

Abstract
The peptide segment of prostatic acid phosphatase (PAP248–286) aggregates to form SEVI (semen‐derived enhancer of virus infection) amyloid fibrils. These are characteristic seminal amyloids that have the ability to promote the effect of HIV infection. In this paper, we explore the binding of sulfonated compounds with PAP248–286 through an in silico study. Three derivatives of suramin, NF110, NF279, and NF340, are selected. All of these sulfonated molecules bind to PAP248–286 and alter the conformation of the peptide, even though they have various structures, sizes, and configurations. The compounds bind with PAP248–286 through multiple interactions, such as hydrogen‐bonding interactions, hydrophobic interactions, π–π stacking interactions, and electrostatic interactions. However, NF110, which has an X‐shaped configuration, has the highest binding affinity of the three derivatives investigated. We also perform surface plasmon resonance and a Congo red assay to validate the results. The interactions between PAP248–286 and the sulfonated compounds are proposed to depend on the orientations of the sulfonate groups and the specific configurations of the compounds instead of the number of sulfonate groups. NF110 molecules occupy the exposed binding sites of PAP248–286, blocking interactions between the peptides. Therefore, these compounds are important in inhibiting the aggregation of PAP248–286. Herein, we provide useful information to develop new efficient microbicides to antagonize seminal amyloid fibrils and to block HIV transmission.
Keywords: amyloid fibrils, HIV, inhibitors, sulfur, viruses
1. Introduction
Acquired immunodeficiency syndrome (AIDS) is known as a serious infectious disease caused by human immunodeficiency virus (HIV), which greatly threatens the safety of the world and the survival of mankind. Sexual transmission has become the main transmission route of HIV. In 2007, Münch et al. found that semen could significantly promote HIV infection.1 They described that semen could enhance infection because the endogenous peptide compositions of semen form amyloid fibrils.1, 2, 3, 4 The most important peptides are 1) prostatic acid phosphatase (PAP) hydrolytic polypeptide PAP248–286 and 2) semenogelin 1 hydrolytic polypeptide SEM86‐107. The former assemble themselves into amyloid fibrils termed SEVI (semen‐derived enhancer of virus infection), and the latter form SEM1 fibrils.1, 2, 3, 4
Amyloid fibrils, which are a kind of highly ordered aggregates with rich β‐sheet structures, are related to a number of neurological diseases.5, 6, 7 Ramamoorthy et al. recently summarized biophysical and immunological studies on SEVI.8 SEVI and SEM1 are proposed to promote the effect of HIV infection on account of the strong cationic characteristics of the amyloid fibrils, which possess large amounts of positively charged amino‐acid residues such as Arg and Lys.4 There are probably two ways to enhance HIV infection: one way is to ensure close proximity of the negatively charged HIV virus with the target cell by reducing repulsion, and the other way is to capture the HIV virus to assist interaction of the virus with the host cell.4, 9, 10, 11, 12, 13, 14 It was reported that PAP248–286 could bind to lipid membranes by adopting a helical structure. This membrane fusion process plays an important role in enhancing interactions between the HIV virus and a target cell.15, 16 In addition, the bacterial curli protein was found to catalyze the assembly of PAP248–286 to form SEVI as a result of an increase in the rate of aggregation and elongation of PAP248–286 cross seeding.17 Therefore, the amyloid fibrils of semen are an important target to inhibit the transmission of HIV infection. As SEVI is the main component of semen fibrils, three strategies have been developed to inhibit HIV infection: one, inhibition of PAP248–286 to form SEVI fibrils at an early stage; two, coat the fibrils to block their infection enhancement properties; three, remodel or degenerate the fibrils.18 In recent years, some active small molecules were found to antagonize semen amyloid fibrils. For instance, inhibitors of metal ions, as well as non‐natural amino acids, were shown to inhibit the formation of SEVI.19, 20 Roan et al. found that surfen molecules could effectively interfere during the combination of SEVI and HIV virus particles with target cells, thus weakening the ability of SEVI to promote viral infection.21 Furthermore, epigallocatechin‐3‐gallate (EGCG) was shown to decompose SEVI fibrils under acidic conditions.22 EGCG interacts with PAP248–286 through two steps and binds to the Lys residue of PAP248–286 specifically in the Lys251–Arg257 and Asn269–Ile277 regions.23
Recently, Münch and co‐workers also found that CLR01 has anionic phosphate groups.18 The compound works as a tweezer to engage the Lys or Arg residues of PAP248–286 (Figure 1 b) to inhibit spontaneous fibrillization of the peptides. Experimental data revealed that the ability of human semen containing the HIV virus to infect cells was diminished greatly.18 The properties of CLR01 to antagonize amyloids as well as their antiviral activities prompted us to seek microbicides that could specifically bind with the target peptides. ADS‐J1 was the first published anionic sulfonated compound that was shown to inhibit the formation of semen‐derived amyloid fibrils and to have high inhibitory activity.5, 14, 24 Unfortunately, however, ADS‐J1 contains an azo group, which has toxic effects. Suramin is also a polysulfonate that has clinical application in the treatment of worm infections and trypanosomiasis.25 A lot of commercial derivatives of suramin have been developed, and all of them have anionic sulfonate groups (Figure 1 a). As these derivatives possess strong anionic characteristics, we predicted that they could specifically bind with PAP248–286 and play a role in inhibiting aggregation and fibrillization. Nevertheless, suramin was previously demonstrated to induce severe polyneuropathy, which was related to its dose.26, 27 In contrast, some suramin derivatives, such as NF110, were reported to inhibit enterovirus A71 infection at lower doses than suramin.27 Thus, we will investigate the interactions between PAP248–286 and the derivatives of suramin in this paper.
Figure 1.
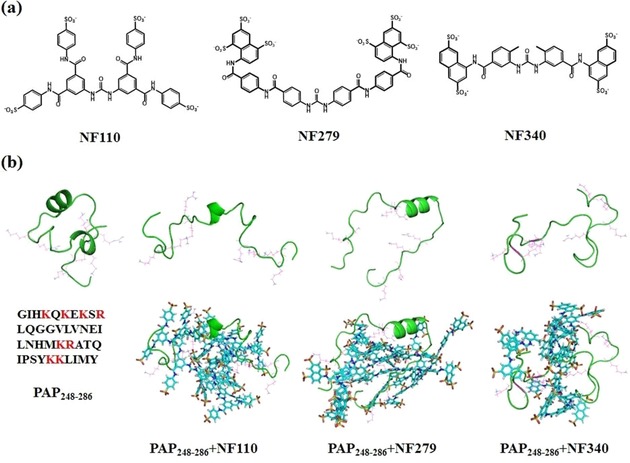
a) Structures of NF110, NF279, and NF340. b) Representative conformations of PAP248–286 alone, P248–286–NF110, PAP248–286–NF279, and PAP248–286–NF340 (from left to right) in the top row, without sulfonated molecules for clarity. The bottom row shows the conformations with sulfonated molecules. The cationic residues of the peptide, such as Lys and Arg, are marked in magenta; sulfonated molecules are displayed in the stick style.
However, the answers to many questions still remain obscure. First, it is unclear whether all of the derivatives bind with PAP248–286. Second, these sulfonated compounds have various sizes, shapes, and spatial configurations. This diversity may affect the interactions and binding affinities between the peptide and molecules. To discriminate the differences, three molecules (i.e. NF110, NF279, and NF340) were selected as representatives (Figure 1 a). Both NF110 and NF340 only have four sulfonate groups, whereas NF279 has six. Moreover, NF110 has an X‐shaped configuration, NF279 has an allosteric strategy (linear to V‐shaped configuration) upon interaction with the target, and NF340 is smaller and shorter than the previous two molecules. In this manuscript, we explore all of the aforementioned questions by studying the interactions between these molecules (i.e. NF110, NF279, and NF340) and PAP248–286 by combining computational and experimental methods. The results provide valuable clues for the development of new molecules that bind with the target peptides specifically to inhibit the aggregation of PAP248–286.
2. Results and Discussion
In this manuscript, we implement molecular dynamics (MD) simulations for four systems to investigate the interactions between PAP248–286 and different sulfonated compounds. The four systems include PAP248–286–NF110, PAP248–286–NF279, PAP248–286–NF340, and PAP248–286 alone. The root‐mean‐square deviations (RMSDs) for the four systems are shown in Figure S1 (see the Supporting Information). The plots reveal that all of the systems reach equilibrium within 100 ns simulations and then remain stable. Therefore, the trajectories in the last 50 ns were utilized for analysis.
2.1. PAP248–286 Changes Its Conformation Upon Binding to Sulfonated Molecules
We first compared the structures of PAP248–286 in the four systems. The timelines for the radius of gyration for PAP248–286 are displayed in Figure 2. The results reveal that the peptide presents various properties of compactness upon interaction with the different sulfonated molecules. Even though NF110, NF279, and NF340 are all sulfonated molecules, the radii of gyration for the peptide decrease in the order PAP248–286–NF110, PAP248–286–NF279, PAP248–286–NF340, and PAP248–286. The average radius of gyration for PAP248–286 in the PAP248–286–NF110 system is 1.60 nm. The radii of gyration are 1.42, 1.35, and 1.05 nm in PAP248–286–NF279, PAP248–286–NF340, and PAP248–286, respectively. The peptide in the PAP248–286–NF340 system becomes tight during the simulation and is more compact than those in PAP248–286–NF110 and PAP248–286–NF279. These changes make PAP248–286 in PAP248–286–NF110 more exposed to the sulfonated molecules. Given that PAP248–286 has eight positive charged residues (Lys251, Lys253, Lys255, Arg257, Lys272, Arg273, Lys281, and Lys282), open PAP248–286 in PAP248–286–NF110 has enough accessible area to interact directly or indirectly with the NF110 molecules (Figure 1 b).
Figure 2.
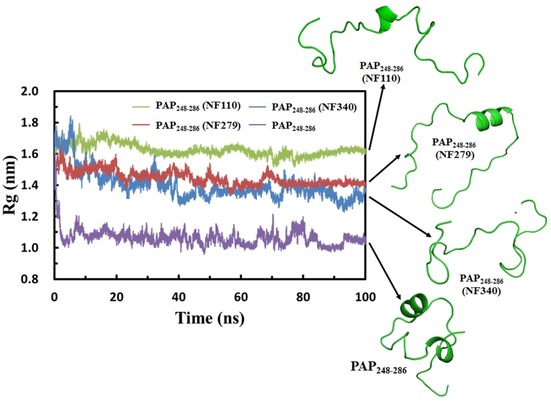
Timelines of the radius of gyration (Rg) for PAP248–286 from PAP248–286–NF110, PAP248–286–NF279, PAP248–286–NF340, and PAP248–286.
We also compared the secondary structures of the peptides in different systems. PAP248–286 alone possesses the highest α helix content; the percentage is as high as 38.5 %. There are two regions of helical structures: Gly261–Asn269 and Ser279–Met285 (Figure 1 b). However, the α helix content decreases in all of the peptides. The α helix content is 15.4 % in PAP248–286–NF110 within the Asn265–His270 segment and 25.6 % in PAP248–286–NF279 within the corresponding Val262–Met271 fragment, whereas PAP248–286–NF340 loses the helical structure completely and turns into a coil. These changes in the secondary structures of the peptide can also be seen from the contact maps in Figure S2. The contact maps reveal the distances of the heavy atoms from the side chains between two amino‐acid residues. Distances less than 0.5 nm can be displayed. The decrease in or complete disappearance of the helical conformation in the peptide structures may be due to the loss of intramolecular interactions within the peptide. The Cα atoms of the residues were calculated by root‐mean‐square fluctuations (RMSFs) and are shown in Figure S3. These data are consistent with the aforementioned results. The peptide in PAP248–286–NF110 has flexible N and C termini. However, the maximum fluctuations of PAP248–286–NF340 are present in the middle part, as the peptide loses the α‐helix character and becomes a random coil.
2.2. Interactions between PAP248–286 and Sulfonated Molecules
PAP248–286, an amyloidogenic peptide fragment of prostatic acid phosphatase, tends to assemble to form amyloid fibrils in semen. These characteristic seminal amyloids have the ability to promote the effect of HIV infection.1, 2, 4, 12 Sulfonated molecules can remodel the peptides and alter their conformations according to previous analysis. These molecules occupy the surrounding sites of PAP248–286 by binding with the peptide, and this interferes with interactions between peptides. Therefore, these molecules are very important in inhibiting the aggregation of PAP248–286. In this section, we investigate interactions between PAP248–286 and different sulfonated molecules and provide detailed information.
Molecules of NF110 bind with PAP248–286 through many interactions, such as hydrogen‐bonding interactions, hydrophobic interactions, π–π stacking interactions, and electrostatic interactions. The structures of PAP248–286 separately bound with NF110, NF279, and NF340 are presented in Figure 1 b. Further illustration of PAP248–286 interacting with one NF110 molecule is shown in Figure 3 a. NF110 is denoted as Mol B, and the other seven NF110 molecules are omitted for clarity. The configuration of NF110 is a distorted X shape owing to the inclusion of several benzene rings, amide bonds, and sulfonate groups, The amide bonds and sulfonate groups of NF110 form hydrogen bonds with the backbones of Gln259 and Gly260 and the side chains of Lys255, Asn269, Lys272, and Arg273 (Figure 3 a and Table 1). Moreover, the residues with cationic side chains, such as the ammonium group of Lys and the guanidinium group of Arg, are attracted by the sulfonate groups of NF110 (Figure 3 a and Table S1). Besides, Mol B forms hydrophobic and CH–π or NH–π interactions with multiple residues of the peptide (Figure 3 a and Table S2). It was reported that π–π interactions affect the fibrillization of amyloid polypeptides.28, 29 EGCG was found to interact with Aβ40 through π–π interactions and to inhibit the formation of amyloid fibrils.30 Besides hydrophobic interactions, oxidized EGCG could covalently bind to the amyloid fibrils to prevent aggregation.31 Details about the interactions between PAP248–286 and other NF110 molecules can be found in Tables 1, S1, and S2. All of the interactions contribute to strong binding between PAP248–286 and NF110. In addition, interactions exist among the NF110 molecules. Figure 3 b reveals that one sulfonate group of Mol B coordinates with Mol F through two hydrogen bonds. This not only helps to bind NF110 more tightly but also allows PAP248–286 to bind with as many NF110 molecules as possible. NF110 molecules are located on almost the entire surface of PAP248–286 and occupy the exposed accessible sites. The whole peptide is buried in a bulky complex. Peptides cannot approach each other, as the NF110 molecules cover the surface of the fibril growth. Furthermore, it is noteworthy that the Gly261–Tyr286 segment was reported to be important for the formation of β‐sheet structures in SEVI fibrils,32 whereas all of these residues interact with the NF110 molecules (Tables 1, S1, and S2). In this way, the NF110 molecules should be able to inhibit the aggregation of PAP248–286 through interactions with PAP248–286 and among themselves.
Figure 3.
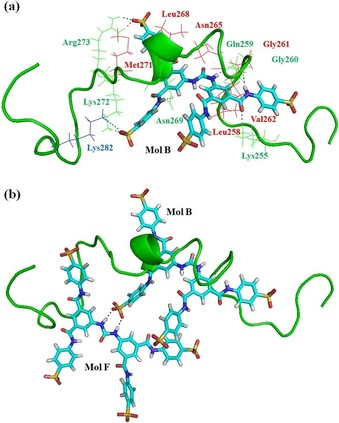
The structure of NF110 binding with PAP248–286: a) PAP248–286 interacts with one NF110 molecule, which is denoted Mol B (the other seven NF110 molecules are omitted for clarity). b) PAP248–286 interacts with two NF110 molecules, labeled Mol B and Mol F (the other six NF110 molecules are not shown for clarity). PAP248–286 is shown as a ribbon. The NF110 molecule is displayed in stick style. The residues of PAP248–286 are marked in various colors according to different interactions: green for hydrogen bonds; red for π–π, CH–π, and NH–π interactions; and blue for electrostatic interactions.
Table 1.
Hydrogen‐bonding interactions between sulfonated compounds and PAP248–286.
| Entry | NF110 molecule | PAP248–286 residue | NF279 molecule | PAP248–286 residue | NF340 molecule | PAP248–286 residue |
|---|---|---|---|---|---|---|
| 1 | B | Lys255 | B | Arg273 | B | Gln252 |
| 2 | Gln259 | C | Lys281 | D | Asn269 | |
| 3 | Gly260 | D | Gln252 | Met271 | ||
| 4 | Asn269 | E | Arg257 | Lys281 | ||
| 5 | Lys272 | Arg257 | E | Gly260 | ||
| 6 | Arg273 | Arg257 | Gly261 | |||
| 7 | Arg273 | Asn265 | G | Arg257 | ||
| 8 | C | Arg257 | Asn269 | Gly248 | ||
| 9 | Arg257 | F | Lys253 | Lys251 | ||
| 10 | Arg273 | Ser256 | H | Arg273 | ||
| 11 | D | Lys251 | Arg257 | Arg273 | ||
| 12 | Ser256 | Lys282 | I | Gln276 | ||
| 13 | Lys272 | G | Lys282 | Met285 | ||
| 14 | F | Lys281 | Leu283 | |||
| 15 | Ser279 | Tyr286 | ||||
| 16 | H | Arg273 | I | Arg257 | ||
| 17 | Arg273 | Ser256 | ||||
| 18 | Lys281 | |||||
| 19 | Leu283 | |||||
| 20 | Tyr286 | |||||
| 21 | I | Lys253 |
NF279, which also comprises a few amide bonds, sulfonate groups, and aromatic rings, has two more sulfonate groups than NF110. Nevertheless, unlike NF110, NF279 has two different configurations: linear and V shaped. Many interactions exist between PAP248–286 and NF279. Figure 4 a reveals one NF279 molecule binding with the peptide. This NF279 molecule, which is expressed as Mol E, possesses various types of interactions with PAP248–286. The side chains of Asn265 and Asn269 from the peptide form hydrogen bonds with one of the sulfonate groups of Mol E (Figure 4 a and Table 1). The V‐shaped configuration allows Mol E to work as a clip to pinch the peptide. As a result, the guanidinium group of Arg257 enters into the crack and interacts with the sulfonate group and the oxygen atom of the amide group of Mol E through three hydrogen bonds. In addition, Arg257 and Arg273 are strongly attracted by the nearby anionic groups of Mol E through electrostatic attractions (Figure 4 a and Table S1). Furthermore, there are hydrophobic and CH–π or NH–π interactions between PAP248–286 and Mol E (Table S3). Because NF279 adopts an allosteric strategy, this molecule has another linear configuration upon interaction with the peptide. Figure 4 b shows the structure of PAP248–286 with two NF279 molecules (Mol E and Mol G). Mol G in the linear conformation is able to contact many residues of the peptide. For instance, Mol G has π–π and CH–π interactions with PAP248–286 from residues Ile249 to Tyr286 (Table S3). The long structure of Mol G makes it more likely to associate with the peptide. As a matter of fact, the linear configuration is predominantly found for NF279 molecules interacting with PAP248–286.
Figure 4.
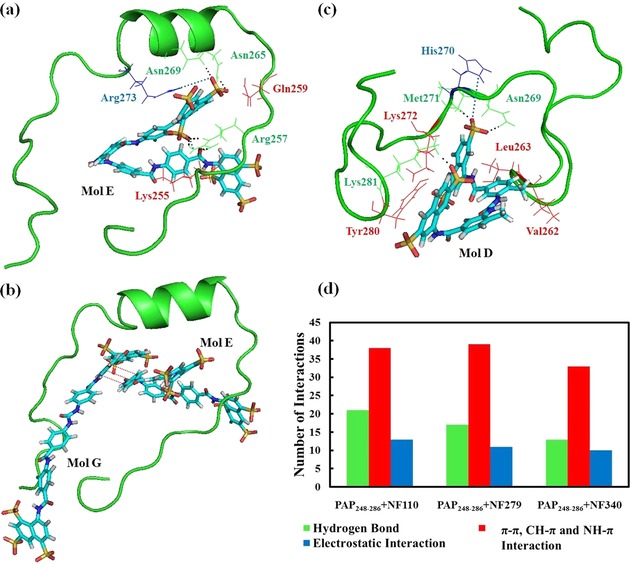
a) PAP248–286 binds with one NF279 molecule expressed as Mol E. b) The structure of PAP248–286 with two NF279 molecules (denoted Mol E and Mol G). c) PAP248–286 interacts with one NF340 molecule labeled as Mol D. All the other molecules are not shown for clarity. The colors of the residues of PAP248–286 represent different interactions with sulfonated molecules: green for hydrogen bonds; red for π–π, CH–π, and NH–π interactions; and blue for electrostatic interactions. d) The number of interactions between PAP248–286 and NF110, NF279, and NF340.
NF340, similar to NF110 and NF279, also contains aromatic rings, amide bonds, and sulfonate groups, but NF340 is smaller and shorter than the previous two molecules. The structures of PAP248–286 interacting with NF340 molecules are observed in Figure 1 b and Figure 4 c. Detailed information of the interactions can be found in Tables 1, S1, and S4. The results indicate that NF110 is the best compound among the three molecules investigated to interact with PAP248–286. The timeline of the number of hydrogen bonds exhibits that the PAP248–286–NF110 complex forms more hydrogen bonds than the other two peptide–compound systems in Figure S4. The number of other interactions of PAP248–286 with the compounds, including electrostatic and π–π interactions, are shown in Figure 4 d.
2.3. Binding of PAP248–286 and Sulfonated Molecules
We know from the aforementioned results that PAP248–286 can interact with NF110, NF279, and NF340 through different kinds of interactions. However, it is still unclear how tightly these compounds bind with PAP248–286. The distance between the nitrogen atom of the ammonium group of Lys or the carbon atom of the guanidinium group of Arg with the nearest sulfur atom of the sulfonated molecules was measured, as the cationic residues of PAP248–286 are attracted by the sulfonate groups of the compounds. Relative to the initial values, most of the distances listed in Table 2 are markedly lower, whereas the distances in the PAP248–286–NF279 and PAP248–286–NF340 complexes are greater than those in the PAP248–286–NF110 complex. It is suggested that the NF110 molecules bind with PAP248–286 more tightly than the other two derivatives. This indication is verified by the binding free energy. Table S5 shows the binding free energy (ΔE b) of the peptide–compound complexes. The ΔE b value of PAP248–286–NF110 is the lowest and is followed by that of PAP248–286–NF279, whereas PAP248–286–NF340 has the highest binding free energy. Consequently, the PAP248–286–NF340 complex has the weakest binding affinity of the three peptide–compound complexes.
Table 2.
Distance between the ammonium group of Lys or the guanidinium group of Arg with the bound sulfur atom of NF110, NF279, and NF340.
| Residue | Distance [nm] | |
|---|---|---|
| Initial structure | Representative structure | |
| PAP248–286–NF110 | ||
| Lys251 | 1.3429±0.0465 | 0.3925±0.0850 |
| Lys253 | 1.0459±0.1169 | 0.3942±0.0691 |
| Lys255 | 0.7601±0.1097 | 0.5684±0.0844 |
| Arg257 | 1.0460±0.0935 | 0.5975±0.0931 |
| Lys272 | 0.4315±0.1184 | 0.3875±0.1130 |
| Arg273 | 1.6453±0.1187 | 0.4186±0.0988 |
| Lys281 | 0.7680±0.1059 | 0.3839±0.1031 |
| Lys282 | 1.2894±0.1076 | 0.5927±0.0927 |
| PAP248–286–NF279 | ||
|---|---|---|
| Lys251 | 2.9181±0.0898 | 0.5450±0.1004 |
| Lys253 | 1.2734±0.0866 | 0.4095±0.1094 |
| Lys255 | 0.8940±0.1082 | 0.5277±0.0150 |
| Arg257 | 0.6510±0.0914 | 0.4558±0.1164 |
| Lys272 | 0.5835±0.0865 | 0.7359±0.0818 |
| Arg273 | 1.0045±0.0156 | 0.6043±0.0841 |
| Lys281 | 1.9025±0.1183 | 0.3734±0.1184 |
| Lys282 | 2.4143±0.0895 | 0.4127±0.1126 |
| PAP248–286–NF340 | ||
|---|---|---|
| Lys251 | 3.5621±0.0407 | 0.3698±0.0913 |
| Lys253 | 2.4326±0.0929 | 0.7635±0.0975 |
| Lys255 | 0.5769±0.0973 | 1.3823±0.0638 |
| Arg257 | 2.0381±0.1154 | 0.4689±0.1146 |
| Lys272 | 0.6953±0.0758 | 0.5733±0.0954 |
| Arg273 | 0.6836±0.1076 | 0.4190±0.0905 |
| Lys281 | 0.7686±0.1143 | 0.3530±0.0723 |
| Lys282 | 2.2401±0.0395 | 0.8556±0.1165 |
We also determined the abilities of these sulfonated compounds to bind PAP248–286 by surface plasmon resonance (SPR). As shown in Figure 5, all three sulfonated compounds bind to PAP248–286 but with different binding affinities. The equilibrium dissociation constant (K D) of NF110 bound to PAP248–286 is 2.11×10−6 m with an association rate constant (k a) of 3.96×103 m −1 s−1. Under the same conditions, NF279 bound to PAP248–286 with a K D of 6.74×10−6 m and k a of 59.8 m −1 s−1, whereas NF340 bound to PAP248–286 with a K D of 4.72×10−5 m. The K D value of NF110 is 22.4‐fold higher than that of NF340. This is in agreement with previous results.
Figure 5.
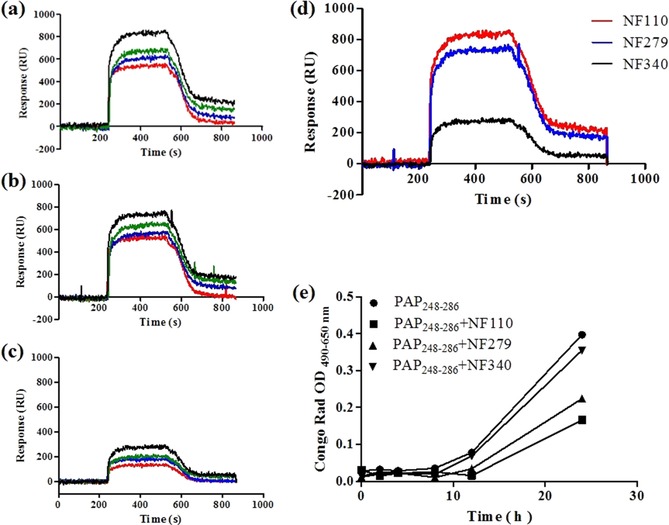
SPR assay. Dose‐dependent binding of a) NF110, b) NF279, and c) NF340 to PAP248–286. PAP248–286 was injected onto the surface at various concentrations of 100, 50, 25, and 12.5 μm (from top to bottom) at a flow rate of 2 μL s−1 for 300 s. d) Response curves obtained from different sulfonated compounds at 10 mm binding to PAP248–286 at 100 μm. e) Congo red binding assay. The data represent mean±SD of triplicate measurements.
2.4. Sulfonated Molecules Inhibit the Aggregation of PAP248–286
NF110, NF279, and NF340 were used to examine their abilities to inhibit PAP248–286 aggregation. We incubated PAP248–286 to detect fibril formation by Congo red staining in the absence and presence of sulfonated molecules. Congo red is a specific dye to detect amyloid, as it can bind amyloid fibrils and induce increased optical absorbance proportional to the level of fibrils. In the absence of sulfonated molecules, PAP248–286 aggregated following a lag phase of around 12 h. The Congo red absorbance signal increased gradually until it reached a plateau after 24 h. In the presence of a twofold excess amount of the sulfonated molecules, NF110, NF279, and NF340 all displayed inhibitory effects on PAP248–286 fibril formation, as indicated by reduced optical absorbance intensity at 48 h (Figure 5 e and Table S6). Relative to the PAP248–286 control, NF110 showed a maximum decrease in absorbance intensity and NF340 showed only a slight decrease in the Congo red signals. Therefore, NF110 exhibited the strongest ability to inhibit PAP248–286 aggregation and NF279 showed a moderate effect on PAP248–286 fibrillization, whereas NF340 weakly inhibited PAP248–286 self‐ assembly.
3. Summary and Conclusions
In this work, we studied the binding of sulfonated compounds with prostatic acid phosphatase (PAP248–286). Three molecules, NF110, NF279, and NF340, were selected from plenty of derivatives of suramin. These sulfonated compounds have different sizes and configurations, although their structures are related, as they all possess amide bonds, sulfonate groups, and aromatic rings. However, many problems remained unsolved, including how these compounds bind with PAP248–286, whether the number of sulfonate groups affects peptide–compound interactions, and how tightly the compounds bind to PAP248–286. Thus, a computational approach would be useful to investigate protein–ligand interactions and to give us a hand to understand the binding of PAP248–286 with these sulfonated compounds.
NF110, having four sulfonate groups, was found to have a distorted X‐shaped configuration upon interaction with PAP248–286. Thus, NF110 has four flexible arms and each arm has one sulfonate group at the end. As a result, NF110 can contact the peptide from four different directions. This accounts for good interaction between the peptide and NF110. NF279 has two more sulfonate groups than NF110, three on one tail of the compound and the other three on the other tail. Moreover, NF279 introduces an allosteric strategy for folding itself into a V shape. V‐shaped NF279 looks like a clip to pinch the peptide. However, NF279 only has sulfonate groups on both ends, regardless of the configuration it adopts. Finally, the smallest compound, NF340, similar to NF110 only has four sulfonate groups. The difference in these two compounds resides in the fact that all of the sulfonate groups of NF340 are distributed equally on both tails. This is similar to that found for NF279. The structural properties of these compounds have a great influence on peptide–compound interactions.
As learned from the results, all of the sulfonated compounds were able to bind with PAP248–286 through a variety of interactions, including hydrogen‐bonding interactions, hydrophobic interactions, π–π stacking interactions, and electrostatic interactions. The NF110 molecules form 21 hydrogen bonds with PAP248–286, whereas the number of hydrogen bonds is 17 in the PAP248–286–NF279 complex and decreases to 13 in the PAP248–286–NF340 complex. The number of π–π or CH–π interactions in PAP248–286–NF110 is comparable to that in PAP248–286–NF279 but is higher than that in PAP248–286–NF340. Electrostatic contributions are crucial for binding due to strong attraction between the negatively charged sulfonate groups of the compounds and the positively charged residues of PAP248–286. From the above, molecules of NF110 form the most interactions with PAP248–286 among the three compounds. This validates previous analyses indicating that the structural properties of the compounds were important. On the basis of the data, the interactions between PAP248–286 and the sulfonated compounds are not determined by the number of sulfonate groups. In contrast, the orientations of the sulfonate groups and the specific configurations of the compounds have a marked effect on binding.
The NF110, NF279, and NF340 compounds occupy binding sites around PAP248–286. In particular, NF110 is located on almost the entire exposed surface of PAP248–286, which leads to an open conformation of the peptide compared to the folded PAP248–286 system. The average radius of gyration for PAP248–286 exhibits the compactness of the peptide. In fact, these values increase for all three systems, and PAP248–286–NF110 has the largest value of 1.60 nm. Changes in the conformations of PAP248–286 also support the idea that the NF110 molecules interact more effectively than the two other compounds with the peptide. In addition, the distances between the cationic residues of PAP248–286 and the anionic sulfonate groups of the compounds are shortened. The PAP248–286–NF110 complex shows the strongest binding affinity of the three peptide–compound complexes due to the fact that it has the most negative binding free energy of all the compounds. The surface plasmon resonance data agree with the results. Moreover, the critical Gly261–Tyr286 fragment of PAP248–286, interacting with the sulfonated compounds, no longer forms β‐sheet structures with other PAP248–286. It blocks the formation of SEVI (semen‐derived enhancer of virus infection) fibrils. A Congo red staining experiment confirmed this analysis.
In consequence, we suggest that NF110 and its analogues would be promising microbicides to antagonize seminal amyloids. The results will give us useful information to develop novel target selective inhibitors with high efficiency to block HIV transmission. However, more biophysical and biochemical studies are essential to provide in‐depth investigation. For example, NMR and HSQC spectra should be included to illustrate the interactions between the compounds and PAP248–286, and circular dichroism analysis, transmission electron microscopy, and a viral infection assay should be applied to confirm the effects of the compounds on the inhibition of fibril formation. In addition, further in vivo studies in rhesus macaques model to examine the anti‐HIV activity would be recommended.
Methods
Computational Methods
The structure of PAP248–286 was gained from the Protein Data Bank (PDB ID 2L3H).33 The NF110, NF279, and NF340 compounds were constructed and optimized by using the Gaussian 09 program package34 without any geometrical constraints.35, 36, 37, 38 Molecular docking procedures were implemented by Autodock Vina 1.5.6 software39 to investigate the binding of NF110, NF279, and NF340 to PAP248–286. The spacing was set to 1.00 Å. There are eight positively charged amino‐acid residues (Lys251, Lys253, Lys255, Arg257, Lys272, Arg273, Lys281, and Lys282) in PAP248–286. Because the Arg and Lys residues of PAP248–286 can attract the anionic sulfonate groups of NF110, NF279, and NF340, we investigated the following four systems: PAP248–286 with 8 NF110 molecules, PAP248–286 with 8 NF279 molecules, PAP248–286 with 8 NF340 molecules, and PAP248–286 alone. The side chains of Lys and Arg were set as flexible. On the basis of the interactions and binding energies, the most promising poses were chosen. The structures of the molecules bound to PAP248–286 were preliminarily energy minimized by YASARA.40
Molecular dynamics (MD) simulations were utilized to investigate the binding of PAP248–286 and the sulfonated compounds. GROMACS 4.5.341 with CHARMM2742, 43 force field were used for all the systems. The Swissparam server produced the force field parameters of the NF110, NF279, and NF340 molecules.44, 45 The peptide–ligand complex was immerged in a cubic box, and the box size was extended 12 Å from the edges of the complex. Then, the box was filled with TIP3P water.46 Sodium and chloride were also added for neutralization and to offer physiological conditions. After that, the system was energy minimized through 3000 steps of steepest descent. The structure produced from the minimization was prepared for MD simulations.
The 100 ns MD simulations were implemented with the NPT ensemble, which was used in our previous paper.47, 48, 49 The bonds and water were constrained by the LINCS algorithm50 and SETTLE algorithm,51 respectively. The Particle‐Mesh Ewald was utilized to measure the electrostatic interactions of long range.52 A pressure of 0.1 MPa and a temperature of 300 K were applied. The time step was 2 fs. We utilized the GROMACS tools to analyze the MD trajectories. YASARA53, 54 and PyMOL55, 56 programs were used in this paper to visualize and prepare the structures. The root‐mean‐square deviations (RMSDs) evaluate the deviation of the backbone of the peptide. Molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA), which was previously employed in many protein–ligand systems, was utilized to study the binding free energy.57, 58, 59, 60
Experiments
The NF110, NF279, and NF340 compounds were bought from Tocris Bioscience. Scilight‐Peptide (Beijing, China) synthesized PAP248–286 (>95 % purity).
SPR Binding Kinetics of PAP248–286–Molecule Interactions
A PlexArray HT system was used to analyze the SPR measurements. The experiment was performed by using the same method as that in our published paper.61 Details can be found in the Supporting Information.
Congo Red Binding Assay
The fibril formation kinetics of PAP248–286 were determined by a Congo red binding assay.62 For the experimental procedures, see to our previous paper63 and the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support from the National Natural Science Foundation of China (21603095); Medical Scientific Research Foundation of Guangdong Province, China (A2016378); Pearl River S&T Nova Program of Guangzhou (201610010132); Scientific Research Foundation of Southern Medical University (LX2016N005); and Special Program for Applied Research on Super Computation of the NSFC‐Guangdong Joint Fund (the second phase) under Grant No.U1501501 is acknowledged.
T. Zhang, H. Yang, Z. Yang, S. Tan, J. Jin, S. Liu, J. Zhang, ChemistryOpen 2018, 7, 447.
Contributor Information
Shuwen Liu, Email: liusw@smu.edu.cn.
Dr. Jiajie Zhang, Email: zhangjj@smu.edu.cn.
References
- 1. Münch J., Rucker E., Standker L., Adermann K., Goffinet C., Schindler M., Wildum S., Chinnadurai R., Rajan D., Specht A., Gimenez-Gallego G., Sanchez P. C., Fowler D. M., Koulov A., Kelly J. W., Mothes W., Grivel J. C., Margolis L., Keppler O. T., Forssmann W. G., Kirchhoff F., Cell 2007, 131, 1059–1071. [DOI] [PubMed] [Google Scholar]
- 2. Usmani S. M., Zirafi O., Muller J. A., Sandi-Monroy N. L., Yadav J. K., Meier C., Weil T., Roan N. R., Greene W. C., Walther P., Nilsson K. P., Hammarstrom P., Wetzel R., Pilcher C. D., Gagsteiger F., Fandrich M., Kirchhoff F., Munch J., Nat. Commun. 2014, 5, 3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim K. A., Yolamanova M., Zirafi O., Roan N. R., Staendker L., Forssmann W. G., Burgener A., Dejucq-Rainsford N., Hahn B. H., Shaw G. M., Greene W. C., Kirchhoff F., Munch J., Retrovirology 2010, 7, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roan N. R., Munch J., Arhel N., Mothes W., Neidleman J., Kobayashi A., Smith-McCune K., Kirchhoff F., Greene W. C., J. Virol. 2009, 83, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xun T., Li W., Chen J., Yu F., Xu W., Wang Q., Yu R., Li X., Zhou X., Lu L., Jiang S., Li L., Tan S., Liu S., Antimicrob. Agents Chemother. 2015, 59, 5123–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nunes R., Sarmento B., das Neves J., J. Controlled Release 2014, 194, 278–294. [DOI] [PubMed] [Google Scholar]
- 7. Baeten J., Celum C., Annu. Rev. Med. 2013, 64, 219–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee Y. H., Ramamoorthy A., Protein Sci. 2018, https://doi.org/10.1002/pro.3395. [Google Scholar]
- 9. Castellano L. M., Shorter J., Biology 2012, 1, 58–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gonzalez-Ortega E., Mena M. P., Permanyer M., Ballana E., Clotet B., Este J. A., Antimicrob. Agents Chemother. 2010, 54, 4487–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roan N. R., Liu H., Usmani S. M., Neidleman J., Muller J. A., Avila-Herrera A., Gawanbacht A., Zirafi O., Chu S., Dong M., Kumar S. T., Smith J. F., Pollard K. S., Fandrich M., Kirchhoff F., Munch J., Witkowska H. E., Greene W. C., J. Virol. 2014, 88, 7221–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roan N. R., Muller J. A., Liu H., Chu S., Arnold F., Sturzel C. M., Walther P., Dong M., Witkowska H. E., Kirchhoff F., Munch J., Greene W. C., Cell Host Microbe 2011, 10, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sheik D. A., Dewhurst S., Yang J., Acc. Chem. Res. 2017, 50, 2159–2166. [DOI] [PubMed] [Google Scholar]
- 14. Yu F., Lu L., Liu Q., Yu X., Wang L., He E., Zou P., Du L., Sanders R. W., Liu S., Jiang S., Biochim. Biophys. Acta 2014, 1838, 1296–1305. [DOI] [PubMed] [Google Scholar]
- 15. Brender J. R., Hartman K., Gottler L. M., Cavitt M. E., Youngstrom D. W., Ramamoorthy A., Biophys. J. 2009, 97, 2474–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brender J. R., Nanga R. P., Popovych N., Soong R., Macdonald P. M., Ramamoorthy A., Biochim. Biophys. Acta 2011, 1808, 1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hartman K., Brender J. R., Monde K., Ono A., Evans M. L., Popovych N., Chapman M. R., Ramamoorthy A., PeerJ 2013, 1, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lump E., Castellano L. M., Meier C., Seeliger J., Erwin N., Sperlich B., Stürzel C. M., Usmani S., Hammond R. M., von Einem J., Gerold G., Kreppel F., Bravo-Rodriguez K., Pietschmann T., Holmes V. M., Palesch D., Zirafi O., Weissman D., Sowislok A., Wettig B., Heid C., Kirchhoff F., Weil T., Klärner F.-G., Schrader T., Bitan G., Sanchez-Garcia E., Winter R., Shorter J., Münch J., eLife 2015, 4, e05397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sheftic S. R., Snell J. M., Jha S., Alexandrescu A. T., Eur. Biophys. J. 2012, 41, 695–704. [DOI] [PubMed] [Google Scholar]
- 20. Sievers S. A., Karanicolas J., Chang H. W., Zhao A., Jiang L., Zirafi O., Stevens J. T., Munch J., Baker D., Eisenberg D., Nature 2011, 475, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roan N. R., Sowinski S., Munch J., Kirchhoff F., Greene W. C., J. Biol. Chem. 2010, 285, 1861–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hauber I., Hohenberg H., Holstermann B., Hunstein W., Hauber J., Proc. Natl. Acad. Sci. USA 2009, 106, 9033–9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Popovych N., Brender J. R., Soong R., Vivekanandan S., Hartman K., Basrur V., Macdonald P. M., Ramamoorthy A., J. Phys. Chem. B 2012, 116, 3650–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang S., Lin K., Zhang L., Debnath A. K., J. Virol. Methods 1999, 80, 85–96. [DOI] [PubMed] [Google Scholar]
- 25. McGeary R. P., Bennett A. J., Tran Q. B., Cosgrove K. L., Ross B. P., Mini-Rev. Med. Chem. 2008, 8, 1384–1394. [DOI] [PubMed] [Google Scholar]
- 26. La Rocca R. V., Meer J., Gilliatt R. W., Stein C. A., Cassidy J., Myers C. E., Dalakas M. C., Neurology 1990, 40, 954–960. [DOI] [PubMed] [Google Scholar]
- 27. Nishimura Y., McLaughlin N. P., Pan J., Goldstein S., Hafenstein S., Shimizu H., Winkler J. D., Bergelson J. M., PLoS Pathog. 2015, 11, e1005184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Azriel R., Gazit E., J. Biol. Chem. 2001, 276, 34156–34161. [DOI] [PubMed] [Google Scholar]
- 29. Cohen T., Frydman-Marom A., Rechter M., Gazit E., Biochemistry 2006, 45, 4727–4735. [DOI] [PubMed] [Google Scholar]
- 30. Wang J., Yamamoto T., Bai J., Cox S. J., Korshavn K. J., Monette M., Ramamoorthy A., Chem. Commun. (Camb.) 2018, 54, 2000–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palhano F. L., Lee J., Grimster N. P., Kelly J. W., J. Am. Chem. Soc. 2013, 135, 7503–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. French K. C., Makhatadze G. I., Biochemistry 2012, 51, 10127–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nanga R. P., Brender J. R., Vivekanandan S., Popovych N., Ramamoorthy A., J. Am. Chem. Soc. 2009, 131, 17972–17979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaussian 09, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian, Inc., Wallingford, CT, 2009.
- 35. Zhang T., Sharma G., Paul T. J., Hoffmann Z., Prabhakar R., J. Chem. Inf. Model. 2017, 57, 1079–1088. [DOI] [PubMed] [Google Scholar]
- 36. Zhang T., Ozbil M., Barman A., Paul T. J., Bora R. P., Prabhakar R., Acc. Chem. Res. 2015, 48, 192–200. [DOI] [PubMed] [Google Scholar]
- 37. Zhang T. T., Zhu X. X., Prabhakar R., Organometallics 2014, 33, 1925–1935. [Google Scholar]
- 38. Zhang T., Zhu X., Prabhakar R., J. Phys. Chem. B 2014, 118, 4106–4114. [DOI] [PubMed] [Google Scholar]
- 39. Trott O., Olson A. J., J. Comput. Chem. 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K., Proteins 2009, 77, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J., J. Comput. Chem. 2005, 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- 42. Bjelkmar P., Larsson P., Cuendet M. A., Hess B., Lindahl E., J. Chem. Theory Comput. 2010, 6, 459–466. [DOI] [PubMed] [Google Scholar]
- 43. A. D. Mackerell, Jr. , Feig M., Brooks C. L., J. Comput. Chem. 2004, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- 44. Bier D., Rose R., Bravo-Rodriguez K., Bartel M., Ramirez-Anguita J. M., Dutt S., Wilch C., Klarner F. G., Sanchez-Garcia E., Schrader T., Ottmann C., Nat. Chem. 2013, 5, 234–239. [DOI] [PubMed] [Google Scholar]
- 45. Zoete V., Cuendet M. A., Grosdidier A., Michielin O., J. Comput. Chem. 2011, 32, 2359–2368. [DOI] [PubMed] [Google Scholar]
- 46. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- 47. Zhang T., Tian Y., Li Z., Liu S., Hu X., Yang Z., Ling X., Liu S., Zhang J., J. Chem. Inf. Model. 2017, 57, 2281–2293. [DOI] [PubMed] [Google Scholar]
- 48. Tian Y., Yu Y., Shen Y., Wan H., Chang S., Zhang T., Wan S., Zhang J., J. Chem. Inf. Model. 2017, 57, 977–987. [DOI] [PubMed] [Google Scholar]
- 49. Song E. S., Ozbil M., Zhang T., Sheetz M., Lee D., Tran D., Li S., Prabhakar R., Hersh L. B., Rodgers D. W., PLoS One 2015, 10, e0133114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M., J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar]
- 51. Miyamoto S., Kollman P. A., J. Comput. Chem. 1992, 13, 952–962. [Google Scholar]
- 52. Darden T., York D., Pedersen L., J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- 53. Krieger E., Koraimann G., Vriend G., Proteins 2002, 47, 393–402. [DOI] [PubMed] [Google Scholar]
- 54. Krieger E., Vriend G., Bioinformatics 2002, 18, 315–318. [DOI] [PubMed] [Google Scholar]
- 55. DeLano W. L., Abstr. Pap. Am. Chem. Soc. 2004, 228, U313–U314. [Google Scholar]
- 56. DeLano W. L., Lam J. W., Abstr. Pap. Am. Chem. Soc. 2005, 230, U1371–U1372. [Google Scholar]
- 57. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A., Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dong M., Li H., Hu D., Zhao W., Zhu X., Ai H., ACS Chem. Neurosci. 2016, 7, 599–614. [DOI] [PubMed] [Google Scholar]
- 59. Dong M., Zhao W., Hu D., Ai H., Kang B., ACS Chem. Neurosci. 2017, 8, 1577–1588. [DOI] [PubMed] [Google Scholar]
- 60. Kumari R., Kumar R., Consortium Open Source Drug Discovery, Lynn A., J. Chem. Inf. Model. 2014, 54, 1951–1962. [DOI] [PubMed] [Google Scholar]
- 61. Li L., He L., Tan S., Guo X., Lu H., Qi Z., Pan C., An X., Jiang S., Liu S., Antimicrob. Agents Chemother. 2010, 54, 1700–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nilsson M. R., Methods 2004, 34, 151–160. [DOI] [PubMed] [Google Scholar]
- 63. Tan S., Lu L., Li L., Liu J., Oksov Y., Lu H., Jiang S., Liu S., PLoS One 2013, 8, e59777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary