

Abstract
Case
Alcoholic ketoacidosis (AKA) usually occurs in patients with a history of prolonged alcohol abuse and recent binge drinking followed by abrupt cessation of alcohol consumption.
Outcome
A 61‐year‐old man was brought to our emergency department. He had a history of eating barbecued beef with beer the previous night. He had unexplained hypoglycemia with high anion gap metabolic acidosis and fatty liver, and we strongly suspected AKA. After hydration with saline solution, dextrose, and thiamine, given i.v., his metabolic acidosis rapidly improved. A history of alcohol abuse and high serum β‐hydroxybutyrate concentration were subsequently confirmed, and the diagnosis of AKA was finally made.
Conclusion
Our case suggests that a high‐fat meal can induce AKA without abrupt cessation of alcohol consumption and that AKA should be considered when encountering patients with unexplained high anion gap metabolic acidosis with hypoglycemia and fatty liver, even if the past history of alcohol abuse is unknown.
Keywords: Alcohol abuse, anion gap, consciousness disturbance, metabolic acidosis, β‐hydroxybutyrate
Introduction
Ketoacidosis often occurs in diabetic patients with poor control resulting in diabetic ketoacidosis (DKA), but it is also known to occur in alcoholic patients as alcoholic ketoacidosis (AKA).1, 2, 3 Usually, AKA develops in patients whose meal intake is insufficient due to alcoholism, and who become unable to ingest alcohol for some reason.3, 4 We experienced one case of AKA in which the patient was brought in for suspected stroke the morning after high fat intake.
Patient
The patient was a 61‐year‐old male with a chief complaint of muscle weakness of limbs and impaired consciousness. His present illness developed while staying at a business hotel, where he noticed a lack of strength in limbs on waking up, and called for an ambulance. Stroke was suspected and the patient was brought to this hospital. According to staff at the hotel he was staying at, the patient checked in the day before for business and during his stay (the night before hospitalization), ingested grilled meat and beer at the hotel's restaurant. The patient was in possession of diltiazem hydrochloride and isosorbide mononitrate tablets, suggesting a history of heart disease. However, he did not have hypoglycemic agents or insulin.
On admission, his level of consciousness was 2 on the Japan Coma Scale, blood pressure was 120/51 mmHg, heart rate was 85 b.p.m., body temperature was 34.7°C, breathing rate was 18/min, and percutaneous oxygen saturation was 98% (room air). Hypothermia was observed and mild impaired consciousness and unclear speech made a detailed examination of medical history impossible. His respiratory pattern was deep Kussmaul breathing and his breath smelled of sweet acetone and slightly of alcohol. He was emaciated with mild tenderness in the lower right abdomen. Muscle weakness of grade 4 to 5 in manual muscle testing was observed in the limbs, but there was no left/right difference and no other clear abnormalities were found in a physical examination. A quick blood sugar test directly after being brought in produced a low value of 38 mg/dL. Blood test on admission (Table 1) revealed an increase in white blood cells, impaired liver function, a rise in urea nitrogen, and high neutral fat levels, but there was no rise in C‐reactive protein and blood sugar levels were low, as indicated by the quick blood sugar test. Blood concentration of free fatty acid was not measured. The urine test revealed that ketone bodies were slightly positive at 2+, and an arterial blood gas analysis found significant metabolic acidosis with an opened anion gap (pH 7.063, PaCO2 30.5 mmHg, PaO2 108.7 mmHg, BE −20.6 mmol/L, HCO3 − 8.5 mmol/L), although lactic acid was slightly raised at 4.44 mmol/L. An electrocardiography test revealed sinus tachycardia, but there were no findings indicative of myocardial ischemia. Imaging tests on admission included echocardiography, which revealed that the left ventricle was functioning normally and that there was no strain on the right side of the heart. Abdominal ultrasonography found no abnormalities apart from severe fatty liver. Although emphysematous changes in the lung field, severe fatty liver in the abdomen, and mild thickening of the wall of the right colon were seen, no abnormalities that could cause consciousness disturbance or metabolic acidosis, such as abscess formation, pancreatitis, or intestinal ischemia, were observed on a full body computed tomography scan (Fig. 1).
Table 1.
Laboratory data of a 61‐year‐old man with muscle weakness of limbs and impaired consciousness on arrival at our emergency department
| Hematology | Biochemistry | Blood gas a | ||||||
| WBC | 20,800 | /μL | TP | 7.3 | g/dL | pH | 7.063 | |
| Neu | 15,184 | /μL | Alb | 3.9 | g/dL | PCO2 | 30.5 | mmHg |
| Ly | 3,120 | /μL | BUN | 24 | mg/dL | PO2 | 108.7 | mmHg |
| Mo | 1,664 | /μL | Cr | 0.88 | mg/dL | SaO2 | 95.8 | % |
| RBC | 422 × 104 | /μL | Na | 145 | mEq/L | HCO3 − | 8.5 | mmol/L |
| MCV | 98.1 | fl | K | 3.2 | mEq/L | BE | −20.6 | mmol/L |
| Hb | 14.4 | g/dL | Cl | 106 | mEq/L | Lactate | 4.44 | mmol/L |
| Ht | 41.1 | % | Ca | 9.3 | mg/dL | |||
| Plt | 17.6 × 104 | /μL | P | 3.0 | mg/dL | Serology | ||
| AST | 448 | IU/L | TPLA | (−) | ||||
| Coagulation | ALT | 207 | IU/L | RPR | (−) | |||
| APTT | 27.0 | s | LDH | 419 | IU/L | HBsAg | (−) | |
| PT | 11.6 | s | ALP | 687 | IU/L | HCVAb | (−) | |
| PT‐% | 111 | % | γGTP | 491 | IU/L | |||
| PT‐INR | 0.97 | T‐Bil | 0.5 | mg/dL | Urinalysis | |||
| BS | 40 | mg/dL | Gravity | 1.018 | ||||
| Thyroid func. | CPK | 85 | IU/L | pH | 5.0 | |||
| Free T4 | 0.73 | ng/dL | AMY | 94 | IU/L | WBC | (−) | |
| TSH | 0.309 | μIU/mL | NH3 | 50 | μg/dL | Protein | (+) | |
| T.chol | 179 | mg/dL | Sugar | (−) | ||||
| TG | 543 | mg/dL | Ketone | (2+) | ||||
| CRP | 0.03 | mg/dL | Blood | (2+) |
Room air. Alb, albumin; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AMY, amylase; APTT, activated partial thromboplastin time; AST, aspartate transaminase; BS, blood sugar; BUN, blood urea nitrogen; CPK, creatine phosphokinase; Cr, creatinine; CRP, C‐reactive protein; func., function; γGTP, γ‐glutamyl transpeptidase; Hb, hemoglobin; HBsAg, hepatitis B surface antigen; HCV Ab, hepatitis C antibody; Ht, hematocrit; LDH, lactate dehydrogenase; Ly, lymphocytes; MCV, mean corpuscular volume; Mo, monocytes; Neu, neutrophils; Plt, platelets; PT, prothrombin time; RBC, red blood cells; RPR, rapid plasma reagin; T4, thyroxine; T‐Bil, total bilirubin; T.chol, total cholesterol; TG, thyroglobulin; TP, total protein; TPLA, Treponema pallidum latex agglutination; TSH, thyroid‐stimulating hormone; WBC, white blood cells.
Figure 1.
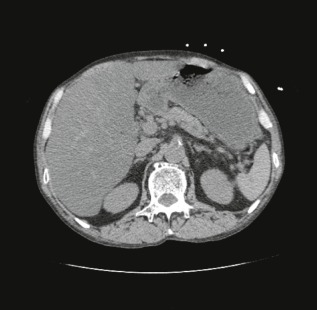
Radiological findings on arrival at our emergency department of a 61‐year‐old man complaining of muscle weakness of limbs and impaired consciousness. Computed tomography scans of the whole body revealed neither the main cause of the metabolic acidosis nor the consciousness disturbance. However, severe fatty liver was observed.
After admission, results of the quick blood test and blood agent analysis prompted immediate i.v. administration of 100 mg thiamine and 40 mL of 20% glucose. As symptoms could not be fully explained by the mild rise in lactic acid levels, thought to be due to hypothermia and circulatory failure, a combination of alcoholic hypoglycemia and AKA was strongly suspected. The reasons for this suspicion were the fact that the general condition was being maintained despite severe metabolic acidosis for which the cause was not clear on imaging, and the concurrent idiopathic hypoglycemia, urine ketones, and significant fatty liver, despite the patient being non‐obese. While heeding refeeding syndrome, fluid resuscitation containing thiamine and sugar solution was initiated while periodically compensating for blood sugar levels and electrolytes, and blood gas analysis findings were tracked. The metabolic acidosis shown in Table 2 completely recovered without administration of bicarbonate, and the patient's condition was almost normalized approximately 10 h after beginning infusion. With normalization of blood sugar levels, level of consciousness and muscle weakness improved, making it possible to obtain the patient's medical history. The patient revealed that he had a history of alcoholism, that his dietary intake had been irregular for the past 2 months, that he drank five glasses of alcohol each day, and that the day before hospitalization, he had consumed a large amount of grilled meat and beer without any carbohydrates. At a later date, vitamin B1 levels returned to within the normal range (61.8 ng/mL) and blood ethanol concentration only rose slightly to 0.7 mg/mL. However, results of blood ketone bodies fractionation showed acetoacetate (AcAc) 244 μmol/L and β‐hydroxybutyrate (OHBA) 4643 μmol/L, indicating a significant rise in OHBA and leading to a definitive diagnosis of concurrent alcoholic hypoglycemia and AKA. The patient experienced delirium from alcohol withdrawal, but there were no other major complications and he was given an ambulatory discharge after satisfactory progress on the sixth hospital day.
Table 2.
Clinical time course of a 61‐year‐old man admitted with muscle weakness of limbs and impaired consciousness, focusing on arterial blood gas analysis
| Time | 11:43 on arrival | 12:10 | 13:30 | 15:00 | 18:00 | 21:00 | 7:00 next day |
|---|---|---|---|---|---|---|---|
| CL (JCS) | 2 | 2 | 1 | clear | clear | clear | clear |
| RR (b.p.m.) | 18 | 24 | 24 | 20 | 16 | 14 | 14 |
| pH | 7.063 | 7.052 | 7.067 | 7.176 | 7.326 | 7.341 | 7.349 |
| PaCO2 (mmHg) | 30.5 | 27.7 | 27.7 | 24.4 | 30.6 | 31.7 | 35.9 |
| PaO2 (mmHg) | 108.7 | 116.8 | 103.4 | 107.2 | 96.4 | 94.2 | 95.8 |
| HCO3 − (mmol/L) | 8.5 | 7.5 | 7.8 | 8.8 | 15.6 | 19.2 | 22.2 |
| BE (mmol/L) | −20.6 | −21.6 | −21.1 | −18.0 | −9.3 | −4.8 | −2.1 |
| AG (mEq/L) | 27.2 | 25.8 | 22.5 | 24.7 | 21.4 | 18.8 | 15.2 |
| Lactate (mmol/L) | 4.44 | 4.53 | 4.01 | N.A. | N.A. | N.A. | 1.09 |
| Glucose (mg/dL) | 38 | 94 | 235 | 204 | 195 | 241 | 120 |
AG, anion gap; BE, base excess; b.p.m., beat per minute; CL, consciousness level; JCS, Japan Coma Scale; N.A., not analyzed; RR, respiratory rate.
Discussion
Ketoacidosis mainly occurs as DKA in diabetes patients with poor control. In DKA, intracellular glucose uptake is blocked by an absolute deficiency of insulin, which enhances fatty acid oxidation (β‐oxidation) that generates alternative energy and results in the production of acetyl‐coenzyme A (acetyl‐CoA). However, this excess acetyl‐CoA is not processed by the tricarboxylic acid cycle (TCA cycle) while glucose catabolism is reduced from lack of insulin, resulting in the synthesis of ketone bodies from acetyl‐CoA and causing DKA.5 This same pathology occurs in alcoholic patients, and is known as AKA.1, 2, 3 Alcoholic ketoacidosis occurs as a result of: (i) reduced glycogen from excessive intake of alcohol, (ii) a relative decrease in insulin activity from lack of carbohydrates, (iii) overproduction of NADH, the reduced form of the coenzyme nicotinamide adenine dinucleotide (NAD) in the constant ethanol metabolism process, which inhibits both gluconeogenesis and the TCA cycles through the rise in the NADH/NAD ratio, (iv) the acetyl‐CoA supply from ethanol metabolic production, (v) dehydration and starvation in patients with factors such as stimulation of fatty acid metabolism by ethanol itself, (vi) production of ketone bodies from fatty acid‐induced enhanced β‐oxidation due to insulin antagonistic hormones such as glucagon for generating alternative energy.2, 3, 4, 6, 7 In other words, AKA typically develops in patients who depend on alcohol for their small supply of carbohydrates due to chronic alcoholism, and occurs when alcohol can no longer be consumed due to symptoms such as nausea, vomiting, or abdominal pain. These digestive symptoms are the main reasons for examinations at medical facilities.3 In addition to this kind of typical medical history and clinical features, diagnoses are made by verifying metabolic acidosis with an open anion gap and excessive ketone body production.2, 3 However, unlike DKA, AKA is characterized by the significant bias towards OHBA of produced ketone bodies compared with AcAc, due to the rising NADH/NAD ratio that occurs for the above‐mentioned reasons.3, 8, 9 β‐Hydroxybutyrate cannot be detected with urine test strips that use the nitroprusside reaction frequently used in routine medical care,10, 11 and even if urinary ketone is negative, AKA cannot be ruled out. In this case, it is necessary to verify the rise in blood OHBA, but it is not always useful in initial diagnoses that are not as fast. That is to say, diagnosis of AKA is highly dependent on medical history and clinical features.
In this case, the alcoholism was revealed after the fact; although this was different from a typical case of AKA since the patient had consumed grilled meat the night before, there was no decrease in oral intake and he was admitted on suspicion of stroke. The reason AKA occurred despite no decrease in oral intake, is that alcoholic patients with a background of fatty acid degradation due to the aforementioned chronic lack of carbohydrates have a large influx of free fatty acids in the blood from ingesting large quantities of a high‐fat diet in a short amount of time.
Acetyl‐CoA was produced through β‐oxidation. Although due to the increased NADH/NAD ratio as a result of alcohol metabolism, the excessive acetyl‐CoA that was not incorporated into gluconeogenesis or the TCA cycle ended up being used in ketone body metabolism (Fig. 2). In other words, this case suggests that even if there is no decrease in oral intake, AKA may develop through the exogenous influx of a large amount of free fatty acids, similar to DKA. Grilled meat consists of not only large amounts of fat but also protein that can be used to incorporate acetyl‐CoA into TCA or used for gluconeogenesis. However, because too much fat compared to protein had the potential to promote excessive acetyl‐CoA, and gluconeogenesis was also impaired by overproduction of NADH, AKA developed in this case. Furthermore, AKA presents with various clinical features besides digestive symptoms3, 4, 7 and impaired consciousness is not unusual at 10–15%.2 Common causes of impaired consciousness are hypoglycemia and hypothermia (as shown), and alcoholic poisoning and infection.2 The metabolic acidosis of this patient was the result of a complex case, including hypothermia and peripheral hypoperfusion. However, the main metabolic event was AKA because lactic acid did not rise markedly and vitamin B1 levels returned to within the normal range, as described above. In alcoholic patients with suppressed gluconeogenesis and depleted glycogen stores, risk of hypoglycemic attack, known as alcoholic hypoglycemia, is high and alcohol‐related diseases are important in differentiating from hypoglycemia in non‐diabetic patients.12
Figure 2.
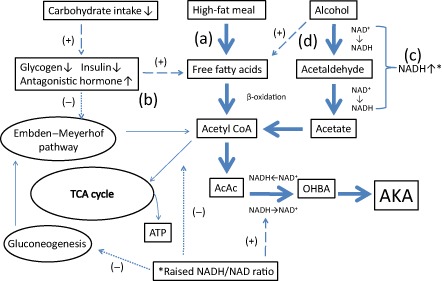
Pathophysiological concept of alcoholic ketoacidosis (AKA) in this case. (a) Instead of acute starvation, intake of a high‐fat meal resulted in increased release of free fatty acids into the circulation, which are subsequently metabolized to ketoacids by the liver. (b) The hormonal profile, such as suppression of insulin with increased secretion of antagonistic hormones reflecting alcohol abuse, further enhances ketogenesis by inhibiting the hepatic metabolism of acetyl coenzyme A (acetyl‐CoA) via the tricarboxylic acid (TCA) cycle. (c) Chronic alcohol metabolism contributes to the ketosis to increase the ratio of the reduced form of nicotinamide adenine dinucleotide (NADH) to nicotinamide adenine dinucleotide NAD. The elevated NADH/NAD ratio inhibits hepatic gluconeogenesis and the TCA cycle. (d) Ethanol directly stimulates lipolysis and releases free fatty acids. Moreover, ethanol is metabolized to acetate, which then partially forms acetyl CoA. Large dashed means (+), small dashed means (‐), and thick dashed indicates this case. AcAc, acetoacetate; ATP, adenosine triphosphate; OHBA, β‐hydroxybutyric acid.
At this facility, excluding external wounds, patients admitted for impaired consciousness account for 10% of emergency admissions, and of these, 9.5% are the result of hypoglycemia. The majority of hypoglycemia is iatrogenic hypoglycemia associated with diabetes treatment, but we sometimes encounter idiopathic hypoglycemia. In patients with impaired consciousness, we often have to begin initial treatment without knowing the patient's medical history because not enough information can be gleaned from the patient. When the history of alcoholism is unknown and the onset is atypical, AKA is not easily suspected. However, it is important to always be aware of alcohol‐related diseases as a cause for impaired consciousness, as well as not overlooking AKA idiopathic hypoglycemia hidden in the background of alcoholic hypoglycemia. This can be done by carefully excluding uremia, drug overdose, and sepsis when idiopathic hypoglycemia is observed, or when inexplicable metabolic acidosis with an open anion gap is observed in combination with fatty liver in non‐obese patients due to increased lactic acid alone. In Japan, awareness of AKA is not high, but it is becoming clear that AKA is not uncommon among emergency admissions, as a result of proactive diagnostic efforts.13 Furthermore, when AKA was overlooked, fulminant cases of pancreatitis, rhabdomyolysis, multiple organ failure, and sudden death have been seen,6 and despite these being associated with 7% of deaths in alcoholic patients,6, 14 the treatment principle for AKA is electrolyte management including sufficient infusion of carbohydrates and phosphorous.3 If appropriate treatment is carried out in a timely manner, lives can be saved even in cases like these.13 In Japan, alcohol consumption is following an increasing trend comparable with other developed countries, and the number of alcoholic patients is increasing proportionally. Therefore, it may be essential for emergency and primary care physicians who handle a high number of initial examinations of alcohol‐related acute illness to become familiar with the concept of AKA.
Conclusion
Good results can be obtained by suspecting AKA early and providing treatment based on the combination of metabolic acidosis with an open anion gap in idiopathic hypoglycemia and fatty liver in non‐obese patients.
Conflict of Interest
None.
This article is based on a case first reported in the Journal of the Japanese Association for Acute Medicine 2010; 21: 792–8.
References
- 1. Dillon ES, Dyer WW, Smelo LS. Ketone acidosis of non‐diabetic adults. Med. Clin. North Am. 1940; 24: 1813–1822. [Google Scholar]
- 2. Wrenn KD, Slovis CM, Minion GE, Rutlowski R. The syndrome of alcoholic ketoacidosis. Am. J. Med. 1991; 91: 119–128. [DOI] [PubMed] [Google Scholar]
- 3. McGuire LC, Cruickshank AM, Munro PT. Alcoholic ketoacidosis. Emerg. Med. J. 2006; 23: 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tanaka M, Miyazaki Y, Ishikawa S, Matsuyama K. Alcoholic ketoacidosis associated with multiple complications: report of 3 cases. Intern. Med. 2004; 43: 955–959. [DOI] [PubMed] [Google Scholar]
- 5. Foster DW, McGarry JD. The metabolic derangements and treatment of diabetic ketoacidosis. N. Engl. J. Med. 1983; 21: 159–169. [DOI] [PubMed] [Google Scholar]
- 6. Yanagawa Y, Sakamoto T, Okada Y. Six cases of sudden cardiac arrest in alcoholic ketoacidosis. Intern. Med. 2008; 47: 113–117. [DOI] [PubMed] [Google Scholar]
- 7. Ngatchu T, Sangwaiya A, Dabiri A, Dhar A, McNeli I, Arnold JD. Alcoholic ketoacidosis with multiple complications: a case report. Emerg. Med. J. 2007; 24: 776–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levy LJ, Duga J, Girgis M, Gordon EE. Ketoacidosis associated with alcoholism in non diabetic subjects. Ann. Intern. Med. 1973; 78: 213–219. [DOI] [PubMed] [Google Scholar]
- 9. Cooperman MT, Davidoff F, Spark R, Pallotta J. Clinical studies of alcoholic ketoacidosis. Diabetes 1974; 23: 433–439. [DOI] [PubMed] [Google Scholar]
- 10. Marliss EB, Ohman JL, Aoki TT, Kozak GP. Altered redox state obscuring ketoacidosis in diabetic patients with lactic acidosis. N. Engl. J. Med. 1970; 283: 978–980. [DOI] [PubMed] [Google Scholar]
- 11. Fulop M, Hoberman HD. Alcoholic ketosis. Diabetes 1975; 24: 785–790. [DOI] [PubMed] [Google Scholar]
- 12. Sporer KA, Ernst AA, Conte R, Nick TG. The incidence of ethanol‐induced hypoglycemia. Am. J. Emerg. Med. 1992; 10: 403–405. [DOI] [PubMed] [Google Scholar]
- 13. Yokoyama M, Hori S, Aoki K et al Alcoholic ketoacidosis and ketosis in the emergency room. Nihon Kyukyu Igakukai Zasshi 2002; 13: 711–717. [Google Scholar]
- 14. Thomsen JL, Simonsen KW, Felby S, Frohlich B. A prospective toxicology analysis in alcoholics. Forensic Sci. Int. 1997; 90: 33–40. [DOI] [PubMed] [Google Scholar]