

Abstract
Acute kidney injury in critically ill patients is common and associated with a substantial increase in morbidity and mortality. Even with aggressive medical care and renal replacement therapy, acute kidney injury remains a significant health care concern. Recent published reports offer new strategies for the prevention and amelioration of acute kidney injury using carbon monoxide. Although considered a toxic environmental gas, carbon monoxide has recently aroused scientific and clinical interest, as its beneficial effects and mechanisms of action have been substantially defined in various in vitro and in vivo experiments. The exogenous application of carbon monoxide can confer cytoprotection by modulating intracellular signaling pathways through its anti‐inflammatory, anti‐apoptotic, vasodilative, antithrombotic and antiproliferative properties. Thus, evidence is accumulating to support the notion of carbon monoxide treatment for acute kidney disease. In this review, we focus on the extensively analyzed advantageous value of treatment with inhaled/soluble carbon monoxide in the context of kidney injury. Mechanisms such as signaling pathways, as well as an expanded view regarding toxicity and side‐effects, are described broadly. In addition, we discuss the clinical applicability of carbon monoxide as a promising therapeutic strategy for the treatment of patients with acute kidney disease based on translating basic experimental findings into clinical application.
Keywords: Acute kidney injury, anti‐inflammation, carbon monoxide, ischemia/reperfusion
Introduction
Acute kidney injury (AKI), previously termed acute renal failure, is clinically characterized by a rapid reduction in kidney function, resulting in failure to maintain fluid, electrolyte, and acid–base homoeostasis. Even with aggressive medical care and renal replacement therapy, morbidity and mortality are substantial. Recent published work offers new strategies for the prevention and amelioration of AKI using therapeutic medical gases, pharmaceutical gaseous molecules used for medical needs.1, 2, 3 In particular, carbon monoxide (CO) is known to have anti‐inflammatory, anti‐apoptotic, and antiproliferative properties; the increasing impact of CO in numerous disease models as a means of protection has presented it as a new and effective therapeutic alternative. In this review, we focus on CO and its therapeutic value in the context of AKI and summarize the functional roles and biological and molecular mechanisms of CO in AKI treatment. We also discuss potential applications in the clinical setting.
Clinical problems of AKI in acute care medicine
Acute kidney injury is a common complication of acute illness and significantly increases morbidity and mortality.4 An estimated 5–20% of critically ill patients experience an episode of AKI during the course of their illness and AKI patients receiving renal replacement therapy have been reported in 4.9% of all admissions to intensive care units.5 Acute kidney injury frequently complicates surgical sepsis and powerfully predicts hospital mortality in severe sepsis and septic shock.6
Ischemia due to hypotension or sepsis is the most common cause of AKI in humans.7 Acute kidney injury from ischemia is initiated by unfavorable changes in renal blood flow as a consequence of vasospasm, alterations in ultrafiltration coefficient, tubular obstruction, and/or back‐leak. The renal medulla is particularly susceptible to renal ischemia because of low oxygen tension.8 Acute kidney injury may lead to a number of complications, including metabolic acidosis, high potassium levels, uremia, changes in body fluid balance, and effects on other organ systems.
Carbon Monoxide: A Novel Therapeutic Gas
Carbon monoxide is an invisible, chemically inert, colorless, and odorless gas commonly viewed as anenvironmental pollutant. It avidly binds to hemoglobin with an affinity 240 times higher than that of oxygen and forms carboxyhemoglobin (COHb), resulting in interference with the oxygen‐carrying capacity of the blood and consequent tissue hypoxia. Interestingly, mammalian cells endogenously generate CO, primarily through the catalysis of heme by hemeoxygenase (HO) (Fig. 1). Hemeoxygenase is induced by various oxidative stresses and is thought to play an important role in the protection of tissues from oxidative injuries. A number of works have explored the importance of HO in renal diseases and they have provided consistent evidence that its overexpression has beneficial effects in AKI.9, 10 Hemeoxygenase catalyzes the oxidative cleavage of heme to yield equimolar amounts of CO, iron, and biliverdin. Biliverdin is subsequently reduced to bilirubin through the action of biliverdin reductase, and iron induces increased ferritin synthesis.11 The production rate of CO is estimated to be 384 μmol/day in the body, with an average concentration in the tissues in the nanomolar range.12 Accumulating evidence has revealed that endogenous CO is an important physiological regulatory factor and exerts anti‐inflammatory, anti‐apoptotic, and organ/cellular protective effects. Potent therapeutic efficacies of CO have been shown using experimental models for many conditions, including paralytic ileus,13 hemorrhagic shock,14, 15 hyperoxic lung injury,16 and endotoxemia,17 supporting the new paradigm that, at low concentrations, CO functions as a signaling molecule that exerts significant cytoprotection. Interestingly, endogenous production of CO was found to be high in critical ill patients with sepsis,18 AKI19, 20 and acute respiratory distress syndrome,21 suggesting that HO may, at least partially, function as an antistress protein by generating CO.
Figure 1.
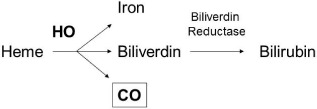
Heme oxygenase (HO) system. Heme oxygenase is the enzyme that catabolizes heme into three byproducts: carbon monoxide (CO), iron, and biliverdin. Biliverdin is further converted to bilirubin through biliverdin reductase.
Mechanism of the Protective Effects of CO
The potent therapeutic efficacy of CO has been shown using clinical and experimental models for many conditions (Fig. 2).
Figure 2.
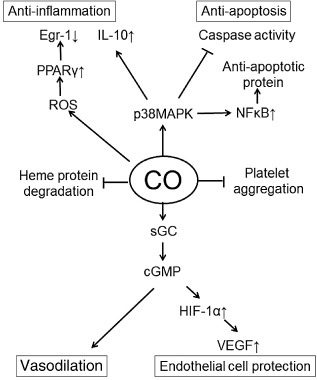
Function of carbon monoxide (CO) in humans. cGMP, cyclic guanosine monophosphate; Egr, early growth response; HIF, hypoxia inducible factor; IL, interleukin; MAPK, mitogen‐activated protein kinase; NFκB, nuclear factor‐κB; PPAR, peroxisome proliferator‐activated receptor; ROS, reactive oxygen species; sGC, soluble guanylyl cyclase; VEGF, vascular endothelial growth factor.
Vasodilation
The major signaling pathway for CO is believed to be mediated by stimulation of soluble guanylyl cyclase, resulting in increases of cyclic guanosine monophosphate (cGMP) levels in target tissues, which dilates blood vessels.22, 23, 24, 25 Carbon monoxide also dilates blood vessels by directly activating potassium channels in vascular smooth muscle cells (SMCs). Stanford et al. reported that CO can inhibit endothelin‐1 release from human pulmonary artery vascular endothelial cells (VECs) and SMCs,26 supporting CO's beneficial effects on endothelin‐1 suppression and inhibition of vasoconstriction.
Protection of vascular endothelial cells
Damage or loss of VECs, disturbance of the microcirculation, activation of potent inflammatory mediators (e.g., cytokines, adhesion molecules, platelet activating factors, eicosanoid products), and inflammatory infiltration are characteristic features of ischemia/reperfusion injury.27 Carbon monoxide protects VECs in vitro in culture systems. Brouard et al. have shown that exogenous CO (10,000 p.p.m.) prevents tumor necrosis factor‐α‐induced apoptosis of cultured VECs.28 Zhang et al. have also shown the protective effects of CO against anoxia–reoxygenation injury in rat primary pulmonary VECs.29 These protective actions of CO are mediated through the activation of p38 mitogen‐activated protein kinase, because SB203580, a selective p38 mitogen‐activated protein kinase inhibitor, abrogates CO's beneficial effects.28 Carbon monoxide regulates hypoxia‐inducible transcription factor‐1 (HIF‐1)‐dependent expression of vascular endothelial growth factor, which is an important angiogenic factor.30
Promotion of stem/progenitor cells in injury site
In terms of myocardial infarction, CO significantly increased the accumulation of stem/progenitor cells into the infarct area and induced formation of new coronary arteries by promoting a substantial differentiation of stem/progenitor cells into vascular SMCs. Furthermore, CO increased proliferation of cardiomyocytes in the infarct border area at 4 weeks post‐infarction. Increased expression of HIF‐1α, stromal cell derived factor‐1α, and vascular endothelial growth factor were found in the infarct areas of CO donor pretreated hearts, suggesting that these factors potentially promoted the migration of stem/progenitor cells into the infarct area and subsequent vasculogenesis and myocardial regeneration by CO. Thus, CO may repair the injured kidney.31, 32
Induction of cytoprotective proteins
Expression of heat shock proteins (HSPs) protects cells from a broad range of cellular stressors such as hypoxia, oxygen radicals, endotoxin, infections, and fever.33 Pretreatment with inhaled CO markedly increased binding of heat shock factor‐1, the central transcription factor in the regulation of HSP expression. This was accompanied by increased expression of HSP‐70, HO‐1, and HO enzyme activity.34
Anti‐inflammation
One of the important cellular targets of CO is the macrophage. Carbon monoxide typically suppresses proinflammatory responses in macrophages, and CO's effects on organ‐infiltrating macrophages may prevent organ dysfunction. Otterbein et al. reported that the lipopolysaccharide (LPS)‐induced inflammatory responses of macrophages, which are associated with induction of tumor necrosis factor‐α and interleukin‐1β, are inhibited by CO; CO increases LPS‐induced expression of the anti‐inflammatory cytokine interleukin‐10.35 Toll‐like receptors (TLRs) are needed for recognition and clearance of bacteria. In macrophages, CO has recently been shown to inhibit signaling by TLR2, TLR4, TLR5, and TLR9 (but not TLR3).36
Inhibition of reactive oxygen species generation
One emerging aspect of CO signaling that may be important to the regulation of sodium reabsorption in the kidney is its effect on the generation of reactive oxygen species (ROS) through cytosolic and mitochondrial sources. Bilban et al. showed that exposure of macrophages to CO in vitro produced a brief burst of mitochondrial‐derived ROS, which led to expression of peroxisome proliferator‐activated receptor γ; this expression was essential for mediating the anti‐inflammatory effects of CO.37 In addition, Chin et al. showed that ROS arising from the mitochondria in response to CO promote rapid activation and stabilization of the transcription factor HIF‐1α, which regulates the expression of genes involved in inflammation, metabolism, and cell survival.38 The increase in HIF‐1α expression induced by CO results in regulated expression of transforming growth factor‐β, a potent anti‐inflammatory cytokine.36, 39 Carbon monoxide acts through the inhibition of cytochrome c oxidase, leading to the generation of low ROS levels that in turn mediate subsequent adaptive signaling.40
Inhibition of platelet aggregation
Platelets play important roles in AKI. Brune et al. reported that CO can inhibit thromboxane release from the platelets in vitro.41 Fujita et al. showed in a lung warm ischemia–reperfusion (I/R) injury model that CO inhalation reduces the deposition of fibrin in the microvasculature and suppresses plasminogen activator inhibitor‐1. Carbon monoxide inhibited platelet aggregation and production of platelet‐derived growth factor and endothelin‐1 by the VECs. These anticoagulative effects may contribute to the protection of transplanted organs.42
Effects of CO on angiotensin II
Angiotensin II is known to increase cellular superoxide production through the stimulation of nicotinamide adenine dinucleotide phosphate oxidase. Increases in superoxide production mediated by nicotinamide adenine dinucleotide phosphate oxidase have also been reported to increase sodium reabsorption in the thick ascending loop of Henle directly and through decrease in the bioavailability of NO.43 Carbon monoxide was found to stimulate the apical 70‐pS K+ channel in isolated cells of the thick ascending loop of Henle.44, 45 Thus, CO mediates antihypertensive actions in models that show increased levels of cytosolic superoxide production, such as angiotensin II‐dependent and deoxycorticosterone acetate–salt hypertension.46
Prevention of heme protein degradation
Recent evidence has indicated that cytochrome P450 (CYP), a large group of heme proteins abundantly present in the kidney, plays critical roles in the renal I/R injury process.47, 48 Susceptible to oxidant stress, CYPs are substantially damaged during renal I/R injury. Damaged CYPs are prone to degrade and release free heme/iron,49, 50 which can directly induce tissue injury by rapidly promoting peroxidation of cell lipid membranes.51 Carbon monoxide possesses a strong affinity to heme moiety of heme proteins. As CYPs are known to play critical roles in I/R injury associated with renal transplantation, CO binding to heme moiety of CYP, in advance, can stabilize and protect CYP from degradation caused by oxidative attack initiated by reperfusion. As the kidney is richly endowed with CYP, particularly in the proximal tubules, CO in preservation solution could effectively bind to CYP during cold storage of the kidney grafts52 (Fig. 3).
Figure 3.
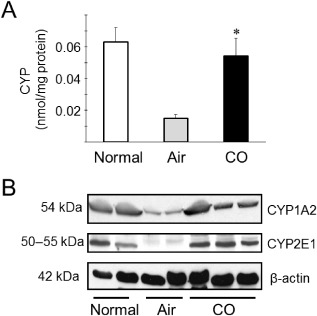
A. Total cytochrome P450 (CYP) contents in the microsomal fraction of kidney grafts 3 h after reperfusion. Total CYP content at 3 h after kidney transplants were significantly decreased in kidney grafts preserved in control UW solution for 24 h. In contrast, kidney grafts stored with carbon monoxide (CO) (soluble CO levels approximately 40 μM) maintained a nearly normal concentration of CYP at the same time point. Data are shown as mean ± standard deviation. *P < 0.05 versus control UW. B. Western blots for typical CYPs CYP1A2 and CYP2E1. Treatment with CO markedly prevented CYP degradation. Representative results are shown from two independent experiments using four samples.
Regulation of calcium‐activated potassium channels
Activation of calcium‐activated potassium channels leads to pore opening and outward potassium (K+) flux, which shifts the membrane to more negative potentials. Carbon monoxide regulates Ca2+‐activated big‐conductance K (BK) channels in the principal cell of the cortical collecting duct. Inhibition of soluble guanylate cyclase failed to abolish the stimulatory effect of CO on BK channels. Moreover, inhibition of cGMP‐dependent protein kinase G did not block the stimulatory effect of CO on the BK channels, suggesting that the stimulatory effect of CO on the BK channels was, at least partially, induced by a cGMP‐independent mechanism.53
Therapeutic Application of CO for AKI
The ability to administer CO by inhalation at a low concentration makes it extremely attractive from a feasibility standpoint for AKI in a human clinical setting. However, safety warrants that CO should be given under the direct care of a physician and the supervision of trained medical staff. The methods and devices for the delivery of CO must be carefully examined. Carbon monoxide used in therapy must be identified with its concentration on a label before safe delivery and precision gas analysis or monitoring.
Increased levels of HO may offer another therapeutic strategy to promote endogenous generation of CO. Pharmacological induction of HO‐1 by hemin is another possible strategy to promote endogenous generation of CO.54, 55 There have been no definitive trials designed to evaluate the efficacy of chemical inducers of HO‐1 as a therapeutic strategy in the clinical setting. Recently, Bharucha et al. showed that hemin treatment increased plasma HO‐1 protein and HO enzymatic activity in healthy human volunteers, demonstrating the ability to induce HO‐1 in humans by pharmacological approaches.56
Inhalation is not the only method for CO delivery. Clinically effective levels of soluble CO can be achieved in the solution for organ perfusion and preservation by bubbling compressed gaseous CO through the solution.52, 57 Another potentially applicable way to deliver CO is by a parenteral injectable drug containing a gas‐releasing moiety. Carbon monoxide‐releasing molecules exert a variety of pharmacological activities through the liberation of controlled amounts of CO into biological systems.58, 59, 60 Many limitations of CO inhalation therapy may be overcome by the use of these molecules. The principal advantage of gases is practicality for non‐invasive clinical use. Furthermore, CO therapy has distinct advantages over pharmaceutical drugs; CO easily penetrates biomembranes and diffuses into the cytosol, mitochondria, and nucleus to reach target tissues.1
Feasibility of CO for AKI
Kidney I/R injury
Renal I/R injury is a common cause of AKI in many clinical transplant settings and compromises renal graft function. Ischemia–reperfusion injury results from multiple factors affecting both the renal tubular epithelium and renal microvasculature.61, 62, 63 Carbon monoxide inhalation has been intensively studied as an anti‐inflammatory therapeutic in experimental organ transplantation.25, 30, 64 A pig kidney allograft model of delayed graft failure has been developed to evaluate the cytoprotective effects of inhaled CO.65 Carbon monoxide reduced acute tubular necrosis, apoptosis, tissue factor expression, and P‐selectin expression and enhanced proliferative repair. In kidney transplantation, a bloodless ex vivo condition occurs during cold storage. In this special situation, binding of CO to hemoglobin is minimized. Therefore, CO could bind efficiently to target molecules or cellular organisms without forming carboxyhemoglobin.52, 57
Acute kidney injury after cardiopulmonary bypass
Postoperative AKI is one of the most serious and frequent complications of cardiac surgery. Previous studies have shown that even small increases in serum creatinine following cardiac surgery are independently associated with increased mortality and longer hospitalization.66, 67 Goebel et al. showed that CO treatment before cardiopulmonary bypass was associated with evidence of renoprotection, indicated by fewer histological injuries and decreased cystatin C concentrations. The findings that the anti‐inflammatory and anti‐apoptotic effects of CO were accompanied by activation of HSP‐70 suggest that renoprotection by pretreatment with inhaled CO before cardiopulmonary bypass is mediated by activation of the renal heat shock response.34
Cisplatin‐induced nephrotoxicity
Although cisplatin is a very effective anticancer drug used in the treatment of cancers of the testis, ovary, head and neck, bladder, lung, cervix, and endometrium, its use is frequently complicated by acute renal injury. Despite various hydration protocols devised to minimize nephrotoxicity, 25–35% of patients experience a significant decline in renal function following cisplatin treatment. Tayem et al. showed that treatment of rats with CO prior to cisplatin treatment prevented increases in plasma creatinine and completely protected the kidney against cisplatin‐induced tubular apoptosis, necrosis, cellular desquamation, and vacuolization.68 The anti‐apoptotic effects of CO in cisplatin nephrotoxicity were mediated by increases in cGMP, as blockade with a soluble guanylyl cyclase inhibitor prevented CO from attenuating caspase‐3 activation in cultured renal tubular cells.68
Clinical Studies
Inhaled CO was administered to healthy human subjects to examine systemic inflammation following experimental endotoxemia. In a randomized, double‐blinded, placebo‐controlled, two‐way cross‐over trial, experimental endotoxemia was induced in healthy volunteers by injection of 2 ng/kg LPS. The potential anti‐inflammatory effects of CO inhalation were investigated by inhalation of 500 p.p.m. CO for 1 h, which resulted in an increase of COHb levels from 1.2% to 7%. Unfortunately, CO had no effect on the inflammatory response as measured by systemic cytokine production.69
The Covox device for CO inhalation (Inkaria, Clinton, NJ, USA) may allow safe CO delivery and monitoring. In the USA, a single‐blind, placebo‐controlled, dose‐escalating Phase 2 study of inhaled CO using the Covox device in patients receiving renal transplants, as well as patients with pulmonary inflammation, is currently being carried out. The primary endpoint of the study is to evaluate the safety and tolerability of increasing CO dose levels consisting of a single 0.7 mg/kg dose or a single 2.0 mg/kg dose when given postoperatively and a single 2.0 mg/kg dose, a single 3.0 mg/kg dose, or a 3.5 mg/kg dose when given intraoperatively as an inhaled gas for 1 h. A European clinical study revealed that inhalation of 100–125 p.p.m. CO by patients with chronic obstructive pulmonary disease in a stable phase was feasible and led to trends in reduction of sputum eosinophils and improvement of responsiveness to methacholine.70 As an alternative approach to the administration of CO gas by inhalation, chemical CO‐releasing molecules have been developed. Alfama (Lisbon, Portugal) has designed and synthesized a number of organic compounds and metallo‐organic complexes that are expected to generate CO without adverse effects on living tissues. The unique approach of Alfama is to search for compounds that are stable in the blood and healthy tissues, but release CO in diseased tissues.
Conclusion
Recent studies have been carried out on the physiological actions of CO in the kidney and potential therapeutic avenues are being developed targeting CO in the kidney, which may be beneficial in AKI. There are currently several limitations that must be overcome in order to convert current knowledge about the beneficial actions of CO in the kidney into routine clinical practice. However, as is clear in this review, CO therapy may have well‐defined benefits in the treatment of AKI.
Conflict of Interest
None.
References
- 1. Nakao A, Sugimoto R, Billiar TR, McCurry KR. Therapeutic antioxidant medical gas. J. Clin. Biochem Nutr. 2009; 44: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang CS, Kawamura T, Toyoda Y, Nakao A. Recent advances in hydrogen research as a therapeutic medical gas. Free Radic. Res. 2010; 44: 971–982. [DOI] [PubMed] [Google Scholar]
- 3. Szabo C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007; 6: 917–935. [DOI] [PubMed] [Google Scholar]
- 4. Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J. Am. Soc. Nephrol. 2005; 16: 3365–3370. [DOI] [PubMed] [Google Scholar]
- 5. Metnitz PG, Krenn CG, Steltzer H et al Effect of acute renal failure requiring renal replacement therapy on outcome in critically ill patients. Crit. Care Med. 2002; 30: 2051–2058. [DOI] [PubMed] [Google Scholar]
- 6. White LE, Hassoun HT, Bihorac A et al Acute kidney injury is surprisingly common and a powerful predictor of mortality in surgical sepsis. J. Trauma Acute Care Surg. 2013; 75: 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xue JL, Daniels F, Star RA et al Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J. Am. Soc. Nephrol. 2006; 17: 1135–1142. [DOI] [PubMed] [Google Scholar]
- 8. Mason J, Torhorst J, Welsch J. Role of the medullary perfusion defect in the pathogenesis of ischemic renal failure. Kidney Int. 1984; 26: 283–293. [DOI] [PubMed] [Google Scholar]
- 9. Correa‐Costa M, Amano MT, Camara NO. Cytoprotection behind heme oxygenase‐1 in renal diseases. World J. Nephrol. 2012; 1: 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase‐1 geneablation and expression. J. Am. Soc. Nephrol. 2000; 11: 965–973. [DOI] [PubMed] [Google Scholar]
- 11. Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl Acad. Sci. U.S.A. 1968; 61: 748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coburn RF. Endogenous carbon monoxide production and body co stores. Acta Med. Scand. Suppl. 1967; 472: 269–282. [DOI] [PubMed] [Google Scholar]
- 13. Moore BA, Otterbein LE, Turler A, Choi AM, Bauer AJ. Inhaled carbon monoxide suppresses the development of postoperative ileus in the murine small intestine. Gastroenterology 2003; 124: 377–391. [DOI] [PubMed] [Google Scholar]
- 14. Kawanishi S, Takahashi T, Morimatsu H et al Inhalation of carbon monoxide following resuscitation ameliorates hemorrhagic shock‐induced lung injury. Mol. Med. Rep. 2013; 7: 3–10. [DOI] [PubMed] [Google Scholar]
- 15. Pannen BH, Kohler N, Hole B, Bauer M, Clemens MG, Geiger KK. Protective role of endogenous carbon monoxide in hepatic microcirculatory dysfunction after hemorrhagic shock in rats. J. Clin. Invest. 1998; 102: 1220–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Otterbein LE, Mantell LL, Choi AM. Carbon monoxide provides protection against hyperoxic lung injury. Am. J. Physiol. 1999; 276: L688–694. [DOI] [PubMed] [Google Scholar]
- 17. Sarady JK, Zuckerbraun BS, Bilban M et al Carbon monoxide protection against endotoxic shock involves reciprocal effects on iNOS in the lung and liver. FASEB J. 2004; 18: 854–856. [DOI] [PubMed] [Google Scholar]
- 18. Takaki S, Takeyama N, Kajita Y et al Beneficial effects of the heme oxygenase‐1/carbon monoxide system in patients with severe sepsis/septic shock. Intensive Care Med. 2010; 36: 42–48. [DOI] [PubMed] [Google Scholar]
- 19. Abraham NG, Cao J, Sacerdoti D, Li X, Drummond G. Heme oxygenase: the key to renal function regulation. Am. J. Physiol. Renal Physiol. 2009; 297: F1137–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nath KA. The role of renal research in demonstrating the protective properties of heme oxygenase‐1. Kidney Int. 2013; 84: 3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mumby S, Upton RL, Chen Y et al Lung heme oxygenase‐1 is elevated in acute respiratory distress syndrome. Crit. Care Med. 2004; 32: 1130–1135. [DOI] [PubMed] [Google Scholar]
- 22. Jin XH, Siragy HM, Carey RM. Renal interstitial cGMP mediates natriuresis by direct tubule mechanism. Hypertension 2001; 38: 309–316. [DOI] [PubMed] [Google Scholar]
- 23. Vera T, Kelsen S, Stec DE. Kidney‐specific induction of heme oxygenase‐1 prevents angiotensin II hypertension. Hypertension 2008; 52: 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li N, Yi F, dos Santos EA, Donley DK, Li PL. Role of renal medullary heme oxygenase in the regulation of pressure natriuresis and arterial blood pressure. Hypertension 2007; 49: 148–154. [DOI] [PubMed] [Google Scholar]
- 25. Neto JS, Nakao A, Kimizuka K et al Protection of transplant‐induced renal ischemia‐reperfusion injury with carbon monoxide. Am. J. Physiol. Renal Physiol. 2004; 287: F979–989. [DOI] [PubMed] [Google Scholar]
- 26. Stanford SJ, Walters MJ, Mitchell JA. Carbon monoxide inhibits endothelin‐1 release by human pulmonary artery smooth muscle cells. Eur. J. Pharmacol. 2004; 486: 349–352. [DOI] [PubMed] [Google Scholar]
- 27. Clavien PA, Harvey PR, Strasberg SM. Preservation and reperfusion injuries in liver allografts. An overview and synthesis of current studies. Transplantation 1992; 53: 957–978. [DOI] [PubMed] [Google Scholar]
- 28. Brouard S, Otterbein LE, Anrather J et al Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000; 192: 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X, Shan P, Otterbein LE et al Carbon monoxide inhibition of apoptosis during ischemia‐reperfusion lung injury is dependent on the p38 mitogen‐activated protein kinase pathway and involves caspase 3. J. Biol. Chem. 2003; 278: 1248–1258. [DOI] [PubMed] [Google Scholar]
- 30. Faleo G, Neto JS, Kohmoto J et al Carbon monoxide ameliorates renal cold ischemia‐reperfusion injury with an upregulation of vascular endothelial growth factor by activation of hypoxia‐inducible factor. Transplantation 2008; 85: 1833–1840. [DOI] [PubMed] [Google Scholar]
- 31. Lakkisto P, Kyto V, Forsten H et al Heme oxygenase‐1 and carbon monoxide promote neovascularization after myocardial infarction by modulating the expression of HIF‐1alpha, SDF‐1alpha and VEGF‐B. Eur. J. Pharmacol. 2010; 635: 156–164. [DOI] [PubMed] [Google Scholar]
- 32. Nakao A, Huang CS, Stolz DB et al Ex vivo carbon monoxide delivery inhibits intimal hyperplasia in arterialized vein grafts. Cardiovasc. Res. 2011; 89: 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ryan M, Levy MM. Clinical review: fever in intensive care unit patients. Crit. Care 2003; 7: 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goebel U, Siepe M, Schwer CI et al Inhaled carbon monoxide prevents acute kidney injury in pigs after cardiopulmonary bypass by inducing a heat shock response. Anesth. Analg. 2010; 111: 29–37. [DOI] [PubMed] [Google Scholar]
- 35. Otterbein LE, Bach FH, Alam J et al Carbon monoxide has anti‐inflammatory effects involving the mitogen‐activated protein kinase pathway. Nat. Med. 2000; 6: 422–428. [DOI] [PubMed] [Google Scholar]
- 36. Nakahira K, Kim HP, Geng XH et al Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS‐induced trafficking of TLRs to lipid rafts. J. Exp. Med. 2006; 203: 2377–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bilban M, Haschemi A, Wegiel B, Chin BY, Wagner O, Otterbein LE. Heme oxygenase and carbon monoxide initiate homeostatic signaling. J. Mol. Med. (Berl.) 2008; 86: 267–279. [DOI] [PubMed] [Google Scholar]
- 38. Chin BY, Jiang G, Wegiel B et al Hypoxia‐inducible factor 1alpha stabilization by carbon monoxide results in cytoprotective preconditioning. Proc. Natl Acad. Sci. U.S.A. 2007; 104: 5109–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bilban M, Bach FH, Otterbein SL et al Carbon monoxide orchestrates a protective response through PPARgamma. Immunity 2006; 24: 601–610. [DOI] [PubMed] [Google Scholar]
- 40. Zuckerbraun BS, Chin BY, Bilban M et al Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007; 21: 1099–1106. [DOI] [PubMed] [Google Scholar]
- 41. Brune B, Ullrich V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol. Pharmacol. 1987; 32: 497–504. [PubMed] [Google Scholar]
- 42. Fujita T, Toda K, Karimova A et al Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat. Med. 2001; 7: 598–604. [DOI] [PubMed] [Google Scholar]
- 43. Ortiz PA, Garvin JL. Superoxide stimulates NaCl absorption by the thick ascending limb. Am. J. Physiol. Renal Physiol. 2002; 283: F957–962. [DOI] [PubMed] [Google Scholar]
- 44. Liu H, Mount DB, Nasjletti A, Wang W. Carbon monoxide stimulates the apical 70‐pS K+ channel of the rat thick ascending limb. J. Clin. Invest. 1999; 103: 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang T, Sterling H, Shao WA et al Inhibition of heme oxygenase decreases sodium and fluid absorption in the loop of Henle. Am. J. Physiol. Renal Physiol. 2003; 285: F484–490. [DOI] [PubMed] [Google Scholar]
- 46. Quan S, Yang L, Shnouda S et al Expression of human heme oxygenase‐1 in the thick ascending limb attenuates angiotensin II‐mediated increase in oxidative injury. Kidney Int. 2004; 65: 1628–1639. [DOI] [PubMed] [Google Scholar]
- 47. Paller MS, Jacob HS. Cytochrome P‐450 mediates tissue‐damaging hydroxyl radical formation during reoxygenation of the kidney. Proc. Natl Acad. Sci. U.S.A. 1994; 91: 7002–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takahashi T, Morita K, Akagi R, Sassa S. Protective role of heme oxygenase‐1 in renal ischemia. Antioxid. Redox Signal. 2004; 6: 867–877. [DOI] [PubMed] [Google Scholar]
- 49. Baliga R, Zhang Z, Shah SV. Role of cytochrome P‐450 in hydrogen peroxide‐induced cytotoxicity to LLC‐PK1 cells. Kidney Int. 1996; 50: 1118–1124. [DOI] [PubMed] [Google Scholar]
- 50. Bysani GK, Kennedy TP, Ky N et al Role of cytochrome P‐450 in reperfusion injury of the rabbit lung. J. Clin. Invest. 1990; 86: 1434–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nath KA, Balla J, Croatt AJ, Vercellotti GM. Heme protein‐mediated renal injury: a protective role for 21‐aminosteroids in vitro and in vivo. Kidney Int. 1995; 47: 592–602. [DOI] [PubMed] [Google Scholar]
- 52. Nakao A, Faleo G, Shimizu H et al Ex vivo carbon monoxide prevents cytochrome P450 degradation and ischemia/reperfusion injury of kidney grafts. Kidney Int. 2008; 74: 1009–1016. [DOI] [PubMed] [Google Scholar]
- 53. Wang Z, Yue P, Lin DH, Wang WH. Carbon monoxide stimulates Ca2+‐dependent big‐conductance K channels in the cortical collecting duct. Am. J. Physiol. Renal Physiol. 2013; 304: F543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ndisang JF, Jadhav A. Hemin therapy improves kidney function in male streptozotocin‐induced diabetic rats: role of the heme oxygenase/atrial natriuretic peptide/adiponectin axis. Endocrinology 2014; 155: 215–229. [DOI] [PubMed] [Google Scholar]
- 55. Kaizu T, Tamaki T, Tanaka M et al Preconditioning with tin‐protoporphyrin IX attenuates ischemia/reperfusion injury in the rat kidney. Kidney Int. 2003; 63: 1393–1403. [DOI] [PubMed] [Google Scholar]
- 56. Bharucha AE, Kulkarni A, Choi KM et al First‐in‐human study demonstrating pharmacological activation of heme oxygenase‐1 in humans. Clin. Pharmacol. Ther. 2010; 87: 187–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yoshida J, Ozaki KS, Nalesnik MA et al Ex vivo application of carbon monoxide in UW solution prevents transplant‐induced renal ischemia/reperfusion injury in pigs. Am. J. Transplant. 2010; 10: 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Caumartin Y, Stephen J, Deng JP et al Carbon monoxide‐releasing molecules protect against ischemia‐reperfusion injury during kidney transplantation. Kidney Int. 2011; 79: 1080–1089. [DOI] [PubMed] [Google Scholar]
- 59. Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010; 9: 728–743. [DOI] [PubMed] [Google Scholar]
- 60. Motterlini R, Mann BE, Foresti R. Therapeutic applications of carbon monoxide‐releasing molecules. Expert Opin. Investig. Drugs 2005; 14: 1305–1318. [DOI] [PubMed] [Google Scholar]
- 61. Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J. Am. Soc. Nephrol. 2003; 14: 2199–2210. [DOI] [PubMed] [Google Scholar]
- 62. Molitoris BA, Sutton TA. Endothelial injury and dysfunction: role in the extension phase of acute renal failure. Kidney Int. 2004; 66: 496–499. [DOI] [PubMed] [Google Scholar]
- 63. Regner KR, Roman RJ. Role of medullary blood flow in the pathogenesis of renal ischemia‐reperfusion injury. Curr. Opin. Nephrol. Hypertens. 2012; 21: 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nakao A, Toyoda Y. Application of carbon monoxide for transplantation. Curr. Pharm. Biotechnol. 2012; 13: 827–836. [DOI] [PubMed] [Google Scholar]
- 65. Hanto DW, Maki T, Yoon MH et al Intraoperative administration of inhaled carbon monoxide reduces delayed graft function in kidney allografts in swine. Am. J. Transplant. 2010; 10: 2421–2430. [DOI] [PubMed] [Google Scholar]
- 66. Loef BG, Epema AH, Smilde TD et al Immediate postoperative renal function deterioration in cardiac surgical patients predicts in‐hospital mortality and long‐term survival. J. Am. Soc. Nephrol. 2005; 16: 195–200. [DOI] [PubMed] [Google Scholar]
- 67. Brown JR, Cochran RP, Dacey LJ et al Perioperative increases in serum creatinine are predictive of increased 90‐day mortality after coronary artery bypass graft surgery. Circulation 2006; 114: I409–413. [DOI] [PubMed] [Google Scholar]
- 68. Tayem Y, Johnson TR, Mann BE, Green CJ, Motterlini R. Protection against cisplatin‐induced nephrotoxicity by a carbon monoxide‐releasing molecule. Am. J. Physiol. Renal Physiol. 2006; 290: F789–794. [DOI] [PubMed] [Google Scholar]
- 69. Mayr FB, Spiel A, Leitner J et al Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am. J. Respir. Crit. Care Med. 2005; 171: 354–360. [DOI] [PubMed] [Google Scholar]
- 70. Bathoorn E, Slebos DJ, Postma DS et al Anti‐inflammatory effects of inhaled carbon monoxide in patients with COPD: a pilot study. Eur. Respir. J. 2007; 30: 1131–1137. [DOI] [PubMed] [Google Scholar]