. 2013. [Cited 19 Aug 2013]. Available from URL: http://www.who.int/healthinfo/global_burden_disease/en/
- 2. Trunkey DD. Trauma. Accidental and intentional injuries account for more years of life lost in the U.S. than cancer and heart disease. Among the prescribed remedies are improved preventive efforts, speedier surgery and further research. Sci. Am. 1983; 249: 28–35. [PubMed] [Google Scholar]
- 3. Wafaisade A, Lefering R, Bouillon B et al Epidemiology and risk factors of sepsis after multiple trauma: an analysis of 29,829 patients from the Trauma Registry of the German Society for Trauma Surgery. Crit. Care Med. 2011; 39: 621–628. [DOI] [PubMed] [Google Scholar]
- 4. Moore FA, Moore EE, Read RA. Postinjury multiple organ failure: role of extrathoracic injury and sepsis in adult respiratory distress syndrome. New Horiz. 1993; 1: 538–549. [PubMed] [Google Scholar]
- 5. Baue AE, Durham R, Faist E. Systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), multiple organ failure (MOF): are we winning the battle? Shock 1998; 10: 79–89. [DOI] [PubMed] [Google Scholar]
- 6. Baue AE. MOF, MODS, and SIRS: what is in a name or an acronym? Shock 2006; 26: 438–449. [DOI] [PubMed] [Google Scholar]
- 7. Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit. Care Med. 1996; 24: 1125–1128. [DOI] [PubMed] [Google Scholar]
- 8. Osuchowski MF, Welch K, Siddiqui J, Remick DG. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J. Immunol. 2006; 177: 1967–1974. [DOI] [PubMed] [Google Scholar]
- 9. Matzinger P. The danger model: a renewed sense of self. Science 2002; 296: 301–305. [DOI] [PubMed] [Google Scholar]
- 10. Harris HE, Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. 2006; 7: 774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr. Opin. Immunol. 2005; 17: 359–365. [DOI] [PubMed] [Google Scholar]
- 12. Manson J, Thiemermann C, Brohi K. Trauma alarmins as activators of damage‐induced inflammation. Br. J. Surg. 2012; 99(Suppl. 1): 12–20. [DOI] [PubMed] [Google Scholar]
- 13. Matzinger P. An innate sense of danger. Ann. N. Y. Acad. Sci. 2002; 961: 341–342. [DOI] [PubMed] [Google Scholar]
- 14. Ogura H, Tanaka H, Koh T et al Priming, second‐hit priming, and apoptosis in leukocytes from trauma patients. J. Trauma 1999; 46: 774–781, discussion 81–3. [DOI] [PubMed] [Google Scholar]
- 15. Hoover L, Bochicchio GV, Napolitano LM et al Systemic inflammatory response syndrome and nosocomial infection in trauma. J. Trauma 2006; 61: 310–316, discussion 6–7. [DOI] [PubMed] [Google Scholar]
- 16. Osuka A, Hanschen M, Stoecklein V, Lederer JA. A protective role for inflammasome activation following injury. Shock 2012; 37: 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mizushima Y, Ueno M, Idoguchi K, Ishikawa K, Matsuoka T. Fever in trauma patients: friend or foe? J. Trauma 2009; 67: 1062–1065. [DOI] [PubMed] [Google Scholar]
- 18. Paterson HM, Murphy TJ, Purcell EJ et al Injury primes the innate immune system for enhanced Toll‐like receptor reactivity. J. Immunol. 2003; 171: 1473–1483. [DOI] [PubMed] [Google Scholar]
- 19. Zang Y, Dolan SM, Ni Choileain N et al Burn injury initiates a shift in superantigen‐induced T cell responses and host survival. J. Immunol. 2004; 172: 4883–4892. [DOI] [PubMed] [Google Scholar]
- 20. Maung AA, Fujimi S, MacConmara MP et al Injury enhances resistance to Escherichia coli infection by boosting innate immune system function. J. Immunol. 2008; 180: 2450–2458. [DOI] [PubMed] [Google Scholar]
- 21. Southard R, Ghosh S, Hilliard J et al Pulmonary contusion is associated with toll‐like receptor 4 upregulation and decreased susceptibility to pseudomonas pneumonia in a mouse model. Shock 2012; 7: 629–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grossman JA, Adams JP, Kunec J. Prophylactic antibiotics in simple hand lacerations. JAMA 1981; 245: 1055–1056. [PubMed] [Google Scholar]
- 23. Fan J, Li Y, Levy RM et al Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1‐TLR4 signaling. J. Immunol. 2007; 178: 6573–6580. [DOI] [PubMed] [Google Scholar]
- 24. Fan J, Kapus A, Li YH, Rizoli S, Marshall JC, Rotstein OD. Priming for enhanced alveolar fibrin deposition after hemorrhagic shock: role of tumor necrosis factor. Am. J. Respir. Cell Mol. Biol. 2000; 22: 412–421. [DOI] [PubMed] [Google Scholar]
- 25. Bone RC. Immunologic dissonance: a continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS). Ann. Intern. Med. 1996; 125: 680–687. [DOI] [PubMed] [Google Scholar]
- 26. Sauaia A, Moore FA, Moore EE, Lezotte DC. Early risk factors for postinjury multiple organ failure. World J. Surg. 1996; 20: 392–400. [DOI] [PubMed] [Google Scholar]
- 27. Ninnemann JL, Fisher JC, Frank HA. Prolonged survival of human skin allografts following thermal injury. Transplantation 1978; 25: 69–72. [DOI] [PubMed] [Google Scholar]
- 28. Lederer JA, Rodrick ML, Mannick JA. The effects of injury on the adaptive immune response. Shock 1999; 11: 153–159. [DOI] [PubMed] [Google Scholar]
- 29. Faist E, Schinkel C, Zimmer S. Update on the mechanisms of immune suppression of injury and immune modulation. World J. Surg. 1996; 20: 454–459. [DOI] [PubMed] [Google Scholar]
- 30. De AK, Kodys KM, Pellegrini J et al Induction of global anergy rather than inhibitory Th2 lymphokines mediates posttrauma T cell immunodepression. Clin. Immunol. 2000; 96: 52–66. [DOI] [PubMed] [Google Scholar]
- 31. Kelly JL, O'Suilleabhain CB, Soberg CC, Mannick JA, Lederer JA. Severe injury triggers antigen‐specific T‐helper cell dysfunction. Shock 1999; 12: 39–45. [DOI] [PubMed] [Google Scholar]
- 32. Murphy T, Paterson H, Rogers S, Mannick JA, Lederer JA. Use of intracellular cytokine staining and bacterial superantigen to document suppression of the adaptive immune system in injured patients. Ann. Surg. 2003; 238: 401–410, discussion 10–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. O'Sullivan ST, Lederer JA, Horgan AF, Chin DH, Mannick JA, Rodrick ML. Major injury leads to predominance of the T helper‐2 lymphocyte phenotype and diminished interleukin‐12 production associated with decreased resistance to infection. Ann. Surg. 1995; 222: 482–490, discussion 90–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mack VE, McCarter MD, Naama HA, Calvano SE, Daly JM. Candida infection following severe trauma exacerbates Th2 cytokines and increases mortality. J. Surg. Res. 1997; 69: 399–407. [DOI] [PubMed] [Google Scholar]
- 35. Guo Z, Kavanagh E, Zang Y et al Burn injury promotes antigen‐driven Th2‐type responses in vivo. J. Immunol. 2003; 171: 3983–3990. [DOI] [PubMed] [Google Scholar]
- 36. Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol. Med. 2007; 13: 108–116. [DOI] [PubMed] [Google Scholar]
- 37. Sakaguchi S, Ono M, Setoguchi R et al Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self‐tolerance and autoimmune disease. Immunol. Rev. 2006; 212: 8–27. [DOI] [PubMed] [Google Scholar]
- 38. Ni Choileain N, MacConmara M, Zang Y, Murphy TJ, Mannick JA, Lederer JA. Enhanced regulatory T cell activity is an element of the host response to injury. J. Immunol. 2006; 176: 225–236. [DOI] [PubMed] [Google Scholar]
- 39. MacConmara MP, Tajima G, O'Leary F et al Regulatory T cells suppress antigen‐driven CD4 T cell reactivity following injury. J. Leukoc. Biol. 2011; 89: 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanschen M, Tajima G, O'Leary F, Ikeda K, Lederer JA. Injury induces early activation of T‐cell receptor signaling pathways in CD4+ regulatory T cells. Shock 2011; 35: 252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Caretto D, Katzman SD, Villarino AV, Gallo E, Abbas AK. Cutting edge: the Th1 response inhibits the generation of peripheral regulatory T cells. J. Immunol. 2010; 184: 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frame JD, Eve MD, Hackett ME et al The toxic shock syndrome in burned children. Burns Incl. Therm. Inj. 1985; 11: 234–241. [DOI] [PubMed] [Google Scholar]
- 43. McAllister RM, Mercer NS, Morgan BD, Sanders R. Early diagnosis of staphylococcal toxaemia in burned children. Burns 1993; 19: 22–25. [DOI] [PubMed] [Google Scholar]
- 44. Maung AA, Fujimi S, Miller ML, MacConmara MP, Mannick JA, Lederer JA. Enhanced TLR4 reactivity following injury is mediated by increased p38 activation. J. Leukoc. Biol. 2005; 78: 565–573. [DOI] [PubMed] [Google Scholar]
- 45. MacConmara MP, Maung AA, Fujimi S et al Increased CD4+ CD25+ T regulatory cell activity in trauma patients depresses protective Th1 immunity. Ann. Surg. 2006; 244: 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Monneret G, Debard AL, Venet F et al Marked elevation of human circulating CD4+CD25+ regulatory T cells in sepsis‐induced immunoparalysis. Crit. Care Med. 2003; 31: 2068–2071. [DOI] [PubMed] [Google Scholar]