

Abstract
In addition to pathogen‐associated molecular patterns from invasive microorganisms, alarmins, which are major components of host defense mechanisms, are involved in the pathophysiology of sepsis. In fact, the magnitude of the insult is defined according to the damage‐associated molecular pattern (DAMP), which is composed of alarmins as well as pathogen‐associated molecular patterns, such as those involving nucleosomes, histones, and DNA. Regarding the antimicrobial mechanism of neutrophils, an alternative non‐phagocytic mechanism was first recognized as “NETosis” in 2004. In this mechanism, microorganisms are trapped and eliminated by neutrophil extracellular traps (NETs). These NETs are composed of histones and DNA that have been expelled from the nucleus as well as antimicrobial proteases, including elastase and myeloperoxidase. NETosis, a cell death pathway reported to be distinct from apoptosis, is an active area of research. As NETs are composed of deleterious substances, they are extremely harmful to the host cells once they are released into the circulating blood. Therefore, the meanings and putative roles of these components in sepsis have attracted much attention.
Keywords: Alarmin, histone, innate immune system, necrosis, neutrophil
Introduction
Excessive inflammatory responses, part of the host defense mechanisms, play important roles in the worsening of the clinical condition in sepsis.1 Research in this area had been focused on microorganisms and their related pathogenic substances named pathogen‐associated molecular patterns (PAMPs) and the host immune response.2 Pathogen‐associated molecular patterns are the exogenous substances associated with pathogens, PAMPs which are recognized by particular receptors, then initiate the production of immune‐related proteins such as pro‐inflammatory cytokines through activation of intracellular signaling.3 In the next stage, the responses of the damaged host cells, including subsequent release of the high mobility group box 1 (HMGB1) and heat shock proteins, have attracted attention. These endogenous proteins that exacerbate the damage became known as “alarmins”.4 According to the European Molecular Biology Organization, alarmins are defined as: (i) being rapidly released by cells that have undergone necrosis; (ii) activating innate immune responses through ligation to receptors; (iii) actively secreted by activated immune cells; (iv) endogenous substances that are associated with repair of tissues damaged by invasion and inflammation, and with maintenance of homeostasis. Additionally, there is increasing interest in sepsis‐associated cell death‐related nucleic substances, such as DNA and histones and, furthermore, in inflammatory mediators from organelles in the cytosol, such as mitochondrial DNA and ATP. Together with alarmins and PAMPs, these substances are known as damage‐associated molecular patterns (DAMPs), and are an intensive focus of research.5, 6 The term “DAMPs” was formerly used in a similar way to alarmins, to mean “danger‐associated molecular patterns”,7 but the meaning was later changed so that it is now used to represent PAMPs, alarmins, and other inflammatory initiators.4
A significant proportion of substances released as necrosis of the host cell takes place promptly ligate to their receptors, named pattern recognition receptors (PRRs).8 A curious point to make is that these components of necrotic cells share the same receptors as those for PAMPs. Neutrophils are particularly important in this case because neutrophil cell death leads to the amplification of inflammation. In contrast to the well understood cell death types apoptosis and necrosis (oncosis), a unique type of cell death known as “NETosis”, which involves the release of neutrophil extracellular traps (NETs),9 has become well publicized. NETosis plays an important role in the removal of pathogens, but when cellular components that have antimicrobial effects, such as histones, myeloperoxidase, and elastase, are dumped into the circulation, they are also harmful to the host cells. This review will focus on the actions of neutrophils during bacterial invasion of the body, including recent findings on NETs, DAMPs, and cell death.
Neutrophil extracellular traps
Neutrophil extracellular traps, released by neutrophils, were first described by Brinkmann et al. in 2004.9 Neutrophils have an important role in the first line of defense against invading microorganisms. Phagocytosis is well known as the process by which neutrophils remove pathogens. Aside from this, a further mechanism named NETosis has become accepted. This is the process whereby NETs are expelled extracellularly and microorganisms are removed through contact with these NETs, which is a network of chromatins (DNA) attached to bactericidal nucleic proteins such as histones, myeloperoxidase, and elastase (Figs. 1, 2).10 Such expulsion of extracellular traps (ETs) was later found to be not limited to neutrophils, with plasma cells, eosinophils, and macrophages behaving in a similar way, and is becoming known by the general term ETosis.11 Alongside pathogens such as bacteria and fungi, PAMPs that originate from pathogen‐derived lipopolysaccharides (LPS) and peptidoglycans, polysaccharides, lipids, nucleic acids, and proteins, are known triggers of NETosis. Additionally, chemicals, phorbol myristate acetate (PMA) for example, are also known to evoke the same response.12 An interesting fact is that receptors for PAMPs, known as PRRs, are common as the receptors for the substances from necrotic cells including nucleosomes and HMGB1, which trigger NETosis.13 As an enormous amount of such stimulating factors are released from the dead cells, it is estimated that they can cause the induction of NETosis.14 With regards to intracellular signaling in NETosis, activation of the Raf/MEK/ERK pathway and protein kinase C is reported,15, 16 but we are yet to uncover the whole picture.
Figure 1.
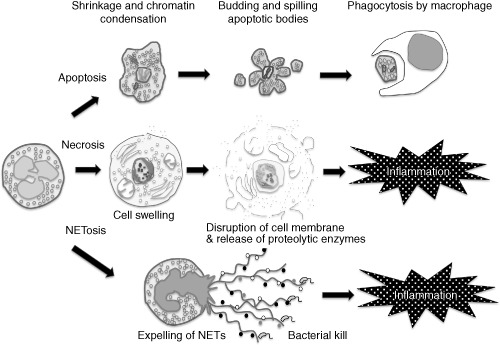
Apoptosis, necrosis, and formation of neutrophil extracellular traps (NETs). Apoptotic neutrophils finally form apoptotic bodies and are phagocytosed by macrophages without eliciting inflammation. In contrast, necrotic neutrophils cause inflammation by releasing proteolytic enzymes. The other form of cell death is NETosis. Neutrophils expose NETs, formed by DNA, histone, and granules, and cause inflammation.
Figure 2.
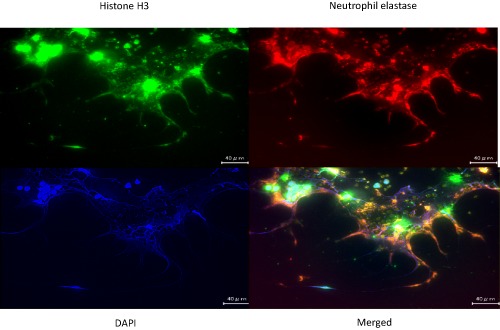
Representative images showing fluorescent immunostaining of neutrophil extracellular traps (NETs). Neutrophils from a healthy volunteer were treated with phorbol myristate acetate for 4 h, then fixed with 4% formalin on the cover glass, and stained with three different colors: green, anti‐histone H3 monoclonal antibody; blue, DNA labeled with 4′,6‐diamidino‐2‐phenylindole (DAPI); red, anti‐elastase monoclonal antibody (objective: ×40). Scale bar = 40 μm. Original data.
The process of NETosis is a powerful means of disposing of pathogens; however, the damage to host cells is also significant, in particular on the vascular endothelium and on pneumocytes.17 Granular proteins such as myeloperoxidase and elastase, and nucleic substances such as histones and nucleosomes are thought to be some of the harmful factors in NETs.
There does not appear to be any organ specificity in the pathogenesis of NETs, but there have been many reports focusing on the lungs, where neutrophils are physiologically highly distributed. In an LPS‐induced acute lung injury model, it was reported that the formation of NETs within the alveoli and bronchoalveolar lavage fluid correlates with the finding of acute lung injury.18 However, based on the total number of neutrophils, evidence suggests that NETosis occurs in only a low proportion of cells, estimated at 10–30%.19
Histones
Histones are the main DNA‐binding proteins in chromatins, and they account for the majority of nucleic proteins. Histones hold DNA together in a double helix structure; traditionally, efficient packing in the nucleus was thought to be the sole function of histones. However, the function of histones expelled extracellularly has attracted much attention in recent years. As introduced in the previous section, histones, along with fragmented DNA, are the main components of NETs. It has been shown that they have a powerful ability to induce cell death and activation in coagulation, which contribute to the host defense mechanism.20 In contrast, histones released into the bloodstream also have an innately damaging effect on tissues (Fig. 3, Table 1).
Figure 3.
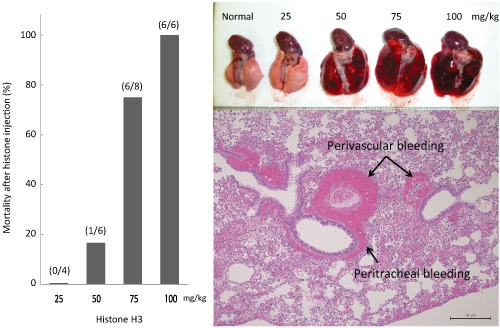
Mortality and histological changes after histone injection. Mice were treated with intravascular injection of different doses of histone H3. As a result, a dose‐dependent increase in mortality was recognized (left panel). Mice were killed 3 h after treatment, and macroscopic findings of the lung showed massive bleeding and edema in a dose‐dependent manner (top right). A representative microscopic image of the lung from animal number 12 shows significant bleeding surrounding the trachea and vessels at 3 h after histone H3 injection (bottom right). Original data.
Table 1.
Histological findings in mice after treatment with histone H3
| Animal number | Control | Histone H3 (25 mg/kg) | Histone H3 (50 mg/kg) | Histone H3 (75 mg/kg) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | |
| Lung | |||||||||||||||
| Peri‐vascular and trachea edema | − | − | − | − | − | − | − | + | ± | ± | + | + | + | + | + |
| Alveolar and interstitial bleeding | − | − | − | − | − | − | − | − | − | ± | + | ± | + | + | + |
| Liver | |||||||||||||||
| Ballooning and vacuolization | − | − | − | − | − | − | − | − | − | − | − | − | + | − | ++ |
| Heart | |||||||||||||||
| − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| Kidney | |||||||||||||||
| Damaged renal tubule | − | − | − | − | − | − | − | − | − | − | − | + | − | − | + |
| Spleen | |||||||||||||||
| − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| Brain | |||||||||||||||
| Congestion | − | − | − | − | − | − | − | − | − | − | − | + | − | − | − |
Microscopic findings in different organs were examined in 16 mice. Edema and bleeding were recognized 3 h after histone H3 injection, in a dose‐dependent manner. These pathologic changes were seen in lung, liver, kidney, and brain, and the changes were most prominent in the lung. −, No change; ±, very mild change; +, mild change; ++, moderate change. Original data.
According to the report by Xu et al.:21 (i) of the core histones, a highly damaging effect on endothelial cells is seen with histones H3 and H4; (ii) when histones are injected intravenously into mice, the accumulation of neutrophils is intense in the lungs, and peri‐bronchoalveolar bleeding and thrombus formation is observed; (iii) when antibody for H4 is given, such damages are minimized; (iv) activated protein C (APC) can degrade histones H3 and H4 and minimize tissue damage, thereby improving survival rates. Semeraro et al.22 reported that histones released from necrotic cells advance thrombus formation in the microvasculature through platelet activation. The same group also reported that histones activate coagulation in a dose‐dependent manner through the inhibition of thrombomodulin‐mediated activation of protein C.23 From these findings, it can be suggested that neutrophils and coagulation play important roles in the amplification of inflammation, and that histones are also important as mediators at the point of microbial invasion. In addition, the report that APC reduces histone‐induced cytotoxicity reemphasizes the importance of the anticoagulant therapy.24 With regard to the connection between invasion and coagulation, it is interesting that the strong coagulative action of histones25 is controlled by the physiological antithrombotic factor, APC.
Toll‐like receptors 2 and 4, typical of PRRs, are hypothesized to act as receptors for histones26 but again this requires a greater amount of information. Research into therapies that target histones have included the use of a synthetic histone mimic,27 attempts to acetylate histones26 or inhibit deacetylation28 in a sepsis model, and research into the role of APC in histone reduction.21
However, there is still a significant limitation to the research in this area, in that there is no reliable procedure to assay the plasma levels of histones. For example, in acute injury, we know that histones are first expelled into the circulation through NETosis and later spilled out from necrotic and apoptotic cells, but the process is yet to be clarified. There are some recent reports that quantify of nucleosomes, which will be reviewed in the next section, refers to circulating histones levels29 but, strictly speaking, this is incorrect. Hence, it would be desirable to find an assay procedure to determine the histone levels in the circulating blood so that their behavior may be elucidated.
Nucleosomes and DNA
Pathogen‐derived DNA and nucleosomes composed of DNA and histones are PAMPs that induce inflammation.30 On a separate point, nucleosomes and DNA released into the circulating blood after host cell death also contribute to the pathogenesis of sepsis as DAMPs.31 The significance of such DAMPs and their usefulness as a severity indicator are points of recent interest.32, 33, 34 The explanations for the suitability of nucleosomes and DNA as severity indicators are as follows. In the absence of microbial invasion, nucleic substances with pro‐inflammatory effects are expelled into the blood by means of cellular apoptosis and are metabolized efficiently giving a short half‐life, thereby keeping their levels low. By contrast, in the event of a serious illness, such a vast number of cells undergo necrosis and apoptosis that it exceeds the clearance capacity, leading to a rise in plasma levels, reflecting the degree of insult.35 However, as both apoptosis and necrosis result in the separation of DNA and nucleosomes into blood, it is necessary to bear in mind that they cannot be distinguished when using them as damage markers. Aside from this, in the LPS‐induced sepsis model, when nucleosomes and HMGB1 were measured 3 h following injection of LPS, a rise in blood levels was detected. At this early stage, indicators of organ failure such as liver enzymes are unchanged, hence in the early stages following microbial invasion, it may be possible to use the elevated levels of nucleosomes and HMGB1 as an indicator of NETosis (Fig. 4).
Figure 4.
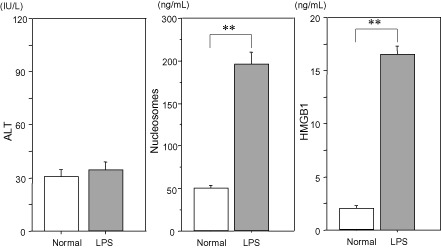
Changes in circulating alanine aminotransferase ALT, nucleosomes, and high mobility group box 1(HMGB1), after lipopolysaccharide (LPS) injection. Plasma levels of ALT, nucleosomes, and HMGB1 were measured 3 h after LPS (8 mg/kg) injection. As cell death usually becomes evident several hours after the insult, significant increases were recognized in nucleosomes and HMGB1 but not in ALT. Therefore, the increases are thought to be mainly from the result of cell death by neutrophil extracellular traps (NETosis). Original data.
Approximately 99% of the circulating DNA following invasion is thought to be derived from the nucleus,31 but in plasma, mitochondria‐derived DNA also exists. Calfee et al.36 reported that, as mitochondria are originally derived from bacteria and as they are taken into, and co‐exist with, the host cells, once they are released into the bloodstream after cell death, they act in a similar way to PAMPS. Indeed, mitochondrial DNA brings about a powerful inflammatory response. Mitochondrial DNA has also been reported as a severity indicator,37 but there can be faults in the accuracy of such data and careful evaluation is required.
Cell death
In the past, two types of cell death, apoptosis and necrosis, were recognized. Apoptosis is a type of “programmed cell death” where the cell shrinks following condensation of the nucleus. DNA is fragmented into the size of nucleosomes and finally enclosed in apoptotic bodies alongside organelles and is phagocytosed by macrophages (Fig. 1). As neutrophils contain a large amount of substances that damage tissues and have a particularly short lifespan, they must be disposed of through apoptosis to avoid spreading inflammation to their surroundings. The anti‐inflammatory effect of the neutrophils undergoing apoptosis is a point of relevant interest. According to the report by Byrne and Reen,38 macrophages that have phagocytosed apoptotic neutrophils produce anti‐inflammatory cytokines such as interleukin (IL)‐10 and transforming growth factor‐β and inhibit local inflammation.
In contrast, necrosis is a type of “unprogrammed death”, where the detoxification of DAMPs‐forming organelles, chromatin and mitochondria, is delayed behind the opening of the membrane channels, which allows calcium ions to influx into the cell leading to the activation of calpains, which is characteristic of necrosis. Following this event, extracellular fluid flows into the cell after breakdown of the cell membrane,39 lysosomal contents also leak out resulting in cellular swelling and local inflammation7 (Fig. 1). Similarly with neutrophils, when they undergo necrosis, inflammatory mediators are also released, bringing about localized inflammatory changes.
In recent years, apart from apoptosis and necrosis, new concepts of cell death such as autophagy and pyroptosis have been proposed.40 Autophagy, where the cell is broken down by lysosomes, is similar to apoptosis in that they are both types of “programmed cell death”, but it is thought that autophagy induces local inflammation.41
Under stress, many types of cell death occur including apoptosis, necrosis, autophagy, and with neutrophils, NETosis, but also their intermediate subtypes also exist. Furthermore, it is known that, with time, complex situations may arise such as those cells under the apoptotic pathway changing to necrosis. With such types of cell death a variety of inflammatory mediators are released into the blood and circulated, contributing to the pathogenesis of sepsis.42 Cell death is seen in all cells but in this review, a specific focus has been placed on the death of neutrophils as they react most quickly in response to an invasion and are closely related to the pathogenesis of acute illness. Neutrophils are released into the blood after having matured in the bone marrow for a few days, then after circulating for approximately 24 h they move to tissues. Following 24–48 h in the tissue they finally undergo apoptosis and are disposed of by macrophages and the reticuloendothelial system.43 The lifespan of neutrophils is tightly controlled through the balance of apoptotic and anti‐apoptotic mechanisms probably to prevent the local inflammatory response as a result of unexpected neutrophil death.44 Cytokines such as IL‐1β, IL‐2, IL‐4, IL‐15, interferon‐γ, and granulocyte colony‐stimulating factor are known to play a part in determining the lifespan of neutrophils, but an interesting fact is that PAMPs, such as LPS, also delay apoptosis.45 In addition, apoptosis of neutrophils is induced by Fas ligand, tumor necrosis factor‐α, or tumor necrosis factor‐related apoptosis‐inducing ligand,46 and also by the process of phagocytosis of pathogens, reported as phagocytosis‐induced cell death.47 In this way, it is considered the rational option to dispose of those pathogens that can be dealt with by phagocytosis alone, by apoptosis; where there are too many pathogens, such that disposal is made challenging, NETosis is thought to be appropriate.48 There are reports that the lethality of systemic inflammatory response syndrome is due to necrosis rather than apoptosis49 and, for the host, it is desirable that neutrophil death occurs by the less invasive method of apoptosis. However, considering the efficiency of microbial disposal, NETosis is more advantageous. As an additional note, the phenomenon where bacteria are disseminated by those neutrophils that have engulfed more than they can metabolize is referred to as “Trojan horse”50 and the purpose of NETosis may also be to prevent such a phenomenon.
Conclusion
It is important to acknowledge that cell death associated with sepsis is not simply a passive action to destroy cells, but that it is a response with a purpose. The body attempts to maintain homeostasis by using different mechanisms of cell death but, depending on the degree of insult, an overreaction is unavoidable. There is potential here for the development of new therapeutic interventions.
Conflict of Interest
None.
Acknowledgments
This paper was initially published in the Journal of Japanese Association for Acute Medicine, in Japanese.
References
- 1. Aikawa N. The role of cytokines in the pathogenetic mechanisms of shock and organ dysfunction. J. Jpn. Soc. Emer. Med. 1994; 5: 641–654. [Google Scholar]
- 2. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011; 30: 16–34. [DOI] [PubMed] [Google Scholar]
- 3. Harris HE, Raucci A. Alarmin(g) news about danger: Workshop on innate danger signals and HMGB1. EMBO Rep. 2006; 7: 774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 2008; 8: 776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seong SY, Matzinger P. Hydrophobicity: An ancient damage‐associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 2004; 4: 469–478. [DOI] [PubMed] [Google Scholar]
- 6. Cinel I, Opal SM. Molecular biology of inflammation and sepsis: A primer. Crit. Care Med. 2009; 37: 291–304. [DOI] [PubMed] [Google Scholar]
- 7. Bianchi ME. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007; 81: 1–5. [DOI] [PubMed] [Google Scholar]
- 8. Denk S, Perl M, Huber‐Lang M. Damage‐ and pathogen‐associated molecular patterns and alarmins: Keys to sepsis? Eur. Surg. Res. 2012; 48: 171–179. [DOI] [PubMed] [Google Scholar]
- 9. Brinkmann V, Reichard U, Goosmann C et al Neutrophil extracellular traps kill bacteria. Science 2004; 303: 1532–1535. [DOI] [PubMed] [Google Scholar]
- 10. Papayannopoulos V, Metzler KD, Hakkim A et al Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010; 191: 677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goldmann O, Medina E. The expanding world of extracellular traps: Not only neutrophils but much more. Front. Immunol. 2012; 3: 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Remijsen Q, Vanden Berghe T, Wirawan E et al Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011; 21: 290–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tadié JM, Bae HB, Jiang S et al HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll like receptor 4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013; 304: L342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang D, de la Rosa G, Tewary P et al Alarmins link neutrophils and dendritic cells. Trends Immunol. 2009; 30: 531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hakkim A, Fuchs TA, Martinez NE et al Activation of the Raf‐MEK‐ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011; 7: 75–77. [DOI] [PubMed] [Google Scholar]
- 16. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140: 805–820. [DOI] [PubMed] [Google Scholar]
- 17. Abrams ST, Zhang N, Manson J et al Circulating histones are mediators of trauma‐associated lung injury. Am. J. Respir. Crit. Care Med. 2013; 187: 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saffarzadeh M, Juenemann C, Queisser MA et al Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS One 2012; 7: e32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fuchs TA, Abed U, Goosmann C et al Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007; 176: 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Esmon CT, Xu J, Lupu F. Innate immunity and coagulation. J. Thromb. Haemost. 2011; 9(Suppl 1): 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu J, Zhang X, Pelayo R et al Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009; 15: 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Semeraro F, Ammollo CT, Morrissey JH et al Extracellular histones promote thrombin generation through platelet‐dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011; 118: 1952–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ammollo CT, Semeraro F, Xu J et al Extracellular histones increase plasma thrombin generation by impairing thrombomodulin‐dependent protein C activation. J. Thromb. Haemost. 2011; 9: 1795–1803. [DOI] [PubMed] [Google Scholar]
- 24. Chaput C, Zychlinsky A. Sepsis: The dark side of histones. Nat. Med. 2009; 15: 1245–1246. [DOI] [PubMed] [Google Scholar]
- 25. Fuchs TA, Brill A, Duerschmied D et al Extracellular DNA traps promote thrombosis. Proc. Natl Acad. Sci. USA 2010; 107: 15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Y, Alam HB. Modulation of acetylation: Creating a pro‐survival and anti‐inflammatory phenotype in lethal hemorrhagic and septic shock. J. Biomed. Biotechnol. 2011; 2011: 523481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu J, Zhang X, Monestier M et al Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J. Immunol. 2011; 187: 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Finkelstein RA, Li Y, Liu B et al Treatment with histone deacetylase inhibitor attenuates MAP kinase mediated liver injury in a lethal model of septic shock. J. Surg. Res. 2010; 163: 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kutcher ME, Xu J, Vilardi RF et al Extracellular histone release in response to traumatic injury: Implications for a compensatory role of activated protein C. J Trauma Acute Care Surg. 2012; 73: 1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pisetsky DS. The origin and properties of extracellular DNA: From PAMP to DAMP. Clin. Immunol. 2012; 144: 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holdenrieder S, Stieber P. Clinical use of circulating nucleosomes. Crit. Rev. Clin. Lab. Sci. 2009; 46: 1–24. [DOI] [PubMed] [Google Scholar]
- 32. Margraf S, Lögters T, Reipen J et al Neutrophil‐derived circulating free DNA (cf‐DNA/NETs): A potential prognostic marker for posttraumatic development of inflammatory second hit and sepsis. Shock 2008; 30: 352–358. [DOI] [PubMed] [Google Scholar]
- 33. Zeerleder S, Stephan F, Emonts M et al Circulating nucleosomes and severity of illness in children suffering from meningococcal sepsis treated with protein C. Crit. Care Med. 2012; 40: 3224–3229. [DOI] [PubMed] [Google Scholar]
- 34. Chen Q, Ye L, Jin Y et al Circulating nucleosomes as a predictor of sepsis and organ dysfunction in critically ill patients. Int. J. Infect. Dis. 2012; 16: e558–564. [DOI] [PubMed] [Google Scholar]
- 35. Huttunen R, Kuparinen T, Jylhävä J et al Fatal outcome in bacteremia is characterized by high plasma cell free DNA concentration and apoptotic DNA fragmentation: A prospective cohort study. PLoS One 2011; 6: e21700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Calfee CS, Matthay MA. Clinical immunology: Culprits with evolutionary ties. Nature 2010; 464: 41–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kung CT, Hsiao SY, Tsai TC et al Plasma nuclear and mitochondrial DNA levels as predictors of outcome in severe sepsis patients in the emergency room. Shock 2012; 38: 337–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Byrne A, Reen DJ. Lipopolysaccharide induces rapid production of IL‐10 by monocytes in the presence of apoptotic neutrophils. J. Immunol. 2002; 168: 1968–1977. [DOI] [PubMed] [Google Scholar]
- 39. Kennedy CL, Smith DJ, Lyras D et al Programmed cellular necrosis mediated by the pore‐forming alpha‐toxin from Clostridium septicum. PLoS Pathog. 2009; 5: e1000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Labbé K, Saleh M. Cell death in the host response to infection. Cell Death Differ. 2008; 15: 1339–1349. [DOI] [PubMed] [Google Scholar]
- 41. Mihalache CC, Yousefi S, Conus S et al Inflammation‐associated autophagy‐related programmed necrotic death of human neutrophils characterized by organelle fusion events. J. Immunol. 2011; 186: 6532–6542. [DOI] [PubMed] [Google Scholar]
- 42. Manson J, Thiemermann C, Brohi K. Trauma alarmins as activators of damage‐induced inflammation. Br. J. Surg. 2012; 99(Suppl 1): 12–20. [DOI] [PubMed] [Google Scholar]
- 43. Summers C, Rankin SM, Condliffe AM et al Neutrophil kinetics in health and disease. Trends Immunol. 2010; 31: 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Duffin R, Leitch AE, Fox S et al Targeting granulocyte apoptosis: Mechanisms, models, and therapies. Immunol. Rev. 2010; 236: 28–40. [DOI] [PubMed] [Google Scholar]
- 45. Fuchs TA, Abed U, Goosmann C et al Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007; 176: 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McGrath EE, Marriott HM, Lawrie A et al TNF‐related apoptosis‐inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J. Leukoc. Biol. 2011; 90: 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kennedy A, DeLeo F. Neutrophil apoptosis and the resolution of infection. Immunol. Res. 2009; 43: 25–61. [DOI] [PubMed] [Google Scholar]
- 48. Yipp BG, Petri B, Salina D et al Infection‐induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012; 18: 1386–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Duprez L, Takahashi N, Van Hauwermeiren F et al RIP kinase‐dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 2011; 35: 908–918. [DOI] [PubMed] [Google Scholar]
- 50. Thwaites GE, Gant V. Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat. Rev. Microbiol. 2011; 9: 215–222. [DOI] [PubMed] [Google Scholar]